
Abstract
Many lipids present in cellular membranes are phosphorylated as part of signaling cascades and participate in the recruitment, localization, and activation of downstream protein effectors. Phosphatidylinositol (3,4,5)-trisphosphate (PtdIns(3,4,5)P3) is one of the most important second messengers and is capable of interacting with a variety of proteins through specific PtdIns(3,4,5)P3 binding domains. Localization and activation of these effector proteins controls a myriad of cellular functions including cell survival, proliferation, cytoskeletal rearrangement, and gene expression. Aberrations in the production and metabolism of PtdIns(3,4,5)P3 have been implicated in many human diseases including cancer, diabetes, inflammation, and heart disease. This chapter provides an overview of the role of PtdIns(3,4,5)P3 in cellular regulation and the implications of PtdIns(3,4,5)P3 dysregulation in human diseases. Additionally, recent attempts at targeting PtdIns(3,4,5)P3 signaling via small molecule inhibitors are summarized.
Robert D. Riehle and Sinziana Cornea contributed equally to this work.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
7.1 Introduction
Phosphoinositides (PIs), phosphorylated derivatives of phosphatidylinositol (PtdIns), have been investigated as agonist-dependent second messengers since their discovery in the mid 1950s by Hokin and Hokin [1]. Initial focus was made on PtdIns(4)P and PtdIns(4,5)P2 and the enzymes responsible for their synthesis and degradation. The discovery of a family of PI3-kinases and its lipid products PtdIns(3)P, PtdIns(3,4)P2 and most importantly phosphatidylinositol (3,4,5)-trisphosphate (PtdIns(3,4,5)P3) uncovered additional complexity to PI-mediated signal transduction [2–4]. Further work lead to the elucidation of the mechanisms of PI3-kinase signaling and the critical importance of this class of lipid kinases in a variety of cellular functions including directional motility, metabolism, growth, and cell survival. Additionally, many diseases can be traced back to aberrations in the PI3-kinase pathways. For this reason, efforts have been made to understand the complex regulation of PIs and PtdIns(3,4,5)P3, in particular. PI3Ks are subdivided into three classes: I, II and III, differing in their structure, products, and regulation. PtdIns(3,4,5)P3 is the product of Class I of PI3-kinases, which will be referred to as PI3K throughout the text.
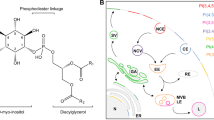
PtdIns(3,4,5)P3 is a member of the PI class of membrane lipids. Typically, PtdIns found in cell membranes account for approximately 5–10 % of the total lipid with only approximately 1 % being phosphorylated, indicating that their primary function is regulatory in nature [5]. PtdIns can be phosphorylated at either the 3′, 4′ or 5′ hydroxyl sites (Fig. 7.1). These hydroxyl groups are phosphorylated alone or in combination to create seven possible PIs with individual stereospecificity and charge. PIs are localized on the endosomal, Golgi, nuclear, and plasma membranes, depending on their structure. Of the 7 PIs only PtdIns(4)P and PtdIns(4,5)P2 are found in relatively high abundance [6]. Unique inositol head groups coupled with extremely low PI concentrations in the cell offer deliberate, specific, sensitive, and localized signaling.

Phosphatidylinositols are composed of long chain fatty acid groups attached to a phosphorylated inositol head group via a glycerol moiety. Fatty acids anchor the molecule in the membrane, while the inositol is exposed to the cytoplasm. Various kinases catalyze the addition or removal of phosphates to the head group at specific locations. Above, PI3K phosphorylates PtdIns(4,5)P2 (shown as PI(4,5)P2) at the 3′ position to form the second messenger PtdIns(3,4,5)P 3 (shown as PI(3,4,5)P3). The phosphatases PTEN and SHIP breakdown PtdIns(3,4,5)P 3 by removing phosphates from the 3′ and 5′ positions respectively
Various kinases and phosphatases regulate the formation and turnover of PIs depending on cellular stimuli [7]. Of the PtdIns present in the cell only 0.25 % are phosphorylated at the 3′ position indicating a highly specific regulatory function [5]. The PI3-kinase family of proteins is responsible for the formation of these PIs and four separate lipid products can be formed: PtdIns-(3)P, PtdIns(3,4)P2, PtdIns(3,5)P2, and PtdIns(3,4,5)P3. PtdIns(3,4,5)P3 is predominately synthesized by the phosphorylation of PtdIns(4,5)P2 by class I PI3K in response to extracellular stimuli [8]. The precursor of PtdIns(3,4,5)P3, PtdIns(4,5)P2, has been shown to have some affinity for various effector proteins and to control the activity of several integral membrane proteins. However, it primarily serves as a pool for formation of second messengers: inositol trisphosphate (Ins(1,4,5)P3), diacylglycerol (DAG), and PtdIns(3,4,5)P3 [9–11]. Though only present at less than 1 % of all plasma membrane phospholipids, PtdIns(4,5)P2 is found at high localized concentrations due to its accumulation into sphingolipid/cholesterol-based rafts [12].
PtdIns(3,4,5)P3 functions both as a protein activator as, for example, in the case with Akt (also known as Protein Kinase B) and as a membrane anchor, recruiting proteins to the plasma membrane where they localize and perform their actions.
7.2 PtdIns(3,4,5)P3 Signaling Pathway
PtdIns(3,4,5)P3 lipid second messengers serve to amplify and propagate signals from receptor tyrosine kinases (RTKs), G protein-coupled receptors (GPCRs), and the small GTPase Ras, as well as to confer signaling specificity by its temporal and spatial distribution in cellular membranes [13]. Tight regulation of PtdIns(3,4,5)P3 levels in the plasma membrane is critical for the prevention of hyper- or hypo-responsiveness to many extracellular stimuli. Since the original discovery of a novel PI3K activity associated with cellular transformation by polyoma middle T antigen in 1985 [2], major insights have been gained into the mechanism of PtdIns(3,4,5)P3 signaling, including initiation and termination of the signal, its effector preferences and its role in cellular regulation.
7.2.1 Signal Initiation and Termination
PtdIns(3,4,5)P3 signaling is initiated by extracellular stimulation of a variety of plasma membrane receptors, including RTKs and GPCRs, leading to the activation of Type I PI3K (Fig. 7.2). Cellular homeostasis is dependent in part on tight regulation of PtdIns(3,4,5)P3 levels in the plasma membrane and is controlled by growth hormones, including epidermal growth factor (EGF), platelet-derived growth factor (PDGF), insulin, and insulin-like growth factor I (IGF-I). PI3Ks specifically phosphorylate the D-3 inositol ring position of PIs in the plasma membrane, generating PtdIns(3)P, PtdIns(3,4)P2, and PtdIns(3,4,5)P3. Synthesis of PtdIns(3,4,5)P3 is the primary cellular function of Type I PI3K, while Type II and III PI3Ks are the key regulators of the cellular PtdIns(3)P pool. Dephosphorylation of PtdIns(3,4,5)P3 by SHIP phosphatases is an important mechanism of PtdIns(3,4)P2 generation [14].

The general model of PtdIns(3,4,5)P3 (shown as PIP3) activation and downstream signaling. Upon growth hormone stimulation of receptors in the plasma membrane, PI3K phosphorylates PtdIns(4,5)P2 to PtdIns(3,4,5)P3, leading to the recruitment of PH-domain containing effector proteins, such as Akt/PKB and PDK1. Activation of these effector proteins leads to signaling cascades controlling a multitude of cellular pathways, including cell survival, metabolism, and cell migration. PTEN and other phosphatases quench PtdIns(3,4,5)P3 signaling
Type I PI3Ks are heterodimers consisting of the p110 catalytic subunit and the p85 regulatory subunit. The two SH2 domains in the p85 subunit recruit the p110 subunit to the cytoplasmic domains of activated receptors at the plasma membrane through a variety of adaptor molecules. These SH2 domains are connected by a coiled coil domain, which constitutively interacts with and stabilizes the p110 subunit. There exist three isoforms of Type IA PI3K (p110α, β and δ) and one Type IB PI3K isoform (p110γ), which is controlled by an alternative regulatory subunit (p101) and was previously believed to be activated exclusively by GPCRs, but more recently was found to respond to RTKs as well [15]. Type IA PI3Ks are mainly activated by growth factor receptors and Ras proteins. p110α and β isoforms are expressed ubiquitously, while p110γ and p110δ are expressed primarily in leukocytes [16].
Under basal conditions, the levels of PtdIns(3,4,5)P3 in the plasma membrane are extremely low and almost undetectable, constituting about 0.0001 % of the plasma membrane’s lipid content. Phosphorylation of PtdIns(4,5)P2 by PI3K results in a rapid, approximately 40-fold increase of PtdIns(3,4,5)P3, to about 10 % of the basal level of PtdIns(4,5)P2, which is relatively abundant in the plasma membrane with an approximately 500-fold greater concentration than basal PtdIns(3,4,5)P3 levels [17]. The low level of PtdIns(3,4,5)P3 reflects its potent effect on key cellular regulatory pathways, requiring a tight control to prevent hyperactivation by extra- and intracellular cues.
Dephosphorylation of PtdIns(3,4,5)P3 is required to prevent its accumulation and constitutive signaling. This is under the control of the phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and SH2-containing inositol 5-phosphatases (SHIPs), which produce PtdIns(4,5)P2 and PtdIns(3,4)P2, respectively [17]. The structural basis of PtdIns(4,5)P2 recognition and membrane binding of PTEN is discussed in detail in Chap. 6. The main function of PTEN is as a lipid phosphatase, dephosphorylating the D-3 position of PtdIns, and negatively regulating the PtdIns(3,4,5)P3 pathway [18]. It is well established to serve as one of the most important tumor suppressors, which is inactivated as frequently as p53 in some forms of cancer. The phosphatase consists of a central phosphatase domain, two C-terminal PEST sequences that serve to stabilize the protein, and an N-terminal tensin- and auxilin-homology region. The gene coding for PTEN is located on human chromosome 10q23, and is often found affected in a multitude of disorders [19]. While SHIP1 is present primarily in hematopoietic cells, SHIP2 is ubiquitously expressed and utilizes PtdIns(3,4,5)P3 as a main substrate [20]. There is some evidence that PTEN plays a more important role in controlling basal levels of PtdIns(3,4,5)P3, while SHIPs counteract stimulus-induced increases [21]. It is also worth noting that another phosphatase, Inositol Polyphosphate 4-phosphatase type II, has emerged more recently as an important tumor suppressor [22]. This enzyme dephosphorylates PtdIns(3,4)P2, which can bind both Akt and PDK1. This lipid is either synthesized de novo from PtdIns(3)P and PtdIns(4)P, or is a result of SHIP dephosphorylation of PtdIns(3,4,5)P3. Therefore, concerted actions of PI3Ks and several phosphatases control signaling by multiple PI species in a highly dynamic fashion in response to growth factor signals.
7.2.2 PH Domains: Primary Sequence vs. Protein Folding and Recognition Preferences
PtdIns(3,4,5)P3 serves to anchor its effector proteins to the membrane surface, as well as to activate, stabilize, and co-localize proteins. The best-known mechanism of action involves direct binding of the PtdIns(3,4,5)P3 inositol head-group to the pleckstrin homology (PH) domain of proteins resulting in their recruitment to the plasma membrane [17]. The effector network of PtdIns(3,4,5)P3 includes only about 40 of the approximately 250 known PH domain-containing proteins, and the members bind PtdIns(3,4,5)P3 with varying degrees of specificity and affinity. The most commonly studied of these include Akt/Protein kinase B, phosphoinositide-dependent kinase-1 (PDK1), guanine nucleotide-exchange factors (GEFs) GRP-1 and ARNO, and Bruton’s tyrosine kinase (Btk), all of which are considered relatively specific for PtdIns(3,4,5)P3.
The PH domain consists of approximately 120 amino acid residues arranged into a bowl-like binding pocket that closely fits the inositol head group of PtdIns(3,4,5)P3. While the tertiary structure of this pocket remains highly conserved between different classes of PtdIns(3,4,5)P3 effector proteins, the primary structure is known to vary significantly. It is believed that this variation contributes to the specificity and affinity for PtdIns(3,4,5)P3 versus other membrane PIs [23]. The binding pocket is a seven-stranded anti-parallel beta-barrel formed by the β2-β3, β4-β5, and β6-β7 loops and capped at one end by an amphipathic α-helix [24] (Fig. 7.3). Basic residues creating a highly positive electrostatic environment capable of attracting negatively charged PtdIns(3,4,5)P3 line the interior of the PH domain. The pattern of phosphate groups on the inositol head group is also very important in binding [17].

Ribbon drawings of PtdIns(3,4,5)P3 bound to the PH domain of Akt. (a) Seven-stranded beta-barrel structure formed from the β2-β3, β4-β5, and β6-β7 loops (blue), capped at one end by an amphipathic α-helix (red), and bound to the inositol (1,3,4,5)-phosphate head group of PtdIns(3,4,5)P3 (purple). The basic residues proposed to bind PtdIns(3,4,5)P3 are drawn in green. (b) Close up of the PtdIns(3,4,5)P3 head group interactions, with dotted lines indicating hydrogen bonds (Reproduced with permission from Ref. [1])
7.2.3 Mechanisms of Effector Activation – Localization and Conformational Change
PtdIns(3,4,5)P3 in cellular membranes serves to recruit effector proteins from the cytoplasm and anchor them by binding to their PH domains. The action of binding to PtdIns(3,4,5)P3 often induces a conformational change, allowing protein activation and propagation of downstream signaling. The induced conformational change, along with membrane localization, both contribute to activation of PtdIns(3,4,5)P3 effectors.
Activation of Akt by PtdIns(3,4,5)P3 has been studied extensively and is best understood. Binding of PtdIns(3,4,5)P3 to Akt produces a conformational change within the protein that allows for its phosphorylation, and full activation, by PDK1 and mammalian target of rapamycin complex 2 (mTORC2) at residues Thr308 and Ser473, respectively. PDK1 is also bound and recruited to the membrane to allow full Akt activation following growth factor signal. Mutation of the PDK1 PH domain inhibits protein kinase B/Akt, leading to small size and insulin resistance in mice [25], by preventing its co-localization with Akt and subsequent activation downstream pathways. Membrane localization is not the only mechanism of Akt activation. Rather, PtdIns(3,4,5)P3 binding also promotes dissociation of the autoinhibitory binding of PH domain to the catalytic domain, thus exposing the catalytic loop residue Thr308 to PDK1 phosphorylation [26, 27].
Regulation of other PH domain-containing proteins is less well characterized. However, complex regulation involving changes in protein conformation and localization, acting in concert, is emerging as a common theme. In the case of PDK1 kinase, initial work has suggested that PtdIns(3,4,5)P3 binding to its PH domain does not affect catalytic activity of the kinase [28]. Hence, inability of the PH domain mutant of PDK1 to promote activation of Akt in response to growth factor signaling primarily reflected defects in membrane localization. The normal cytosolic phosphorylation of S6K by PDK1 in PDK1 PH domain mutants indicates that they retained its kinase activity [29]. However, further detailed analysis suggested a complex role of the PH domain in the regulation of PDK1 activity in the cells. The PH domain has been proposed to both promote Ser241 autophosphorylation, which is required for activity, and to autoinhibit the catalytic activity of phosphorylated kinase under basal conditions, creating a pool of primed kinase [30]. Binding of PtdIns(3,4,5)P3 releases PH domain-dependent inhibition, allowing PDK1 to affect its substrates.
In case of another PH-domain containing factor, Grp1, elements proximal to PH domain have been shown to potently autoinhibit the Sec7 exchange domain of this Arf6 GEF. Membrane localization through PtdIns(3,4,5)P3 binding in concert with complex formation with membrane-targeted Arf6-GTP was shown to result in full catalytic activation of Grp1 towards Arf6-GDP molecules [31].
7.3 Cellular Processes Controlled by PtdIns(3,4,5)P3
PtdIns(3,4,5)P3 is directly involved in a variety of processes within mammalian cells, including cell cycle progression, regulation of cell death, cytoskeleton rearrangement, chemotaxis, and metabolic control. The role of PtdIns(3,4,5)P3 in both plasma and nuclear membranes has been identified. As new cutting edge cell imaging techniques have become widely available, increasingly intricate roles and mechanisms of action for PtdIns(3,4,5)P3 are being elucidated. It is clear that tight regulation of PtdIns(3,4,5)P3 is fundamental to cell function, and deregulation along any of its effector pathways leads to pathogenesis, including cancer, autoimmune disease, cardiovascular problems, and diabetes.
7.3.1 Cellular Growth, Proliferation, and Apoptosis
Regulation of cell growth is one of the key functions of PtdIns(3,4,5)P3 pathway. The growth of cells requires coordinated regulation of proliferation, cell death, and metabolism, all processes involving signaling from PI3K.
7.3.1.1 Progression Through the Cell Cycle
During interphase of the cell cycle, cells increase in size, synthesize lipids and proteins, and replicate DNA, before the cell can enter the division phase, also known as mitosis (or meiosis in the case of reproductive cells). PtdIns(3,4,5)P3 is involved in the regulation of the critical factors (cyclins and cyclin-dependent kinase (CDK) inhibitors) involved in the regulation of the progression through interphase. The necessary conditions for a cell to transition from the G1 checkpoint to the S phase of interphase are dependent in part on the activity of Forkhead-box Class O (FoxO) transcription factors in the nucleus, which are responsible for the repression of cyclins D1 and D2 as well as the transcription of CDK inhibitors, including p21Cip1 and p27Kip1 [32]. Cyclins make up of a family of proteins that oscillate throughout the cell cycle, controlling its progression via interactions with CDKs. Cyclin D is G1/S-specific molecule and is a regulatory subunit of CDK4 and CDK6 [33]. FoxOs also promote the synthesis of cyclin G2, an atypical cyclin highly expressed in quiescent cells.
The binding of growth factors activates the PI3K/PtdIns(3,4,5)P3/Akt pathway, followed by a translocation of active Akt to the nucleus, where it phosphorylates FoxO and promotes its exclusion from the nuclei [34]. Phosphorylation of different FoxOs by Akt occurs at the conserved T1, S1, and S2 residues and promoting the coordinated action of nuclear export machinery (Crm1, Ran GTPase) and 14-3-3 regulatory proteins resulting in the export FoxO into the cytosol and preventing the phosphorylated protein from re-entering the nucleus. Absence of FoxO proteins from the nuclei releases inhibition of cyclin D synthesis, while also decreasing levels of CDK inhibitors (p21, p27, etc.), thus, triggering a G1-S phase transition [35].
Another mechanism involved in the promotion of proliferation by Akt is its direct phosphorylation of p21Cip1 at Thr145 [36]. Interestingly, Akt signaling has been recently shown to also increase expression of cell cycle inhibitor p57Kip2, limiting proliferative responses of breast epithelial cells to insulin and IGF-1, thus, highlighting a system of checks and balances established by Akt in controlling proliferative responses by growth factors [37].
7.3.1.2 Cell Growth
The majority of cell types spend most of their cell cycle growing in size and preparing to proliferate. Nutrients and growth factors in the extracellular matrix initiate the PI3K/PtdIns(3,4,5)P3 pathway, leading to increased biosynthesis of lipids and proteins, which are required for rapid cell growth.
PtdIns(3,4,5)P3-activated Akt phosphorylates and inhibits the action of tuberin, which binds to hamartin, generating a protein complex known as the tuberous sclerosis complex (TSC). The TSC complex suppresses activity of Complex 1 of mTOR (mTORC1) [38]. Two of the best studied downstream effectors of mTORC1 are p70S6Ks, AGC kinases required for cell growth and progression through G1, and 4EBP1, which are inactivated in the absence of mTORC1 activity through the binding to eukaryotic initiation factor 3 (eIF3) [39]. Activated mTORC1 binds to eIF3 and phosphorylates p70S6K and 4EBP1, relieving their inhibition. p70S6K, in turn, phosphorylates multiple effectors required for cell growth and protein biosynthesis including ribosomal S6, promoting translation of a subset of mRNAs containing an oligopyrimidine tract at the 5′ end [40]. Another important target of mTORC1 is 4EBP1, which dissociates from its target eIF4E upon phosphorylation, leading to an increase in cap-dependent translation.
Though regulation of translation is an important function of mTORC1 on cell growth, it is not the only contribution. It has also been linked to the control of the availability of endogenously produced amino acids needed for biosynthesis, mitochondrial biogenesis, and de novo lipogenesis [41, 42].
Cellular metabolism is also altered in the transition of cells from quiescence to rapid proliferation. Activation of PI3K/PtdIns(3,4,5)P3/Akt pathway plays a major role in these changes as well, activating glucose uptake, promoting aerobic glycolysis, and suppressing fatty acid oxidation, thus preserving a source of lipids for rapid membrane expansion [41, 42].
7.3.1.3 Cell Survival and Apoptosis
PI3K, acting through a PtdIns(3,4,5)P3-dependent activation of Akt kinases, provides one of the most important growth and survival signaling circuits in a wide range of cell types. Overactivation of PI3K/PtdIns(3,4,5)P3/Akt signaling plays a central role in the enhanced proliferation and reduced cell death of tumor cells (see below). PI3K signaling also plays a key role in mediating growth factor-dependent “tonic” survival signals in many lineages of normal cells. For example, important early insights into the growth and survival signaling by PI3K and Akt were obtained in the models of interleukin-dependent survival of hematopoietic cells [43–46].
Akt has many well-characterized connections to the basic apoptotic machinery. Early findings suggested that Akt phosphorylates the pro-apoptotic BH3-only Bcl-2 family member Bad, leading to its retention in the cytosol through the binding to 14-3-3 and blocking translocation to mitochondria. Thus, interaction with other Bcl-2 family members is prevented resulting in the inhibition of the mitochondrial step in apoptosis activation [44, 47]. Notably, Akt phosphorylation of Ser-136 residues of Bad, acts in concert with other pro-survival kinases, such as A-kinase anchor proteins, p90 ribosomal S6 kinase and Protein kinase A, to control pro-apoptotic activity of Bad [47–52]. Another critical role of Akt in apoptosis is mediated by the phosphorylation of FoxO transcription factors, including ubiquitously expressed FoxO1, FoxO3, and FoxO4 [53–55]. FoxOs promote transcription and synthesis of a variety of proteins involved in apoptosis, including TNFα family members TRAIL and Fas-ligand, and pro-apoptotic BH3-only family members BIM and PUMA [32].
Glycogen synthase kinase 3 (GSK3) is another class of direct Akt target, playing important and complex roles in the regulation of cell death as well as many other cellular functions. Energy metabolism, transcription, microtubule dynamics and Wnt and Hedgehog pathways are all influenced by GSK3 activity [56]. Akt phosphorylates GSK3α/β on Ser-21/9 residues respectively, targeting them for ubiquitination and degradation. Akt has also been shown to directly promote nuclear factor (NF)-κB activation in response to growth factors and TNFα activation through association with IκB kinase (IKK) complex and subsequent phosphorylation of the Thr23 residue of IKKα [57, 58].
Akt is not the only PtdIns(3,4,5)P3 target involved in the control of cell growth and survival. Bruton’s tyrosine kinase (Btk), expressed in B lymphocytes, is recruited into activated B-cell receptor “signalosome” in a PtdIns(3,4,5)P3-dependent manner where it undergoes activating phosphorylation by the Src family Lyn and Syk kinases [59]. Btk, in turn, activates phospholipase C γ (PLCγ), which regulates Ca2+ mobilization and activation of mitogen-activated protein kinase (MAPK) and NF-κB pathways [60]. Activation of NF-κB in a protein kinase C β (PKCβ)-dependent manner is one of the more important mechanisms of cell survival regulation by Btk [61]. Activity of Btk is required for cell survival during development and provides “tonic” pro-survival signaling in mature resting B cells [62, 63]. Btk has been also reported to promote survival macrophages, but only following stimulation with lipopolysaccharide (LPS) or interferon-γ (IFNγ)[64].
PDK1 is another PtdIns(3,4,5)P3 binding kinase critically involved in the regulation of cell survival [65]. PDK1 plays a key role in the survival of cancer cells with perturbed PI3K signaling, which is mediated by Serum/glucocorticoid regulated kinase 3 (SGK3), rather than Akt activation [66]. In general, PDK1 regulates a large number of downstream kinases linked to the regulation of growth and survival, including Akt, several PKC isoforms, p70S6Ks and SGKs. However, only Akt and SGK3 have been linked to date to PDK1 signaling in plasma membrane, indicating the involvement of PtdIns(3,4,5)P3.
7.3.2 Cytoskeleton Rearrangement and Chemotaxis
Human cell migration is an important process beginning with conception and continuing throughout adulthood. Cell migration is a critical process in a variety of biological responses including cell renewal and tissue repair, immune and inflammatory responses, and angiogenesis. Aberrant changes in cell migration directly contribute to a variety of pathologies. Most importantly, tumor vascularization, aberrant immune responses, osteoporosis, chronic inflammatory diseases, and loss of correct neuronal network formation are all characterized by dysregulation of cellular migration. In most cells, cell migration is highly dependent on the actin cytoskeleton [67]. PtdIns(3,4,5)P3 along with other regulators such as cAMP and cGMP, is the key signaling molecule contributing to the regulation of polymerization and rearrangement of the actin cytoskeleton that occurs during chemotaxis, with a marked enrichment of PtdIns(3,4,5)P3 at the leading edge of migrating cells [68].
The appropriate direction of migration is established by receptor molecules at the cell surface, which sense the locations and intensities of extracellular signals, subsequent activation of the receptors and signal propagation leads to a rearrangement of the actin cytoskeleton. This rearrangement is regulated by Rho GTPases, a subfamily of small signaling G-proteins in the Ras superfamily, including Rho, Rac, and Cdc42 [69]. Activation of PI3K by extracellular cues initiates binding of PtdIns(3,4,5)P3 to GEFs, which, in turn, activate Rho GTPases. Formation of protrusions at the front of the migrating cell, specifically lamellipodia and filopodia, are stimulated by Rac and Cdc42 respectively, while Rho is responsible for the cells posterior retraction [67]. Activation of Cdc42 and Rac has been shown to decrease the activity of Rho at the leading edge, as well as enhance PI3K activity. Cdc42 is also implicated in microtubule growth and recruitment of vesicles to the leading edge, a process dependent on the localization of the microtubule-organizing center and the Golgi apparatus between the nucleus and the front of the cell [70].
The final downstream effectors of Rho GTPase signaling are the Wiscott-Aldrich syndrome protein (WASP) and the WASP verprolin homologous proteins (WAVE), which form links between the GTPases and the actin cytoskeleton. PtdIns(4,5)P2-activated WASP binds via its newly exposed VCA (verprolin homology, cofilin homology and acidic) region to the actin-related protein (Arp)2/3 complex, leading to rapid actin polymerization [67]. Of the three WAVE isoforms present in human tissues, WAVE2 is ubiquitously expressed, localizes to the plasma membrane, possibly by specific binding to PtdIns(3,4,5)P3 to its basic region (rather than through a canonical PH domain), and has been shown to be critical for lamellipodia formation at the leading edge of the cell [69]. More recent evidence indicated that PtdIns(3,4,5)P3 acts in concert with the microtubule binding complex of EB1 and stathmin in promoting localization of WAVE2 to the leading edge of the cell [71]. Furthermore, another PtdIns(3,4,5)P3 target IRSp53, rather than WAVE2 itself, may directly interact with PtdIns(3,4,5)P3 and is responsible for membrane localization of WAVE2, its constitutive binding partner [71].
Regulation of actin polymerization is not the only contribution of PtdIns(3,4,5)P3 to directional migration. The actin motor myosin has also been found to interact with PtdIns(3,4,5)P3 and contributes to lamellipodia formation. Non-muscle myosin IIA [72], several Class I myosins in Dictyostelium and Class IF myosin in mammalian cells are all able to bind PtdIns(3,4,5)P3 through their tail homology domain 1 [73].
An impressive aspect of cell migration is the ability of cells to respond to very shallow chemical gradients, at times experiencing less than 10 % variation in concentration between the leading and trailing edge of the cell. It is thought that this is possible due to positive feedback loops that amplify the signal at the leading edge of the cell, as well as sequestration of PTEN to the sides and rear of the cell, resulting in an anterior focusing the Rho GTPase signaling [70].
It is also worth noting that Akt, PDK1, and PTEN are all also implicated in the regulation of cell migration and motility. For example, Akt1 has been shown to inhibit cell migration through phosphorylation of actin-binding factor, palladin, as well as through the regulation of nuclear factors of activated T-cells, Erk, and TSC2. Alternatively, Akt2-specific regulation of β1 integrin can promote cell migration [74]. Pdk1 −/− cells display defects in cell migration [75]. The mechanisms of this regulation have been proposed to involve direct binding of PDK1 to ROCK1 and PKN kinases, the latter being a direct substrate of PDK1 [76, 77]. However, the role of PtdIns(3,4,5)P3 in the regulation of this function of PDK1 has not been investigated. Curiously, while PTEN certainly contributes to the regulation of migration through the control of PtdIns(3,4,5)P3, its lipid-independent protein phosphatase activity towards Focal adhesion kinase (FAK) and SHC-transforming protein 1 (Shc) has also been proposed to play an important role in the regulation of cell migration and metastasis [78]. These data establish very profound, but complex roles of PtdIns(3,4,5)P3 and its effectors in the regulation of cell migration, motility, chemotaxis, and metastasis.
7.3.3 Nuclear Function
The discovery of the existence of PtdIns(3,4,5)P3 in cellular membranes other than the plasma membrane has led to an investigation of the role of this lipid in other cellular compartments, especially in the nucleus. Components of the PI3K signaling cascade, including PI3K itself, Akt, and PDK1, have been detected in nuclear membranes. In one reported mechanism, nerve growth factor (NGF) stimulation was found to activate PI3K through the activity of nuclear GTPase, PIKE, acting as an equivalent to the cytoplasmic PI3K activator, Ras. PtdIns(3,4,5)P3 in the nuclear membrane plays a critical role in several processes, including cell survival, cell cycle regulation, and DNA repair [79]. Similar to the plasma membrane pool, dephosphorylation of PtdIns(3,4,5)P3 is coordinated by PTEN and SHIP phosphatases, although the nuclear pool of PtdIns(3,4,5)P3 may be less sensitive to PTEN compared to that observed at the plasma membrane [80]. It is interesting to note that while PTEN is found localized to the nucleus in many primary cells, its nuclear pool is dramatically reduced in a number of tumor cells, including exocrine pancreatic tumors and melanomas [81].
It has been established that following growth factor stimulation at the plasma membrane, activated Akt detaches from the plasma membrane and translocates to the nucleus. Nuclear Akt appears to retain activity independently of PtdIns(3,4,5)P3, and was shown to promote cell survival, in part by direct interaction with the nuclear PtdIns(3,4,5)P3 target, nucleophosmin (NPM)/B23. NGF-stimulation promotes PtdIns(3,4,5)P3 binding to B23, recruiting the protein from the nucleoli to the nucleosomes, where it can interact with and stabilize nuclear Akt [82]. Nuclear PtdIns(3,4,5)P3, Akt, and B23 have been shown to protect PC12 cells from apoptosis via the formation of a complex capable of inhibiting DNA fragmentation activity of caspase-activating DNAse (CAD). It is also clear that the spatial and temporal dynamics of PtdIns(3,4,5)P3 following NGF stimulation are critical to the interaction between B23 and Akt, and deregulation of these dynamics leads to impaired biological function [83].
Class I PI3Kβ is also involved in the DNA damage response (DDR) that eukaryotic cells utilize to repair double-stranded breaks (DSB), which are among the most disastrous lesions that affect the human genome. Experiments carried out on irradiated NIH 3 T3 cells revealed that PI3Kβ and PtdIns(3,4,5)P3 localize to damaged DNA sites, and are necessary for the early detection of DSB. PI3Kβ functions to promote recruitment of the Mre11-Rad50-NbsI (MRN) complex, of which Nbs1 is considered the earliest sensor of DNA damage, and is critical for the proper association of the complex. It was also shown that PI3Kβ is necessary for the activation of G2/M cell cycle arrest, during which DNA damage is assessed and repaired, or else apoptosis is induced. Loss of PI3Kβ function results in an impairment of MRN recruitment and inefficient G2/M arrest, resulting in an accumulation of DSB in irradiated cells. While the exact role of local PtdIns(3,4,5)P3 accumulation to DSB sites is not yet completely understood, it is thought that besides its normal role in protein recruitment, it may serve to stabilize DNA in an open conformation in order to facilitate the DDR, either by repelling the negatively charged DNA strands, or by recruiting positively charged histones to chromatin [84].
Finally, a role of the nuclear PtdIns(3,4,5)P3 pool in the differentiation of myeloid cells through a mechanism involving association of the p85 subunit of PI3K with tyrosine phosphorylated Vav1 GEF has also been proposed [85]. Notably, Vav1 is a PH domain-containing protein, which can be activated by PtdIns(3,4,5)P3 in vitro [86, 87]. Novel nuclear PtdIns(3,4,5)P3 targets continue to emerge, making it clear that it has important functions in the control of nuclear processes.
7.3.4 Neuronal Development and Function
The typical characteristic morphology of a neuronal cell consists of one long axonal process, through which the signal is transmitted, and several shorter tapered dendrites, through which neighboring cells activate the neuron. Once a neuronal progenitor cell attaches to the substratum, a five-stage process of neuronal differentiation is initiated [88]. The first stage involves the formation of several lamellipodia along the surface of the cell, which in stage 2 develops into immature neurites of approximately equal length. During the third stage, the neuron becomes polarized with one of the immature neurites lengthening rapidly into a long neurite possessing axonal characteristics (i.e., growth cone). Following development of the axon, the neuron enters the fourth stage, in which the remaining neurites transform into dendrites and all processes continue to mature. In the fifth and final stage, the neuron forms synaptic connections with surrounding neurons and a neuronal network is established [89].
While the exact trigger for the development of a single neurite into a mature axon remains largely unknown, it has been shown that PI3K/PtdIns(3,4,5)P3/Akt signaling is involved in this process, as well as in the maintenance of neuronal polarity [88]. Akt regulates neuronal polarity by inhibiting GSK3β. Inhibition of GSK3β occurs equally among all the neurites during the first two stages of development. During the third stage, Akt localizes to the cell body (the soma) and to tip of the neurite newly designated as the axon. This results in the inhibition of GSK3β primarily at the axonal tip. It has been experimentally shown that the expression of constitutively active Akt throughout the neuron leads to the formation of multiple axons [90] and inhibiting GSK3β in existing dendrites causes their conversion to axons [88]. Thus, spatially-specific accumulation of PtdIns(3,4,5)P3 and activation of Akt is critical for neuronal polarization and development of neuronal networks.
The activation of Akt at the distal tip of neurites is dependent on the local accumulation of PtdIns(3,4,5)P3. Transport of PtdIns(3,4,5)P3 to the neurite tip occurs via guanylate kinase-associated kinesin (GAKIN), a member of the kinesin-3 family of microtubule-based motor proteins, which binds the PtdIns(3,4,5)P3 binding protein (PtdIns(3,4,5)P3-BP), also called centaurin-α, via its forkhead-associated (FHA) domain. PtdIns(3,4,5)P3-BP exhibits ADP-ribosylating factor GTPase activating protein (Arf GAP) activity, and contains two PH domains through which they specifically interact with PtdIns(3,4,5)P3. Using in vitro motility assays, it has been shown that the accumulation of PtdIns(3,4,5)P3 at the distal tip of developing neurons occurs at least in part via transport of PtdIns(3,4,5)P3-containing vesicles bound to PtdIns(3,4,5)P3-BP complexed to GAKIN [91].
7.4 Implications in Disease
As discussed in detail above, PtdIns(3,4,5)P3 mediated signaling is a highly ordered and conserved pathway in a variety of tissues and cells. The relatively low abundance and precise control over the levels of PtdIns(3,4,5)P3 ensure ordered and deliberate activation of downstream targets, which contribute to many core functions in the cells. It is therefore not surprising that a significant number of human diseases can be linked to perturbations in PtdIns(3,4,5)P3 signaling. In fact, approximately 15–30 % of human cancers have been shown to have disruptions in this pathway [92]. Additionally, diabetes, cardiovascular diseases and various inflammatory disorders have also been linked to PtdIns(3,4,5)P3 signaling abnormalities.
In normal cells, proper signaling is reliant on low basal levels of PtdIns(3,4,5)P3 with rapid and controlled production by PI3K in response to extracellular signals. Additionally, a return to basal levels requires the activity of either PTEN or SHIP phosphatases. Activating mutations in PI3K (or its downstream activators) or the loss of PTEN result in elevated cellular levels of PtdIns(3,4,5)P3. Promiscuous and constitutively present PtdIns(3,4,5)P3 is a trademark of tumorigenesis in cells of many tissue types [92]. Alternatively, a loss of PI3K function has been implicated in insulin resistance and the resulting diabetic phenotype [93]. A discussion of specific alterations in PtdIns(3,4,5)P3 signaling and their implications in diseases such as cancer, inflammation, cardiac diseases, and diabetes are presented below.
7.4.1 Cancer
Cancer is a complex disease requiring genetic alterations in the cell resulting in diverse changes including enhanced proliferation, uncontrolled growth, resistance to apoptotic signals, and invasion of surrounding tissues [94]. PtdIns(3,4,5)P3 signaling is implicated in these oncogenic traits and is demonstrably overactivated in a wide range of tumor types [95]. Many excellent in-depth reviews, detailing the role of PI3K signaling pathway in cancer, are available [93, 95, 96], therefore just the major points will be outlined.
As previously discussed, PtdIns(3,4,5)P3 production results in the activation of various downstream proteins, most notably Akt, and is responsible for enhanced cell growth and proliferation, survival, and motility. Upon PtdIns(3,4,5)P3 production and Akt activation, multiple downstream processes promote cell survival and resistance to apoptosis [97]. This occurs through both activation of cellular signaling and transcriptional changes. Akt phosphorylates BAD, thus inhibiting its association with downstream apoptosis factors and decreasing apoptotic tone [98]. Additionally, Akt has been shown to activate NF-κB resulting in transcription of pro-survival and proliferation genes [58, 99]. The FoxO family of transcription factors is phosphorylated by Akt, leading to their sequestration in the cytosol and attenuation of transcription of pro-apoptotic proteins including BIM and Fas-ligand [100], thus, any increase in Akt activity has major implications on the ability of cells to undergo apoptosis. Tumorigenic effects of Akt are not limited to the regulation of cell survival. Activation of Akt pathway contributes to major remodeling of cellular metabolism promoting aerobic glycolysis, reducing β-oxidation of fatty acids and maximizing lipid biosynthesis, all critical for rapid proliferation of cancer cells [101, 102]. Akt is also a major mediator of mTORC1 regulation downstream from growth factor and oncogenic signals, providing a critical link to the robust requirement for protein biosynthesis during proliferation. Inhibition of the p21Cip1 and p27Kip1 CDK inhibitors, through direct phosphorylation by Akt, as well as transcriptional regulation of p27 and Cyclin D1 contribute to increased cellular proliferation [103]. More recent evidence indicates a critical role of Akt2 in epithelial-mesenchymal transition through the regulation of specific miRNAs [104]. These are some of the key examples of the multitude of pro-oncogenic mechanisms controlled by overactivated Akt pathway in cancer cells [95].
Multiple factors contribute to the overactivation of PtdIns(3,4,5)P3 synthesis in cancer cells. Genetic mutations resulting in increased expression and/or activity of RTKs, PI3K, or loss of PTEN activity are some of the most common alterations of the PtdIns(3,4,5)P3 pathway noted to drive tumorigenesis. These mutations can often occur redundantly [92]. RTKs have been repeatedly shown to be dysregulated in a wide variety of cancers [105]. The epidermal growth factor receptor (EGFR) in particular is commonly overexpressed or mutated in gliomas and non-small cell lung cancer [106, 107]. HER2 is overexpressed in approximately 25 % of breast cancer patients and is commonly associated with high recurrence rates and increased mortality [108]. Gain-of-function mutations in the tyrosine-protein kinase c-Kit are evident in gastrointestinal-stromal tumors, acute myeloid leukemia, mast cell leukemia, and melanoma [109, 110]. These are examples of some of the frequent RTK mutations leading to an increased PI3K activation and subsequent PtdIns(3,4,5)P3 production.
Since the discovery of cancer-specific mutations in the PI3KCA gene encoding the p110α subunit of PI3K, oncogenic mechanisms of mutated PI3K has been intensely investigated [111]. Mutations in PI3KCA gene have been implicated in head and neck, squamous cell lung carcinoma, gastric, and cervical cancers [93]. Three hot spot, non-synonymous, missense mutations of amino acids in the helical (E542K and E545K) or kinase (H1047R) domains make up approximately 80 % of the mutations in the PI3KCA gene [112–114]. PtdIns(3,4,5)P3 generation is amplified several fold as a result of increased PI3K kinase activity [115–120]. Increase in PtdIns(3,4,5)P3 concentration is the primary driver of oncogenic signaling [121]. Additionally, constitutively active PI3K uncouples PtdIns(3,4,5)P3 production from growth factor-induced RTK activation. Several mutations in PI3KR1, the gene encoding p85, have also been shown to induce oncogenic transformation in the cell [122]. These mutations typically occur in regions of the protein responsible for p110 binding, thus preventing inhibitory effects on the catalytic activity of p110 [123].
Since the discovery of its role as a potent tumor suppressor in 1997, decreased PTEN function has been correlated with a number of human cancers [124, 125]. Subsequent studies of Pten knockout mice confirmed PTEN tumor suppressor functions in a variety of tissues [126–128]. Additional studies of Pten +/− mice revealed that PTEN functions as a haploinsufficient tumor suppressor gene [129, 130]. Thus, any inhibitory effect on the expression or activity of PTEN has implications in cancer with PTEN levels inversely correlating with disease severity [131, 132]. Additionally, many molecular mechanisms have been associated with decreased expression and/or activity of PTEN, making any correlations of simple step-wise changes in the expression of Pten on cancer progression incomplete [133]. For example, allelic loss, epigenetic silencing, functional mutations, miRNA silencing, and post-translational modifications have all been demonstrated to decrease PTEN expression and/or function. These discoveries have led to the proposal of a “continuum model of tumor suppression” relating overall level and function of PTEN with tumor suppression [131, 134]. The obvious role of PTEN as an inhibitor of PtdIns(3,4,5)P3 signaling is well established, though novel functions of PTEN are continuously being elucidated [133]. For example, nuclear localization of PTEN is associated with tumor suppression independent of its phosphatase activity [80, 135, 136]. Additionally, PTEN has been implicated in fibroblast-mediated shaping of the tumor microenvironment [137, 138].
While Akt plays a critical role in PtdIns(3,4,5)P3 contribution to tumorigenesis, other mechanisms contribute as well. The contribution of PtdIns(3,4,5)P3 to the increased cell motility through regulation of the activity of small GTPases and actin cytoskeleton remodeling should not be discounted. Recent siRNA screens revealed, unexpectedly, that PDK1 may be an even more critical driver than Akt in PI3KCA tumors. This has been linked to the Akt-independent regulation SGK3 as mentioned above [66].
The obvious role of PtdIns(3,4,5)P3 signaling in cancer has lead to a dramatic increase in research into the mechanisms of increased PtdIns(3,4,5)P3 production and expression, while therapies targeting this pathway are concomitantly being developed. Several drugs are currently on the market targeting RTK activity upstream of PtdIns(3,4,5)P3 production and there is a race to develop isoform specific inhibitors of PI3K. Additionally, increased PtdIns(3,4,5)P3 signaling has been associated with drug resistance in tumors, therefore, drugging this pathway is extremely important clinically [139, 140]. Further discussion of these efforts will be presented below.
7.4.2 Inflammation
There are four different isoforms of Class I PI3K, depending on the identity of the catalytic subunit. Class IA PI3Kα and PI3Kβ isoforms are ubiquitously expressed, while class IA PI3Kδ and class IB PI3Kγ are predominately expressed in the hematopoietic cells [141]. As a result of their restricted expression, it has been proposed that these isoforms are important regulators in different populations of immune cells including neutrophils, macrophages, mast cells, eosinophils, T-cells, and B-cells. Current research indicates that PI3Kδ and PI3Kγ-mediated production of PtdIns(3,4,5)P3 is an important druggable target in inflammatory diseases [142]. In the immune system, PtdIns(3,4,5)P3 signaling is initiated in response to antigen, cytokine, and chemokine receptors [19]. Increased PtdIns(3,4,5)P3 signaling results in differing phenotypes depending on the cell type.
Neutrophils and macrophages respond to microbial invasion and are crucial players in inflammatory reactions. In response to inflammatory signals, these cells migrate from the circulation to the site on injury through a process known as chemotaxis [143]. Extracellular chemokine signaling results in the local activation of cellular receptors translating into dramatic localized increases in PtdIns(3,4,5)P3. PtdIns(3,4,5)P3 then recruits and activates GEFs for Rac and Arf GTPases, promoting actin cytoskeletal rearrangement and directional cell movement [144]. Several studies have indicated that this process is dependent on PI3Kγ [145–147]. The precise role of PI3Kδ is still unclear. An isoform specific inhibitor of PI3Kδ was shown to inhibit chemotaxis in neutrophils [148]. On the other hand, subsequent studies have supported the function of PI3Kγ as the primary isoform responsible for cellular migration in response to chemoattractants [149]. PTEN stability and activity is essential for proper inflammatory cell migration, further illustrating the importance of PtdIns(3,4,5)P3 signaling [150]. In addition to chemotaxis, the production of reactive oxygen species (ROS) at the site of inflammation also depends on PtdIns(3,4,5)P3 signaling [145–147, 151]. PDK1 and Akt are both implicated in the phosphorylation of various NADPH oxidase proteins leading to their assembly, subsequent production, and release of ROS [152]. Increased neutrophil and macrophage activation has been associated with atherosclerosis, lupus, and rheumatoid arthritis indicating PtdIns(3,4,5)P3 signaling as a potential druggable target in these inflammatory diseases [153–155].
Mast cells are an additional player in mediating inflammatory activities in response to infection or parasites and are important in amplifying adaptive immunity [156]. These cells are perhaps best known for their detrimental effects in allergic diseases [146]. In response to allergens, the high-affinity IgE receptor is crosslinked and immunoreceptor tyrosine-based activation motifs are phosphorylated. Class I PI3Ks are then activated upon binding to the phosphorylated immunoreceptor tyrosine-based activation motifs. PtdIns(3,4,5)P3 production results in the activation of Btk and, subsequently, PLCγ, ultimately initiating the opening of plasma membrane Ca2+ channels and granule release [157]. Initial degranulation and release of cytotoxic effector molecules is dependent on PI3Kδ activity while subsequent waves of degranulation are dependent on PI3Kγ [158, 159]. In addition, shRNA knockdown or genetic deletion of PTEN increased PtdIns(3,4,5)P3 signaling in mast cells leading to mastocytosis and heightened allergic responses in mice [160, 161]. Shenker et al. have demonstrated that a chimeric toxin containing a PtdIns(3,4,5)P3 phosphatase was able to inhibit mast cell degranulation, further illustrating the importance of this pathway in mast cell pro-inflammatory responses [162]. Increased PtdIns(3,4,5)P3 signaling in mast cells has implications in a variety of immune disorders including allergic disease, asthma, anaphylaxis, autoimmunity, and mastocytosis [163].
T-cells and B-cells play a central role in the adaptive immune responses. T-cells are responsible for cell-mediated immunity while B-cells produce antibodies and comprise humoral immunity. Class I PI3K have been shown to be activated in response to T-cell receptors, cytokines such as IL-2, IL-4, IL-7 and IFNγ, and CD28 signaling [16]. PI3Ks are also essential for proper lymphocyte development [164, 165]. Okkenhaug and Fruman review the importance of PI3K in the development of lymphocytes, in particular, illustrating the importance of p110δ [165]. Thymocyte development is dependent on both class IA and class IB PI3Ks. Loss of class IA PI3Kδ results in aberrant pre-T-cell receptor signaling, though it has no effect on overall thymocyte numbers [166]. PI3Kγ knockout mice display an increase in thymocyte apoptosis and double knockout (PI3Kδ/γ negative) mice display a dramatic reduction in thymocyte numbers [167, 168]. Additionally, PI3Kδ regulates the differentiation and expansion of helper T-cells [169]. The role of PtdIns(3,4,5)P3 in directional lymphocyte migration is still a matter of debate, but it has been suggested that PI3Kγ mediates chemotaxis in T-cells while PI3Kδ is responsible for B-cell migration [170, 171]. Notably, PI3Kγ has not been implicated in B-cell development or survival and all PtdIns(3,4,5)P3 signaling has been shown to be dependent on PI3Kδ [172].
Counterintuitively, both increases and decreases in PtdIns(3,4,5)P3 signaling have been implicated in the development of autoimmunity [173–175]. This is likely due to the alterations in the attenuation of peripheral immune functions mediated by regulatory T-cells. Several studies have linked the contribution of PtdIns(3,4,5)P3 signaling in autoimmunity to its regulation of functions of regulatory T-cells (reviewed in [19]).
The evidence outlined above gives plenty of weight to the importance of PtdIns(3,4,5)P3 signaling in immune function and disease. Current research in this area is focused on elucidating the role and importance of PI3K isoforms and effects of PTEN/SHIP activity on the immune system, in particular, defining the areas where drug intervention could provide the most benefit, given the complex role of PI3K signaling in immune regulation as well as in many other tissues [157, 176]. Targeting the correct PI3K isoform is critical and several isoform-specific inhibitors are currently under clinical investigation for various inflammatory disorders [142].
7.4.3 Cardiovascular Disease
PtdIns(3,4,5)P3 plays an important role in cardiac physiology and pathophysiology of heart disease. PI3K and PTEN are expressed throughout the heart including cardiomyocytes, fibroblasts, endothelial cells, and vascular smooth muscle cells [177]. Cardiac PtdIns(3,4,5)P3 signaling has been shown to have an important role in cell survival, hypertrophy, contractility, metabolism, and mechanotransduction [178]. If normal PtdIns(3,4,5)P3 signaling is enhanced through either overactivation of PI3K or a loss of PTEN, myocardial hypertrophy and decreased contractility can result, thus impairing normal cardiac function. In contrast, a decrease in PtdIns(3,4,5)P3 signaling results in increased areas myocardial infraction and prevents myocardial preconditioning.
Class IA and class IB PI3Ks have both been demonstrated to have functions in cardiac PtdIns(3,4,5)P3 signaling. Class IA PI3K catalytic isoforms p110α and p110β are expressed in the heart and vasculature [179–181] while the class IB isoform p110γ is found in cardiomyocytes, cardiac fibroblasts, vascular smooth muscle cells, and endothelial cells [177, 182, 183]. Each isoform has a distinct role in cardiac regulation and function. Class IA isoforms are thought to regulate physiologic growth mediated by activation of RTKs [177, 179, 184]. Class IB γ isoform is required for proper contractility of the myocardium [179, 185]. Termination of PtdIns(3,4,5)P3 signaling in the heart is dependent on the function of PTEN. Loss of PTEN leads to increased PtdIns(3,4,5)P3 signaling ultimately resulting in various pathologies including hypertrophy and decreased contractility [177, 179, 186, 187]. The downstream targets of PtdIns(3,4,5)P3 signaling described above are similarly involved in cardiac regulation. In particular, Akt/GSK3 pathway plays an important role in heart disease [188, 189].
All three Akt isoforms are expressed in the heart, though Akt1 and Akt2 are the most prevalent [179, 190, 191]. Normal Akt activation in the myocardium is required for cell proliferation, metabolism and inhibition of apoptosis [192]. A transgenic mouse model with cardiac specific knockdown of Akt1 resulted in a reduced number of cardiomyocytes and smaller overall heart sizes, though there was no effect seen on contractility [193]. At the same time, overexpression or increased Akt activation can result in cardiac hypertrophy and abnormal contractility [193–195].
Both GSK3 isoforms, GSK3α and GSK3β are expressed in the heart though more emphasis has been placed on the impact of GSK3β [189]. GSKβ is a constitutively active enzyme, phosphorylating and inactivating glycogen synthase and, thus, inhibiting glycogen synthesis [196]. Upon PtdIns(3,4,5)P3 production and Akt activation, GSK3β is phosphorylated and rendered inactive, thus promoting glycogen synthesis. GSK3β is also shown to be a regulator of important transcription factors, in particular, nuclear factors of activated T-cells, β-catenin, CREB, and the Jun family of proteins [189]. Overall, constituently active GSK3β has been shown to act as a hypertrophic restraint in the heart [197], while induction of hypertrophy by PtdIns(3,4,5)P3 has been linked to the inhibition of GSK3β [189].
As discussed above, PtdIns(3,4,5)P3 signal alteration has variable effects on the physiology of the myocardium. PI3Kα signaling in response to exercise has been shown to provide protective effects in patients with heart failure [198]. Notably, myocardial ischemic preconditioning, a process whereby a brief period of ischemia is able to protect the heart from further ischemic events, has been linked to the changes in PI3K pathway [199–201]. This occurs through the reduction in apoptosis and increased proliferation of preconditioned cells. Activation of PtdIns(3,4,5)P3 signaling pathways, or loss of PTEN, has been shown to mimic the cardio-protective nature of ischemic preconditioning [200, 202–204]. Additional heart pathologies such as diabetic cardiomyopathy [205], adriamycin-induced cardiomyopathy [206], chronic β-adrenergic receptor stimulation [184], or pressure overload induced hypertrophy [207] have all been shown to involve altered PI3K/Akt signaling.
Overall, similar to immune regulation, PtdIns(3,4,5)P3 signaling pathway has multiple effects on the regulation and pathophysiology of the cardiac system. There is still much work to be done to unravel the exact role of each PI3K isoform in the pathophysiology of different heart diseases. Particular care should be taken to avoid cardiovascular toxicity following prolonged exposure to various isoform specific PI3K inhibitors [208].
7.4.4 Diabetes
A number of tissues are involved in the complex process of metabolic regulation. Liver, skeletal muscle, adipose tissue, pancreatic beta cells, and several CNS neuronal populations are all involved in glucose sensing and regulating insulin signaling. Insulin receptors (IR) are coupled to downstream effects in part by inducing PI3K production of PtdIns(3,4,5)P3 [93]. Recent research has implicated the generation and breakdown of PtdIns(3,4,5)P3 as a prime mediator of insulin receptor signaling and alterations in this pathway have been shown to result in metabolic syndromes and the diabetic phenotype [209]. Potential inhibition of glucose metabolism is one of the most important considerations in developing PI3K inhibitors.
Insulin receptors belong to the RTK class of receptors. Upon activation by their ligands and subsequent dimerization, IR and IGF receptors phosphorylate insulin receptor substrate (IRS) proteins [210]. Several IRS proteins displaying differences in tissue expression have been identified [211]. Tyrosine phosphorylated IRS proteins bind to the SH2 domains of the regulatory p85 subunit of Class IA PI3Ks. The p110α catalytic subunit is thought to be the primary subunit involved in IR/PI3K metabolic signaling as inactivation of this subunit results in embryonic death. Heterozygote mice display small body size, insulin resistance, and glucose intolerance [212]. Similarly, isoform-selective p110α inhibitors blocked IR/PI3K signaling, while p110β inhibitors showed little effect [213]. On the other hand, emerging evidence indicates p110β may still play a role by changing the kinetics of PtdIns(3,4,5)P3 production, prolonging insulin induced signaling, resulting in the prolonged Akt activation [214].
Peripheral insulin resistance is thought to be the product of disrupted PI3K signaling in the effector cells [210, 215]. Any mechanism resulting in decreased PtdIns(3,4,5)P3 production or expression can result in diabetic phenotypes. Mice lacking either p110α or p110β die in early embryogenesis while mice heterozygous for p110α and p110β display glucose insensitivity [212, 216]. In contrast, mice lacking PTEN expression in either muscle, adipose, or liver tissue leads to an increase in insulin sensitivity and glucose tolerance [217, 218]. Additionally, studies of SHIP2 heterozygous deletion mice have been shown to result in increased insulin sensitivity indicating SHIP2 is a key regulator of glucose homeostasis [219].
Several reports suggested that decreased expression of p85 regulatory subunit of PI3K resulted in enhanced insulin sensitivity in mice [220]. Subsequent investigation uncovered an IR/PI3K inhibitory function of p85 [221]. p85 is present in excess to p110 in many cell types and free p85 has been shown to sequester activated IRS1 in the cytoplasm, preventing IRS1 from interacting with PI3K at the membrane [222]. Notably, elevated levels of p85 have been found in pregnancy-induced diabetes in women and in skeletal muscles of type-2 diabetic patients [223, 224]. Interestingly, the levels of p110 were not altered indicating the importance of the p85-p110 ratio. At the same time, a complete loss of p85 in either the muscle or liver resulted in compromised insulin signaling in those tissues due to its critical role in linking PI3K to IR [225, 226]. Additionally, chronic inflammation present in adipose tissue of obese patients leads to release of TNFα, which can activate JNK and block IRS thus decreasing PtdIns(3,4,5)P3 signaling [227, 228]. p70S6K activation downstream of mTOR is also able to inhibit PtdIns(3,4,5)P3 signaling by phosphorylation of IRS-1 thus providing a negative feedback and contributing to insulin resistance [229, 230].
Following activation of IR/PI3K and production of PtdIns(3,4,5)P3, a variety of downstream events are regulated by Akt, including GLUT4-mediated glucose uptake [231], glycogen synthase activation through GSK3 inhibition [196], and inhibition of FoxO-mediated gene transcription [232]. In particular, Akt2 has been shown to play a key role in glucose homeostasis. Disruption of Akt2 in mice results in the diabetic phenotype [233]. In addition, full activation of Akt in response to insulin signaling is reliant on PtdIns(3,4,5)P3-dependent phosphorylation by PDK1. Genetic mutation of the PH domain of PDK1 in mice resulted in glucose and insulin intolerance, indicating the critical role of PtdIns(3,4,5)P3 in the connection between PDK1 and Akt [25].
PtdIns(3,4,5)P3 presence in the membrane is able to both propagate IR/PI3K signaling and initiate events leading to the attenuation of the signal. O-linked β-N-acetylglucosamine (O-GlcNAc) transferase (OGT) has been recently shown to bind PtdIns(3,4,5)P3 and to translocate to the membrane upon insulin induced PtdIns(3,4,5)P3 production [234]. Once at the membrane, OGT is able to catalyze the addition of GlcNAc to the key insulin signaling intermediates downstream of PtdIns(3,4,5)P3 such as ribosomal proteins. It also competes with other PH-containing proteins for PtdIns(3,4,5)P3 binding at the membrane, resulting in attenuation of their signal [235]. O-GlcNAc levels were also shown to be elevated under hyperglycemic conditions. Another protein with affinity for PtdIns(3,4,5)P3 is prohibitin, a protein shown to bind PtdIns(3,4,5)P3 and modulate insulin signaling in vitro [236].
Although there is ample evidence to implicate perturbed PtdIns(3,4,5)P3 signaling in diabetes, it remains a difficult pathway to directly target therapeutically. The complex pathophysiology of diabetes covers several tissue types, each with individual expression of PI3K isoforms and unique pathway elements. Furthermore, any attempts to increase PtdIns(3,4,5)P3 signaling could have oncogenic implications, as this pathway is highly associated with tumorigenesis. Conversely, any PI3K pathway inhibitors should be analyzed for their potential effects on metabolic signaling.
7.5 Approaches to Target PtdIns(3,4,5)P3 Signaling
The importance of PtdIns(3,4,5)P3 regulation has been described in detail above. Dysregulation of the pathway is implicated in a wide variety of disorders and, therefore, targeting the pathway pharmacologically represents a great promise. Thus, it is not surprising that the development of small-molecule inhibitors of various components in the PtdIns(3,4,5)P3 signaling cascade is a hot topic, especially in the fields of cancer and inflammation. At present there are over 100 clinical trials underway of PI3K inhibitors alone (http://www.clinicaltrials.gov). This does not include clinical trials on drugs targeting other proteins in the PtdIns(3,4,5)P3 pathway, such as mTOR, Akt, or PDK1, or drugs inhibiting upstream targets, such as activity of RTKs. Discussion of upstream inhibition of PtdIns(3,4,5)P3 pathway by targeting RTKs such as EGFR or IGFR has been extensively reviewed elsewhere and will not be covered here [237, 238].
7.5.1 PI3K Inhibitors
The most straightforward method to inhibit PtdIns(3,4,5)P3 signaling is to prevent the PI3K mediated conversion of PtdIns(4,5)P2 to PtdIns(3,4,5)P3. Many small molecule PI3K inhibitors have been developed with several advancing to clinical trials (Fig. 7.4) [96, 239]. Wortmannin, a sterol like fungal metabolite, was the first PI3K inhibitor and one of the most extensively studied. It was initially discovered to have an inhibitory effect on respiratory burst in neutrophils [240]. Subsequent studies illustrated wortmannin functions as an irreversible inhibitor of PI3K [241]. Nonspecific inhibition of other protein kinases, poor stability and high toxicity were all limiting factors preventing its use in the clinic [242, 243]. The first synthetic PI3K inhibitor was developed by Eli Lily in 1994 [244]. LY294002 has been most widely utilized as a research tool delineating PI3K signaling in cells. The clinical development of LY294002 has been limited by low potency and poor aqueous solubility [35, 245].

Structures of several PI3K inhibitors
Second generation PI3K inhibitors based on the structures of wortmannin and LY294002 have been developed and are currently undergoing clinical trials. PX-866, in development by Oncothyreon, is an irreversible PI3K inhibitor based on the structure of wortmannin [243]. It has shown to have good oral bioavailability and an improved toxicity profile in a phase I trial. SF-1126 developed by Semaphore Pharmaceuticals is a LY294002 pro-drug with improved pharmacological properties and tumor targeting [245]. This compound is currently in clinical trials as well.
An emerging strategy in PI3K inhibition is to design isoform specific inhibitors. Previous discussion has demonstrated the importance of individual isoforms in various disorders. By selectively inhibiting the isoform implicated in disease, an improved therapeutic index is possible [142]. Initial efforts focused on specific inhibition of p110α and p110β due to their role in tumorigenesis. However, these isoforms are ubiquitously expressed and play critical roles in normal homeostasis, such as glucose metabolism. Therefore, development of p110δ and p110γ inhibitors has attracted increasing attention [246]. p110δ is an important isoform present in immune cells and is commonly overexpressed in inflammatory disorders and lymphomas. IC-87114 (ICOS Corporation), CAL-101 and CAL-263 (Calistoga Pharmaceuticals) are all specific p110δ inhibitors with 40–300 fold increased potency versus other isoforms [148, 247, 248]. CAL-101 in particular has shown promising clinical results in patients with various hematologic malignancies [249]. CAL-101 and CAL-263 have both entered clinical trials for the treatment of allergic rhinitis, potentially indicating utility in inflammatory diseases. p110γ specific inhibitors were initially thought to be effective against several inflammatory disorders [250]. AS-605240 (Merck Serono) was tested for isoform specific inhibition of p110γ with some success [251]. However, subsequent studies have been disappointing as selectivity to p110γ is insufficient to overcome off-target effects [142].
7.5.2 Inhibitors of Downstream Targets of PtdIns(3,4,5)P3
Targeting specific isoforms of PI3K is only one way to improve therapeutic index and tolerability. Several downstream activators of PtdIns(3,4,5)P3 signaling have also been targeted for inhibition. It is thought that targeting the critical components specifically contributing to particular pathologies could lead to improved efficacy and reduced toxicity compared broad PI3K inhibition. Akt and mTOR are the two most commonly targeted proteins downstream of PtdIns(3,4,5)P3 [252], although other proteins, such as PDK1, have been targeted as well.
7.5.2.1 Akt Inhibitors
Akt is one of the primary downstream effectors of PtdIns(3,4,5)P3 signaling contributing towards increased cell survival, proliferation and metabolic alterations in cancer cells. Several inhibitors have been developed that target all three isoforms of Akt (Fig. 7.5). Both Perifosine and triciribine inhibit Akt activation by binding to its PH domain, thus preventing the membrane localization and activation by PDK1 [253, 254]. Both of these agents have seen limited success in the clinic due to poor response rates and adverse events, though there remains potential for use as part of combination therapies [252]. The allosteric inhibitor MK-2206 (Merck) showed promising results in combination therapy in xenograft models and is currently in Phase I trials [239, 255]. GlaxoSmithKlein has also developed two ATP competitive inhibitors of Akt, GSK-690693 and GSK-2141795 [256–258]. GSK-2141795 in particular has shown promising results in several phase I trials.

Structures of several Akt inhibitors
7.5.2.2 mTOR Inhibitors
Rapamycin (Wyeth) is a relatively selective mTORC1 inhibitor originally investigated as an antifungal agent. It has now been clinically approved as an immunosuppressant used to prevent rejection in organ transplantation. Analogs of rapamycin (known as rapalogs) such as temsirolimus (Wyeth) and everolimus (Novartis) have been approved for the treatment of renal cell carcinoma (Fig. 7.6) [259, 260]. Other cancers have lower response rates following treatment of rapalogs, thus indicating the heterogeneity of mTOR signaling in human cancers [252, 260]. mTORC1 is responsible for several but not all of mTOR-dependent functions. It has therefore been proposed that targeting the kinase activity of mTOR is a more effective means of PtdIns(3,4,5)P3 signal inhibition [261, 262]. Very potent class ATP-competitive mTOR inhibitors (Torin-1 and Torin-2) have shown promise in pre-clinical development [263, 264]. Other ATP-competitive inhibitors, AZD-8055 (AstraZeneca), INK-128 (Intellikine), and OSI-027 (OSI Pharmaceuticals) are also under development and currently undergoing clinical trials [252, 265].

Structures of several mTOR inhibitors
7.5.2.3 Dual PI3K/mTOR Inhibitors
Many PI3K inhibitors under development are also active against structurally similar proteins, including mTOR [265]. mTOR is activated by both nutrient signaling and PI3K dependent growth factor signaling. Therefore, inhibition of PI3K does not completely abolish mTOR activity. Additionally, mTOR inhibition can increase PI3K activity through suppression of p70S6K-dependent negative feedback loop [96]. Therefore, strategies targeting both PI3K and mTOR have been pursued and several dual PI3K/mTOR inhibitors have been described (Fig. 7.7) [266]. PI-103 in particular was developed as a dual inhibitor with much preclinical success [267, 268]. Despite its potency, PI-103 has poor solubility and rapid clearance. Therefore several analogs, GDC-0980 and GDC-0941, have been developed by Genentech and are currently in clinical trials [252, 265]. Two other PI3K/mTOR inhibitors are being developed by Novartis, BEZ-235 and BGT-226. BEZ-235 is an orally available ATP-competitive inhibitor with low nanomolar IC50 values against both PI3K and mTOR [269, 270]. Phase I trials have indicated this compound is well tolerated with significant activity against breast cancer cells with high levels of PI3K signaling [271]. BGT-226 has also showed promising results both in vitro and in vivo and the clinical development of this compound is also underway [272, 273].

Structures of several dual PI3K/mTOR inhibitors
7.5.2.4 Btk Inhibitors
Another important target in the treatment of B-cell malignancies and autoimmune arthritis is the PtdIns(3,4,5)P3-dependent Btk. As discussed above, Btk is a member of the Tec family of cytosolic tyrosine kinases and is activated upon recruitment to the inner leaflet of the membrane through interaction with PtdIns(3,4,5)P3 and its PH domain. Two inhibitors of note have been developed targeting Btk, primarily for the treatment of B-cell malignancies, Pharmacyclics’ PCI-32765 and Avila’s AVL-292 (recently purchased by Celgene) [274]. PCI-32765 in particular has advanced through the clinic with positive Phase II results and is also being investigated as a treatment in autoimmune arthritis [275].
7.5.3 PH Domain Inhibitors
Another approach to inhibit PtdIns(3,4,5)P3 signaling involves preventing the interaction of various proteins with membrane associated PtdIns(3,4,5)P3. Blocking this interaction prevents the proper localization and activation of downstream effectors. Research into the structure and binding properties of the PH domain of Akt has provided insights into PH-PtdIns(3,4,5)P3 interaction and development of inhibitors [17, 24, 276]. Inhibiting PtdIns(3,4,5)P3 signaling at this step should universally block downstream activation of PI3K mediators.
PtdIns(3,4,5)P3-PH inhibition can be achieved either by lipid-based antagonists or by non-lipid small molecule antagonists (Fig. 7.8) [265]. Initially, modified water-soluble head groups of PIs, inositol polyphosphates, were investigated for their PtdIns(3,4,5)P3-PH inhibition [277, 278]. Later optimization revealed a compound capable of increased specificity for PDK1 and inhibition of mTOR [279]. The previously discussed perifosine is another lipid based PtdIns(3,4,5)P3-PH inhibitor with specific activity against Akt, preventing recruitment to the membrane. Non-lipid antagonists under investigation include metabolite of triciribine (triciribine-phosphate), NSC-348900, PHT-427 and API-1 [265]. Additionally, a novel class of compounds known as PITENINs has been developed [280, 281]. Chemical modification of the lead compound DM-PIT-1 has resulted in more potent and selective inhibition of Akt-PtdIns(3,4,5)P3 interaction [281, 282].

Structures of several inhibitors of PtdIns(3,4,5)P3 /PH interactions
7.5.4 Drug Resistance
Many in vitro studies have demonstrated that PtdIns(3,4,5)P3 signaling is associated with decreased response to therapy and drug resistance. Constitutively active Akt signaling is implicated in both paclitaxel and TNFα-related apoptosis inducing ligand resistance [140, 283]. Additionally, breast cancer cell lines demonstrating activated HER2/PI3K/Akt signaling were less susceptible to chemotherapeutic apoptosis and inhibiting either PI3K or Akt restored drug sensitivity [284]. Furthermore, clinical evidence implicates PI3K and PTEN mutations with Herceptin resistance in breast cancer [285, 286]. Increased PtdIns(3,4,5)P3 signaling has also been linked to colon cancer resistance to EGFR inhibition [287]. As a result of the increasing evidence implicating PtdIns(3,4,5)P3 signaling in drug resistance, combination therapy strategies involving the targeted inhibition of the PtdIns(3,4,5)P3 pathway and the Raf/MEK/ERK and BRAF pathways are being pursued [288, 289]. Several clinical trials are underway investigating the utility of PtdIns(3,4,5)P3 pathway inhibition in avoiding drug resistance.
7.6 Conclusion
PtdIns(3,4,5)P3 has emerged as an important second messenger in many cellular processes with implications for a wide range of diseases. Genetic studies in mice have illustrated the complexity of PtdIns(3,4,5)P3 formation with various PI3K isoforms uniquely contributing to cellular signal propagation. PTEN expression and regulation has also emerged as an important mediator of PtdIns(3,4,5)P3 activity. Following the formation of PtdIns(3,4,5)P3, effector proteins such as Akt, PDK1, Btk, and others are able to bind to PtdIns(3,4,5)P3 via PH domains and assemble at the inner plasma membrane where they are able to exert their effects resulting in cell survival, proliferation, cytoskeletal rearrangement, and gene expression. To better understand the effects of PtdIns(3,4,5)P3 signaling in human diseases, additional elucidation of PI3K isoform activities needs to be performed. The ubiquitous expression of PI3K in healthy tissue necessitates the use of therapies targeting specific isoforms to reduce off target effects. Additionally, while a lot of progress has been made in understanding the regulation of key PtdIns(3,4,5)P3 effectors, the contribution of many of the putative targets remains to be fully characterized.
As we begin to understand the complex relationships between upstream receptors, PI3K and downstream effectors, new small molecule inhibitors targeted at specific components are being developed with several successfully advancing through the clinic. Development of key biomarkers indicating which elements of the pathway to target will be a key step in the advancement of PtdIns(3,4,5)P3 pathway inhibitors.
References
Hokin MR, Hokin LE (1953) Enzyme secretion and the incoroporation of P32 into phospolipides of pancreas slices. J Biol Chem 203(2):967–977
Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM (1985) Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 315(6016):239–242
Traynor-Kaplan AE, Harris AL, Thompson BL, Taylor P, Sklar LA (1988) An inositol tetrakisphosphate-containing phospholipid in activated neutrophils. Nature 334(6180):353–356
Whitman M, Downes CP, Keeler M, Keller T, Cantley L (1988) Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 332(6165):644–646
Rameh LE, Cantley LC (1999) The role of phosphoinositide 3-kinase lipid products in cell function. J Biol Chem 274(13):8347–8350
Rusten TE, Stenmark H (2006) Analyzing phosphoinositides and their interacting proteins. Nat Method 3(4):251–258
Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD (2001) Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem 70(1):535
Hawkins PT, Jackson TR, Stephens LR (1992) Platelet-derived growth factor stimulates synthesis of Ptdlns(3,4,5)P3 by activating a Ptdlns(4,5)P2 3-OH kinase. Nature 358(6382):157–159
Kavran JM, Klein DE, Lee A, Falasca M, Isakoff SJ, Skolnik EY, Lemmon MA (1998) Specificity and promiscuity in phosphoinositide binding by pleckstrin homology domains. J Biol Chem 273(46):30497–30508
Kwiatkowska K (2010) One lipid, multiple functions: how various pools of PI(4,5)P3 are created in the plasma membrane. Cell Mol Life Sci 67(23):3927–3946
Lemmon MA, Ferguson KM, O’Brien R, Sigler PB, Schlessinger J (1995) Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc Natl Acad Sci U S A 92(23):10472–10476
Martin TFJ (2001) PI(4,5)P2 regulation of surface membrane traffic. Curr Opin Cell Biol 13(4):493–499
Castellano E, Santos E (2011) Functional specificity of Ras isoforms. Genes Cancer 2(3):216–231
Erneux C, Edimo WE, Deneubourg L, Pirson I (2011) SHIP2 multiple functions: a balance between a negative control of PtdIns(3,4,5)P(3) level, a positive control of PtdIns(3,4)P(2) production, and intrinsic docking properties. J Cell Biochem 112(9):2203–2209
Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, Acevedo LM, Manglicmot JRE, Song X, Wrasidlo W, Blair SL, Ginsberg MH, Cheresh DA, Hirsch E, Field SJ, Varner JA (2011) Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3K, a single convergent point promoting tumor inflammation and progression. Cancer Cell 19(6):715–727
Koyasu S (2003) The role of PI3K in immune cells. Nat Immunol 4(4):313–319
Rosen SAJ, Gaffney PRJ, Spiess B, Gould IR (2012) Understanding the relative affinity and specificity of the pleckstrin homology domain of protein kinase B for inositol phosphates. Phys Chem Chem Phys 14(2):929–936
Sun H, Lesche R, Li D-M, Liliental J, Zhang H, Gao J, Gavrilova N, Mueller B, Liu X, Wu H (1999) PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A 96(11):6199–6204
Kashiwada M, Lu P, Rothman P (2007) PIP3 pathway in regulatory T cells and autoimmunity. Immunol Res 39(1):194–224
Wisniewski D, Strife A, Swendeman S, Erdjument-Bromage H, Geromanos S, Kavanaugh WM, Tempst P, Clarkson B (1999) A novel SH2-containing phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase (SHIP2) is constitutively tyrosine phosphorylated and associated with src homologous and collagen gene (SHC) in chronic myelogenous leukemia progenitor cells. Blood 93(8):2707–2720
Blero D, Payrastre B, Schurmans S, Erneux C (2007) Phosphoinositide phosphatases in a network of signalling reactions. Pflugers Arch 455(1):31–44
Agoulnik IU, Hodgson MC, Bowden WA, Ittmann MM (2011) INPP4B: the new kid on the PI3K block. Oncotarget 2(4):321–328
He J, Haney RM, Vora M, Verkhusha VV, Stahelin RV, Kutateladze TG (2008) Molecular mechanism of membrane targeting by the GRP1 PH domain. J Lipid Res 49(8):1807–1815
Thomas CC, Deak M, Alessi DR, van Aalten DMF (2002) High-resolution structure of the pleckstrin homology domain of protein kinase B/Akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr Biol 12(14):1256–1262
Bayascas JR, Wullschleger S, Sakamoto K, García-Martínez JM, Clacher C, Komander D, van Aalten DMF, Boini KM, Lang F, Lipina C, Logie L, Sutherland C, Chudek JA, van Diepen JA, Voshol PJ, Lucocq JM, Alessi DR (2008) Mutation of the PDK1 PH domain inhibits protein kinase B/Akt, leading to small size and insulin resistance. Mol Cell Biol 28(10):3258–3272
Vr C, Alcor D, Laguerre M, Park J, Vojnovic B, Hemmings BA, Downward J, Parker PJ, Larijani B (2007) Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol 5(4):e95
Milburn CC, Deak M, Kelly SM, Price NC, Alessi DR, Van Aalten DMF (2003) Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem J 375(Pt 3):531–538
Currie RA, Walker KS, Gray A, Deak M, Casamayor A, Downes CP, Cohen P, Alessi DR, Lucocq J (1999) Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem J 337(Pt 3):575–583
McManus EJ, Collins BJ, Ashby PR, Prescott AR, Murray-Tait V, Armit LJ, Arthur JSC, Alessi DR (2004) The in vivo role of PtdIns(3,4,5)P3 binding to PDK1 PH domain defined by knockin mutation. EMBO J 23(10):2071–2082
Gao X, Harris TK (2006) Role of the PH domain in regulating in vitro autophosphorylation events required for reconstitution of PDK1 catalytic activity. Bioorg Chem 34(4):200–223
DiNitto JP, Delprato A, Gabe Lee M-T, Cronin TC, Huang S, Guilherme A, Czech MP, Lambright DG (2007) Structural basis and mechanism of autoregulation in 3-phosphoinositide-dependent Grp1 family Arf GTPase exchange factors. Mol Cell 28(4):569–583
van der Vos KE, Coffer PJ (2011) The extending network of FOXO transcriptional target genes. Antioxid Redox Signal 14(4):579–592
Lew DJ, Dulic V, Reed SI (1991) Isolation of three novel human cyclins by rescue of G1 cyclin (Cln) function in yeast. Cell 66(6):1197–1206
Furukawa-Hibi Y, Kobayashi Y, Chen C, Motoyama N (2005) FOXO transcription factors in cell-cycle regulation and the response to oxidative stress. Antioxid Redox Signal 7(5–6):752–760
Marone R, Cmiljanovic V, Giese B, Wymann MP (2008) Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta -Proteins Proteomics 1784(1):159–185
Zhou BP, Liao Y, Xia W, Spohn B, Lee M-H, Hung M-C (2001) Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol 3(3):245–252
Worster DT, Schmelzle T, Solimini NL, Lightcap ES, Millard B, Mills GB, Brugge JS, Albeck JG (2012) Akt and ERK control the proliferative response of mammary epithelial cells to the growth factors IGF-1 and EGF through the cell cycle inhibitor p57Kip2. Sci Signal 5(214):ra19
Manning BD, Cantley LC (2003) United at last: the tuberous sclerosis complex gene products connect the phosphoinositide 3-kinase/Akt pathway to mammalian target of rapamycin (mTOR) signalling. Biochem Soc Trans 31(Pt 3):573–578
Yang Q, Guan KL (2007) Expanding mTOR signaling. Cell Res 17(8):666–681
Fenton TR, Gout IT (2011) Functions and regulation of the 70 kDa ribosomal S6 kinases. Int J Biochem Cell Biol 43(1):47–59
Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21(3):297–308
Ward PS, Thompson CB (2012) Signaling in control of cell growth and metabolism. Cold Spring Harb Perspect Biol 4(7)
Johnson DE (1998) Regulation of survival pathways by IL-3 and induction of apoptosis following IL-3 withdrawal. Front Biosci 3:d313–d324
del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G (1997) Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278(5338):687–689
Songyang Z, Baltimore D, Cantley LC, Kaplan DR, Franke TF (1997) Interleukin 3-dependent survival by the Akt protein kinase. Proc Natl Acad Sci U S A 94(21):11345–11350
Ahmed NN, Grimes HL, Bellacosa A, Chan TO, Tsichlis PN (1997) Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc Natl Acad Sci U S A 94(8):3627–3632
Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME (2000) 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell 6(1):41–51
Lizcano JM, Morrice N, Cohen P (2000) Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem J 349(Pt 2):547–557
Tan Y, Demeter MR, Ruan H, Comb MJ (2000) BAD Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell survival. J Biol Chem 275(33):25865–25869
Harada H, Becknell B, Wilm M, Mann M, Huang LJ, Taylor SS, Scott JD, Korsmeyer SJ (1999) Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol Cell 3(4):413–422
Tan Y, Ruan H, Demeter MR, Comb MJ (1999) p90(RSK) blocks bad-mediated cell death via a protein kinase C-dependent pathway. J Biol Chem 274(49):34859–34867
Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286(5443):1358–1362
Tzivion G, Dobson M, Ramakrishnan G (2011) FoxO transcription factors; regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta 1813(11):1938–1945
Zhang X, Tang N, Hadden TJ, Rishi AK (2011) Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta 1813(11):1978–1986
Hay N (2011) Interplay between FOXO, TOR, and Akt. Biochim Biophys Acta 1813(11):1965–1970
Forde JE, Dale TC (2007) Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol Life Sci 64(15):1930–1944
Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB (1999) NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 401(6748):82–85
Romashkova JA, Makarov SS (1999) NF-kappa B is a target of AKT in anti-apoptotic PDGF signalling. Nature 401(6748):86–90
Genevier HC, Hinshelwood S, Gaspar HB, Rigley KP, Brown D, Saeland S, Rousset F, Levinsky RJ, Callard RE, Kinnon C et al (1994) Expression of Bruton’s tyrosine kinase protein within the B cell lineage. Eur J Immunol 24(12):3100–3105
Buggy JJ, Elias L (2012) Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol 31(2):119–132
Anderson JS, Teutsch M, Dong Z, Wortis HH (1996) An essential role for Bruton’s [corrected] tyrosine kinase in the regulation of B-cell apoptosis. Proc Natl Acad Sci U S A 93(20):10966–10971
Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K (2004) Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell 117(6):787–800
Niiro H, Clark EA (2002) Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol 2(12):945–956
Khare A, Viswanathan B, Gund R, Jain N, Ravindran B, George A, Rath S, Bal V (2011) Role of Bruton’s tyrosine kinase in macrophage apoptosis. Apoptosis 16(4):334–346
Raimondi C, Falasca M (2011) Targeting PDK1 in cancer. Curr Med Chem 18(18):2763–2769
Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, Dunn IF, Schinzel AC, Sandy P, Hoersch S, Sheng Q, Gupta PB, Boehm JS, Reiling JH, Silver S, Lu Y, Stemke-Hale K, Dutta B, Joy C, Sahin AA, Gonzalez-Angulo AM, Lluch A, Rameh LE, Jacks T, Root DE, Lander ES, Mills GB, Hahn WC, Sellers WR, Garraway LA (2009) AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 16(1):21–32
Fernando HS, Kynaston HG, Jiang WG (2009) WASP and WAVE proteins: vital intrinsic regulators of cell motility and their role in cancer. Int J Mol Med 23(2):141–148
Wong W (2012) Focus issue: a cell’s sense of direction. Sci Signal 5(213):eg3
Oikawa T, Yamaguchi H, Itoh T, Kato M, Ijuin T, Yamazaki D, Suetsugu S, Takenawa T (2004) PtdIns(3,4,5)P3 binding is necessary for WAVE2-induced formation of lamellipodia. Nat Cell Biol 6(5):420–426
Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR (2003) Cell migration: integrating signals from front to back. Science 302(5651):1704–1709
Takahashi K, Suzuki K (2010) WAVE2 targeting to phosphatidylinositol 3,4,5-triphosphate mediated by insulin receptor substrate p53 through a complex with WAVE2. Cell Signal 22(11):1708–1716
Morimura S, Suzuki K, Takahashi K (2011) Nonmuscle myosin IIA is required for lamellipodia formation through binding to WAVE2 and phosphatidylinositol 3,4,5-triphosphate. Biochem Biophys Res Commun 404(3):834–840
Chen C-L, Wang Y, Sesaki H, Iijima M (2012) Myosin I links PIP3 signaling to remodeling of the actin cytoskeleton in chemotaxis. Sci Signal 5(209):ra10
Chin YR, Toker A (2011) Akt isoform-specific signaling in breast cancer: uncovering an anti-migratory role for palladin. Cell Adh Migr 5(3):211–214
Primo L, di Blasio L, Roca C, Droetto S, Piva R, Schaffhausen B, Bussolino F (2007) Essential role of PDK1 in regulating endothelial cell migration. J Cell Biol 176(7):1035–1047
Dong LQ, Landa LR, Wick MJ, Zhu L, Mukai H, Ono Y, Liu F (2000) Phosphorylation of protein kinase N by phosphoinositide-dependent protein kinase-1 mediates insulin signals to the actin cytoskeleton. Proc Natl Acad Sci U S A 97(10):5089–5094
Pinner S, Sahai E (2008) PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat Cell Biol 10(2):127–137
Schneider E, Keppler R, Prawitt D, Steinwender C, Roos FC, Thuroff JW, Lausch E, Brenner W (2011) Migration of renal tumor cells depends on dephosphorylation of Shc by PTEN. Int J Oncol 38(3):823–831
Ananthanarayanan B, Ni Q, Zhang J (2005) Signal propagation from membrane messengers to nuclear effectors revealed by reporters of phosphoinositide dynamics and Akt activity. Proc Natl Acad Sci U S A 102(42):15081–15086
Lindsay Y, McCoull D, Davidson L, Leslie NR, Fairservice A, Gray A, Lucocq J, Downes CP (2006) Localization of agonist-sensitive PtdIns(3,4,5)P3 reveals a nuclear pool that is insensitive to PTEN expression. J Cell Sci 119(24):5160–5168
Lian Z, Di Cristofano A (2005) Class reunion: PTEN joins the nuclear crew. Oncogene 24(50):7394–7400
Ahn JY, Liu X, Cheng D, Peng J, Chan PK, Wade PA, Ye K (2005) Nucleophosmin/B23, a nuclear PI(3,4,5)P(3) receptor, mediates the antiapoptotic actions of NGF by inhibiting CAD. Mol Cell 18(4):435–445
Kwon IS, Lee KH, Choi JW, Ahn JY (2010) PI(3,4,5)P3 regulates the interaction between Akt and B23 in the nucleus. BMB Rep 43(2):127–132
Kumar A, Fernandez-Capetillo O, Carrera AC (2010) Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc Natl Acad Sci U S A 107(16):7491–7496
Bertagnolo V, Brugnoli F, Marchisio M, Celeghini C, Carini C, Capitani S (2004) Association of PI 3-K with tyrosine phosphorylated Vav is essential for its activity in neutrophil-like maturation of myeloid cells. Cell Signal 16(4):423–433
Aoki K, Nakamura T, Fujikawa K, Matsuda M (2005) Local phosphatidylinositol 3,4,5-trisphosphate accumulation recruits Vav2 and Vav3 to activate Rac1/Cdc42 and initiate neurite outgrowth in nerve growth factor-stimulated PC12 cells. Mol Biol Cell 16(5):2207–2217
Vedham V, Phee H, Coggeshall KM (2005) Vav activation and function as a rac guanine nucleotide exchange factor in macrophage colony-stimulating factor-induced macrophage chemotaxis. Mol Cell Biol 25(10):4211–4220
Jiang H, Guo W, Liang X, Rao Y (2005) Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3β and its upstream regulators. Cell 120(1):123–135
Dotti CG, Sullivan CA, Banker GA (1988) The establishment of polarity by hippocampal neurons in culture. J Neurosci 8(4):1454–1468
Yan D, Guo L, Wang Y (2006) Requirement of dendritic Akt degradation by the ubiquitin-proteasome system for neuronal polarity. J Cell Biol 174(3):415–424
Horiguchi K, Hanada T, Fukui Y, Chishti AH (2006) Transport of PIP3 by GAKIN, a kinesin-3 family protein, regulates neuronal cell polarity. J Cell Biol 174(3):425–436
Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27(41):5497–5510
Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7(8):606–619
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2(7):489–501
Engelman JA (2009) Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 9(8):550–562
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129(7):1261–1274
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91(2):231–241
Bader AG, Kang S, Zhao L, Vogt PK (2005) Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer 5(12):921–929
Tzivion G, Dobson M, Ramakrishnan G (2011) FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta (BBA) – Mol Cell Res 1813(11):1938–1945
Plas DR, Thompson CB (2005) Akt-dependent transformation: there is more to growth than just surviving. Oncogene 24(50):7435–7442
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7(1):11–20
Liang J, Slingerland JM (2003) Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2(4):339–345
Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, Tsichlis PN (2009) MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci Signal 2(92):ra62
Lemmon MA, Schlessinger J (2010) Cell signaling by receptor tyrosine kinases. Cell 141(7):1117–1134
Arteaga CL (2006) EGF receptor mutations in lung cancer: from humans to mice and maybe back to humans. Cancer Cell 9(6):421–423
Narita Y, Nagane M, Mishima K, Huang H-JS, Furnari FB, Cavenee WK (2002) Mutant epidermal growth factor receptor signaling down-regulates p27 through activation of the phosphatidylinositol 3-kinase/Akt pathway in glioblastomas. Cancer Res 62(22):6764–6769
Ross JS, Slodkowska EA, Symmans WF, Pusztai L, Ravdin PM, Hortobagyi GN (2009) The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist 14(4):320–368
Corless CL, Heinrich MC (2008) Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol Mech 3(1):557–586
Eggermont AMM, Robert C (2012) Melanoma in 2011: a new paradigm tumor for drug development. Nat Rev Clin Oncol 9(2):74–76
Samuels Y, Velculescu VE (2004) Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 3(10):1221–1224
McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ et al (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455(7216):1061–1068
Samuels Y, Diaz LA, Schmidt-Kittler O, Cummins JM, DeLong L, Cheong I, Rago C, Huso DL, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE (2005) Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 7(6):561–573
Zhao L, Vogt PK (2010) Hot-spot mutations in p110α of phosphatidylinositol 3-kinase (PI3K): differential interactions with the regulatory subunit p85 and with RAS. Cell Cycle 9(3):596–600
Carson JD, Van Aller G, Lehr R, Sinnamon RH, Kirkpatrick RB, Auger KR, Dhanak D, Copeland RA, Gontarek RR, Tummino PJ, Luo L (2008) Effects of oncogenic p110α subunit mutations on the lipid kinase activity of phosphoinositide 3-kinase. Biochem J 409(2):519–524
Chaussade C, Cho K, Mawson C, Rewcastle GW, Shepherd PR (2009) Functional differences between two classes of oncogenic mutation in the PIK3CA gene. Biochem Biophys Res Commun 381(4):577–581
Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, Imamura J, Ohta M, Jazag A, Guleng B, Tateishi K, Asaoka Y, Matsumura M, Kawabe T, Omata M (2005) Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res 65(11):4562–4567
Kang S, Bader AG, Vogt PK (2005) Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A 102(3):802–807
Sugita H, Dan S, Kong D, Tomida A, Yamori T (2008) A new evaluation method for quantifying PI3K activity by HTRF assay. Biochem Biophys Res Commun 377(3):941–945
Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM (2005) The oncogenic properties of mutant p110α and p110β phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A 102(51):18443–18448
Denley A, Gymnopoulos M, Kang S, Mitchell C, Vogt PK (2009) Requirement of phosphatidylinositol(3,4,5)trisphosphate in phosphatidylinositol 3-kinase-induced oncogenic transformation. Mol Cancer Res 7(7):1132–1138
Vadas O, Burke JE, Zhang X, Berndt A, Williams RL (2011) Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal 4(195):re2
Miled N, Yan Y, Hon W-C, Perisic O, Zvelebil M, Inbar Y, Schneidman-Duhovny D, Wolfson HJ, Backer JM, Williams RL (2007) Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 317(5835):239–242
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275(5308):1943–1947
Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV (1997) Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 15(4):356–362
Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP (1998) Pten is essential for embryonic development and tumour suppression. Nat Genet 19(4):348–355
Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R (1999) Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A 96(4):1563–1568
Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, Barrantes IB, Ho A, Wakeham A, ltie A, Khoo W, Fukumoto M, Mak TW (1998) High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol:CB 8(21):1169–1178
Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP (1999) Impaired Fas response and autoimmunity in Pten+/− mice. Science 285(5436):2122–2125
Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP (2001) Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet 27(2):222–224
Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, Salmena L, Sampieri K, Haveman WJ, Brogi E, Richardson AL, Zhang J, Pandolfi PP (2010) Subtle variations in Pten dose determine cancer susceptibility. Nat Genet 42(5):454–458
Salmena L, Carracedo A, Pandolfi PP (2008) Tenets of PTEN tumor suppression. Cell 133(3):403–414
Song MS, Salmena L, Pandolfi PP (2012) The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 13(5):283–296
Berger AH, Knudson AG, Pandolfi PP (2011) A continuum model for tumour suppression. Nature 476(7359):163–169
Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y (2007) Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128(1):157–170
Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M, Pandolfi PP (2011) Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 144(2):187–199
Bronisz A, Godlewski J, Wallace JA, Merchant AS, Nowicki MO, Mathsyaraja H, Srinivasan R, Trimboli AJ, Martin CK, Li F, Yu L, Fernandez SA, Pecot T, Rosol TJ, Cory S, Hallett M, Park M, Piper MG, Marsh CB, Yee LD, Jimenez RE, Nuovo G, Lawler SE, Chiocca EA, Leone G, Ostrowski MC (2012) Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol 14(2):159–167
Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, Thompson JC, Caserta E, Wang H, Chong J-L, Naidu S, Wei G, Sharma SM, Stephens JA, Fernandez SA, Gurcan MN, Weinstein MB, Barsky SH, Yee L, Rosol TJ, Stromberg PC, Robinson ML, Pepin F, Hallett M, Park M, Ostrowski MC, Leone G (2009) Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 461(7267):1084–1091
Hafsi S, Pezzino FM, Candido S, Ligresti G, Spandidos DA, Soua Z, McCubrey JA, Travali S, Libra M (2012) Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drug-resistance. Int J Oncol 40(3):639–644
Xu J, Zhou J-Y, Wei W-Z, Wu GS (2010) Activation of the Akt survival pathway contributes to TRAIL resistance in cancer cells. PLoS One 5(4):e10226
Okkenhaug K, Vanhaesebroeck B (2003) PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol 3(4):317–330
Blunt MD, Ward SG (2012) Targeting PI3K isoforms and SHIP in the immune system: new therapeutics for inflammation and leukemia. Curr Opin Pharmacol 12(4):444–451
Van Haastert PJM, Devreotes PN (2004) Chemotaxis: signalling the way forward. Nat Rev Mol Cell Biol 5(8):626–634
Hawkins PT, Stephens LR, Suire S, Wilson M (2010) PI3K signaling in neutrophils. Curr Top Microbiol Immunol 346:183–202
Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP (2000) Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287(5455):1049–1053
Williams CMM, Galli SJ (2000) The diverse potential effector and immunoregulatory roles of mast cells in allergic disease. J Allergy Clin Immunol 105(5):847–859
Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, Joza N, Mak TW, Ohashi PS, Suzuki A, Penninger JM (2000) Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science 287(5455):1040–1046
Sadhu C, Masinovsky B, Dick K, Sowell CG, Staunton DE (2003) Essential role of phosphoinositide 3-kinase δ in neutrophil directional movement. J Immunol 170(5):2647–2654
Ferrandi C, Ardissone V, Ferro P, Ruckle T, Zaratin P, Ammannati E, Hauben E, Rommel C, Cirillo R (2007) Phosphoinositide 3-kinase γ inhibition plays a crucial role in early steps of inflammation by blocking neutrophil recruitment. J Pharmacol Exp Ther 322(3):923–930
Vemula S, Shi J, Hanneman P, Wei L, Kapur R (2010) ROCK1 functions as a suppressor of inflammatory cell migration by regulating PTEN phosphorylation and stability. Blood 115(9):1785–1796
Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K, Vanhaesebroeck B, Turner M, Webb L, Wymann MP, Hirsch E, Ruckle T, Camps M, Rommel C, Jackson SP, Chilvers ER, Stephens LR, Hawkins PT (2005) Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood 106(4):1432–1440
Perisic O, Wilson MI, Karathanassis D, Jn B, Pacold ME, Ellson CD, Hawkins PT, Stephens L, Williams RL (2004) The role of phosphoinositides and phosphorylation in regulation of NADPH oxidase. Adv Enzyme Regul 44(1):279–298
Fung-Leung W-P (2011) Phosphoinositide 3-kinase delta (PI3Kδ) in leukocyte signaling and function. Cell Signal 23(4):603–608
Weber C, Noels H (2011) Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 17(11):1410–1422
Kaplan MJ (2011) Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol 7(12):691–699
Wedemeyer J, Tsai M, Galli SJ (2000) Roles of mast cells and basophils in innate and acquired immunity. Curr Opin Immunol 12(6):624–631
Rommel C, Camps M, Ji H (2007) PI3Kδ and PI3Kγ: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol 7(3):191–201
Ali K, Bilancio A, Thomas M, Pearce W, Gilfillan AM, Tkaczyk C, Kuehn N, Gray A, Giddings J, Peskett E, Fox R, Bruce I, Walker C, Sawyer C, Okkenhaug K, Finan P, Vanhaesebroeck B (2004) Essential role for the p110δ phosphoinositide 3-kinase in the allergic response. Nature 431(7011):1007–1011
Laffargue M, Calvez R, Finan P, Trifilieff A, Barbier M, Altruda F, Hirsch E, Wymann MP (2002) Phosphoinositide 3-kinase γ is an essential amplifier of mast cell function. Immunity 16(3):441–451
Furumoto Y, Brooks S, Olivera A, Takagi Y, Miyagishi M, Taira K, Casellas R, Beaven MA, Gilfillan AM, Rivera J (2006) Cutting edge: lentiviral short hairpin RNA silencing of PTEN in human mast cells reveals constitutive signals that promote cytokine secretion and cell survival. J Immunol 176(9):5167–5171
Furumoto Y, Charles N, Olivera A, Leung WH, Dillahunt S, Sargent JL, Tinsley K, Odom S, Scott E, Wilson TM, Ghoreschi K, Kneilling M, Chen M, Lee DM, Bolland S, Rivera J (2011) PTEN deficiency in mast cells causes a mastocytosis-like proliferative disease that heightens allergic responses and vascular permeability. Blood 118(20):5466–5475
Shenker BJ, Boesze-Battaglia K, Zekavat A, Walker L, Besack D, Ali H (2010) Inhibition of mast cell degranulation by a chimeric toxin containing a novel phosphatidylinositol-3,4,5-triphosphate phosphatase. Mol Immunol 48(1‚Äì3):203–210
Shenker BJ, Ali H, Boesze-Battaglia K (2011) PIP3 regulation as promising targeted therapy of mast-cell-mediated diseases. Curr Pharm Des 17(34):3815–3822
Fruman DA, Bismuth G (2009) Fine tuning the immune response with PI3K. Immunol Rev 228(1):253–272
Okkenhaug K, Fruman DA (2010) PI3Ks in lymphocyte signaling and development. Curr Top Microbiol Immunol 346:57–85
Fayard E, Moncayo G, Hemmings BA, Hollander GA (2010) Phosphatidylinositol 3-kinase signaling in thymocytes: the need for stringent control. Sci Signal 3(135):re5
Swat W, Montgrain V, Doggett TA, Douangpanya J, Puri K, Vermi W, Diacovo TG (2006) Essential role of PI3Kδ and PI3Kγ in thymocyte survival. Blood 107(6):2415–2422
Webb LMC, Vigorito E, Wymann MP, Hirsch E, Turner M (2005) Cutting edge: T cell development requires the combined activities of the p110γ and p110δ catalytic isoforms of phosphatidylinositol 3-kinase. J Immunol 175(5):2783–2787
Okkenhaug K, Patton DT, Bilancio A, Garcon F, Rowan WC, Vanhaesebroeck B (2006) The p110δ isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J Immunol 177(8):5122–5128
Nombela-Arrieta C, Lacalle RA, Montoya MC, Kunisaki Y, Megias D, Marques M, Carrera AC, Manes S, Fukui Y, Martinez-A C, Stein JV (2004) Differential requirements for DOCK2 and phosphoinositide-3-kinase γ during T and B lymphocyte homing. Immunity 21(3):429–441
Reif K, Okkenhaug K, Sasaki T, Penninger JM, Vanhaesebroeck B, Cyster JG (2004) Cutting edge: differential roles for phosphoinositide 3-kinases, p110γ and p110δ, in lymphocyte chemotaxis and homing. J Immunol 173(4):2236–2240
Werner M, Hobeika E, Jumaa H (2010) Role of PI3K in the generation and survival of B cells. Immunol Rev 237(1):55–71
Oak JS, Deane JA, Kharas MG, Luo J, Lane TE, Cantley LC, Fruman DA (2006) Sjogren’s syndrome-like disease in mice with T cells lacking class 1A phosphoinositide-3-kinase. Proc Natl Acad Sci U S A 103(45):16882–16887
Ohashi PS (2002) T-cell signalling and autoimmunity: molecular mechanisms of disease. Nat Rev Immunol 2(6):427–438
Okkenhaug K, Bilancio A, Gr F, Priddle H, Sancho S, Peskett E, Pearce W, Meek SE, Salpekar A, Waterfield MD, Smith AJH, Vanhaesebroeck B (2002) Impaired B and T cell antigen receptor signaling in p110δ PI 3-kinase mutant mice. Science 297(5583):1031–1034
So L, Fruman DA (2012) PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem J 442(3):465–481
Oudit GY, Sun H, Kerfant B-G, Crackower MA, Penninger JM, Backx PH (2004) The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J Mol Cell Cardiol 37(2):449–471
Oudit GY, Penninger JM (2009) Cardiac regulation by phosphoinositide 3-kinases and PTEN. Cardiovasc Res 82(2):250–260
Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng H-YM, Rybin VO, Lembo G, Fratta L, Oliveira-dos-Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH, Penninger JM (2002) Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell 110(6):737–749
Northcott CA, Poy MN, Najjar SM, Watts SW (2002) Phosphoinositide 3-kinase mediates enhanced spontaneous and agonist-induced contraction in aorta of deoxycorticosterone acetate-salt hypertensive rats. Circ Res 91(4):360–369
Wymann MP, Zvelebil M, Laffargue M (2003) Phosphoinositide 3-kinase signalling – which way to target? Trends Pharmacol Sci 24(7):366–376
Chavakis E, Carmona G, Urbich C, Göttig S, Henschler R, Penninger JM, Zeiher AM, Chavakis T, Dimmeler S (2008) Phosphatidylinositol-3-kinase γ is integral to homing functions of progenitor cells. Circ Res 102(8):942–949
Cieslik K, Abrams CS, Wu KK (2001) Up-regulation of endothelial nitric-oxide synthase promoter by the phosphatidylinositol 3-kinase γ/Janus kinase 2/MEK-1-dependent pathway. J Biol Chem 276(2):1211–1219
Oudit GY, Crackower MA, Eriksson U, Sarao R, Kozieradzki I, Sasaki T, Irie-Sasaki J, Gidrewicz D, Rybin VO, Wada T, Steinberg SF, Backx PH, Penninger JM (2003) Phosphoinositide 3-kinase gamma-deficient mice are protected from isoproterenol-induced heart failure. Circulation 108(17):2147–2152
McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM, Izumo S (2003) Phosphoinositide 3-kinase(p110α) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci U S A 100(21):12355–12360
Huang J, Kontos CD (2002) PTEN modulates vascular endothelial growth factor-mediated signaling and angiogenic effects. J Biol Chem 277(13):10760–10766
Schwartzbauer G, Robbins J (2001) The tumor suppressor gene PTEN can regulate cardiac hypertrophy and survival. J Biol Chem 276(38):35786–35793
Chaanine AH, Hajjar RJ (2011) AKT signalling in the failing heart. Eur J Heart Fail 13(8):825–829
Sugden PH, Fuller SJ, Weiss SC, Clerk A (2008) Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. Br J Pharmacol 153(S1):S137–S153
DeBosch B, Sambandam N, Weinheimer C, Courtois M, Muslin AJ (2006) Akt2 regulates cardiac metabolism and cardiomyocyte survival. J Biol Chem 281(43):32841–32851
DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, Muslin AJ (2006) Akt1 is required for physiological cardiac growth. Circulation 113(17):2097–2104
Hers I, Vincent EE, Tavare JM (2011) Akt signalling in health and disease. Cell Signal 23(10):1515–1527
Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC, Izumo S (2002) Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol 22(8):2799–2809
Condorelli G, Drusco A, Stassi G, Bellacosa A, Roncarati R, Iaccarino G, Russo MA, Gu Y, Dalton N, Chung C, Latronico MVG, Napoli C, Sadoshima J, Croce CM, Ross J (2002) Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci U S A 99(19):12333–12338
Cook SA, Matsui T, Li L, Rosenzweig A (2002) Transcriptional effects of chronic Akt activation in the heart. J Biol Chem 277(25):22528–22533
Cohen P, Frame S (2001) The renaissance of GSK3. Nat Rev Mol Cell Biol 2(10):769–776
Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, Richardson JA, Hill JA, Olson EN (2002) Activated glycogen synthase-3β suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A 99(2):907–912
McMullen JR, Amirahmadi F, Woodcock EA, Schinke-Braun M, Bouwman RD, Hewitt KA, Mollica JP, Zhang L, Zhang Y, Shioi T, Buerger A, Izumo S, Jay PY, Jennings GL (2007) Protective effects of exercise and phosphoinositide 3-kinase(p110α) signaling in dilated and hypertrophic cardiomyopathy. Proc Natl Acad Sci U S A 104(2):612–617
Ban K, Cooper AJ, Samuel S, Bhatti A, Patel M, Izumo S, Penninger JM, Backx PH, Oudit GY, Tsushima RG (2008) Phosphatidylinositol 3-kinase γ is a critical mediator of myocardial ischemic and adenosine-mediated preconditioning. Circ Res 103(6):643–653
Murphy E, Tong H, Steenbergen C (2003) Preconditioning: is the Akt-ion in the PI3K pathway? J Mol Cell Cardiol 35(9):1021–1025
Yellon DM, Downey JM (2003) Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev 83(4):1113–1151
Gross ER, Hsu AK, Gross GJ (2004) Opioid-induced cardioprotection occurs via glycogen synthase kinase β inhibition during reperfusion in intact rat hearts. Circ Res 94(7):960–966
Cai Z, Semenza GL (2005) PTEN activity is modulated during ischemia and reperfusion. Circ Res 97(12):1351–1359
Siddall H, Warrell C, Yellon D, Mocanu M (2008) Ischemia-reperfusion injury and cardioprotection: investigating PTEN, the phosphatase that negatively regulates PI3K, using a congenital model of PTEN haploinsufficiency. Basic Res Cardiol 103(6):560–568
Poornima IG, Parikh P, Shannon RP (2006) Diabetic cardiomyopathy. Circ Res 98(5):596–605
Kim K-H, Oudit GY, Backx PH (2008) Erythropoietin protects against doxorubicin-induced cardiomyopathy via a phosphatidylinositol 3-kinase-dependent pathway. J Pharmacol Exp Ther 324(1):160–169
Carnevale D, Lembo G (2012) PI3Kγ in hypertension: a novel therapeutic target controlling vascular myogenic tone and target organ damage. Cardiovasc Res 95(4):403–408
McMullen JR, Jay PY (2007) PI3K(p110α) inhibitors as anti-cancer agents: minding the heart. Cell Cycle 6(8):910–913
Foukas LC, Withers DJ (2011) Phosphoinositide signalling pathways in metabolic regulation. In: Rommel C, Vanhaesebroeck B, Vogt PK (eds) Phosphoinositide 3-kinase in health and disease, vol 346, Current topics in microbiology and immunology. Springer, Berlin/Heidelberg, pp 115–141
Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414(6865):799–806
Taniguchi CM, Emanuelli B, Kahn CR (2006) Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7(2):85–96
Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E, Sancho S, Smith AJH, Withers DJ, Vanhaesebroeck B (2006) Critical role for the p110α phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 441(7091):366–370
Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM (2006) A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell 125(4):733–747
Ciraolo E, Iezzi M, Marone R, Marengo S, Curcio C, Costa C, Azzolino O, Gonella C, Rubinetto C, Wu H, Dastru W, Martin EL, Silengo L, Altruda F, Turco E, Lanzetti L, Musiani P, Ruckle T, Rommel C, Backer JM, Forni G, Wymann MP, Hirsch E (2008) Phosphoinositide 3-kinase p110β activity: key role in metabolism and mammary gland cancer but not development. Sci Signal 1(36):ra3
Shulman GI (2004) Unraveling the cellular mechanism of insulin resistance in humans: new insights from magnetic resonance spectroscopy. Physiology 19(4):183–190
Brachmann SM, Ueki K, Engelman JA, Kahn RC, Cantley LC (2005) Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol Cell Biol 25(5):1596–1607
Kurlawalla-Martinez C, Stiles B, Wang Y, Devaskar SU, Kahn BB, Wu H (2005) Insulin hypersensitivity and resistance to streptozotocin-induced diabetes in mice lacking PTEN in adipose tissue. Mol Cell Biol 25(6):2498–2510
Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim Y-J, Sherwin R, Devaskar S, Lesche R, Magnuson MA, Wu H (2004) Live-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity. Proc Natl Acad Sci U S A 101(7):2082–2087
Suwa A, Kurama T, Shimokawa T (2010) SHIP2 and its involvement in various diseases. Expert Opin Ther Targets 14(7):727–737
Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD (2001) Cellular function of phosphoinositide 3-kinases: implications for development, immunity, homeostasis, and cancer. Annu Rev Cell Dev Biol 17(1):615–675
Ueki K, Fruman DA, Yballe CM, Fasshauer M, Klein J, Asano T, Cantley LC, Kahn CR (2003) Positive and negative roles of p85α and p85β regulatory subunits of phosphoinositide 3-kinase in insulin signaling. J Biol Chem 278(48):48453–48466
Luo J, Field SJ, Lee JY, Engelman JA, Cantley LC (2005) The p85 regulatory subunit of phosphoinositide 3-kinase down-regulates IRS-1 signaling via the formation of a sequestration complex. J Cell Biol 170(3):455–464
Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM (2005) Increased p85/55/50 expression and decreased phosphotidylinositol 3-kinase activity in insulin-resistant human skeletal muscle. Diabetes 54(8):2351–2359
Kirwan JP, Varastehpour A, Jing M, Presley L, Shao J, Friedman JE, Catalano PM (2004) Reversal of insulin resistance postpartum is linked to enhanced skeletal muscle insulin signaling. J Clin Endocrinol Metab 89(9):4678–4684
Luo J, Sobkiw CL, Hirshman MF, Logsdon MN, Li TQ, Goodyear LJ, Cantley LC (2006) Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab 3(5):355–366
Taniguchi CM, Kondo T, Sajan M, Luo J, Bronson R, Asano T, Farese R, Cantley LC, Kahn CR (2006) Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKCλ/ζ. Cell Metab 3(5):343–353
Meshkani R, Adeli K (2009) Hepatic insulin resistance, metabolic syndrome and cardiovascular disease. Clin Biochem 42(13‚Äì14):1331–1346
Popa C, Netea MG, van Riel PLCM, van der Meer JWM, Stalenhoef AFH (2007) The role of TNF-α in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res 48(4):751–762
Um SH, D’Alessio D, Thomas G (2006) Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab 3(6):393–402
Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G (2004) Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431(7005):200–205
Thong FSL, Dugani CB, Klip A (2005) Turning signals on and off: GLUT4 traffic in the insulin-signaling highway. Physiology 20(4):271–284
Barthel A, Schmoll D, Unterman TG (2005) FoxO proteins in insulin action and metabolism. Trends Endocrinol Metab 16(4):183–189
Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ (2001) Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB β). Science 292(5522):1728–1731
Yang X, Ongusaha PP, Miles PD, Havstad JC, Zhang F, So WV, Kudlow JE, Michell RH, Olefsky JM, Field SJ, Evans RM (2008) Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 451(7181):964–969
Hanover JA, Krause MW, Love DC (2010) The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim Biophys Acta – Gen Subs 1800(2):80–95
Ande SR, Mishra S (2009) Prohibitin interacts with phosphatidylinositol 3,4,5-triphosphate (PIP3) and modulates insulin signaling. Biochem Biophys Res Commun 390(3):1023–1028
Pollak M (2012) The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer 12(3):159–169
Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK (2012) Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets 16(1):15–31
Liu P, Cheng H, Roberts TM, Zhao JJ (2009) Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 8(8):627–644
Baggiolini M, Dewald B, Schnyder J, Ruch W, Cooper PH, Payne TG (1987) Inhibition of the phagocytosis-induced respiratory burst by the fungal metabolite wortmannin and some analogues. Exp Cell Res 169(2):408–418
Arcaro A (1993) Wortmannin is a potent phosphatidylinostitol 3-kinase inhibitor: the role of phosphatidylinositol 3, 4, 5-trisphosphate in neutrophil responses. Biochem J 296:297–301
Cleary J, Shapiro G (2010) Development of phosphoinositide-3 kinase pathway inhibitors for advanced cancer. Curr Oncol Rep 12(2):87–94
Ihle NT, Williams R, Chow S, Chew W, Berggren MI, Paine-Murrieta G, Minion DJ, Halter RJ, Wipf P, Abraham R, Kirkpatrick L, Powis G (2004) Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther 3(7):763–772
Vlahos CJ, Matter WF, Hui KY, Brown RF (1994) A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 269(7):5241–5248
Garlich JR, De P, Dey N, Su JD, Peng X, Miller A, Murali R, Lu Y, Mills GB, Kundra V, Shu HK, Peng Q, Durden DL (2008) A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res 68(1):206–215
Rommel C (2010) Taking PI3Kδ and PI3Kγ one step ahead: dual active PI3Kdelta/gamma inhibitors for the treatment of immune-mediated inflammatory diseases. Curr Top Microbiol Immunol 346:279–299
Hoellenriegel J, Meadows SA, Sivina M, Wierda WG, Kantarjian H, Keating MJ, Giese N, O’Brien S, Yu A, Miller LL, Lannutti BJ, Burger JA (2011) The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood 118(13):3603–3612
Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, Druker BJ, Puri KD, Ulrich RG, Giese NA (2011) CAL-101, a p110δ selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 117(2):591–594
Flinn IW, Schreeder MT, Coutre SE, Leonard J, Wagner-Johnston ND, De Vos S, Boccia RV, Holes L, Peterman S, Miller LL, Yu AS (2011) A phase I study of CAL-101, an isoform-selective inhibitor of phosphatidylinositol 3-kinase P110δ, in combination with anti-CD20 monoclonal antibody therapy and/or bendamustine in patients with previously treated B-cell malignancies. ASCO Meet Abstr 29(15 suppl):3064
Ruckle T, Schwarz MK, Rommel C (2006) PI3Kγ inhibition: towards an ‘aspirin of the 21st century’? Nat Rev Drug Discov 5(11):903–918
Camps M, Ruckle T, Ji H, Ardissone V, Rintelen F, Shaw J, Ferrandi C, Chabert C, Gillieron C, Francon B, Martin T, Gretener D, Perrin D, Leroy D, Vitte PA, Hirsch E, Wymann MP, Cirillo R, Schwarz MK, Rommel C (2005) Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med 11(9):936–943
Sheppard K, Kinross KM, Solomon B, Pearson RB, Phillips WA (2012) Targeting PI3 kinase/AKT/mTOR signaling in cancer. Crit Rev Oncog 17(1):69–95
Berndt N, Yang H, Trinczek B, Betzi S, Zhang Z, Wu B, Lawrence NJ, Pellecchia M, Schonbrunn E, Cheng JQ, Sebti SM (2010) The Akt activation inhibitor TCN-P inhibits Akt phosphorylation by binding to the PH domain of Akt and blocking its recruitment to the plasma membrane. Cell Death Differ 17(11):1795–1804
Hilgard P, Klenner T, Stekar J, Nossner G, Kutscher B, Engel J (1997) D-21266, a new heterocyclic alkylphospholipid with antitumour activity. Eur J Cancer 33(3):442–446
Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan B-S, Kotani H (2010) MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9(7):1956–1967
Burris HA, Siu LL, Infante JR, Wheler JJ, Kurkjian C, Opalinska J, Smith DA, Antal JM, Gauvin JL, Gonzalez T, Adams LM, Bedard P, Gerecitano JF, Kurzrock R, Moore KN, Morris SR, Aghajanian C (2011) Safety, pharmacokinetics (PK), pharmacodynamics (PD), and clinical activity of the oral AKT inhibitor GSK2141795 (GSK795) in a phase I first-in-human study. ASCO Meet Abstr 29(15 suppl):3003
Kurzrock R, Patnaik A, Rosenstein L, Fu S, Papadopoulos KP, Smith DA, Falchook GS, Chambers G, Gauvin JL, Naing A, Smith LS, Gonzalez T, Tsimberidou AM, Mays TA, Cox DS, Hong DS, DeMarini DJ, Le NT, Morris SR, Tolcher AW (2011) Phase I dose-escalation of the oral MEK1/2 inhibitor GSK1120212 (GSK212) dosed in combination with the oral AKT inhibitor GSK2141795 (GSK795). ASCO Meet Abstr 29(15 suppl):3085
Rhodes N, Heerding DA, Duckett DR, Eberwein DJ, Knick VB, Lansing TJ, McConnell RT, Gilmer TM, Zhang SY, Robell K, Kahana JA, Geske RS, Kleymenova EV, Choudhry AE, Lai Z, Leber JD, Minthorn EA, Strum SL, Wood ER, Huang PS, Copeland RA, Kumar R (2008) Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res 68(7):2366–2374
Atkins MB, Hidalgo M, Stadler WM, Logan TF, Dutcher JP, Hudes GR, Park Y, Liou SH, Marshall B, Boni JP, Dukart G, Sherman ML (2004) Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol 22(5):909–918
Faivre S, Kroemer G, Raymond E (2006) Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov 5(8):671–688
Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM (2009) Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol 7(2):e38
Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS (2009) An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 284(12):8023–8032
Liu Q, Chang JW, Wang J, Kang SA, Thoreen CC, Markhard A, Hur W, Zhang J, Sim T, Sabatini DM, Gray NS (2010) Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benz o[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J Med Chem 53(19):7146–7155
Liu Q, Wang J, Kang SA, Thoreen CC, Hur W, Ahmed T, Sabatini DM, Gray NS (2011) Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2(1H)-one (Torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. J Med Chem 54(5):1473–1480
McNamara CR, Degterev A (2011) Small – molecule inhibitors of the PI3K signaling network. Future Med Chem 3(5):549
Emerling BM, Akcakanat A (2011) Targeting PI3K/mTOR signaling in cancer. Cancer Res 71(24):7351–7359
Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D, Shokat KM, Weiss WA (2006) A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 9(5):341–349
Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, Henley A, Di-Stefano F, Ahmad Z, Guillard S, Bjerke LM, Kelland L, Valenti M, Patterson L, Gowan S, de Haven BA, Hayakawa M, Kaizawa H, Koizumi T, Ohishi T, Patel S, Saghir N, Parker P, Waterfield M, Workman P (2007) Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res 67(12):5840–5850
Serra V, Markman B, Scaltriti M, Eichhorn PJA, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J, Baselga J (2008) NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res 68(19):8022–8030
Maira S-M, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chone P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C (2008) Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 7(7):1851–1863
Burris H, Rodon J, Sharma S, Herbst RS, Tabernero J, Infante JR, Silva A, Demanse D, Hackl W, Baselga J (2010) First-in-human phase I study of the oral PI3K inhibitor BEZ235 in patients with advanced solid tumors. ASCO Meet Abstr 28(15 suppl):3005
Markman B, Tabernero J, Krop I, Shapiro GI, Siu L, Chen LC, Mita M, Melendez Cuero M, Stutvoet S, Birle D, Anak N, Hackl W, Baselga J (2012) Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol 23(9):2399–2408
Chang K-Y, Tsai S-Y, Wu C-M, Yen C-J, Chuang B-F, Chang J-Y (2011) Novel phosphoinositide 3-kinase/mTOR dual inhibitor, NVP-BGT226, displays potent growth-inhibitory activity against human head and neck cancer cells in vitro and in vivo. Clin Cancer Res 17(22):7116–7126
Robak T, Robak E (2012) Tyrosine kinase inhibitors as potential drugs for B-cell lymphoid malignancies and autoimmune disorders. Expert Opin Investig Drugs 21(7):921–947
Chang BY, Huang MM, Francesco M, Chen J, Sokolove J, Magadala P, Robinson WH, Buggy JJ (2011) The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res Ther 13(4):R115
Ferguson KM, Kavran JM, Sankaran VG, Fournier E, Isakoff SJ, Skolnik EY, Lemmon MA (2000) Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol Cell 6(2):373–384
Berrie C, Falasca M (2000) Patterns within protein/polyphosphoinositide interactions provide specific targets for therapeutic intervention. FASEB J 14(15):2618–2622
Falasca M, Chiozzotto D, Godage HY, Mazzoletti M, Riley AM, Previdi S, Potter BVL, Broggini M, Maffucci T (2010) A novel inhibitor of the PI3K/Akt pathway based on the structure of inositol 1,3,4,5,6-pentakisphosphate. Br J Cancer 102(1):104–114
Falasca M, Selvaggi F, Buus R, Sulpizio S, Edling CE (2011) Targeting phosphoinositide 3-kinase pathways in pancreatic cancer – from molecular signalling to clinical trials. Anti Cancer Agents Med Chem 11(5):455–463
Miao B, Skidan I, Yang J, Lugovskoy A, Reibarkh M, Long K, Brazell T, Durugkar KA, Maki J, Ramana CV, Schaffhausen B, Wagner G, Torchilin V, Yuan J, Degterev A (2010) Small molecule inhibition of phosphatidylinositol-3,4,5-triphosphate (PIP3) binding to pleckstrin homology domains. Proc Natl Acad Sci U S A 107(46):20126–20131
Miao B, Skidan I, Yang J, You Z, Fu X, Famulok M, Schaffhausen B, Torchilin V, Yuan J, Degterev A (2011) Inhibition of cell migration by PITENINs: the role of ARF6. Oncogene 31(39):4317–4332
Riehle RD, Cornea S, Degterev A, Torchilin VMicellar formulations of pro-apoptotic DM-PIT-1 analogs and TRAIL in vitro and in vivo. Drug Deliv 20(2):78–85
Kim SH, Juhnn YS, Song YS (2007) Akt involvement in paclitaxel chemoresistance of human ovarian cancer cells. Ann N Y Acad Sci 1095(1):82–89
Knuefermann C, Lu Y, Liu B, Jin W, Liang K, Wu L, Schmidt M, Mills GB, Mendelsohn J, Fan Z (2003) HER2/PI-3 K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene 22(21):3205–3212
Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R (2007) A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 12(4):395–402
Nagata Y, Lan K-H, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung M-C, Yu D (2004) PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6(2):117–127
Jhawer M, Goel S, Wilson AJ, Montagna C, Ling Y-H, Byun D-S, Nasser S, Arango D, Shin J, Klampfer L, Augenlicht LH, Soler RP, Mariadason JM (2008) PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res 68(6):1953–1961
Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN, Dickerson SH, Laquerre SG, Liu L, Gilmer TM (2012) Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther 11(4):909–920
McCubrey JA, Steelman LS, Kempf CR, Chappell WH, Abrams SL, Stivala F, Malaponte G, Nicoletti F, Libra M, Bäsecke J, Maksimovic-Ivanic D, Mijatovic S, Montalto G, Cervello M, Cocco L, Martelli AM (2011) Therapeutic resistance resulting from mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways. J Cell Physiol 226(11):2762–2781
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Riehle, R.D., Cornea, S., Degterev, A. (2013). Role of Phosphatidylinositol 3,4,5-Trisphosphate in Cell Signaling. In: Capelluto, D. (eds) Lipid-mediated Protein Signaling. Advances in Experimental Medicine and Biology, vol 991. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6331-9_7
Download citation
DOI: https://doi.org/10.1007/978-94-007-6331-9_7
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6330-2
Online ISBN: 978-94-007-6331-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)