
Abstract
Fueled by new technologies that allow rapid and inexpensive assessment of fine scale individual genomic variation, researchers are making transformational discoveries at the interface between genomes and biological complexity. Here we review genomic research in Heliconius butterflies – a radiation characterized by extraordinary phenotypic diversity in warningly colored wing patterns and composed of a continuum of taxa across the stages of speciation. These characteristics, coupled with a 50-year legacy of ecological and behavioral research, offer exceptional prospects for genomic studies into the nature of adaptive differences and the formation of new species. Research in Heliconius provides clear connections between genotype, phenotype, and fitness of wing color patterns shown to underlie adaptation and speciation. This research is challenging our perceptions about how speciation occurs in the presence of gene flow and the role of hybridization in generating adaptive novelty. With the release of the first Heliconius genome assembly, emerging genomic studies are painting a dynamic picture of the evolving species boundary. As the field of speciation genomics moves beyond describing patterns, towards a more integrated understanding of the process of speciation, groups such as Heliconius, where there is a clear speciation continuum and the traits underlying adaptation and speciation are known, will provide a roadmap for identifying variation crucial in the origins of biodiversity.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Adaptation
- Association mapping
- Genomic divergence
- Hybridization
- Introgression
- Phenotypic evolution
- Speciation
13.1 Introduction
Over 150 years ago, Henry Walter Bates published his first observations of butterfly diversity in the New World Tropics (Bates 1862). Exploring deep within the Amazon basin for over a decade, Bates documented extraordinary cases of mimicry in the vivid wing patterns of distantly related butterfly species. His writings provided Darwin with some of the most visually stunning examples of evolution by natural selection and the best evidence of a link between natural selection and speciation. Thanks to Bates and Fritz Müller, who arrived in Brazil a few years after Bates, butterflies, arguably more than any other group, contributed to the early establishment and acceptance of evolutionary theory (Carroll et al. 2009). Research on butterflies continues to be as relevant today as it was 150 years ago. Using modern technologies, there is an active research community encompassing most areas of ecology and evolution, ranging from the molecular details of vision to the analysis of human impact on biodiversity (Boggs et al. 2003; Kotiaho et al. 2005; Briscoe et al. 2010). This includes a vibrant genomics research community working on a number of different species, including passion-vine butterflies (Heliconius spp., Heliconius Genome Consortium 2012), monarchs (Danaus plexippus, Zhan et al. 2011), swallowtails (Papilio spp., O’Neil et al. 2010), the Glanville fritillary (Melitae acinxia, Hanski 2011), Bicyclus anynana (Brakefield et al. 2009), and Lycaeides (Gompert et al. 2012). The past several years have seen remarkable progress in the development of genomic resources in these species, culminating in the publication of the first two butterfly reference genomes (Zhan et al. 2011; Heliconius Genome Consortium 2012), with a number of additional genomes scheduled to be released in the coming year.
In this review, we examine emerging ecological and evolutionary genomic research addressing adaptation and speciation in Heliconiusbutterflies. Genomic research is ongoing in several butterfly species, but genomic studies on Heliconius are arguably further developed. Research on Heliconius provides a foundation to discuss larger issues relating to the nature of adaptive differences and the formation of new species. We begin with an overview of the Heliconius radiation – a radiation that has produced an extraordinary evolutionary continuum composed of divergent races and species at different stages of speciation (Mallet et al. 1998; Mallet 2008; Merrill et al. 2011a). Using this continuum as a backdrop, we review recent progress to (i) identify functional variation in the group and reconstruct the history of adaptive alleles, (ii) understand the importance of hybridization in speciation, and (iii) explore the genomic architecture that allows speciation to proceed in the face of gene flow. We conclude with a discussion of how to move beyond patterns of genomic variation to gain a deeper understanding of the processes that drive divergence and speciation in nature.
13.2 The Heliconius Radiation: A Primer
The butterfly subtribe Heliconiina (Lepidoptera: Nymphalidae: Heliconiinae) is restricted to the New World tropics and has a host of life history, ecological, and phenotypic traits that have long fascinated biologists and naturalists. Heliconiines get their common name, passion-vine butterfly, from their strong association with the passion flower family (Passifloraceae). Passion vines are protected by a diverse arsenal of cyanogenic compounds that are likely a by-product of an evolutionary arms race with heliconiines (Spencer 1988). Heliconiines have adopted this defensive tactic–evolving the ability to make and, in some cases sequester, cyanogenic glycosides (Engler et al. 2000; Engler-Chaouat and Gilbert 2007). These compounds render the bearer highly distasteful and avian predators quickly learn to associate a wing color pattern with unpalatability (Chai 1986).
Within the subtribe, the genus Heliconius is characterized by an ecological shift to pollen feeding. Unlike most butterflies, which feed only on fluids (e.g. nectar, decomposing animals and fruits, and dung), Heliconius actively collect and process pollen (Gilbert 1972). The origin of pollen feeding is associated with subtle changes in morphology (Gilbert 1972; Krenn and Penz 1998; Eberhard et al. 2009), coupled with more profound changes in a host of life history and behavioral traits. In particular, the transition to pollen feeding is hypothesized to be important in the butterflies’ ability to synthesize toxic compounds and to enable a very long adult life – one of the longest recorded for a butterfly (Gilbert 1972). Pollen feeding is also associated with a rapid increase in brain size (Sivinski 1989), the evolution of slower and more maneuverable flight, the development of large eyes with accentuated ultraviolet color vision (Briscoe et al. 2010), and the evolution of a suite of complex behaviors, including trap-line feeding, gregarious roosting, and elaborate mating strategies (Brown 1981).
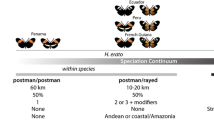
The Heliconius genus is best known for the extraordinary mimicry-related wing pattern diversity seen among its 43 species (Fig. 13.1). With over 400 distinct color pattern varieties, the group represents one of the most striking adaptive radiations in the animal kingdom. Repeated convergent and divergent evolution creates a colorful tapestry where distantly related species often look identical and closely related species or races can look strikingly different (Fig. 13.1). The complexity of this tapestry is exemplified by the parallel radiations of H. erato and H. melpomene, which, although phylogenetically distant and unable to hybridize, have converged on 25 different mimetic color patterns across the Neotropics (Fig. 13.2). Most of the diversity in these two species can be partitioned into two major phenotypic groups: (i) “postman” phenotypes, which have red on their forewing and either possess or lack a yellow hindwing bar; and (ii) “rayed” phenotypes, which have a yellow forewing band, a red patch on the proximal area of the forewing, and red rays on the hindwing. Variations on these themes generate the abundant pattern diversity we see in nature (Fig. 13.2b). In addition to the postman and rayed phenotypes, there are also a number of tiger striped Heliconius. For example, H. numata shows numerous sympatric color patterns races that are likely a result of strong selection pressure to mimic distantly related Melinaea species (Nymphalidae: Ithomiinae) (Brown and Benson 1974; Joron et al. 1999), which can vary dramatically in abundance over small spatial and temporal scales. Geographic variation and convergent evolution are common across Heliconius and the wing patterns of most species converge onto a handful of common color patterns, so called mimicry rings, which coexist locally (Mallet and Gilbert 1995). This convergence between species led to the original hypothesis of mimicry (Bates 1862) and Heliconius is now a classic example of Müllerian mimicry, where distantly related, but similarly distasteful, species converge on the same warningly coloredpattern.

Nature’s palette – the Heliconius radiation. This representation of the major Heliconius clades incorporates over half of the 43 different Heliconius species and a large portion of wing pattern variation. Note the reuse of phenotypes across the phylogeny and the extreme phenotypic variation that exists within and between closely related species. The origins of several major ecological and behavioral innovations are indicated, including pollen feeding, pupal mating, and gregarious larvae. Pupal mating is the reproductive behavior where males search and guard pupae and mate with females as they eclose (Deinert et al. 1994). Pollen feeding and pupal mating likely evolved only once in the radiation. In contrast, gregarious aggregations of larvae evolved several times including the lineage containing the primitive Heliconius. Branch length does not necessarily reflect divergence time (Beltrán et al. 2007)

Variations on a theme – parallel color pattern radiations in Heliconius erato and Heliconius melpomene . (a) Geographic distributions of the major phenotypes of H. erato and H. melpomene, showing the “rayed” phenotype throughout the Amazon basin and disjoint populations of the two variations of the “postman” phenotype. (b) Each row shows some of the color pattern divergence within the two concordant radiations, classified by the two major phenotypes (“postman” and “rayed”). Comparing color pattern phenotypes between the two species reflects the convergent color pattern evolution
Divergence in wing color pattern is also associated with speciation due to the dual role of color pattern in signaling to predators and in mate selection. For example, H. melpomene and H. cydno are very closely related, broadly sympatric, and occasionally hybridize in nature. In this case, speciation is associated with, and reinforced by, divergence in mimetic color patterns. Heliconius melpomene is generally black with red and yellow markings and mimics H. erato (Flanagan et al. 2004) (Fig. 13.2); whereas, H. cydno is black with white or yellow markings and typically mimics H. sapho and H. eleuchia (see Fig. 13.1). Mate recognition involves visual attraction of males to females, which leads to strong color-pattern based assortative mating and disruptive sexual selection against hybrids (Jiggins et al. 2001b; Naisbit et al. 2001). Furthermore, there is ecological post-mating isolation that results from increased predation on hybrids due to their non-mimetic wing patterns (Merrill et al. 2012). Species boundaries are often associated with mimetic color pattern shifts, highlighting the pervasive role that color pattern evolution plays in reproductive isolation across the radiation (Mallet et al. 1998).
13.3 The Genetics of Color Pattern in Heliconius
Given the visually dynamic nature and clear importance of coloration in the Heliconius radiation, much effort has focused on characterizing the genetic basis of wing pattern variation in the group. The spectrum of pattern diversity in Heliconius is largely controlled by variation at a handful of loci of large phenotypic effect (Sheppard et al. 1985; Joron et al. 2006, 2011; Reed et al. 2011; Martin et al. 2012; Papa et al. 2013). These loci are phylogenetically conserved “hotspots” of phenotypic evolution, responsible for color pattern evolution in both convergent and divergent Heliconius species. Allelic variation at these loci produce complex phenotypic changes that show spatial variation across wing surfaces (Fig. 13.3). The identification and characterization of these color pattern alleles has been a major step forward in understanding the evolution and development of lepidopteran wing patterns and provides a unique glimpse into the developmental genetic architecture of pattern evolution in nature.

Hotspots of phenotypic evolution. Although more than two dozen loci have been described, color pattern variation across Heliconius is largely regulated by four major effect loci – two color “switch” loci and two melanin “shutter” loci. These loci interact to create the natural diversity of Heliconius color patterns. The “red switch” controls various red elements across the wing surfaces and was originally described as multiple loci (Sheppard et al. 1985). The “yellow switch” changes forewing and hindwing pattern elements between white and yellow. The “forewing melanin shutter” controls the distribution of black/melanic wing scale cells on the forewing to either expose or cover white or yellow pattern elements and generates variation in forewing band shape, size and position. This locus also originally described as at least five distinct loci in different Heliconius species. Finally, the “global melanin shutter” is similar to the forewing melanin shutter, but it acts more broadly across the wing. It was similarly described as a number of distinct loci in different Heliconius species. Allelic variation at this locus can have multiple phenotypic effects across the wing including: (i) creating the white fringes on the fore wing and hindwing, (ii) causing the presence or absence of the yellow hindwing bar, and (iii) changing the shape and color of the forewing band in some species, including H. himera and H. heurippa. In addition, allelic variation at this locus, in the form of a series of localized inversions, controls all color and pattern variation in H. numata (see Joron et al. 2011)
The nomenclature surrounding the genetics of color pattern variation in Heliconius that has developed over the past 50 years is complicated; however, most described color pattern variation is due to the combined effects of four major loci. All four loci contain functional variation that affects distinct color pattern elements. For simplicity, we will refer to them as the “red switch”, the “yellow switch”, the “forewing melanin shutter”, and the “global melanin shutter” (Fig. 13.3). The red switch controls the presence of several discrete red elements, including the forewing band, the proximal forewing “dennis” patch, and the hindwing rays (Fig. 13.3). The yellow switch causes a loss of yellow ommochrome pigments across both wing surfaces, resulting in a switch from yellow to white pattern elements (Linares 1997; Naisbit et al. 2003) (Fig. 13.3). This locus has largely been described from crosses of H. cydno, but probably underlies pattern differences in races of H. sapho and H. eleuchia as well. The two “shutters” (sensu Gilbert 2003) modulate the complex distribution of black/melanin across both wing surfaces. The forewing melanin shutter acts predominately in the central portion of the forewing, generating variation in the shape, position, and size of the forewing band (Fig. 13.3). The global melanin shutter affects the distribution of melanin across both wing surfaces to create variation in a number of red, yellow, and white pattern elements (Fig. 13.3). Due to the broad phenotypic effects of these color pattern loci, most were originally described as supergenes (Mallet 1989), or clusters of linked genes that facilitate co-segregation of adaptive variation (sensu Mather 1950).
The above synthesis is an oversimplification in several ways. First, there are other loci that have moderate size effects that contribute to pattern variation in important ways (Papa et al. 2013). This includes completely unexplored loci that alter the nanostructures of wing scale surfaces, causing light to scatter, which results in iridescent wing surfaces. Second, all of the major loci have pleiotropic effects that extend beyond the neat categorizations described above. Furthermore, most interact epistatically with each other to generate additional pattern diversity. For example, in H. melpomene the global melanin shutter interacts with the red switch and the forewing melanin shutter to finely control the color, size, and position of the forewing band. Finally, the magnitude of color pattern variation generated by allelic differences within these loci can be extreme. This is best exemplified in H. numata, where the entire spectrum of wing pattern variation is controlled by variation at a single locus – the global melanin shutter (Fig. 13.3) (Joron et al. 2006).
13.4 Genomic Divergence Across the Speciation Continuum
A major strength of Heliconius as a genomic model system is that the radiation has produced a continuum of divergent races and species, which provide an excellent opportunity to study taxa at different stages of speciation (Fig. 13.4). At one end of the continuum, many divergent color pattern races freely hybridize, showing only weak reproductive isolation from each other. These racial boundaries are maintained primarily by selection on wing color patterns, with free gene flow across the rest of the genome. This heterogeneity in gene flow across the genome is ideal for identifying functional variation, as the genomes of divergent, hybridizing races should only differ at regions responsible for phenotypic differences. For some pairs of Heliconius taxa, speciation has progressed further. For example, the hybrid zone between H. erato and H. himera in Ecuador is characterized by the evolution of strong pre-mating isolation through assortative mating, but there is no evidence for hybrid inviability or sterility (McMillan et al. 1997). Both prezygotic and postzygotic isolation have been shown to contribute to isolation in other Heliconius hybrid zones, including the Colombian hybrid zone between H. e. venus and H. e. chestertonii (Arias et al. 2008). Species pairs such as these permit analysis of the earliest stages of speciation, where speciation has begun, but reproductive isolation is not complete, with hybrids making up a small proportion of the population in narrow areas of overlap (McMillan et al. 1997; Arias et al. 2008). Further along the continuum, there are many closely related, sympatric species where hybridization occurs, but it is rare (Mallet et al. 2007; Mallet 2009). For example, H. melpomene and H. cydno are broadly sympatric in Central America and northern South America, coexisting as a result of several ecological differences, including their mimetic association, host plant association, and habitat preference (Naisbit 2001). There are also more distantly related species in sympatry, such as H. melpomene and H. elevatus, where the occurrence of very rare hybridization has facilitated adaptive introgression of color pattern alleles and the spread of mimetic patterns across the genus (Heliconius Genome Consortium 2012). Finally, there are non-hybridizing species, such as H. erato and H. melpomene, which are ecologically and behaviorally distinct, yet share identical mimetic wing patterns. These species provide a comparative framework for exploring the repeatability of evolution and for gaining a more general understanding of how genomic changes influence developmental pathways, phenotype, and ultimately fitness.

Across the speciation continuum. The Heliconius radiation provides an exceptional opportunity to study taxa at different stages of speciation. The continuum, from freely hybridizing populations to non-hybridizing species, provides insight into a variety of key areas of research related to speciation. The genomes of freely hybridizing races should diverge only at genomic regions driving phenotypic divergence, allowing identification of important functional variation. Species pairs at the earliest stages of speciation give insights into how reproductive isolation is established. As reproductive isolation increases, but low levels of hybridization remain, patterns of divergence across the genome reflect the complex interplay between evolutionary forces and genome structure. Studying mimetic species pairs that do not hybridize gives insights into how genomes diverge with complete reproductive isolation and how distantly related species can converge on nearly identical phenotypes
13.4.1 Identifying Functional Variation
Decades of research in Heliconius has shown that wing color patterns are under strong natural selection (Benson 1972; Mallet and Barton 1989; Mallet et al. 1990; Kapan 2001). In one of the best studied Heliconius hybrid zones, the transition between divergent postman and rayed color pattern races of H. erato in Northeastern Peru is sharp and occurs across a narrow 10 km transect (Fig. 13.5a). Strong natural selection on color pattern was demonstrated experimentally on either side of the hybrid zone by releasing individuals with the postman pattern within the rayed population, and vice versa, and estimating survivorship (Mallet and Barton 1989). On both sides of the hybrid zone, recapture rates were significantly lower for butterflies with the foreign color pattern, yielding an estimated overall selection coefficient of 0.52. This estimate was comparable to indirect measures of selection based on fitting linkage disequilibria and cline theory models to extensive hybrid zone data (Mallet et al. 1990).

Linking genotype to phenotype to fitness in Heliconius . (a) Allele frequency variation at the red switch locus across the Peruvian hybrid zone reflects strong natural selection on color pattern. (b) optix is the only gene in the red switch locus showing strong and significant differential expression between the red and yellow forewing bands of divergent red phenotypes. (c) In situ hybridizations show that spatial expression of optix exactly prefigures red color pattern elements in pupal wings 60 hours after pupation. (d) Sliding window genomic divergence between two major H. erato phenotypes (“postman” and “rayed”) from three hybrid zones across the red switch locus. There are two peaks of divergence, one near optix and one at a 65 kb region 3’ of optix, indicating potentially functional regions. The 65 kb peak also contains numerous SNPs perfectly associated with phenotype. The divergent peak stands in marked contrast to the lack of differentiation at regions unlinked to color pattern \( \left({\protect\widehat{\theta}}_s=-0.07\right) \) (Figure modified from Mallet and Barton 1989; Reed et al. 2011; Supple et al. 2013)
Recent research has focused on identifying and understanding the architecture of color pattern loci in order to connect genotype to phenotype and fitness. Using a combination of traditional genetic and emerging genomic approaches, the genomic regions containing the four major color pattern loci have been localized to small intervals and, in two cases, very narrow genomic regions, with specific genes being strongly implicated (Joron et al. 2006; Counterman et al. 2010; Baxter et al. 2010; Ferguson et al. 2010; Reed et al. 2011; Joron et al. 2011; Martin et al. 2012; Heliconius Genome Consortium 2012; Supple et al. 2013). The most progress has been made in understanding red color pattern variation, with research on the red switch locus serving as an exemplar of how to identify functionally important variation. Allelic variation at the red switch controls multiple distinct red color pattern elements that vary between the two divergent color pattern phenotypes (Fig. 13.3). The genomic interval containing this switch was localized to a 400 kb region on chromosome 18 that contained more than a dozen predicted genes (Counterman et al. 2010). Within this region, microarray expression studies, using probes tiled across this region, identified optix, a homeobox transcription factor, as the only gene that was consistently differently expressed between divergent color pattern races – showing high upregulation in regions of the pupal wing fated to become red in the adult wing (Fig. 13.5b). Furthermore, beginning at approximately 60 hours after pupation, optix expression perfectly prefigures red pattern elements (Fig. 13.5c) – even reflectingsubtlepatterndifferences between co-mimics (Reed et al. 2011). The optix amino acid sequence is highly conserved within Heliconius, which suggests that the control of red pattern variation is due to allelic variation in cis-regulatory elements (Hines et al. 2011; Reed et al. 2011).
The prediction that cis-regulatory variation modulates red color pattern variation is supported by population genomic analyses of individuals collected across narrow hybrid zones between divergent color pattern phenotypes. These hybrid zones contain individuals representing many generations of recombination and phenotypically distinct individuals collected within them differ only at genomic regions responsible for the those differences (Counterman et al. 2010; Baxter et al. 2010; Nadeau et al. 2012). Analysis of three replicated hybrid zones between postman and rayed H. erato races shows a sharp peak of genomic divergence in a region approximately 100 kb 3′ of optix, in a “gene desert” containing no predicted genes nor any transcriptional activity. The peak of divergence is approximately 65 kb wide and contains numerous SNPs perfectly associated with color pattern across the broader H. erato pattern radiation (Fig. 13.5d) (Supple et al. 2013).
Relative to other regions of the genome, there is extensive linkage disequilibrium (LD), reduced heterozygosity, reduced nucleotide diversity, and high levels of population differentiation across the 65 kb peak (Supple et al. 2013)—hallmarks of a history of strong selection. A compelling hypothesis is that this region contains a series of modular enhancers that regulate the spatial expression of optix, similar to the architecture recently described for genes responsible for morphological variation in melanin and trichome patterns in Drosophila (Bickel et al. 2011; Frankel et al. 2011). This model is consistent with phenotypic recombinants occasionally collected in postman/rayed hybrid zones that disassociate the proximal red patch on the forewing from the red hindwing rays, which are red color pattern elements that typically occur together (Mallet 1989). In fact, in both co-mimics, H. erato and H. melpomene, there is a single race found in a narrow geographic region in the Guianas that have the red forewing patch, but lack the hindwing rays. Additionally, one of the polymorphic forms of H. timareta in Ecuador also has a recombinant phenotype – showing hindwing rays, but no red forewing patch (Mallet 1999).
The genome scans described above are exceptionally powerful for localizing functional regions. However, the ability to more finely characterize these regions and to identify functional elements or functional changes using population genomic approaches will ultimately depend on what is causing the strong LD seen across the region. One possibility is that the region contains an inversion that suppresses recombination and locks loci into specific allelic combinations. Inversions have been shown to be important in maintaining a series of major effect color alleles in H. numata (Joron et al. 2011). However, we see no evidence for inversions in the red switch locus in H. erato and H. melpomene pair-end sequence data (Supple et al. 2013), nor were inversions evident in analysis of different color pattern races of H. melpomene (Nadeau et al. 2012). Moreover, fine scale analysis of haplotype structure across this region suggests that recombination occurs (Supple et al. 2013). Given the lack of evidence of an inversion and the presence of recombinant individuals, it is most likely that the observed LD is a result of strong natural selection. Strong selection can establish extensive LD among loci, even among unlinked color loci (Mallet et al. 1990). In this case, the presence of recombination raises the possibility that more extensive population and taxon sampling will further refine the boundaries of the functional elements. However, in order to fully understand how variation in this region modulates pattern evolution, population genomic approaches must be coupled with other strategies, including exploring protein-DNA interactions with DNA foot printing (Cai and Huang 2012) and confirming functional mutations with transgenic manipulations (Merlin et al. 2013; Cong et al. 2013).
There has been similar progress in identifying a candidate gene for the forewing melanin shutter. Linkage mapping, gene expression analysis, and pharmacological treatments all indicate that the WntA ligand modulates variation of the forewing band (Martin et al. 2012). The spatial pattern of WntA expression corresponds to the black forewing pattern in multiple species across the Heliconius radiation. As with optix, the WntA protein is highly conserved and variation in cis-regulatory regions is likely responsible for pattern diversity. WntA is a signaling ligand that creates a morphogen gradient across the developing wing tissue and is the type of molecule predicted to underlie pattern formation in theoretical models of wing color pattern development (Kondo and Miura 2010; Nijhout 1991). WntA is expressed earlier in pattern formation than optix and may act as a negative regulator of optix, with the interaction between the two genes being responsible for establishing black versus non-black wing pattern boundaries. This is only the second report of a morphogen involved in pattern generation (see Werner et al. 2010), but it is the first that directly links change in a patterning molecule to the evolution of a highly variable trait with clear adaptive significance (Martin et al. 2012).
The yellow switch and the global melanin shutter have similarly been positionally cloned (Joron et al. 2006; M. Kronforst, pers. comm.). Nonetheless, specific candidate genes and/or functional elements have yet to be identified. The global melanin shutter, in particular, has proven difficult to characterize. It was the first color pattern locus to be positionally cloned and it has the broadest range of phenotypic effects of any of the Heliconius color loci. Moreover, this region has recently been shown to underlie pattern change in several Lepidoptera species, including eyespot size in Bicyclus (Saenko et al. 2010) and the classic case of industrial melanism in the British peppermoth, Biston betularia (van’t Hof et al. 2011), underscoring the flexibility and broad evolutionary importance of the Heliconius patterning loci. In Heliconius, this locus has been hypothesized to be a “supergene” composed of a number of distinct co-adapted protein coding loci. Support for this comes from recent work on H. numata that showed that allelic variants at this locus are actually a set of different chromosomal inversions across a region containing at least 18 genes (Joron et al. 2011). However, ongoing expression and genome scan studies in H. erato and H. melpomene indicate that, similar to the red switch and the forewing melanin shutter, only a single protein coding region at this locus may underlie the global variation in melanin pattern across Heliconius wings (C. Jiggins, pers. comm.).
13.4.2 The Origins of Novel Phenotypes
Natural selection can explain why wing patterns of different Heliconius species should converge – strong selection against rare color patterns promotes mimicry (Müller 1879). Natural selection can also explain the maintenance of existing wing pattern diversity – strong frequency-dependent selection removes non-mimetic individuals creating sharp transition zones between divergent phenotypes (Mallet et al. 1998). However, natural selection cannot easily explain the origin and spread of new phenotypes in Heliconius. This is a complex issue at the core of the mimicry paradox – the frequency – dependent selection that stabilizes existing patterns is the same force that eliminates novel forms, yet pattern divergence is rampant (Mallet and Gilbert 1995; Turner and Mallet 1996; Joron and Mallet 1998). To begin to explain this paradox, we first need to understand the evolutionary dynamics of the genomic regions that cause pattern change. An essential first question to ask is whether novel phenotypes arise once and spread within and between species or are there multiple, independent origins of the same phenotype? It has only been with the identification of the regions that modulate phenotypic differences that we can begin to address this question. The answer seems to be a bit of both – phenotypic evolution within races and species with even low levels of hybridization occurs by sharing uniquely derived color pattern alleles; while convergent phenotypes evolve independently in more distantly related co-mimics.
Analyses of the genomic region responsible for color pattern diversity support a single origin for major red color pattern phenotypes within species. For both H. erato and H. melpomene, variation around the red switch locus sorts by color pattern phenotype (Hines et al. 2011; Supple et al. 2013). In both species, individuals possessing a rayed phenotype cluster together to the exclusion of individuals possessing the postman phenotype (Fig. 13.6). Rayed patterns are found in the Amazon basin and are co-mimetic with several other Heliconius species and day flying moths; whereas, the postman phenotypes are largely unique to H. erato and H. melpomene and are found in multiple disjunct regions around the periphery of the Amazon and in Central America (Fig. 13.2). The pattern of genetic variation around the red switch locus supports the hypothesis that one rayed phenotype evolved in each species and spread quickly, fragmenting the geographic distribution of the older postman phenotypes. This phylogenetic signal is restricted to a region containing the 65 kb divergence peak identified in the hybrid zone comparisons (Fig. 13.5d). As you move away from this region, the phylogenetic signal increasingly reflects a history of recent gene flow, with variation clustering by geographic proximity and biogeographic boundaries, regardless of color pattern (Fig. 13.6). This pattern of clustering by geography is the same pattern that is observed across regions unlinked to color pattern, which previously led to the incorrect conclusion that similar color pattern phenotypes evolved multiple times within each species (Brower 1994; Flanagan et al. 2004; Quek et al. 2010). This discordance demonstrates how inferences drawn from a specific subset of the genome can be misleading (Hines et al. 2011).

The origins of an adaptive radiation – phylogenetic analysis of the red switch locus in H. erato . Phylogenetic analysis of optimal topological partitions highlight a region around the gene optix as clustering samples by phenotype, rather than geographic proximity. The tree topologies are shown, with phenotypes represented by branch color and geographic regions by terminal node color. Trees were generated from SNPs determined by aligning short sequencing reads to a reference genome. The grey scale bar is colored by the general history inferred. Around optix, samples are clustered by phenotype (black bar), while the farthest partition from optix clusters by geography (light grey bar), and the regions in between are intermediate, clustering by a mix of geography and phenotype (dark grey bar). Gene annotations, with the gene optix indicated, are shown below (Figure modified from Supple et al. 2013)
Mimetic convergence between distantly related species, in contrast, likely occurs by independent evolution. For example, population genetic and phylogenetic analyses of the H. erato and H. melpomene radiations, using variation within color pattern intervals, consistently clusters individuals by species designation, which is similar to the groupings obtained at loci unlinked to color pattern. Thus, although the same genomic region regulates mimetic color pattern variation, the changes responsible for mimetic convergence likely arose independently in the two species. The independent origin of similar color patterns in H. erato and H. melpomene is perhaps not unexpected, given that the two species diverged from each other over 15 million years ago (Pohl et al. 2009) and do not hybridize.
It is less clear how often more closely related species “borrow” color pattern alleles to acquire a mimetic wing color pattern. A cursory review of a phylogeny of the Heliconius radiation shows the high frequency that similar wing patterns are shared by species across the tree (Fig. 13.1). For example, within the melpomene/cydno/silvaniform (MCS) clade, the rayed and postman phenotypes occur within three of the four major lineages (Fig. 13.1). These species are all closely related and many are known to hybridize in the wild (Mallet et al. 2007) and in greenhouses (Gilbert 2003). These observations lead to a proposed model whereby Heliconius mimicry evolved by repeated interspecific transfer of color patterning alleles (Gilbert 2003). Adaptive introgression, which is the spread of beneficial variation through interspecific hybridization, may have provided the genetic raw material for both accelerated adaptation and speciation due to the dual role of color patterns in mimicry and mating behavior.
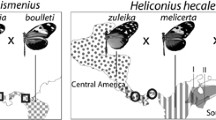
Compelling evidence for hybridization and introgression, particularly around color pattern loci, comes from analyses of closely related Heliconius species that share similar mimetic patterns. Phylogenetic analysis of genetic variation from the region identified as crucial to red color pattern differences, clusters populations and species by red pattern phenotype across species boundaries, rather than by phylogenetic relationship (Heliconius Genome Consortium 2012; Pardo-Diaz et al. 2012) (Fig. 13.7). This clustering by phenotype includes H. elevatus, which is the only rayed species in the more distantly related silvaniform clade, which usually have orange, black, and yellow tiger patterns and are known to hybridize, albeit rarely, with H. melpomene. Additional support comes from genome-wide tests that attempt to distinguish shared ancestral polymorphism from shared polymorphism resulting from recent gene flow (Green et al. 2010; Durand et al. 2011). This analysis shows a statistically significant excess of shared polymorphisms between sympatric co-mimics than would be expected from random sorting of ancestral polymorphisms, with a particularly strong signal at known color pattern loci. Although this test has been shown to be biased by population structure and to be sensitive to genotyping errors (Durand et al. 2011), the pattern of shared polymorphisms combined with the traditional phylogenetic analyses paints a dramatic picture of the adaptive spread of color pattern alleles across species boundaries.

Evolution by adaptive introgression. Phylogenetic analysis across the red switch locus shows introgression between sympatric, mimetic species in the genomic region believed to control red color pattern variation. A single 50 kb region clusters all rayed samples together, including H. elevatus, a proposed hybrid species. Windows farthest from this region generate the expected species tree (Figure modified from Heliconius Genome Consortium 2012)
13.4.3 Wing Color Patterns as a “Magic Trait” Promoting Speciation
As we are beginning to understand the role that adaptive introgression plays in the spread of mimetic color pattern alleles, it is becoming equally clear that divergent color pattern alleles in Heliconius likewise play a profound role in speciation. Disruptive selection on an ecological trait, such as Heliconus wing patterns, imposes a barrier to gene flow (Schluter 2009; Nosil 2012). We see this clearly in the signature of differentiation across hybrid zones between color pattern races of H. erato (Fig. 13.5d). However, in the absence of strong assortative mating, even with this barrier to gene flow, intermediate phenotypes will be continually produced and recombination will prevent speciation. This antagonism is the principal reason why the idea of speciation with gene flow remains extremely controversial (Felsenstein 1981). One way around this difficulty is if the trait under disruptive selection also contributes to nonrandom mating. In this case, there is a clear path to speciation (Dieckmann and Doebeli 1999; Gavrilets 2005) and such traits have become known as “magic traits”. Rather than the term “magic trait”, which implies a trait encoded by a “magic gene”, a better term would be “multiple-effect trait”. A multiple-effect trait is simply a trait that has multiple functions – it is under disruptive selection and contributes to non-random mating. This definition is of more value because it does not presuppose any particular underlying genetic architecture (c.f. Servedio et al. 2011).
The wing patterns of Heliconius provide one of the best experimental systems to study how “magic” or “multiple-effect” traits can generate biodiversity. Research on how these traits can promote speciation is most progressed in H. melpomene and H. cydno, where experimental manipulation demonstrates the importance of wing color patterns in both natural and sexual selection (Naisbit et al. 2001; Jiggins et al. 2001b; Merrill et al. 2011b, 2012). Recent field and cage experiments demonstrate that wing color patterns are under disruptive natural selection – F1 hybrids, whose wing color patterns show an intermediate forewing phenotype, were attacked more frequently than either parental species (Merrill et al. 2012). Mate choice experiments demonstrate both color pattern based assortative mating between the two species and disruptive sexual selection against hybrids (Jiggins et al. 2001b; Naisbit et al. 2001; Merrill et al. 2011b). In addition, assortative mating is much higher in populations where H. melpomene and H. cydno are sympatric, as compared to populations where H. melpomene does not encounter H. cydno. This is consistent with the expectation of the reinforcement hypothesis – selection against hybrids lead to the evolution of stronger pre-mating isolation (Jiggins et al. 2001b). It is important to point out that, in addition to color pattern based mating, there are other forms of isolation between the pair, including host plant preferences, microhabitat usage, and sterility barriers. The sterility barriers occur because the F1 offspring follow Haldane’s rule – the homogametic males are fertile and the heterogametic females are sterile. However, the strength of selection against hybrids that results from female sterility is only about as strong as mimicry selection and not nearly as strong as pre-mating isolation due to assortative mating (Naisbit et al. 2002).
In Heliconius, ongoing research is beginning to make the connection between multiple-effect traits and the loci that underlie these traits. A series of recent studies have demonstrated that the loci causing color pattern differences and the loci responsible for color pattern based mating preference are physically linked in Heliconius (Kronforst et al. 2006a; Merrill et al. 2011b). Physical linkage between a color pattern locus and a male mating preference was demonstrated first in H. pachinus and H. cydno galantus – mating was strongly assortative by white versus yellow color and variation in male mating preference mapped to the yellow switch locus (Kronforst et al. 2006a). A similar association was demonstrated between H. cydno and H. melpomene. In this species pair, male approach and courtship behavior was also highly assortative by coloration and strong male preference for red pattern mapped to the red switch locus (Merrill et al. 2011b). This is a very intriguing finding given that the gene that controls the distribution of red on a Heliconius wing, optix, also plays a role in compound eye development (Seimiya and Gehring 2000). This raises the possibility for a direct link between the perception and transmission of color pattern cues. Ongoing research, including experiments to create introgression lines that differ primarily around the regions responsible for red pattern variation, seeks to better understand the nature of the observed association and its role in promoting the radiation of Heliconius butterflies.
In addition to facilitating speciation by divergent natural selection, physical linkage between color pattern traits and the mating preference for those traits can promote the formation of new species through hybridization (Arnold 2006). Hybrid speciation results when hybridization produces novel genotypes that are reproductively isolated from the parental species. In this regard, hybrid genotypes that confer an ecological advantage and influence assortative mating (sensu Smith 1966) could quickly result in the origin of a novel hybrid population that is reproductively isolated from the parental species. This process has been termed hybrid trait speciation (Jiggins et al. 2008). Although hybrid speciation is thought to be rare in animals, hybrid trait speciation may provide a route for hybridization to play a role in animal diversification.
In Heliconius there are some of the most compelling Heliconius examples of hybrid trait speciation in the animal kingdom (Mavárez et al. 2006; Salazar et al. 2010; Heliconius Genome Consortium 2012). For example, evidence from a number of independent datasets suggest Heliconius heuripa arose through hybrid speciation: (i) the observation of regions in Venezuela where hybrids between the proposed parental species commonly occur (Mavárez et al. 2006), (ii) laboratory crosses demonstrating a clear path to the H. heurippa phenotype (Mavárez et al. 2006), (iii) molecular genetic analysis showing that the H. heurippa genome is a mosaic of pieces from the parental species (Salazar et al. 2010), and (iv) mate choice experiments demonstrating incipient reproductive isolation from the parental species via assortative mating (Mavárez et al. 2006; Melo et al. 2009). Heliconius elevatus is another interesting example of a putative hybrid species, as speciation potentially involves both color pattern mimicry and mate choice (Heliconius Genome Consortium 2012). The hypothesis is that hybridization between H. pardalinus and H. melpomene resulted in adaptive introgression of color pattern alleles, followed by reproductive isolation due to assortative mating on wing color pattern. Genomic data strongly support adaptive introgression of the H. melpomene rayed color pattern allele into a H. pardalinus genome (Heliconius Genome Consortium 2012). The prediction of reproductive isolation secondary to adaptive introgression remains untested. It is predicted that the new rayed H. pardalinus population became reproductively isolated from other H. pardalinus and H. melpomene due to assortative mating based on color pattern and perhaps other signals, such as short-range chemical cues (Estrada and Jiggins 2008), resulting in a new species – H. elevatus. Although the genomic, ecological, and behavioral evidence for these examples are impressive, further studies are needed as alternative speciation scenarios have been proposed for these species that do not invoke introgression and hybridization (see Brower 2013). A whole genome perspective should help shed light on this debate, but it requires a more fundamental understanding of how genomes change during speciation and what signature hybridization and introgression would leave on expected patterns of genomic divergence.
13.4.4 Genomic Heterogeneity at the Species Boundary
Although hybridization between closely related species is common in nature (Mallet 2005; Rieseberg 2009), the idea that divergence and speciation can occur in the face of ongoing gene flow remains contentious. Nonetheless, over the last decade the debate has largely shifted from questions about the geographic context of speciation towards gaining an understanding of the processes and mechanisms that can generate biodiversity in the face of gene flow (Nosil 2012). More recently, next-generation sequencing technologies have matured to permit a whole-genome perspective on divergence during speciation. These data promise to advance our understanding of the origins of reproductive isolation by moving research towards the processes that shape patterns of divergence across whole genomes (Feder et al. 2012).
With the publication of the first Heliconius genome (Heliconius Genome Consortium 2012), a number of studies have used whole genome resequencing to explore the genomic landscape of divergence and speciation along an evolutionary continuum of hybridizing taxa. These studies have focused on the melpomene/cydno/silvaniform clade and use the H. melpomene genome as a reference to layer resequence data and to characterize individual variation across the genome (Kronforst et al. 2013; Martin et al. 2013). These studies join several recent studies in other organisms (Hohenlohe et al. 2010; Lawniczak et al. 2010; Ellegren et al. 2012; Gagnaire et al. 2013) to provide the first full genome perspectives on speciation.
Genomic analyses across the Heliconius speciation continuum highlight some characteristics that are emerging from these early speciation genomics studies. At the early stages in the speciation continuum, recently diverged populations freely hybridize but show strong divergence at regions of the genome known to be under strong selection. The result is differentiation that is restricted to few areas of the genome. For example, hybridizing races within both H. erato and H. melpomene showed clear regions of divergence around known mimicry loci, with little divergence evident elsewhere in the genome (Figs. 13.5d and 13.8). It is notable just how restricted differentiation is, even under conditions of very strong natural selection when patterns of divergence are expected to extend well beyond functional sites. Despite strong frequency dependent selection on color pattern, genomic divergence is limited to sharp and narrow peaks tightly linked to color pattern loci. These divergence peaks have long tails that extend about 1 Mb, but differentiation is only slightly above background levels. This observation is interesting given that differences in a number of important ecological traits, including host plant preference and larval survival, map to color pattern regions (Merrill et al. 2013).

Genomic architecture of speciation – empirical data. The empirical divergence data from the melpomene/cydno clade, with arrows pointing to known color pattern loci. Hybridizing races of H. melpomene show islands of divergence at the color pattern loci. Comparisons between closely related, sympatric species, show heterogeneous patterns of divergence across the whole genome. Allopatric species pairs also show high heterogeneity, but with elevated divergence across the genome (Figure modified from Martin et al. 2013)
As speciation progresses, selection, genetic hitchhiking, and the accumulation of neutral mutations during these latter stages of speciation result in highly heterogeneous patterns of genomic divergence across the genome. This pattern of heterogeneous divergence is evident across the continuum from incipient species with pre-mating isolation, to species with strong pre-mating and post-mating isolation. For example, H. cydno and H. pachinus are two closely related species that differ in color pattern and show strong color pattern based assortative mating (Kronforst et al. 2006a), yet hybridize occasionally in narrow regions of overlap in Costa Rica (Kronforst et al. 2006b). In addition to peaks of divergence at known color pattern loci, there are over a dozen regions of the genome that are more divergent than expected by chance and may harbor previously unidentified ecologically important variation. The heterogeneous patterns of divergence persist as the evolutionary distance increases to closely-related species with stronger reproductive isolation, such as H. melpomene versus H. timareta and H. cydno, (Martin et al. 2013) (Fig.13.8).
Heterogeneity is emerging as a common feature of genomic divergence in a number of recent studies. For example, studies of differentiation in Anopheles mosquitoes, Ficedula flycatchers, and Coregonus whitefish also showed highly heterogeneous patterns of differentiation (Lawniczak et al. 2010; Ellegren et al. 2012; Gagnaire et al. 2013). In Ficedula and Coregonus, the patterns are thought to be the result of recent admixture following allopatric divergence. In contrast, the Heliconius and Anopheles patterns are thought to have emerged as a result of speciation without periods of allopatry. Both speciation with gene flow and allopatric divergence with secondary contact can generate genomic heterogeneity. However, finer analysis of the patterns should allow one to distinguish the two scenarios. A commonly used measure of the extent and timing of gene flow is the number and distribution of shared polymorphisms between species. The basic principle is that (i) longer periods of gene flow should result in a greater proportion of shared polymorphic alleles between older species, (ii) recent gene flow will reduce differentiation and increase the proportion of shared alleles among sympatric populations, as compared to allopatric populations, and (iii) recent gene flow should initially result in strong linkage disequilibrium between shared alleles at linked sites, which would breakdown over time if gene flow was ongoing for longer periods. Various methods for comparing the numbers and distribution of shared polymorphisms are being developed and have recently been used to study the role of gene flow during speciation in the handful of organisms with population genomic data available (Kulathinal et al. 2009; Ellegren et al. 2012), including humans and neandertals (Green et al. 2010).
The melpomene/cydno/silvaniform clade provides an ideal opportunity to explore the genomics of speciation with gene flow versus allopatric divergence with secondary contact in a rich comparative framework. This is because the clade includes many allopatric and sympatric/parapatric races and species with varying degrees of known phylogenetic relatedness that can be used to compare patterns of shared polymorphisms and test different speciation models. For example, the observed increase of shared polymorphisms across the genome at increasing phylogenetic depths is suggestive of a long history of gene flow during speciation (Martin et al. 2013). Interestingly, a similar conclusion was reached using a completely different approach that involved modeling introgression rates in a community assessment of genomic differentiation among Costa Rican Heliconius species (Kronforst et al. 2013). However, extreme caution in interpreting these patterns is warranted. High heterogeneity in genomic divergence is indicative of the complex interplay between a diverse array of ecological and demographic factors, including selection, gene flow, and population history, as well as intrinsic genomic features such as variation in recombination rate.
With these new data, we are beginning to appreciate the complexity and the challenges of identifying genomic regions responsible for adaptive divergence and reproductive isolation and understanding how they affect genome-wide patterns of divergence throughout the speciation process. This is a serious challenge, yet systems with replicated examples of adaptation or speciation, such as Heliconius, sticklebacks, and whitefish, can be extremely powerful for inferring the functional importance of regions of divergence and understanding the history of gene flow between species.
13.5 From Patterns to Process
Moving forward requires a much better understanding of the how genomes diverge. As genomic technologies advance, empirical descriptions of genomic divergence will be layered onto one another to describe how the genomes of species change through space and time. The accumulating genomic data are already revealing extremely heterogeneous patterns of divergence that result from complex interactions between selection and gene flow. The research is quickly transitioning to identifying systems that have the most promise to provide insights into the process of genomic divergence. In this respect, new model systems, such as Heliconius, which (i) have replicated examples of adaptation, (ii) are composed of taxa representing distinct stages of the speciation process, and (iii) have traits that are known to contribute to adaptation and speciation, will provide an important framework to determine the processes that drive ecological divergence and speciation from the patterns of genomic divergence.
Genomic data in Heliconius highlight the ability to identify the genomic regions that are known targets of selection and show how divergence around these regions changes when populations are increasingly isolated from each other. However, divergent races and incipient Heliconius species differ by more than wing color patterns. They show differences in mating preference (McMillan et al. 1997; Jiggins et al. 2001b; Chamberlain et al. 2009; Merrill et al. 2011a), hybrid sterility (Jiggins et al. 2001a; Naisbit et al. 2002), host plant choice (Brown 1981; Estrada and Jiggins 2002), and physiology (Davison et al. 1999) – all of which may play key roles in speciation. Leveraging the extraordinary radiation for broader insights into the origins of diversity requires that we better utilize genomic datasets. We need to identify regions under divergent selection and understand how they contribute to differences in survival or otherwise cause a reduction in gene flow between incipient forms. This challenge is not unique to Heliconius – the overarching goal of ecological and speciation genomic research is to identify and characterize regions of the genome under divergent selection and to understand what role they play in speciation.
To reach this goal, we need new theory that will “transform current predictions concerning genetic divergence into more dynamic recreations of how genomic differentiation unfolds through time during speciation” (Feder et al. 2012). Presently, the analysis of genomic landscapes is largely descriptive. Formal models that explain how selection and genetic hitchhiking can drive patterns of genomic divergence are beginning to emerge (Smadja et al. 2008; Feder and Nosil 2010; Feder et al. 2012), but presently there is no standardized procedure to rigorously delimit the shape, size, and distribution of divergent regions of the genomes, and more importantly, to model how they change through time (Feder et al. 2012). Even among Heliconius studies, different strategies were used to identify the location and size of divergent regions and to estimate the degree and timing of gene flow. Without common tools and practices, it becomes very difficult to compare patterns of genomic divergence and to identify general patterns emerging from genome-wide studies of speciation. However, just as new and better datasets will be generated, new and better theories will be developed. The field needs to (i) establish better understandings of the genomic architectures of the traits under divergent selection and influencing reproductive isolation, including the number of loci, their size effect on isolation, and their relative contribution in the speciation process; (ii) investigate how mutation and recombination rates vary locally across the genome and between populations, particularly for those regions of the genome that influence isolation; and (iii) develop increasingly complex models of speciation history and understand how heterogeneous patterns of divergence evolve as species diverge with and without gene flow.
When studying adaptation and speciation, we speculate on the specific historical events that generated the extant genomic patterns that we observe. As such, we have to be very careful to temper our enthusiasm (Nielsen 2009; Barrett and Hoekstra 2011). Molecular “spandrels” (sensu Gould and Lewontin 1979) abound in the genomes of all organisms and establishing direct links between genotype, phenotype, form, and fitness requires integrated datasets. Identifying highly divergent regions of the genome is a starting point for building an integrative understanding of the nature of variation between taxa. For some species, experiments can be designed that measure the success of individuals under specific ecological conditions. In these cases, researchers can actually follow genomic change forward in time. Experimental genomics is moving beyond the laboratory to directly testing hypotheses about how selection causes genome-wide change (Barrett et al. 2008) and provides a powerful approach that can be used in a number of emerging model systems (Barrett and Hoekstra 2011). For other groups, a combination of traditional genetic and functional genomic approaches, coupled with functional manipulation experiments, remains the best strategy to identify functionally important variation. In either case, the combination of technological and analytical advances ensures that genomic exploration will continue to transform our understanding of the origins of biodiversity.
Reference
Arias CF, Muñoz AG, Jiggins CD, Mavárez J, Bermingham E, Linares M (2008) A hybrid zone provides evidence for incipient ecological speciation in Heliconius butterflies. Mol Ecol 17(21):4699–4712
Arnold ML (2006) Evolution through genetic exchange. Oxford University Press, Oxford
Barrett RD, Hoekstra HE (2011) Molecular spandrels: tests of adaptation at the genetic level. Nat Rev Genet 12(11):767–780
Barrett RD, Rogers SM, Schluter D (2008) Natural selection on a major armor gene in threespine stickleback. Science 322(5899):255–257
Bates HW (1862) Contributions to an insect fauna of the Amazon valley.–Lepidoptera:–Heliconinæ. J Proc Linnean Soc Lond Zool 6(22):73–77
Baxter SW, Nadeau NJ, Maroja LS, Wilkinson P, Counterman BA, Dawson A, Beltran M, Perez-Espona S, Chamberlain N, Ferguson L, Clark R, Davidson C, Glithero R, Mallet J, McMillan WO, Kronforst M, Joron M, ffrench Constant RH, Jiggins CD (2010) Genomic hotspots for adaptation: the population genetics of Müllerian mimicry in the Heliconius melpomene clade. PLoS Genet 6(2):e1000794
Beltrán M, Jiggins CD, Brower AV, Bermingham E, Mallet J (2007) Do pollen feeding, pupal-mating and larval gregariousness have a single origin in Heliconius butterflies? Inferences from multilocus DNA sequence data. Biol J Linn Soc 92(2):221–239
Benson WW (1972) Natural selection for Müllerian mimicry in Heliconius erato in Costa Rica. Science 176(4037):936–939
Bickel RD, Kopp A, Nuzhdin SV (2011) Composite effects of polymorphisms near multiple regulatory elements create a major-effect QTL. PLoS Genet 7(1):e1001275
Boggs CL, Watt WB, Ehrlich PR (2003) Butterflies: ecology and evolution taking flight. University of Chicago Press, Chicago, IL, USA
Brakefield PM, Beldade P, Zwaan BJ (2009) The African butterfly Bicyclus anynana: a model for evolutionary genetics and evolutionary developmental biology. Cold Spring Harb Protoc 2009(5): pdb–emo122
Briscoe AD, Bybee SM, Bernard GD, Yuan F, Sison-Mangus MP, Reed RD, Warren AD, Llorente-Bousquets J, Chiao CC (2010) Positive selection of a duplicated UV-sensitive visual pigment coincides with wing pigment evolution in Heliconius butterflies. Proc Natl Acad Sci USA 107(8):3628–3633
Brower AV (1994) Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc Natl Acad Sci USA 91(14):6491–6495
Brower AV (2013) Introgression of wing pattern alleles and speciation via homoploid hybridization in Heliconius butterflies: a review of evidence from the genome. Proc R Soc B 280(1752):20122302
Brown KS (1981) The biology of Heliconius and related genera. Annu Rev Entomol 26(1):427–457
Brown KS, Benson WW (1974) Adaptive polymorphism associated with multiple Müllerian mimicry in Heliconius numata (Lepid. Nymph). Biotropica 6:205–228
Cai YH, Huang H (2012) Advances in the study of protein – DNA interaction. Amino Acids 43:1141–1146
Carroll SB, Grenier J, Weatherbee S (2009) From DNA to diversity: molecular genetics and the evolution of animal design. Wiley-Blackwell, Hoboken, NJ, USA
Chai P (1986) Field observations and feeding experiments on the responses of rufous-tailed jacamars (Galbula ruficauda) to free-flying butterflies in a tropical rainforest. Biol J Linn Soc 29(3):161–189
Chamberlain NL, Hill RI, Kapan DD, Gilbert LE, Kronforst MR (2009) Polymorphic butterfly reveals the missing link in ecological speciation. Science 326(5954):847–850
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marrafini LA et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823
Counterman BA, Araujo-Perez F, Hines HM, Baxter SW, Morrison CM, Lindstrom DP, Papa R, Ferguson L, Joron M, ffrench Constant RH, Smith CP, Nielsen DM, Chen R, Jiggins CD, Reed RD, Halder G, Mallet J, McMillan WO (2010) Genomic hotspots for adaptation: the population genetics of Müllerian mimicry in Heliconius erato. PLoS Genet 6(2):e1000796
Davison A, McMillan WO, Griffin AS, Jiggins CD, Mallet JLB (1999) Behavioral and physiological differences between two parapatric Heliconius species. Biotropica 31(4):661–668
Deinert E, Longino J, Gilbert L (1994) Mate competition in butterflies. Nature 370(6484):23–24
Dieckmann U, Doebeli M (1999) On the origin of species by sympatric speciation. Nature 400(6742):354–357
Durand EY, Patterson N, Reich D, Slatkin M (2011) Testing for ancient admixture between closely related populations. Mol Biol Evol 28(8):2239–2252
Eberhard SH, Nemeschkal HL, Krenn HW (2009) Biometrical evidence for adaptations of the salivary glands to pollen feeding in Heliconius butterflies (Lepidoptera: nymphalidae). Biol J Linn Soc 97(3):604–612
Ellegren H, Smeds L, Burri R, Olason PI, Backström N, Kawakami T, Künstner A, Mäkinen H, Nadachowska-BrzyskaK, Qvarnström A, et al (2012) The genomic landscape of species divergence in Ficedula flycatchers. Nature 491(7426):756–760
Engler HS, Spencer KC, Gilbert LE (2000) Insect metabolism: preventing cyanide release from leaves. Nature 406(6792):144–145
Engler-Chaouat HS, Gilbert LE (2007) De novo synthesis vs. sequestration: negatively correlated metabolic traits and the evolution of host plant specialization in cyanogenic butterflies. J Chem Ecol 33(1):25–42
Estrada C, Jiggins CD (2002) Patterns of pollen feeding and habitat preference among Heliconius species. Ecol Entomol 27(4):448–456
Estrada C, Jiggins CD (2008) Interspecific sexual attraction because of convergence in warning colouration: is there a conflict between natural and sexual selection in mimetic species? J Evol Biol 21(3):749–760
Feder JL, Nosil P (2010) The efficacy of divergence hitchhiking in generating genomic islands during ecological speciation. Evolution 64(6):1729–1747
Feder JL, Egan SP, Nosil P (2012) The genomics of speciation-with-gene-flow. Trends Genet 28:342–350
Felsenstein J (1981) Skepticism towards Santa Rosalia, or why are there so few kinds of animals? Evolution 35(1):124–138
Ferguson L, Lee SF, Chamberlain N, Nadeau N, Joron M, Baxter S, Wilkinson P, Papanicolaou A, Kumar S, KEE TJ, et al (2010) Characterization of a hotspot for mimicry: assembly of a butterfly wing transcriptome to genomic sequence at the HmYb/Sb locus. Mol Ecol 19(Suppl 1):240–254
Flanagan NS, Tobler A, Davison A, Pybus OG, KapanDD, Planas S, Linares M, Heckel D, McMillanWO (2004) Historical demography of Müllerian mimicry in the neotropical Heliconius butterflies. Proc Natl Acad Sci USA 101(26):9704–9709
Frankel N, Erezyilmaz DF, McGregor AP, Wang S, Payre F, Stern DL (2011) Morphological evolution caused by many subtle-effect substitutions in regulatory DNA. Nature 474(7353):598–603
Gagnaire PA, Pavey SA, Normandeau E, Bernatchez L (2013) The genetic architecture of reproductive isolation during speciation-with-gene-flow in lake whitefish species pairs assessed by RAD-sequencing. Evolution
Gavrilets S (2005) “Adaptive speciation” – it is not that easy: reply to Doebeli et al. Evolution 59(3):696–699
Gilbert LE (1972) Pollen feeding and reproductive biology of Heliconius butterflies. Proc Natl Acad Sci USA 69(6):1403–1407
Gilbert L (2003) Adaptive novelty through introgression in Heliconius wing patterns: evidence for shared genetic “tool box” from synthetic hybrid zones and a theory of diversification. In: Ecology and evolution taking flight: butterflies as model systems. University of Chicago Press, Chicago, pp 281–318
Gompert Z, Lucas LK, Nice CC, Buerkle CA (2012) Genome divergence and the genetic architecture of barriers to gene flow between Lycaeides idas and L. melissa. Evolution 67:2498–2514
Gould SJ, Lewontin RC (1979) The spandrels of San Marco and the Panglossian paradigm: a critique of the adaptationist programme. Proc R Soc B 205(1161):581–598
Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MHY, et al (2010) A draft sequence of the Neandertal genome. Science 328(5979):710–722
Hanski IA (2011) Eco-evolutionary spatial dynamics in the Glanville fritillary butterfly. Proc Natl Acad Sci USA 108(35):14,397–14,404
Heliconius Genome Consortium (2012) Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487:94–98
Hines HM, Counterman BA, Papa R, Albuquerque de Moura P, Cardoso MZ, Linares M, Mallet J, Reed RD, Jiggins CD, Kronforst MR, McMillan WO (2011) Wing patterning gene redefines the mimetic history of Heliconius butterflies. Proc Natl Acad Sci USA 108(49):19666–19671
Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA (2010) Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet 6(2):e1000862
Jiggins CD, Linares M, Naisbit RE, Salazar C, Yang ZH, Mallet J (2001a) Sex-linked hybrid sterility in a butterfly. Evolution 55(8):1631–1638
Jiggins CD, Naisbit RE, Coe RL, Mallet J (2001b) Reproductive isolation caused by colour pattern mimicry. Nature 411:302–305
Jiggins CD, Salazar C, Linares M, Mavarez J (2008) Hybrid trait speciation and Heliconius butterflies. Phil Trans R Soc B Biol Sci 363(1506):3047–3054
Joron M, Mallet JL (1998) Diversity in mimicry: paradox or paradigm? Trends Ecol Evol 13(11):461–466
Joron M, Wynne IR, Lamas G, Mallet J (1999) Variable selection and the coexistence of multiple mimetic forms of the butterfly Heliconius numata. Evol Ecol 13(7):721–754
Joron M, Papa R, Beltrán M, Chamberlain N, Mavárez J, Baxter S, Abanto M, Bermingham E, Humphray SJ, Rogers J, et al (2006) A conserved supergene locus controls colour pattern diversity in Heliconius butterflies. PLoS Biol 4(10):e303
Joron M, Frezal L, Jones RT, Chamberlain NL, Lee SF, Haag CR, Whibley A, Becuwe M, Baxter SW, Ferguson L, Wilkinson PA, Salazar C, Davidson C, Clark R, Quail MA, Beasley H, Glithero R, Lloyd C, Sims S, Jones MC, Rogers J, Jiggins CD, ffrench Constant RH (2011) Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477(7363):203–206
Kapan DD (2001) Three-butterfly system provides a field test of müllerian mimicry. Nature 409:338–340
Kondo S, Miura T (2010) Reaction-diffusion model as a framework for understanding biological pattern formation. Science 329(5999):1616–1620
Kotiaho JS, Kaitala V, Komonen A, Päivinen J (2005) Predicting the risk of extinction from shared ecological characteristics. Proc Natl Acad Sci USA 102(6):1963–1967
Krenn HW, Penz CM (1998) Mouthparts of Heliconius butterflies (Lepidoptera: nymphalidae): a search for anatomical adaptations to pollen-feeding behavior. Int J Insect Morphol Embryol 27(4):301–309
Kronforst MR, Young LG, Kapan DD, McNeely C, O’Neill RJ, Gilbert LE (2006) Linkage of butterfly mate preference and wing color preference cue at the genomic location of wingless. Proc Natl Acad Sci USA 103(17):6575–6580
Kronforst MR, Hansen MEB, Crawford NG, Gallant JR, Zhang W, Kulathinal RJ, Kapan DD, Mullen SP (2013) Hybridization reveals the evolving genomic architecture of speciation. Cell Reports
Kulathinal RJ, Stevison LS, Noor MA (2009) The genomics of speciation in Drosophila: diversity, divergence, and introgression estimated using low-coverage genome sequencing. PLoS Genet 5(7):e1000550
Lawniczak M, Emrich S, Holloway A, Regier A, Olson M, White B, Redmond S, Fulton L, Appelbaum E, Godfrey J et al (2010) Widespread divergence between incipient Anopheles gambiae species revealed by whole genome sequences. Science 330(6003):512–514
Linares M (1997) Origin of neotropical mimetic biodiversity from a threeway hybrid zone of Heliconius cydno butterflies. In: Ulrich H (ed) Tropical biodiversity and systematics. Proceedings of the international symposium on biodiversity and systematics in tropical ecosystems. Zoologisches Forschungsinstitut und Museum Alexander Koenig, Bonn, pp 93–108
Mallet J (1989) The genetics of warning colour in Peruvian hybrid zones of Heliconius erato and H. Melpomene. Proc R Soc B 236(1283):163–185
Mallet J (1999) Causes and consequences of a lack of coevolution in Müllerian mimicry. Evol Ecol 13(7–8):777–806
Mallet J (2005) Hybridization as an invasion of the genome. Trends Ecol Evol 20(5):229–237
Mallet J (2008) Hybridization, ecological races and the nature of species: empirical evidence for the ease of speciation. Phil Trans R Soc B Biol Sci 363(1506):2971
Mallet J (2009) Rapid speciation, hybridization and adaptive radiation in the Heliconius melpomene group. In: Butlin R, Bridle J, Schluter D (eds) Speciation and patterns of diversity. Cambridge University Press, Cambridge, UK, pp 177–194
Mallet J, Barton NH (1989) Strong natural selection in a warning-color hybrid zone. Evolution 43:421–431
Mallet J, Gilbert LE (1995) Why are there so many mimicry rings? Correlations between habitat, behaviour and mimicry in Heliconius butterflies. Biol J Linn Soc 55(2):159–180
Mallet J, Barton N, Lamas G, Santisteban J, Muedas M, Eeley H (1990) Estimates of selection and gene flow from measures of cline width and linkage disequilibrium in Heliconius hybrid zones. Genetics 124(4):921–936
Mallet J, McMillan WO, Jiggins CD (1998) Mimicry and warning color at the boundary between races and species. In: Endless forms: species and speciation. Oxford University Press, New York, pp 390–403
Mallet J, Beltrán M, Neukirchen W, Linares M (2007) Natural hybridization in heliconiine butterflies: the species boundary as a continuum. BMC Evol Biol 7(1):28
Martin A, Papa R, Nadeau NJ, Hill RI, Counterman BA, Halder G, Jiggins CD, Kronforst MR, Long AD, McMillan WO, Reed RD (2012) Diversification of complex butterfly wing patterns by repeated regulatory evolution of a Wnt ligand. Proc Natl Acad Sci USA 109:12632–12637
Martin SH, Dasmahapatra KK, Nadeau NJ, Salazar C, Walters JR, Simpson F, Blaxter MD, Manica A, Mallet J, Jiggins CD (2013) Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res 23(11)
Mather K (1950) The genetical architecture of heterostyly in Primula sinensis. Evolution 4:340–352
Mavárez J, Salazar CA, Bermingham E, Salcedo C, Jiggins CD, Linares M (2006) Speciation by hybridization in Heliconius butterflies. Nature 441(7095):868–871
McMillan WO, Jiggins CD, Mallet J (1997) What initiates speciation in passion-vine butterflies? Proc Natl Acad Sci USA 94(16):8628–8633
Melo MC, Salazar C, Jiggins CD, Linares M (2009) Assortative mating preferences among hybrids offers a route to hybrid speciation. Evolution 63(6):1660–1665
Merlin C, Beaver LE, Taylor OR, Wolfe SA, Reppert SM (2013) Efficient targeted mutagenesis in the monarch butterfly using zinc finger nucleases. Genome Res 23:159–168
Merrill RM, Gompert Z, Dembeck LM, Kronforst MR, McMillan WO, Jiggins CD (2011a) Mate preference across the speciation continuum in a clade of mimetic butterflies. Evolution 65(5):1489–1500
Merrill RM, Van Schooten B, Scott JA, Jiggins CD (2011b) Pervasive genetic associations between traits causing reproductive isolation in Heliconius butterflies. Proc R Soc B 278(1705):511–518
Merrill RM, Wallbank RW, Bull V, Salazar PC, Mallet J, Stevens M, Jiggins CD (2012) Disruptive ecological selection on a mating cue. Proc R Soc B 279(1749):4907–4913
Merrill RM, Naisbit RE, Mallet J, Jiggins CD (2013) Ecological and genetic factors influencing the transition between host-use strategies in sympatric Heliconius butterflies. J Evol Bio 26(9):1959--1967
Müller F (1879) Ituna and Thyridia: a remarkable case of mimicry in butterflies. Trans Entomol Soc Lond 1879:20–29
Nadeau NJ, Whibley A, Jones RT, Davey JW, Dasmahapatra KK, Baxter SW, Quail MA, Joron M, ffrench Constant RH, Blaxter ML, Mallet J, Jiggins CD (2012) Genomic islands of divergence in hybridizingHeliconius butterflies identified by large-scale targeted sequencing. Phil Trans R Soc B Biol Sci 367(1587):343–353
Naisbit RE (2001) Ecological divergence and speciation in Heliconius cydno and H. melpomene. PhD thesis, University of London
Naisbit RE, Jiggins CD, Mallet J (2001) Disruptive sexual selection against hybrids contributes to speciation between Heliconius cydno and Heliconius melpomene. Proc R Soc B 268(1478):1849–1854
Naisbit RE, Jiggins CD, Linares M, Salazar C, Mallet J (2002) Hybrid sterility, Haldane’s rule and speciation in Heliconius cydno and H. Melpomene. Genetics 161(4):1517–1526
Naisbit RE, Jiggins CD, Mallet J (2003) Mimicry: developmental genes that contribute to speciation. Evol Dev 5(3):269–280
Nielsen R (2009) Adaptionis – 30 years after Gould and Lewontin. Evolution 63(10):2487–2490
Nijhout HF (1991) The development and evolution of butterfly wing patterns. Smithsonian Institution Press, Washington, DC, USA
Nosil P (2012) Ecological speciation. OUP, Oxford
O’Neil ST, Dzurisin JD, Carmichael RD, Lobo NF, Emrich SJ, Hellmann JJ (2010) Population-level transcriptome sequencing of nonmodel organisms Erynnis propertius and Papilio zelicaon. BMC Genomics 11(1):310
Papa R, Kapan DD, Counterman BA, Maldonado K, Lindstrom DP, Reed R, Nijhout HF, Hrbek T, McMillan WO (2013) Multi-allelic major effect genes interact with minor effect QTLs to control adaptive color pattern variation in Heliconius erato. PLoS One. 8(3):e57033
Pardo-Diaz C, Salazar C, Baxter SW, Merot C, Figueiredo-Ready W, Joron M, McMillan WO, Jiggins CD (2012) Adaptive introgression across species boundaries in Heliconius butterflies. PLoS Genet 8(6):e1002752
Pohl N, Sison-Mangus M, Yee E, Liswi S, Briscoe A (2009) Impact of duplicate gene copies on phylogenetic analysis and divergence time estimates in butterflies. BMC Evol Biol 9(1):99
Quek SP, Counterman BA, Albuquerque de Moura P, Cardoso MZ, Marshall CR, McMillan WO, Kronforst MR (2010) Dissecting comimetic radiations in Heliconius reveals divergent histories of convergent butterflies. Proc Natl Acad Sci USA 107(16):7365–7370
Reed RD, Papa R, Martin A, Hines HM, Counterman BA, Pardo-Diaz C, Jiggins CD, Chamberlain NL, Kronforst MR, Chen R, Halder G, Nijhout HF, McMillan WO (2011) optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science 333(6046):1137–1141
Rieseberg LH (2009) Evolution: replacing genes and traits through hybridization. Curr Biol 19(3):R119–R122
Saenko SV, Brakefield PM, Beldade P (2010) Single locus affects embryonic segment polarity and multiple aspects of an adult evolutionary novelty. BMC Biol 8(1):111
Salazar C, Baxter SW, Pardo-Diaz C, Wu G, Surridge A, Linares M, Bermingham E, Jiggins CD (2010) Genetic evidence for hybrid trait speciation in Heliconius butterflies. PLoS Genet 6(4):e1000930
Schluter D (2009) Evidence for ecological speciation and its alternative. Science 323(5915):737–741
Seimiya M, Gehring WJ (2000) The Drosophila homeobox gene optix is capable of inducing ectopic eyes by an eyeless-independent mechanism. Development 127(9):1879–1886
Servedio MR, Doorn G, Kopp M, Frame AM, Nosil P (2011) Magic traits in speciation: ‘magic’ but not rare? Trends Ecol Evol 26(8):389–397
Sheppard PM, Turner JRG, Brown KS, Benson WW, Singer MC (1985) Genetics and the evolution of Muellerian mimicry in Heliconius butterflies. Philos Trans R Soc Lond Ser B Biol Sci 308(1137):433–610
Sivinski J (1989) Mushroom body development in nymphalid butterflies: a correlate of learning? J Insect Behav 2(2):277–283
Smadja C, Galindo J, Butlin R (2008) Hitching a lift on the road to speciation. Mol Ecol 17(19):4177–4180
Smith JM (1966) Sympatric speciation. Am Nat 100:637–650
Spencer KC (1988) Glycosides: the interface between plant secondary and insect primary metabolism. In: ACS symposium series. ACS Publications, Washington, DC, USA, vol 380, pp 403–416
Supple MA, Hines HM, Dasmahapatra KK, Nielsen DM, Lavoie C, Ray DA, Salazar C, McMillan WO, Counterman BA (2013) Genomic architecture of adaptive color pattern divergence and convergence in Heliconius butterflies. Genome Res. 23(8):1248--1257
Turner JR, Mallet JL (1996) Did forest islands drive the diversity of warningly coloured butterflies? Biotic drift and the shifting balance. Philos Trans R Soc Lond Ser B Biol Sci 351(1341):835–845
van’t Hof AE, Edmonds N, Dalková M, Marec F, Saccheri IJ (2011) Industrial melanism in British peppered moths has a singular and recent mutational origin. Science 332(6032):958–960
Werner T, Koshikawa S, Williams TM, Carroll SB (2010) Generation of a novel wing colour pattern by the Wingless morphogen. Nature 464(7292):1143–1148
Zhan S, Merlin C, Boore JL, Reppert SM (2011) The monarch butterfly genome yields insights into long-distance migration. Cell 147(5):1171–1185
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Supple, M., Papa, R., Counterman, B., McMillan, W.O. (2014). The Genomics of an Adaptive Radiation: Insights Across the Heliconius Speciation Continuum. In: Landry, C., Aubin-Horth, N. (eds) Ecological Genomics. Advances in Experimental Medicine and Biology, vol 781. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-7347-9_13
Download citation
DOI: https://doi.org/10.1007/978-94-007-7347-9_13
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-7346-2
Online ISBN: 978-94-007-7347-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)