
Abstract
Pancreatic β-cells are often referred to as “fuel sensors” as they continually monitor and respond to dietary nutrients, under the modulation of additional neurohormonal signals, in order to secrete insulin to best meet the needs of the organism. β-Cell nutrient sensing requires metabolic activation, resulting in production of stimulus-secretion coupling signals that promote insulin biosynthesis and release. The primary stimulus for insulin secretion is glucose, and islet β-cells are particularly responsive to this important nutrient secretagogue. It is important to consider individual effects of different classes of nutrient or other physiological or pharmacological agents on metabolism and insulin secretion. However, given that β-cells are continually exposed to a complex milieu of nutrients and other circulating factors, it is important to also acknowledge and examine the interplay between glucose metabolism and that of the two other primary nutrient classes, the amino acids and fatty acids. It is the mixed nutrient sensing and outputs of glucose, amino and fatty acid metabolism that generate the metabolic coupling factors (MCFs) involved in signaling for insulin exocytosis. Primary MCFs in the β-cell include ATP, NADPH, glutamate, long chain acyl-CoA and diacylglycerol and are discussed in detail in this article.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Pancreatic β-cells
- Insulin secretion
- Nutrient metabolism
- Incretins
- Signal transduction
- Stimulus-secretion coupling
- Gene expression
- Desensitization
Overview of β-Cell Stimulus-Secretion Coupling
Pancreatic β-cells are often referred to as “fuel sensors,” continually monitoring and responding to circulating nutrient levels, under the modulation of additional neurohormonal signals, in order to secrete insulin to best meet the needs of the organism. β-cell nutrient sensing involves notable metabolic activation, resulting in production of coupling signals that promote insulin biosynthesis and secretion. The primary stimulus of insulin secretion is glucose, and islet β-cells are particularly responsive to this important nutrient secretagogue, coupling metabolic and other stimuli with the insulin-secretory machinery. In writing this chapter we are fully aware that most of the studies cited have utilized rat-, mouse- or hamster-derived insulinoma β-cell lines to study function in vitro. This is due to the inherent difficulty in maintaining primary rodent islet β-cell mass and function for more than a few days in vitro and of course the scarcity of human islets for research purposes. The generation of functional and stable human β-cell lines has also proved difficult. Recently, three novel insulin-secreting human β-cell lines have been generated and deposited at European Collection of Cell Cultures (ECACC) although their full characterization is still in its infancy (McCluskey et al. 2011; Guo-Parke et al. 2012). Nevertheless, the major rodent β-cell lines have provided substantial data and insights into cell function in normal or pathogenic situations. The most widely used cell lines include INS 1, MIN 6, RINm5F and BRIN-BD11. It is important to state that in vivo intact islet structures (comprising α, β and δ-cells, which secrete glucagon, insulin and somatostatin, respectively) are required to maintain appropriate and pulsatile hormone secretion in response to nutrient stimuli.
Elevation in blood glucose concentrations results in rapid rises in intracellular glucose levels as glucose is transported across the β-cell plasma membrane. Glucose uptake and metabolism are two essential steps in the so-called “glucose-stimulated insulin secretion” (GSIS) pathway. GSIS represents the increase in insulin secretion over basal release in response to increased extracellular, and ultimately intracellular, glucose. As illustrated in Fig. 1, glucose rapidly enters β-cells, through specific glucose transporters (GLUT-1 in humans; GLUT-2 in rodents), after which it is swiftly phosphorylated by the enzyme glucokinase, which has a high Km for glucose. These primary steps, particularly glucokinase, determine the rate of glucose utilization by the β-cell over a range of physiological glucose levels (3–20 mM) and the combination of transport and phosphorylation determines metabolic flux through glycolysis.

Metabolic stimulus-secretion coupling in the β-cell. Glucose metabolism results in an enhanced cytoplasmic ATP/ADP ratio, which prompts closure of ATP-sensitive K+ (K ATP ) channels in the plasma membrane evoking membrane depolarization, and subsequent opening of voltage-gated Ca2+ channels. This culminates in an increase in cellular Ca2+ influx – a primary driver of the GSIS mechanism. Ca2+ and vesicle docking and fusion events can also be modulated by agents acting through the phospholipase C (PLC)/protein kinase C (PKC) or adenylate cyclase (AC)/protein kinase A (PKA) pathways
Increased β-cell glycolytic flux results in a rapid increase in production of reducing equivalents, an increased activity of shuttle mechanisms (responsible for transferring electrons to the mitochondrial matrix), and TCA cycle activity, leading to increased ATP production in mitochondria. The outcome is an enhanced cytoplasmic ATP to ADP ratio, which prompts closure of ATP-sensitive K+ (KATP) channels in the plasma membrane evoking membrane depolarization, and subsequent opening of voltage-gated Ca2+ channels (Fig. 1). This culminates in an increase in cellular Ca2+ influx – a primary driver of the GSIS mechanism (Straub and Sharp 2002). Ca2+ and vesicle docking and fusion events can also be modulated by agents acting through phospholipase C (PLC)/protein kinase C (PKC) or adenylate cyclase (AC)/protein kinase A (PKA) pathways as shown in Fig. 1.
Importantly, nutrients (including amino acids and lipids), insulinotropic drugs (including the sulphonylureas), or neurohormonal signals (including incretin hormones and autonomic innervation) can markedly affect glucose-stimulated insulin secretion (Fig. 2). Much interest has revolved around acute enhancement of β-cell function by the two incretin hormones, glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), currently being hailed as important new therapeutics for type 2 diabetes (for review (Green and Flatt 2007)). These two new classes of therapeutic agent, along with their receptor agonists (e.g. exenatide, liraglutide and GIP fatty acid derivatives), could offer considerable advantages over sulphonylureas and other insulinotropic drugs, as their insulin-secretory action is glucose- and/or nutrient-dependent (Green and Flatt 2007; Peterson 2012). The incretin mimetics may also play a role in maintaining β-cell mass in the hostile type 2 diabetes environment (Green and Flatt 2007). Moreover, there is a parallel strategy to overcome the rapid local degradation and short plasma half-lives of GLP-1 and GIP through development of therapeutic dipeptidyl peptidase-4 (DPP IV) inhibitors that prevent DPP IV-mediated cleavage of GLP-1 and GIP. However, the latter strategy relies on the endogenous production of GLP-1 and GIP to elicit the insulinotropic response, which is perhaps less elegant than the use of stable engineered incretin mimetics. Also, GLP-1 agonists would appear to stimulate a greater reduction in postprandial glucose, body weight and glycated haemoglobin (A1C) than DPP IV inhibitors alone, and modulation of their structure with lipophilic chains extends plasma half-life by allowing interaction with serum albumin (Peterson 2012; Reid 2012). So, while GLP-1, GIP and related-receptor agonists possess great potential as a new generation of anti-diabetic agents, as yet they are not considered primary therapies for maintenance of glycaemic control in the majority of diabetic individuals (Reid 2012).

Modulation of insulin secretion by insulinotropic drugs and incretins . In the presence of glucose, insulinotropic drugs (including the sulphonylureas), or neurohormonal signals (including the two incretin hormones, glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) and autonomic innervation) can markedly affect insulin secretion via modulation of signal transduction and/or ion channel activity. Sulphonylurea drugs mainly act via promoting closure of the KATP channel and thus membrane depolarization. GLP-1 and GIP mediate their effects through G-protein-coupled receptors and associated signal transduction pathways which include serine/threonine kinase activation and phosphorylation of proteins associated with the molecular mechanism of exocytosis
It is convenient to also consider individual effects of different classes of nutrient or other physiological or pharmacological agents on metabolism and insulin secretion. However, given that β-cells are continually exposed to a complex milieu of nutrients and other circulating factors, it is important to also acknowledge and examine the interplay between glucose metabolism and that of the two other primary nutrient classes, the amino acids and fatty acids. Cumulatively, it is the mixed nutrient sensing and outputs of glucose, amino and fatty acid metabolism that generate the metabolic coupling factors (MCFs) involved in signaling for insulin exocytosis (Charles and Henquin 1983; Newsholme et al. 2007a). Primary MCFs in the β-cell include ATP, NADPH, glutamate, long-chain acyl-CoA and diacylglycerol (DAG) and are discussed further below.
Phases and Pulsatility of Insulin Secretion
Tight regulation of insulin secretion is necessary for glucose homeostasis, where disturbances are associated with glucose intolerance and diabetes. However, glucose stimulated insulin secretion is under stimulatory and inhibitory control by hormones and neurotransmitters and regular oscillations of circulating insulin in normal subjects can even occur without accompanying changes in plasma glucose – a response related to the so-called “pacemaker” function of the pancreas (for review (Tengholm and Gylfe 2009)). Pulsatile insulin secretion from individual islets appears to follow a dominating pancreatic frequency, where a rhythmic variation in islet secretion is synchronized with oscillations in β-cell cytoplasmic Ca2+. In clusters of β-cells exposed to intermediate stimulatory concentrations of glucose, synchronized oscillations can spread to silent cells as the glucose concentration is increased (Tengholm and Gylfe 2009). This demonstrates β-cell recruitment and intracellular coupling in glucose regulation of insulin secretion.
While foetal islets demonstrate a monophasic (first phase) secretory response, in mature adult islets, insulin secretion occurs very rapidly after glucose administration, and is reported to occur with precise and biphasic kinetics (Fig. 3). Over the years there has been debate as to the underlying mechanisms regulating this biphasic pattern of insulin secretion, which has been proposed to involve at least two signaling pathways, the so-called KATP channel-dependent pathway, noted above, and another KATP channel-independent pathway. While both phases would appear to be critically dependent on Ca2+ influx, and can be modulated by various agents (including sulphonylureas and incretin hormones; Fig. 2), they affect different pools of insulin-secretory granules. It is understood that whereas the KATP-dependent pathway prompts exocytosis of an “immediately releasable pool” of granules that elicits and represents the first phase response, the KATP channel-independent pathway, working in synergy with the KATP-dependent pathway plays an important role the second phase response (see review (Straub and Sharp 2002)).

Biphasic insulin secretion. Insulin secretion from islet β-cells occurs very rapidly after glucose administration, and is reported to occur with precise and biphasic kinetics. (i) First phase insulin secretion which is dependent on ATP generation and a rise in intracellular Ca2+. (ii) Second phase insulin secretion which is dependent on mitochondrial metabolism and a rise in intracellular Ca2+
Primary Metabolic Factors Regulating Glucose-Stimulated Insulin Secretion
Clearly glucose metabolism plays a central role in the regulation of β-cell function, and glucose-derived carbons are understood to be metabolized following three main pathways generating MCFs for activation of insulin exocytosis: (i) glycolysis followed by TCA cycle-dependent glucose oxidation; (ii) anaplerosis; (iii) provision of glycerol-3-phosphate (Gly3P) for glycerolipid/fatty acid (GL/FA) cycling (see review (Nolan and Prentki 2008)). While the first two of these pathways are linked to GSIS, there is less clarity regarding the latter, though Gly3P is believed to be incorporated into GL and GL/FA cycling which could produce lipid-signaling MCFs for insulin secretion.
Products of glucose metabolism can activate isoforms of PLC, promoting the generation of 1,4,5 inositol-triphosphate (IP3) and a plasma membrane associated pool of DAG, a potent activator of specific isoforms of protein kinase C (Newsholme et al. 2007a; Nolan and Prentki 2008), which can help mediate insulin-secretory granule trafficking and exocytosis (Newsholme et al. 2007a; Nolan and Prentki 2008) (Fig. 1). Inositol-triphosphate (IP3) stimulates Ca2+ efflux from the endoplasmic reticulum and increases Ca2+ concentration in the cytosol, also favouring activation of the secretory mechanism. Other glucose-derived MCFs such as ATP, LC-acylCoA and DAG can also amplify insulin secretion, and Gly3P metabolism through GL/FA cycling can produce nutrient-derived MCFs not dependent on mitochondrial metabolism. It has been suggested that extracellular externalization of ATP within insulin vesicles stimulates localized plasma membrane accumulation of DAG that is spatially restricted (Wuttke et al. 2013). Since DAG interacts with protein kinase C as outlined above, external ATP levels generated by stimulated β-cells may lead to sustained release of insulin, via a feedback mechanism involving association with the purinoreceptor P2Y1, and this may occur in either a paracrine, or possibly autocrine fashion (Wuttke et al. 2013). In addition, various amino acids and their metabolic products may also impact on GSIS by a combination of enhancement of glucose oxidation, anaplerosis, and direct plasma membrane depolarization effects (Brennan et al. 2002; Smith et al. 1997; Dixon et al. 2003; Sener and Malaisse 1980).
Pancreatic β-cell glucose metabolism also increases arachidonic acid (AA) production, mainly by activation of phospholipase A2 (Keane and Newsholme 2008). The AA metabolites, prostaglandins (PGs) and leukotrienes, would appear to provide respective positive and negative modulation of glucose-stimulated insulin secretion (Keane and Newsholme 2008). While it would appear that free fatty acids do not stimulate insulin secretion in the absence of glucose, there is a substantial body of evidence to indicate that they are essential for GSIS (Salehi et al. 2005). Recent reports utilizing human islets suggest that non-esterified AA is critical for normal pancreatic β-cell function. Inhibition of the release of endogenous AA by inhibiting PLA2 activity resulted in a significant reduction of GSIS perhaps acting via G-protein-coupled receptor(s) (Persaud et al. 2007). Furthermore, AA demonstrated a regulatory and protective role in the BRIN-BD11 β-cell line (Keane et al. 2011). It increased expression of genes involved in cell proliferation and fatty acid metabolism (e.g. cyclo-oxygenase I and II), while reducing the expression of pro-inflammatory factors induced by the saturated fatty acid palmitate. These included iNOS (inducible NO synthase), NF-kB (nuclear factor k B) and NOX (NADPH oxidase), suggesting that that AA may prove to be beneficial in a lipotoxic environment such as is observed in diabetic patients.
Increases and oscillations in the intracellular Ca2+ concentration associated with the mechanism of GSIS can stimulate mitochondrial generation of ROS (via electron transport chain activity), whereas Ca2+, via PKC activation and subsequent phosphorylation/translocation of the cytosolic regulatory subunit P47phox, may enhance β-cell NOX-dependent generation of ROS (Kruman et al. 1998; Yu et al. 2006; Morgan et al. 2007). The O2 − and H2O2 so produced, acutely stimulate (Morgan et al. 2009), but chronically induce, inhibitory effects on β-cell metabolic pathways, and can promote KATP channel opening, with resulting inhibitory effects on insulin secretion (Nakazaki et al. 1995).
Metabolically-active pancreatic β-cells have inherently relatively low levels of free radical detoxifying and redox-regulating enzymes, such as, glutathione reductase, glutathione peroxidase, catalase and thioredoxin, rendering them vulnerable to damage and destruction. The consequence of limited scavenging systems is that upon Ca2+ stimulation of mitochondrial and NADPH oxidase systems, ROS concentrations in β-cells may increase rapidly, and to high levels. Given this, the β-cell, while utilizing necessary and positive aspects of ROS production for insulin production and release (Morgan et al. 2009; Newsholme et al. 2012), is susceptible to the damaging effects of unregulated ROS generation and accumulation. The following sections give an overview of both positive actions of a range of important nutrient regulators of β-cells together with insights into mechanisms underlying nutrient-induced desensitization and toxicity associated with prolonged exposure.
Investigating Nutrient Regulation of β-Cell Metabolism and Insulin Secretion
Many insights into the mechanisms regulating insulin production and secretion have been gleaned from studies of freshly isolated islets, constituent β-cells and increasingly bioengineered β-cell lines. While early insulin-secreting cell lines, such as RINm5F and HIT-T15, represented rather crude β-cell models, advances in molecular biology and emerging bioengineering technologies offer considerable opportunities to improve and establish more appropriate clonal β-cells (see (McClenaghan 2007)). Indeed, bioengineered pancreatic β-cells, such as the popular glucose-responsive pancreatic BRIN-BD11 cells (McClenaghan et al. 1996a), have helped facilitate studies of the mechanisms of β-cell metabolism, insulin secretion, cell dysfunction and destruction. Combining the attributes of long-term functional stability of BRIN-BD11 cells with state-of-the-art NMR approaches have enabled the authors to unravel complexities, and provide novel insights into the relationships between glucose, fatty acid and amino acid handling and insulin secretion (Brennan et al. 2002, 2003; McClenaghan 2007). The following sections give a brief overview of the complex mechanisms regulating nutrient-stimulated insulin secretion and gene expression by pancreatic β-cells in response to various stimuli, utilizing isolated islet β-cells and other insulin-secreting cells.
Mechanisms Underlying β-Cell Actions of Glucose
As noted earlier, glucose is a primary physiological β-cell fuel, stimulating insulin secretion as a result of its metabolism and generation of MCFs. As illustrated in Fig. 4, after internalization through membrane-associated transporters, glucose is rapidly metabolized to pyruvate, following initial phosphorylation by glucokinase (GK) to glucose 6-phosphate, and subsequent glycolytic reactions. The third reaction in glycolysis is catalysed by phosphofructokinase (PFK), itself a key β-cell metabolic control site, and fluctuations in its activity result in oscillations in glycolytic flux (Nielsen et al. 1997, 1998; Westermark and Lansner 2003). The end product of glycolysis, pyruvate, is metabolized by either pyruvate dehydrogenase (PDH; the glucose oxidation pathway) or pyruvate carboxylase (PC; the anaplerosis/cataplerosis pathway) to acetyl-CoA or oxaloacetate, respectively, resulting in enhanced mitochondrial tricarboxycylic acid (TCA) cycle activity (Fig. 4).

Mechanisms of glucose and fatty acid enhanced mitochondrial activity and ATP generation. The end product of glycolysis – pyruvate – is metabolized by either pyruvate dehydrogenase (PDH; so committing glucose to the oxidation pathway) or pyruvate carboxylase (PC; so committing glucose to the anaplerosis/cataplerosis pathway). The products will be acetyl-CoA or oxaloacetate, respectively, which will contribute to enhanced mitochondrial tricarboxycylic acid (TCA) cycle activity. The malate-aspartate shuttle transfers cytosolic NADH to the mitochondrial matrix, a process which requires aspartate-glutamate exchange across the mitochondrial inner membrane by Aralar1. The generation of ATP and the increase in intracellular Ca2+ drives insulin secretion. Fatty acids may potentiate insulin secretion via generation of LC acyl-CoA and stimulation of signal transducing events
Among the enzymes responsible for glucose metabolism, GK, PC and PDH appear to play particularly important regulatory roles in the insulin-secretory pathway (Fig. 4). In β-cells, PC activity is high even though the cell does not participate in gluconeogenesis (MacDonald 1995a), which suggests this enzyme exerts anaplerotic functions. Note that β-cells lack phosphoenolpyruvate carboxykinase (an essential enzyme for gluconeogenesis, converting oxaloacetate to phosphoenol pyruvate) (MacDonald 1995b). A recent study has highlighted that siRNA targeted to PC resulted in a reduction of insulin secretion from INS-1 cells (Xu et al. 2008), consistent with the observation that PC activity may be reduced in type 2 diabetes (MacDonald et al. 1996). Furthermore, these researchers showed that PC expression was elevated in the islets of mildly hyperglycaemic mice, while it was reduced in severely hyperglycaemic mice. The authors suggested that the increased levels of PC may possibly correlate to an adaptive response of β-cells to insulin resistance, with reduced PC expression corresponding to β-cell failure (Han and Liu 2010). Interestingly, overexpression of PC in INS-1 cells resulted in increased insulin release (Xu et al. 2008), again supporting an important role for this enzyme in the maintenance of GSIS. However, inhibition of PDH by overexpression of PDH kinase 4 in INS-1 cells did not result in a decrease of insulin secretion (Xu et al. 2008), although it is important not to over-interpret this observation by dismissing an important regulatory role of pyruvate dehydrogenase.
Transfer of electrons from TCA cycle to the mitochondrial electron transport chain is mediated by NADH and FADH2 formation, resulting in ATP generation (Fig. 4). The increase in intracellular ATP to ADP ratio leads to the characteristic closure of KATP channels (Cook and Hales 1984), membrane depolarization, opening of voltage gated Ca2+ channels and rapid rise in intracellular Ca2+ concentration, leading to mobilization and ultimately fusion of insulin-containing granules with the plasma membrane and insulin release (Fig. 4) (Tarasov et al. 2004; Wiederkehr and Wollheim 2006). The primary actions of glucose are mediated by potentiation of ATP concentration by enhanced TCA cycle substrate (oxidative and anaplerotic) supply. Generation of other additive factors derived from glucose metabolism might also be promoted by mitochondrial Ca2+ elevation (Maechler et al. 1997).
Pyruvate may be converted in the β-cell to both acetyl-CoA and oxaloacetate, as discussed above (Fig. 4). A number of possibilities for mitochondrial metabolism of pyruvate exist: (i) generation of CO2 via TCA cycle activity; (ii) export from the mitochondria as glutamate (due to 2-oxoglutarate conversion to glutamate via transamination or glutamate dehydrogenase activity); (iii) export from the mitochondria as malate to be converted back to pyruvate by NADP+-dependent malic enzyme; and (iv) export from the mitochondria as citrate to be acted on by ATP citrate lyase and subsequently acetyl CoA carboxylase (Carpentier et al. 2000) to form malonyl-CoA which is an inhibitor of carnitine palmitoyl transferase-1 and thus an inhibitor of fatty acid oxidation (malonyl-CoA can subsequently be used for fatty acid synthesis via the action of fatty acid synthase).
One of these pathways, the so-called pyruvate-malate cycle, predicts a role for malate in insulin secretion via generation of the stimulus–secretion coupling factor NADPH (Jensen et al. 2008). Flow of the cycle requires oxaloacetate derived from pyruvate (via PC) to be converted to malate via a reversal of the malate dehydrogenase reaction, consuming NADH and generating NAD+ in the mitochondrial matrix. Following this, malate is exported to the cytosol, converted to pyruvate via NADP+-dependent malate dehydrogenase, generating NADPH (Newsholme et al. 2007b). Glucose stimulation of β-cells or isolated rodent islet cells increases malate levels (Jensen et al. 2006; Macdonald 2003) and while the workings of the malate–pyruvate cycle (recently reviewed (Jensen et al. 2008)) are known, concerns have been raised as to the operation and impact of this cycle under physiologic conditions.
Normal TCA cycle activity ensures the malate → oxaloacetate direction of flux, thus generating NADH in the mitochondrial matrix. In addition, malate → oxaloacetate conversion forms part of the malate-aspartate shuttle, which has a high activity in the β-cell, and is essential for transfer of cytosolic NADH to the mitochondrial matrix (for review (Bender et al. 2006)). In β-cells, reducing equivalents may be transported to the mitochondrial matrix by either the glycerol-phosphate or the malate-aspartate shuttle (Eto et al. 1999). Inhibition of the malate–aspartate shuttle by amino–oxyacetate (which acts on transamination reactions and inhibits cytosolic NADH reoxidation) has been demonstrated to attenuate the secretory response to nutrients, thus highlighting the dominance of this latter shuttle in the β-cell. In addition, Aralar1 (see Fig. 4), a mitochondrial aspartate–glutamate carrier which takes part in the malate–aspartate shuttle, has been demonstrated to play an important role in glucose-induced insulin secretion, as its deletion in INS-1 cells leads to a complete loss of malate–aspartate shuttle activity in mitochondria, and to a 25 % decrease of insulin release in response to glucose (Marmol et al. 2009). Moreover, overexpression of Aralar1 in BRIN-BD11 cells, enhances GSIS and amino acid-stimulated insulin secretion, while increasing glycolytic capacity (Bender et al. 2009).
One key constituent of the malate-aspartate NADH shuttle is the mitochondrial aspartate–glutamate transporter, with its two Ca2+-sensitive isoforms, Citrin and Aralar1, expressed in excitatory tissues (Rubi et al. 2004; del Arco and Satrustegui 1998). However, Aralar1 is the dominant aspartate–glutamate transporter isoform expressed in β-cells (Rubi et al. 2004), and the function of this transporter in the malate-aspartate shuttle is illustrated in Fig. 4. Adenoviral-mediated overexpression of Aralar1 in INS-1E β-cells and rat pancreatic islets enhanced glucose-evoked NAD(P)H generation, electron transport chain activity and mitochondrial ATP formation, and Aralar1 was demonstrated to exert its effect on insulin secretion upstream of the TCA cycle (Rubi et al. 2004). Indeed, the capacity of the aspartate-glutamate transporter appeared to limit NADH shuttle activity and subsequent mitochondrial metabolism. Thus, it is highly improbable that a malate-pyruvate cycle is active and important to insulin secretion, if the malate–aspartate shuttle is indeed a key component of stimulus–secretion coupling.
An alternative pyruvate-cycling pathway has been proposed, where generation of citrate from condensation of oxaloacetate (OAA) and acetyl-CoA occurs in the TCA cycle, followed by export of citrate from the mitochondria via the citrate–isocitrate carrier, cleavage of citrate by ATP citrate lyase to OAA and acetyl-CoA, and recycling to pyruvate via a cytosolic malate dehydrogenase and NADP+-dependent malic enzyme (Jensen et al. 2008). In this proposed cycle, OAA to malate formation occurs in the cytosol, similar to the malate–aspartate shuttle. Acetyl-CoA can also serve a substrate for acetyl-CoA carboxylase, leading to formation of long-chain acyl-CoA accumulation in the cytosol via malonyl-CoA and in β-cells, glucose stimulation increases malonyl-CoA levels before insulin release (Corkey et al. 1989), and addition of long-chain acyl-CoA results in a stimulation of insulin secretion (Deeney et al. 2000).
However, the evidence used to refute a role of fatty acid synthesis is not compelling. Suppression of citrate lyase mRNA levels by 92 % and citrate lyase protein levels by 75 % by adenovirus-mediated siRNA delivery did not affect GSIS in 832/13 β-cells compared with cells treated with a control adenovirus (Joseph et al. 2007). Also, citrate lyase suppression in primary islet preparations using recombinant adenovirus technology to suppress citrate lyase expression by 65 % reported no impact on GSIS (Joseph et al. 2007). It is possible to reinterpret these findings if we consider that citrate lyase is expressed at very high levels in β-cells (Roche et al. 1998), so suppression of protein expression, even by 65–75 %, would not be expected to be sufficient to alter the synthesis of key LC-acyl-CoA species.
Mechanisms Underlying β-Cell Actions of Lipids
Fatty acids appear to freely diffuse into pancreatic β-cells through the plasma membrane (Hamilton and Kamp 1999). As illustrated in Fig. 4, inside β-cells, fatty acids are transformed in long-chain acyl-CoA, by acyl-CoA synthase (ACS), and enter the mitochondria via Carnitine Palmitoyl Transferase 1 (CPT-1), so β-oxidation can occur when glucose levels are low. The resulting acetyl-CoA is subsequently oxidized in the TCA cycle and under these conditions, ATP generation is sufficient for β-cell survival, and to maintain basal levels of insulin secretion (Fig. 4). When the extracellular glucose concentration is increased, fatty acid oxidation is inhibited, due to formation of malonyl-CoA by acetyl-CoA carboxylase (Carpentier et al. 2000). Malonyl-CoA under glucose stimulatory conditions is derived from glucose carbon, via formation of citrate. Malonyl-CoA inhibits CPT-1, thus blocking transport of long-chain acyl-CoA into the mitochondria (Prentki et al. 2002) (Fig. 4). Accumulation of long-chain acyl-CoA in the cytosol leads to an increase of intracellular Ca2+ levels and to changes in acylation state of proteins involved both in regulation of ion channel activity and exocytosis (Keane and Newsholme 2008; Yaney and Corkey 2003; Haber et al. 2006). In addition, long-chain acyl-CoA can also enhance fusion of insulin-secretory vesicles with plasma membrane and insulin release (Deeney et al. 2000).
However, effects of fatty acids on glucose-induced insulin secretion are directly correlated with chain length and the degree of unsaturation, where long-chain fatty acids (such as palmitate or linoleate) acutely improve, but chronically reduce insulin release in response to glucose stimulation (Newsholme et al. 2007a). It is possible that chronic elevated synthesis of triacylglycerol species such as tripalmitin is detrimental to β-cell function due to adverse morphological changes (Moffitt et al. 2005) but it is more likely that apoptosis is triggered by lipid-specific signaling pathways and/or endoplasmic reticulum stress-activated pathways, so resulting in β-cell failure and death (reviewed in (Newsholme et al. 2007a)). A study by the authors demonstrated that 24 h culture of BRIN-BD11 cells with the polyunsaturated fatty acid, arachidonic acid (AA), increased insulin secretion in response to the amino acid l-alanine. On the other hand, 24 h exposure of BRIN-BD11 cells to saturated fatty acid palmitic acid in culture inhibited l-alanine-induced insulin secretion (Dixon et al. 2004). Interestingly, AA exhibited a protective function in the BRIN-BD11 β-cell line by preventing the detrimental effects of palmitic acid (Keane et al. 2011).
A recent advance in the understanding of the mechanism(s) by which non-esterified fatty acids (NEFAs) modulate insulin secretion in vivo was the discovery of high levels of expression of the membrane-bound G-protein-coupled receptor GPR40, a putative NEFA receptor in human and animal islet β-cell preparations (Tomita et al. 2006). GPR40 mRNA levels positively correlated with the insulinogenic index (Tomita et al. 2006). Furthermore, it has also been demonstrated that omega-3 fatty acids can interact with the GPR120 receptor, and mediate insulin-sensitisation and anti-inflammatory effects in obese mice models (Oh et al. 2010). Other G-protein-coupled receptors that may be important in islet physiology are GPR41 and GPR119 (Oh and Lagakos 2011; Nguyen et al. 2012). However, while the potential signaling mechanism(s) by which G-protein-coupled receptors regulates insulin secretion are still under investigation, it appears likely that they involve changes in intracellular Ca2+ mobilization (Salehi et al. 2005; Itoh and Hinuma 2005; Shapiro et al. 2005; Newsholme and Krause 2012).
Mechanisms Underlying β-Cell Actions of Amino Acids
Under appropriate conditions, amino acids enhance insulin secretion from primary islet cells and β-cell lines (Charles and Henquin 1983; Brennan et al. 2002; Smith et al. 1997; Dixon et al. 2003; Sener and Malaisse 1980). In vivo, l-glutamine and l-alanine are quantitatively the most abundant amino acids in blood and extracellular fluids, closely followed by the branched chain amino acids (Blau et al. 2003). However, individual amino acids do not evoke insulin-secretory responses in vitro when added at physiological concentrations, rather, combinations of physiological concentrations of amino acids or high concentrations of individual amino acids are much more effective. In vivo, amino acids derived from dietary proteins and those released from intestinal epithelial cells, in combination with glucose, stimulate insulin secretion, thereby leading to protein synthesis and amino acid transport in target tissues such as skeletal muscle (Keane and Newsholme 2008).
While amino acids can potentially affect a number of aspects of β-cell function, a relatively small number of amino acids promote or synergistically enhance insulin release from pancreatic β-cells (Fajans et al. 1967; McClenaghan et al. 1996b). As illustrated in Fig. 5, the mechanisms by which amino acids enhance insulin secretion are understood to primarily rely on: (i) direct depolarization of the plasma membrane (e.g., cationic amino acid, l-arginine); (ii) metabolism (e.g., glutamine, leucine); and (iii) co-transport with Na+ and cell membrane depolarization (e.g., l-alanine). Notably, partial oxidation, e.g., l-alanine (Brennan et al. 2002) may also initially increase the cellular content of ATP impacting on KATP channel closure prompting membrane depolarization, Ca2+ influx and insulin exocytosis.

Common mechanisms of nutrient-stimulated insulin secretion. Glucose metabolism is essential for stimulation of insulin secretion. The mechanisms by which amino acids enhance insulin secretion are understood to primarily rely on (i) direct depolarization of the plasma membrane (e.g., cationic amino acid, l-arginine); (ii) metabolism (e.g., alanine, glutamine, leucine); and (iii) co-transport with Na+ and cell membrane depolarization (e.g., alanine). Notably, rapid partial oxidation may also initially increase both the cellular content of ATP (impacting on KATP channel closure prompting membrane depolarization) and other stimulus–secretion coupling factors. In the absence of glucose, fatty acids may be metabolized to generate ATP and maintain basal levels of insulin secretion
Additional mitochondrial signals that affect insulin secretion may also be generated (Fig. 6) (Dukes et al. 1994; Malaisse-Lagae et al. 1982; Maechler 2002), and in β-cells, the mTOR-signaling pathway acts in synergy with growth factor/insulin signaling to stimulate mitochondrial function and insulin secretion (Kwon et al. 2004). At present, the mechanism by which amino acids activate the mTOR complex has not been fully elucidated, and recent publications have suggested that amino acids can activate both mTOR1 and mTOR2 complexes (Tato et al. 2011). Furthermore, recent data suggests that amino acids regulate mTOR signaling via class I PI3K enzymes, in addition to the already established class III PI3K enzyme (hVps34). This novel signaling mechanism may impact on a variety of conditions that display differences in nutrient processing such as that observed in diabetes and cancer (Tato et al. 2011). However, given that amino acid nutrients may play a role in the pathophysiological of many disorders, it is interesting to speculate involvement of kinase stimulation or inhibition of a phosphatase utilizing mTOR as a substrate (Kwon et al. 2004; McDaniel et al. 2002; Briaud et al. 2003).

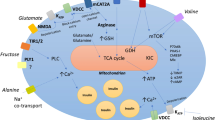
Glucose, alanine, glutamine, leucine and arginine are the major nutrient drivers of insulin secretion. Metabolism of glucose, alanine and glutamine result in enhanced TCA cycle activity and generation of metabolic secretion coupling factors including ATP, Ca2+ and glutamate. Leucine may enhance glutamine oxidation via activation of glutamate dehydrogenase (GDH). Arginine may depolarize the plasma membrane by net import of positive charge thus causing opening of voltage-gated Ca2+ channels. The key sites of metabolic control in the β-cell are indicated; glucokinase (GK), pyruvate dehydrogenase (PDH), pyruvate carboxylase (PC), glutamate dehydrogenase (GDH)
Arginine: This amino acid stimulates insulin release through electrogenic transport into the β-cell via the mCAT2A amino acid transporter (Fig. 6), thereby increasing membrane depolarization, rise in intracellular Ca2+ through opening of voltage-gated Ca2+ channels and insulin secretion (Sener et al. 2000). However, in some situations, arginine principally through its metabolism is understood to exert a negative effect on β-cell insulin release.
The potentially detrimental effect of arginine metabolism hinges on arginine-derived nitric oxide (NO) through the action of inducible nitric oxide synthase (iNOS) (McClenaghan et al. 2009). High levels of NO are known to interfere with β-cell mitochondrial function and generation of key stimulus–secretion coupling factors, which could lead to a reduction in cellular insulin output (Newsholme and Krause 2012; McClenaghan et al. 2009).
Glutamine: Among the amino acids, glutamine is considered one of the most important, playing an essential role in promotion and maintenance of functionality of various organs and cells, including pancreatic β-cells (Curi et al. 2005). Both rat islets and BRIN-BD11 cells consume glutamine at high rates (Dixon et al. 2003), but notably while glutamine can potentiate GSIS and interact with other nutrient secretagogues, it does not initiate an insulin-secretory response (McClenaghan et al. 1996b). In rat islets, glutamine is converted to y-amino butyric acid (GABA) and aspartate (Fig. 6), and in the presence of leucine oxidative metabolism is increased. Previously, it was shown that the potential glutamine synthetase inhibitor – methionine sulfoximide – completely abolished GSIS in normal mouse islets (Li et al. 2004), a phenomenon reversed by addition of glutamine or a non-metabolizable analogue. However, it is important to note that this inhibitor may block a number of glutamate-utilizing enzymes and so the outcome cannot be interpreted to arise as a result of a specific action on glutamine synthetase.
Glutamate: The ability of glutamate to stimulate insulin secretion and its actions in β-cells has been hotly debated. Intracellular generation of l-glutamate has been proposed to participate in nutrient-induced stimulus-secretion coupling as an additive factor in the amplifying pathway of GSIS (Maechler and Wollheim 1999). During glucose stimulation, total cellular glutamate levels have been demonstrated to increase in human, mouse and rat islets, as well as clonal β-cells (Brennan et al. 2002; Dixon et al. 2003; Maechler and Wollheim 1999; Broca et al. 2003), whereas other studies have reported no change (Danielsson et al. 1970; MacDonald and Fahien 2000). The observation that mitochondrial activation in permeabilized β-cells directly stimulates insulin exocytosis (Maechler et al. 1997) pioneered the identification of glutamate as a putative intracellular messenger (Maechler and Wollheim 1999; Hoy et al. 2002). However, in recent years, the role of l-glutamate in direct actions on insulin secretion has been challenged (MacDonald and Fahien 2000; Bertrand et al. 2002). For example, stimulatory (16.7 mM) glucose did not increase intracellular l-glutamate concentrations in rat islets in one study (MacDonald and Fahien 2000), and while l-glutamine (10 mM) increased the l-glutamate concentration tenfold, this was not accompanied by a stimulation of insulin release. In a separate study, incubation with glucose resulted in a significant increase in l-glutamate concentration in depolarized mouse and rat islets, but l-glutamine while increasing l-glutamate content did not alter insulin secretion (Bertrand et al. 2002). Additionally, in this latter study, BCH-induced activation of GDH lowered l-glutamate levels, but increased insulin secretion. However, it is probable that experimental conditions in which l-glutamine is used as l-glutamate precursor may lead to saturating concentrations of l-glutamate without necessarily activating the KATP-dependent pathway and associated increase in insulin secretion (Broca et al. 2003). It is likely that during enhanced glucose metabolism, the concentration of the key TCA cycle intermediate α-ketoglutarate (2-oxoglutarate) is elevated and a proportion of this metabolite is subsequently transaminated to glutamate (Brennan et al. 2003). It is the opinion of the authors that the glutamate so formed may indirectly stimulate insulin secretion through additive actions on the malate-aspartate shuttle (as glutamate is a substrate for the mitochondrial membrane aspartate/glutamate carrier 1, thus may increase the capacity of the shuttle, see Fig. 4) or by contribution to glutathione synthesis (as glutamate is one of the three amino acids required for glutathione synthesis) and subsequent positive effects on cellular redox state and mitochondrial function (for further detail see (Brennan et al. 2003)). As glutamate is not readily taken up into β-cells it is difficult to design robust experiments considering intracellular actions of this metabolizable nutrient. Indeed, glutamate release from β-cells has recently been reported (Kiely et al. 2007), adding complexity to this story and offering the intriguing possibility of other β-cell actions. Some of the latest data has shown that glutamate can be transported into insulin-containing vesicle while inside the cell (Gammelsaeter et al. 2011). This may enhance Ca2+-dependent insulin secretion through glutamate receptors. Currently, this and other aspects of β-cell glutamate signaling and actions are under investigation by the authors.
Leucine: Prolonged exposure of rat islets to leucine increases ATP, cytosolic Ca2+, and potentiates glucose-stimulated insulin secretion. In addition, chronic exposure to leucine leads to an increase in both ATP synthase and glucokinase, which can sensitize pancreatic β-cells to glucose-induced insulin secretion (Yang et al. 2006). Leucine-induced insulin secretion involves allosteric activation of glutamate dehydrogenase (GDH) leading to an increase in glutamine → glutamate → 2-oxoglutarate flux, elevated mitochondrial metabolism and an increase in ATP production leading to a membrane depolarization (Fig. 6). Additionally transamination of leucine to α-ketoisocaproate (KIC) and entry into TCA cycle via acetyl-CoA can contribute to ATP generation by increasing the oxidation rate of the amino acid and thus stimulation of insulin secretion. Moreover, it has been reported that α-keto acids (including KIC) can directly block KATP channel activity and exert additional KATP channel-independent effects thereby inducing insulin secretion (Heissig et al. 2005; McClenaghan and Flatt 2000). Notably, a recent study reported patients with mutations in the regulatory (GTP binding) site of GDH had increased β-cell responsiveness to leucine, presenting with hypoglycaemia after a protein rich meal (Heissig et al. 2005; Hsu et al. 2001). In addition, mice harbouring a β-cell-specific GDH deletion exhibit a marked decrease (37 %) in glucose-induced insulin secretion, supporting an essential role of GDH in insulin release (Carobbio et al. 2009).
Alanine: Effects of l-alanine have been studied in BRIN-BD11 cells and primary rat islet cells, which consume high rates of this amino acid (Dixon et al. 2003). Moreover, l-alanine is known to potentiate GSIS by enhancing glucose utilization and metabolism (Brennan et al. 2002), and numerous studies have highlighted l-alanine as a potent initiator of insulin release. The authors have utilized BRIN-BD11 cells to study the actions of l-alanine on β-cells demonstrating an influence on GSIS by electrogenic Na+ transport, and exploited13C nuclear magnetic resonance technologies to trace l-alanine metabolism, demonstrating generation of glutamate, aspartate and lactate. Interestingly, the authors have also developed an integrated mathematical model to determine the effect of l-alanine metabolism and l-alanine-mediated Ca2+-handling on GSIS and amino acid-stimulated insulin secretion (Salvucci et al. 2013). Here, using BRIN-BD11 cells to validate the model in vitro, the authors found that elevated intracellular ATP and Ca2+ levels were required for complete insulin secretory responses. Furthermore, this model confirmed that l-alanine-associated Na+ co-transport acted in synergy with membrane depolarization leading to K+ ATP-independent Ca2+ influx and insulin secretion (Salvucci et al. 2013). In addition, l-alanine metabolism to pyruvate followed by oxidation via the TCA cycle could increase ATP levels and thus promote insulin secretion via the K+ ATP-dependent mechanism, and could also generate putative stimulus-secretion factors such as intracellular l-glutamate or citrate that may result in increased insulin secretion (Salvucci et al. 2013). Other studies using the respiratory poison oligomycin have also illustrated the importance of metabolism and oxidation of alanine for its ability to stimulate insulin secretion (Brennan et al. 2002).
Overview of Nutrient Regulation of β-Cell Gene Expression
Glucose can impact on insulin secretion and pancreatic β-cell function by regulating gene expression, enabling mammals to adapt metabolic activity to changes in nutrient supply. In pancreatic β-cells, in addition to a fundamental role in the regulation of insulin secretion and pancreatic β-cell function, glucose serves as a principal physiological regulator of insulin gene expression (Poitout et al. 2006). Glucose is known to control transcription factor recruitment, level of transcription, alternative splicing and stability of insulin mRNA (Bensellam et al. 2009). To cover all aspects of the diverse actions of glucose and other key nutrients on β-cell gene expression is certainly outside the scope of this chapter, but the following gives an overview of some notable aspects of this complex area of study.
In β-cells, three transcriptional factors bind to insulin promoter to regulate insulin gene expression: pancreatic and duodenal homeobox 1 (Pdx-1), neurogenic differentiation 1 (NeuroD1) and V-maf musculoaponeurotic fibrosarcoma oncogene homolog A (MafA), acting in synergy and stimulating insulin gene expression in response to increasing plasma glucose (Andrali et al. 2008). However, consistent with detrimental β-cell actions of prolonged exposure to high glucose concentrations, impairments of Pdx-1 and MafA binding to the insulin promoter have been noted, in turn leading to decreased insulin biosynthesis, content and capacity for secretion. Similarly, prolonged exposure to high fatty acid levels can impair insulin gene expression, this time accompanied by an accumulation of triglycerides in β-cells – particularly palmitate – where the negative effect may be attributable to ceramide formation (Kelpe et al. 2003). Moreover, palmitate is known to induce a decrease in binding activity of transcriptional factors on the insulin promoter, where both Pdx-1 translocation to the nucleus and MafA expression are affected (Hagman et al. 2005).
An important role of amino acids on gene expression has recently been highlighted (Newsholme et al. 2006). In an Affymetrix microarray study utilizing BRIN-BD11 cells, prolonged (24 h) exposure to alanine and glutamine upregulated β-cell gene expression, particularly genes involved in metabolism, signal transduction and oxidative stress (Cunningham et al. 2005; Corless et al. 2006). This upregulation could be due to alanine metabolism, provision of amino acid stimulus-secretion coupling factors and lipid metabolites (such as long-chain acyl-CoAs), and leading to an alteration of cellular redox state (Brennan et al. 2002; Dixon et al. 2003). Interestingly, 24 h exposure of BRIN-BD11 cells to glutamine strongly increased calcineurin catalytic and regulatory subunit mRNA expression (Corless et al. 2006) and this Ca2+-binding protein has been reported to play a role in the somatostatin induced inhibition of exocytosis in mouse pancreatic β-cells (Renstrom et al. 1996). Glutamine can also increase Pdx-1 and acetyl-CoA carboxylase mRNA expression. Of the amino acids, alanine and glutamine appear to play particularly important roles in the regulation of gene expression (Newsholme et al. 2006; Corless et al. 2006) and further study of the precise mechanisms underlying these actions should help understanding of β-cell responses to nutrient supply, metabolism and secretory and functional integrity.
Nutrient-Induced β-Cell Desensitization, Dysfunction, and Toxicity
Persistently elevated fuel supply such as glucose, amino acids, fatty acids (or a mixture) is known to exert detrimental effects on a number of cells, and can induce insulin resistance in muscle – perhaps as a first line protective adaptation to fuel overload (Tremblay et al. 2007). The β-cell does not protect itself by blocking uptake of excess nutrients and thus is vulnerable to potential excess activation of mitochondrial metabolism, ROS production, elevated intracellular Ca2+ and cell injury (Nolan and Prentki 2008; Morgan et al. 2007; Newsholme et al. 2007c). While expansion of β-cell mass can offer part of a compensatory response, desensitization may also help reduce the burden on β-cells. Desensitization is commonly observed in eukaryotic cells, is believed to have an underlying role in cell protection (McClenaghan 2007), and may be defined as a readily induced and reversible state of cellular refractoriness attributed to repeated or prolonged exposure to high concentrations of a stimulus.
While acute exposure to glucose generally promotes increased metabolism and generation of MCFs, as well as changes in insulin gene transcription and translation, chronic exposure to high levels of this sugar has been associated with β-cell deterioration, with glucose desensitization in the first instance progressing to glucotoxicity likely arising from oxidative stress. Likewise, while the acute β-cell actions of fatty acids are usually positive, chronic exposure can exert substantive changes to nutrient metabolism and so-called lipotoxicity, and both the hyperglycaemia and hyperlipidemia of diabetes can alter insulin secretion and β-cell function. However, while experimental glucotoxicity and lipotoxicity can be independently demonstrated, it is clear that these two are interrelated adverse forces on the β-cell (Prentki et al. 2002). Some characteristics of this so-called “glucolipotoxicity” (Prentki et al. 2002) are: (i) impaired glucose oxidation, resulting in ACC inhibition (due to an increase in cellular AMP levels as ATP generation decreases, subsequent activation of AMP kinase, and phosphorylation of ACC, so inhibiting generation of malonyl-CoA and LC acyl-CoA), (ii) promotion of fatty acid oxidation due to relief of CPT-1 inhibition and (iii) enhanced FFA esterification and lipid accumulation with respect to the excess FFA that are not oxidized. These combined effects lead to a decrease in glucose-induced insulin secretion, impaired insulin gene expression and an increase in β-cell failure and even cell death (Newsholme et al. 2007a).
Although mechanisms by which chronic exposure to high levels of glucose and/or lipids damage β-cells have been the subject to intense clinical and experimental investigation, much less attention has been directed to other diet-derived factors, including the other major nutrient class, the amino acids. As noted earlier, prolonged exposure to amino acids such as alanine or glutamine may (at least in the first instance) upregulate gene expression of certain metabolic and signal transduction elements, and can also offer enhanced protection against cytokine-induced apoptosis (Cunningham et al. 2005). However, these primary observations also indicated an alteration in β-cell responsiveness, later studied by the authors in more detail (McClenaghan et al. 2009). These latter studies demonstrate for the first time that the desensitization phenomenon previously reported with other pharmacological and physiological agents (see review (McClenaghan 2007)) may extend to the amino acids, where 18 h exposure to l-alanine resulted in reversible alterations in metabolic flux (a reduction in flux), Ca2+ handling (reduced level of intracellular Ca2+) and insulin secretion (reduction in insulin secretion).
More intriguing evidence for detrimental β-cell actions of amino acids relate to the reported effects of acute and chronic exposure to homocysteine (Patterson et al. 2006, 2007). Interestingly, elevated circulating homocysteine and hyperhomocysteinemia have emerged as important risk factors for cardiovascular disease and other diseases of the metabolic syndrome, including type 2 diabetes. Studies of prolonged effects of alanine and homocysteine in the authors’ laboratories represent compelling evidence for the existence of β-cell amino acid desensitization. While these data prompt further study, it is interesting to speculate that nutrient-induced desensitization may be a first line compensatory mechanism to over-nutrition. However, if observations on the “toxic” effects of glucose/lipids also extend to amino acids, this would support the view that prolonged over-nutrition generally results in adverse β-cell events which may contribute to the pathogenesis of diabetes.
Conclusion
Pancreatic β-cells are well equipped to respond as metabolic fuel sensors, and additionally possess inherent mechanisms to adapt to nutrient overconsumption in order to preserve glucose homeostasis. Glucose signaling is of primary importance in the β-cell, and as discussed both fatty acids and amino acids can interface with central signaling pathways to help regulate insulin secretion. Inherently, the metabolic sensing ability of the β-cell comes at the expense of its protection and islet β-cells are more vulnerable than other cells in the body to excess fuel supply. However, as illustrated in Fig. 7, β-cells play a key role in countering nutrient over-consumption through hyperinsulinemia and β-cell expansion as initial attempts to curb the characteristic hyperglycaemia of impaired glucose tolerance (IGT) and type 2 diabetes. Ultimately it is the interplay between nutrient handling by β-cells and other insulin-sensitive cells such as skeletal muscle, adipocytes and liver that dictates whole body nutrient homeostasis (Fig. 7). It would seem that β-cell failure due to excess nutrients is dominant in the pathogenesis of type 2 diabetes with a significant underlying genetic or environmental susceptibility defect, contributing to the process (Nolan and Prentki 2008). However, the alarming epidemic rise in diabesity only serves to highlight how precious and important β-cells are to the maintenance of whole body metabolism. This also prompts further efforts to understand the complexities of β-cell function, demise and destruction, and indeed novel targets and treatments for diabetes, obesity and the metabolic syndrome.

Interplay between β-cells and insulin-sensitive tissues in the pathogenesis of type 2 diabetes. Key interplay between nutrient handling by insulin secreting β-cells and insulin-sensitive cells such as skeletal muscle, adipocytes and liver regulates whole body nutrient homeostasis. Defective insulin secretion (due to excessive nutrient-induced desensitization of the β-cell, see main text) will result in high plasma levels of glucose. Insulin resistance in muscle and adipose tissue will result in reduced glucose uptake. Insulin resistance in the liver will result in enhanced glucose release into the blood, compounding hyperglycaemia. Insulin resistance in the adipose tissue will result in elevated fatty acid release and pro-inflammatory factor release, contributing to insulin resistance due to impairment of insulin-signaling pathways and also reduced insulin secretion from the β-cell due to impairment of regulation of nutrient metabolism
Abbreviations
- ACC:
-
Acetyl-CoA carboxylase
- CPT-1:
-
Carnitine Palmitoyl Transferase 1
- DAG:
-
Diacylglycerol
- FFA:
-
Free fatty acid
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- GLP-1:
-
Glucagon-like peptide-1
- Gly3P:
-
Glycerol-3-phosphate
- GSIS:
-
Glucose-stimulated insulin secretion
- LC-acyl:
-
CoA long-chain acyl-CoA
- MCF:
-
Metabolic coupling factors
- PI3K:
-
Phosphatidylinositide-3-kinases
- PKA:
-
Protein kinase A
- PKC:
-
Protein kinase C
- PLC:
-
Phospholipase C
References
Andrali SS, Sampley ML, Vanderford NL, Ozcan S (2008) Glucose regulation of insulin gene expression in pancreatic β-cells. Biochem J 415:1–10
Bender K, Newsholme P, Brennan L, Maechler P (2006) The importance of redox shuttles to pancreatic β-cell energy metabolism and function. Biochem Soc Trans 34:811–814
Bender K, Maechler P, McClenaghan NH, Flatt PR, Newsholme P (2009) Overexpression of the malate-aspartate NADH shuttle member Aralar1 in the clonal β-cell line BRIN-BD11 enhances amino-acid-stimulated insulin secretion and cell metabolism. Clin Sci (Lond) 117:321–330
Bensellam M, Van Lommel L, Overbergh L, Schuit FC, Jonas JC (2009) Cluster analysis of rat pancreatic islet gene mRNA levels after culture in low-, intermediate- and high-glucose concentrations. Diabetologia 52:463–476
Bertrand G, Ishiyama N, Nenquin M, Ravier MA, Henquin JC (2002) The elevation of glutamate content and the amplification of insulin secretion in glucose-stimulated pancreatic islets are not causally related. J Biol Chem 277:32883–32891
Blau N, Duran M, Blaskovics M, Gibson K (2003) Amino acid analysis. Physician’s guide to the laboratory diagnosis of metabolic diseases, 2nd edn. Springer, New York, pp 11–26
Brennan L, Shine A, Hewage C, Malthouse JP, Brindle KM, McClenaghan N, Flatt PR, Newsholme P (2002) A nuclear magnetic resonance-based demonstration of substantial oxidative l-alanine metabolism and l-alanine-enhanced glucose metabolism in a clonal pancreatic β-cell line: metabolism of l-alanine is important to the regulation of insulin secretion. Diabetes 51:1714–1721
Brennan L, Corless M, Hewage C, Malthouse JP, McClenaghan NH, Flatt PR, Newsholme P (2003)13C NMR analysis reveals a link between l-glutamine metabolism, d-glucose metabolism and gamma-glutamyl cycle activity in a clonal pancreatic β-cell line. Diabetologia 46:1512–1521
Briaud I, Lingohr MK, Dickson LM, Wrede CE, Rhodes CJ (2003) Differential activation mechanisms of Erk-1/2 and p70(S6K) by glucose in pancreatic β-cells. Diabetes 52:974–983
Broca C, Brennan L, Petit P, Newsholme P, Maechler P (2003) Mitochondria-derived glutamate at the interplay between branched-chain amino acid and glucose-induced insulin secretion. FEBS Lett 545:167–172
Carobbio S, Frigerio F, Rubi B, Vetterli L, Bloksgaard M, Gjinovci A, Pournourmohammadi S, Herrera PL, Reith W, Mandrup S, Maechler P (2009) Deletion of glutamate dehydrogenase in β cells abolishes part of the insulin secretory response not required for glucose homeostasis. J Biol Chem 284:921–929
Carpentier A, Mittelman SD, Bergman RN, Giacca A, Lewis GF (2000) Prolonged elevation of plasma free fatty acids impairs pancreatic β-cell function in obese non-diabetic humans but not in individuals with type 2 diabetes. Diabetes 49:399–408
Charles S, Henquin JC (1983) Distinct effects of various amino acids on 45Ca2+ fluxes in rat pancreatic islets. Biochem J 214:899–907
Cook DL, Hales CN (1984) Intracellular ATP directly blocks K+ channels in pancreatic β-cells. Nature 311:271–273
Corkey BE, Glennon MC, Chen KS, Deeney JT, Matschinsky FM, Prentki M (1989) A role for malonyl-CoA in glucose-stimulated insulin secretion from clonal pancreatic β-cells. J Biol Chem 264:21608–21612
Corless M, Kiely A, McClenaghan NH, Flatt PR, Newsholme P (2006) Glutamine regulates expression of key transcription factor, signal transduction, metabolic gene, and protein expression in a clonal pancreatic β-cell line. J Endocrinol 190:719–727
Cunningham GA, McClenaghan NH, Flatt PR, Newsholme P (2005) l-Alanine induces changes in metabolic and signal transduction gene expression in a clonal rat pancreatic β-cell line and protects from pro-inflammatory cytokine-induced apoptosis. Clin Sci (Lond) 109:447–455
Curi R, Lagranha CJ, Doi SQ, Sellitti DF, Procopio J, Pithon-Curi TC, Corless M, Newsholme P (2005) Molecular mechanisms of glutamine action. J Cell Physiol 204:392–401
Danielsson A, Hellman B, Idahl LA (1970) Levels of α-ketoglutarate and glutamate in stimulated pancreatic β-cells. Horm Metab Res 2:28–31
Deeney JT, Gromada J, Hoy M, Olsen HL, Rhodes CJ, Prentki M, Berggren PO, Corkey BE (2000) Acute stimulation with long chain acyl-CoA enhances exocytosis in insulin-secreting cells (HIT T-15 and NMRI β-cells). J Biol Chem 275:9363–9368
del Arco A, Satrustegui J (1998) Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J Biol Chem 273:23327–23334
Dixon G, Nolan J, McClenaghan N, Flatt PR, Newsholme P (2003) A comparative study of amino acid consumption by rat islet cells and the clonal β-cell line BRIN-BD11 – the functional significance of l-alanine. J Endocrinol 179:447–454
Dixon G, Nolan J, McClenaghan NH, Flatt PR, Newsholme P (2004) Arachidonic acid, palmitic acid and glucose are important for the modulation of clonal pancreatic β-cell insulin secretion, growth and functional integrity. Clin Sci (Lond) 106:191–199
Dukes ID, McIntyre MS, Mertz RJ, Philipson LH, Roe MW, Spencer B, Worley JF 3rd (1994) Dependence on NADH produced during glycolysis for β-cell glucose signaling. J Biol Chem 269:10979–10982
Eto K, Tsubamoto Y, Terauchi Y, Sugiyama T, Kishimoto T, Takahashi N, Yamauchi N, Kubota N, Murayama S, Aizawa T, Akanuma Y, Aizawa S, Kasai H, Yazaki Y, Kadowaki T (1999) Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science 283:981–985
Fajans SS, Floyd JC Jr, Knopf RF, Conn FW (1967) Effect of amino acids and proteins on insulin secretion in man. Recent Prog Horm Res 23:617–662
Gammelsaeter R, Coppola T, Marcaggi P, Storm-Mathisen J, Chaudhry FA, Attwell D, Regazzi R, Gundersen V (2011) A role for glutamate transporters in the regulation of insulin secretion. PLoS One 6:e22960
Green BD, Flatt PR (2007) Incretin hormone mimetics and analogues in diabetes therapeutics. Best Pract Res Clin Endocrinol Metab 21:497–516
Guo-Parke H, McCluskey JT, Kelly C, Hamid M, McClenaghan NH, Flatt PR (2012) Configuration of electrofusion-derived human insulin-secreting cell line as pseudoislets enhances functionality and therapeutic utility. J Endocrinol 214:257–265
Haber EP, Procopio J, Carvalho CR, Carpinelli AR, Newsholme P, Curi R (2006) New insights into fatty acid modulation of pancreatic β-cell function. Int Rev Cytol 248:1–41
Hagman DK, Hays LB, Parazzoli SD, Poitout V (2005) Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans. J Biol Chem 280:32413–32418
Hamilton JA, Kamp F (1999) How are free fatty acids transported in membranes? Is it by proteins or by free diffusion through the lipids? Diabetes 48:2255–2269
Han J, Liu YQ (2010) Reduction of islet pyruvate carboxylase activity might be related to the development of type 2 diabetes mellitus in Agouti-K mice. J Endocrinol 204:143–152
Heissig H, Urban KA, Hastedt K, Zunkler BJ, Panten U (2005) Mechanism of the insulin-releasing action of α-ketoisocaproate and related α-keto acid anions. Mol Pharmacol 68:1097–1105
Hoy M, Maechler P, Efanov AM, Wollheim CB, Berggren PO, Gromada J (2002) Increase in cellular glutamate levels stimulates exocytosis in pancreatic β-cells. FEBS Lett 531:199–203
Hsu BY, Kelly A, Thornton PS, Greenberg CR, Dilling LA, Stanley CA (2001) Protein-sensitive and fasting hypoglycaemia in children with the hyperinsulinism/hyperammonemia syndrome. J Pediatr 138:383–389
Itoh Y, Hinuma S (2005) GPR40, a free fatty acid receptor on pancreatic β cells, regulates insulin secretion. Hepatol Res 33:171–173
Jensen MV, Joseph JW, Ilkayeva O, Burgess S, Lu D, Ronnebaum SM, Odegaard M, Becker TC, Sherry AD, Newgard CB (2006) Compensatory responses to pyruvate carboxylase suppression in islet β-cells. Preservation of glucose-stimulated insulin secretion. J Biol Chem 281:22342–22351
Jensen MV, Joseph JW, Ronnebaum SM, Burgess SC, Sherry AD, Newgard CB (2008) Metabolic cycling in control of glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab 295:E1287–E1297
Joseph JW, Odegaard ML, Ronnebaum SM, Burgess SC, Muehlbauer J, Sherry AD, Newgard CB (2007) Normal flux through ATP-citrate lyase or fatty acid synthase is not required for glucose-stimulated insulin secretion. J Biol Chem 282:31592–31600
Keane D, Newsholme P (2008) Saturated and unsaturated (including arachidonic acid) non-esterified fatty acid modulation of insulin secretion from pancreatic β-cells. Biochem Soc Trans 36:955–958
Keane DC, Takahashi HK, Dhayal S, Morgan NG (2011) Curi, Newsholme P. Arachidonic acid actions on functional integrity and attenuation of the negative effects of palmitic acid in a clonal pancreatic β-cell line. Clin Sci (Lond) 120:195–206
Kelpe CL, Moore PC, Parazzoli SD, Wicksteed B, Rhodes CJ, Poitout V (2003) Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J Biol Chem 278:30015–30021
Kiely A, McClenaghan NH, Flatt PR, Newsholme P (2007) Pro-inflammatory cytokines increase glucose, alanine and triacylglycerol utilization but inhibit insulin secretion in a clonal pancreatic β-cell line. J Endocrinol 195:113–123
Kruman I, Guo Q, Mattson MP (1998) Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J Neurosci Res 51:293–308
Kwon G, Marshall CA, Pappan KL, Remedi MS, McDaniel ML (2004) Signaling elements involved in the metabolic regulation of mTOR by nutrients, incretins, and growth factors in islets. Diabetes 53(Suppl 3):S225–S232
Li C, Buettger C, Kwagh J, Matter A, Daikhin Y, Nissim IB, Collins HW, Yudkoff M, Stanley CA, Matschinsky FM (2004) A signaling role of glutamine in insulin secretion. J Biol Chem 279:13393–13401
MacDonald MJ (1995a) Influence of glucose on pyruvate carboxylase expression in pancreatic islets. Arch Biochem Biophys 319:128–132
MacDonald MJ (1995b) Feasibility of a mitochondrial pyruvate malate shuttle in pancreatic islets. Further implication of cytosolic NADPH in insulin secretion. J Biol Chem 270:20051–20058
Macdonald MJ (2003) Export of metabolites from pancreatic islet mitochondria as a means to study anaplerosis in insulin secretion. Metabolism 52:993–998
MacDonald MJ, Fahien LA (2000) Glutamate is not a messenger in insulin secretion. J Biol Chem 275:34025–34027
MacDonald MJ, Tang J, Polonsky KS (1996) Low mitochondrial glycerol phosphate dehydrogenase and pyruvate carboxylase in pancreatic islets of Zucker diabetic fatty rats. Diabetes 45:1626–1630
Maechler P (2002) Mitochondria as the conductor of metabolic signals for insulin exocytosis in pancreatic β-cells. Cell Mol Life Sci 59:1803–1818
Maechler P, Wollheim CB (1999) Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature 402:685–689
Maechler P, Kennedy ED, Pozzan T, Wollheim CB (1997) Mitochondrial activation directly triggers the exocytosis of insulin in permeabilized pancreatic β-cells. Embo J 16:3833–3841
Malaisse-Lagae F, Sener A, Garcia-Morales P, Valverde I, Malaisse WJ (1982) The stimulus-secretion coupling of amino acid-induced insulin release. Influence of a non-metabolized analogue of leucine on the metabolism of glutamine in pancreatic islets. J Biol Chem 257:3754–3758
Marmol P, Pardo B, Wiederkehr A, del Arco A, Wollheim CB, Satrustegui J (2009) Requirement for Aralar and its Ca2+-binding sites in Ca2+ signal transduction in mitochondria from INS-1 clonal β-cells. J Biol Chem 284:515–524
McClenaghan NH (2007) Physiological regulation of the pancreatic β-cell: functional insights for understanding and therapy of diabetes. Exp Physiol 92:481–496
McClenaghan NH, Flatt PR (2000) Metabolic and KATP channel-independent actions of keto acid initiators of insulin secretion. Pancreas 20:38–46
McClenaghan NH, Barnett CR, Ah-Sing E, Abdel-Wahab YH, O'Harte FP, Yoon TW, Swanston-Flatt SK, Flatt PR (1996a) Characterization of a novel glucose-responsive insulin-secreting cell line, BRIN-BD11, produced by electrofusion. Diabetes 45:1132–1140
McClenaghan NH, Barnett CR, O'Harte FP, Flatt PR (1996b) Mechanisms of amino acid-induced insulin secretion from the glucose-responsive BRIN-BD11 pancreatic β-cell line. J Endocrinol 151:349–357
McClenaghan NH, Scullion SM, Mion B, Hewage C, Malthouse JP, Flatt PR, Newsholme P, Brennan L (2009) Prolonged l-alanine exposure induces changes in metabolism, Ca2+ handling and desensitization of insulin secretion in clonal pancreatic β-cells. Clin Sci (Lond) 116:341–351
McCluskey JT, Hamid M, Guo-Parke H, McClenaghan NH, Gomis R, Flatt PR (2011) Development and functional characterization of insulin-releasing human pancreatic β cell lines produced by electrofusion. J Biol Chem 286:21982–21992
McDaniel ML, Marshall CA, Pappan KL, Kwon G (2002) Metabolic and autocrine regulation of the mammalian target of rapamycin by pancreatic β-cells. Diabetes 51:2877–2885
Moffitt JH, Fielding BA, Evershed R, Berstan R, Currie JM, Clark A (2005) Adverse physiochemical properties of tripalmitin in β cells lead to morphological changes and lipotoxicity in vitro. Diabetologia 48:1819–1829
Morgan D, Oliveira-Emilio HR, Keane D, Hirata AE, Santos da Rocha M, Bordin S, Curi R, Newsholme P, Carpinelli AR (2007) Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal β cell line. Diabetologia 50:359–369
Morgan D, Rebelato E, Abdulkader F, Graciano MF, Oliveira-Emilio HR, Hirata AE, Rocha MS, Bordin S, Curi R, Carpinelli AR (2009) Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic β cells. Endocrinology 150(5):2197–2201
Nakazaki M, Kakei M, Koriyama N, Tanaka H (1995) Involvement of ATP-sensitive K+ channels in free radical-mediated inhibition of insulin secretion in rat pancreatic β-cells. Diabetes 44:878–883
Newsholme P, Krause M (2012) Nutritional regulation of insulin secretion: implications for diabetes. Clin Biochem Rev 33:35–47
Newsholme P, Brennan L, Bender K (2006) Amino-acid metabolism, β cell function and diabetes. Diabetes 55(Suppl 2):S39–S47
Newsholme P, Keane D, Welters HJ, Morgan NG (2007a) Life and death decisions of the pancreatic β-cell: the role of fatty acids. Clin Sci (Lond) 112:27–42
Newsholme P, Bender K, Kiely A, Brennan L (2007b) Amino acid metabolism, insulin secretion and diabetes. Biochem Soc Trans 35:1180–1186
Newsholme P, Haber EP, Hirabara SM, Rebelato EL, Procopio J, Morgan D, Oliveira-Emilio HC, Carpinelli AR, Curi R (2007c) Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J Physiol 583:9–24
Newsholme P, Rebelato E, Abdulkader F, Krause M, Carpinelli A, Curi R (2012) Reactive oxygen and nitrogen species generation, antioxidant defenses, and β-cell function: a critical role for amino acids. J Endocrinol 214:11–20
Nguyen CA, Akiba Y, Kaunitz JD (2012) Recent advances in gut nutrient chemosensing. Curr Med Chem 19:28–34
Nielsen K, Sorensen PG, Hynne F (1997) Chaos in glycolysis. J Theor Biol 186:303–306
Nielsen K, Sorensen PG, Hynne F, Busse HG (1998) Sustained oscillations in glycolysis: an experimental and theoretical study of chaotic and complex periodic behaviour and of quenching of simple oscillations. Biophys Chem 72:49–62
Nolan CJ, Prentki M (2008) The islet β-cell. Fuel responsive and vulnerable. Trends Endocrinol Metab 19:285–291
Oh DY, Lagakos WS (2011) The role of G-protein-coupled receptors in mediating the effect of fatty acids on inflammation and insulin sensitivity. Curr Opin Clin Nutr Metab Care 14:322–327
Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM (2010) GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142:687–698
Patterson S, Flatt PR, Brennan L, Newsholme P, McClenaghan NH (2006) Detrimental actions of metabolic syndrome risk factor, homocysteine, on pancreatic β-cell glucose metabolism and insulin secretion. J Endocrinol 189:301–310
Patterson S, Scullion SM, McCluskey JT, Flatt PR, McClenaghan NH (2007) Prolonged exposure to homocysteine results in diminished but reversible pancreatic β-cell responsiveness to insulinotropic agents. Diabetes Metab Res Rev 23:324–334
Persaud SJ, Muller D, Belin VD, Kitsou-Mylona I, Asare-Anane H, Papadimitriou A, Burns CJ, Huang GC, Amiel SA, Jones PM (2007) The role of arachidonic acid and its metabolites in insulin secretion from human islets of langerhans. Diabetes 56:197–203
Peterson G (2012) Current treatments and strategies for type 2 diabetes: can we do better with GLP-1 receptor agonists? Ann Med 44:338–349
Poitout V, Hagman D, Stein R, Artner I, Robertson RP, Harmon JS (2006) Regulation of the insulin gene by glucose and fatty acids. J Nutr 136:873–876
Prentki M, Joly E, El-Assaad W, Roduit R (2002) Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in β-cell adaptation and failure in the etiology of diabetes. Diabetes 51(Suppl 3):S405–S413
Reid T (2012) Choosing GLP-1 receptor agonists or DPP-4 inhibitors: weighing the clinical trial evidence. Clin Diab 30:3–12
Renstrom E, Ding WG, Bokvist K, Rorsman P (1996) Neurotransmitter-induced inhibition of exocytosis in insulin-secreting β cells by activation of calcineurin. Neuron 17:513–522
Roche E, Farfari S, Witters LA, Assimacopoulos-Jeannet F, Thumelin S, Brun T, Corkey BE, Saha AK, Prentki M (1998) Long-term exposure of β-INS cells to high glucose concentrations increases anaplerosis, lipogenesis, and lipogenic gene expression. Diabetes 47:1086–1094
Rubi B, del Arco A, Bartley C, Satrustegui J, Maechler P (2004) The malate-aspartate NADH shuttle member Aralar1 determines glucose metabolic fate, mitochondrial activity, and insulin secretion in β cells. J Biol Chem 279:55659–55666
Salehi A, Flodgren E, Nilsson NE, Jimenez-Feltstrom J, Miyazaki J, Owman C, Olde B (2005) Free fatty acid receptor 1 (FFA(1)R/GPR40) and its involvement in fatty-acid-stimulated insulin secretion. Cell Tissue Res 322:207–215
Salvucci M, Neufeld Z, Newsholme P (2013) Mathematical model of metabolism and electrophysiology of amino acid and glucose stimulated insulin secretion: in vitro validation using a β-cell line. PLoS One 8:e52611
Sener A, Malaisse WJ (1980) l-Leucine and a non-metabolized analogue activate pancreatic islet glutamate dehydrogenase. Nature 288:187–189
Sener A, Best LC, Yates AP, Kadiata MM, Olivares E, Louchami K, Jijakli H, Ladriere L, Malaisse WJ (2000) Stimulus-secretion coupling of arginine-induced insulin release: comparison between the cationic amino acid and its methyl ester. Endocrine 13:329–340
Shapiro H, Shachar S, Sekler I, Hershfinkel M, Walker MD (2005) Role of GPR40 in fatty acid action on the β cell line INS-1E. Biochem Biophys Res Commun 335:97–104
Smith PA, Sakura H, Coles B, Gummerson N, Proks P, Ashcroft FM (1997) Electrogenic arginine transport mediates stimulus-secretion coupling in mouse pancreatic β-cells. J Physiol 499(Pt 3):625–635
Straub SG, Sharp GW (2002) Glucose-stimulated signaling pathways in biphasic insulin secretion. Diabetes Metab Res Rev 18:451–463
Tarasov A, Dusonchet J, Ashcroft F (2004) Metabolic regulation of the pancreatic β-cell ATP-sensitive K+ channel: a pas de deux. Diabetes 53(Suppl 3):S113–S122
Tato I, Bartrons R, Ventura F, Rosa JL (2011) Amino acids activate mammalian target of rapamycin complex 2 (mTORC2) via PI3K/Akt signaling. J Biol Chem 286:6128–6142
Tengholm A, Gylfe E (2009) Oscillatory control of insulin secretion. Mol Cell Endocrinol 297:58–72
Tomita T, Masuzaki H, Iwakura H, Fujikura J, Noguchi M, Tanaka T, Ebihara K, Kawamura J, Komoto I, Kawaguchi Y, Fujimoto K, Doi R, Shimada Y, Hosoda K, Imamura M, Nakao K (2006) Expression of the gene for a membrane-bound fatty acid receptor in the pancreas and islet cell tumours in humans: evidence for GPR40 expression in pancreatic β cells and implications for insulin secretion. Diabetologia 49:962–968
Tremblay F, Lavigne C, Jacques H, Marette A (2007) Role of dietary proteins and amino acids in the pathogenesis of insulin resistance. Annu Rev Nutr 27:293–310
Westermark PO, Lansner A (2003) A model of phosphofructokinase and glycolytic oscillations in the pancreatic β-cell. Biophys J 85:126–139
Wiederkehr A, Wollheim CB (2006) Mini-review: implication of mitochondria in insulin secretion and action. Endocrinology 147:2643–2649
Wuttke A, Idevall-Hagren O, Tengholm A (2013) P2Y1 receptor-dependent diacylglycerol signaling microdomains in β cells promote insulin secretion. Faseb J 27:1610–1620
Xu J, Han J, Long YS, Epstein PN, Liu YQ (2008) The role of pyruvate carboxylase in insulin secretion and proliferation in rat pancreatic β cells. Diabetologia 51:2022–2030
Yaney GC, Corkey BE (2003) Fatty acid metabolism and insulin secretion in pancreatic β cells. Diabetologia 46:1297–1312
Yang J, Wong RK, Park M, Wu J, Cook JR, York DA, Deng S, Markmann J, Naji A, Wolf BA, Gao Z (2006) Leucine regulation of glucokinase and ATP synthase sensitizes glucose-induced insulin secretion in pancreatic β-cells. Diabetes 55:193–201
Yu JH, Kim KH, Kim H (2006) Role of NADPH oxidase and calcium in cerulein-induced apoptosis: involvement of apoptosis-inducing factor. Ann N Y Acad Sci 1090:292–297
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media Dordrecht
About this entry
Cite this entry
Newsholme, P., Keane, K., Gaudel, C., McClenaghan, N. (2015). (Dys)Regulation of Insulin Secretion by Macronutrients. In: Islam, M. (eds) Islets of Langerhans. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6686-0_4
Download citation
DOI: https://doi.org/10.1007/978-94-007-6686-0_4
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6685-3
Online ISBN: 978-94-007-6686-0
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences