
Abstract
Tight junctions (TJs) are multi-protein complexes whose principal function is to mediate cell-cell adhesion between epithelial or endothelial cells. While once thought to participate solely as passive effectors of adhesion, it is increasingly being recognised that TJs are dynamic structures which regulate many aspects of cellular function and physiology. Accordingly, dysregulation of TJ-based adhesion or signalling is emerging as an intriguing contributor to several pathophysiologies including cancer. This review will attempt to summarise the current state of knowledge about molecular aspects which regulate, and are regulated by, TJs. The first section will outline selected physiological processes known to influence TJ structure or function, under the headings of cell adhesion/polarity, cell-matrix signalling, ion transport, hormone effects, pro-inflammatory cytokines and hypoxia. The second section will describe selected functional behaviours within the pathophysiology of cancer which TJs have been demonstrated to influence, encompassing cell proliferation and apoptosis, migration and invasion, cell fate and differentiation, metastasis across the blood brain barrier and finally angiogenesis. Collectively, these sections illustrate that a wealth of mechanistic information can be gained from interrogating the contribution of TJs to normal physiology. In turn they highlight how TJ-based disturbances can promote some of the functional behaviours associated with cancer, and thereby offer insight into new TJ-based targets that may offer pharmacological promise in halting tumour progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Tight junction
- Epithelium
- Barrier function
- Cancer
- Polarity
- Tumour progression
- Metastasis
- Cell-Cell adhesion
- Cell migration
1.1 Introduction
Externally, epithelial cells of the skin form a selective physical barrier between an organism and environmental insults including allergens and chemicals. Internally, the epithelial cells lining most visceral organs in conjunction with endothelial cells lining the vasculature also function as barriers to prevent absorption of pathogens and harmful substances from their external surfaces.
Tight junctions (TJs), adhesion complexes which connect the lateral membranes of adjacent epithelial or endothelial cells close to their external surfaces, are responsible for sealing the intracellular space and thereby creating separate apical and basolateral compartments (Schneeberger and Lynch 2004; Tsukita et al. 2001). This intracellular TJ seal performs several crucial functions. Firstly, TJ proteins constitute a molecular barrier, which controls paracellular permeability and transport of ions, solutes and even cells. Secondly, homo- and hetero-typic binding of TJ proteins between neighboring cells aids in the establishment and maintenance of cell polarity by effectively linking polarity complexes with the underlying cytoskeletal structure of individual cells. Finally, TJ proteins can facilitate transmission of signals culminating in processes such as cell differentiation, growth, migration and invasion.
The molecular components of tight junctions can be broadly split into three main groups reviewed in detail in (Brennan et al. 2010): (1) integral transmembrane proteins including occludin, claudins, junctional adhesion molecules (JAMs), crumbs; (2) peripheral or plaque adaptor proteins which generally contain PDZ domains that facilitate protein-protein interactions, such as the Zona Occludens (ZO) family, Par3, Par6, Afadin; (3) associated regulatory/signalling proteins including cingulin and Rho-GTPases.
Deficits in tight junction function which lead to increased paracellular permeability have been linked to several pathologies including blistering skin diseases (Simon et al. 1999) and inflammatory bowel diseases (IBD) such as ulcerative colitis (Schulzke et al. 2009) and Crohn’s disease (Hollander 1988) (reviewed in detail in (Marchiando et al. 2010a)). In addition, patients with IBD have a significantly higher risk of developing colitis-associated cancer, suggesting that efficient TJ barrier function may play a crucial role in preventing cancer development. Indeed, a wealth of evidence has recently associated alterations in several TJ proteins with many solid tumours including breast, lung colorectal, and gastric (Martin et al. 2011).
Common putative mechanisms of TJ dysregulation in cancer include aberrant microRNA regulation of gene expression (e.g. JAM-A in breast cancer (Gotte et al. 2010)), aberrant methylation control (e.g. Claudin 6 in breast cancer (Osanai et al. 2007)) and protein mislocalisation (e.g. ZO-1 in pancreatic cancer (Prat et al. 2010)). Furthermore, common dysregulation patterns are evident across multiple cancer types, such as widespread down-regulation of claudin 1 and loss of ZO protein expression and localization (Martin et al. 2011). Intriguingly, genetic classifications of human breast cancer subtypes describe a highly-aggressive subtype, the claudin-low subtype, which is characterized by reduced expression of claudins 3, 4 and 7 and accompanied by increased expression of proteins involved in epithelial to mesenchymal transition (EMT) (Prat et al. 2010). Taken together, this wealth of data suggests that alterations in TJ functions may in fact play a causal role in cancer. The easiest interpretations are that simple downregulation of TJ proteins leading to barrier breakdown and concomitant loss of adhesion would disrupt cell polarity and promote cancer cell dissemination; or that upregulation or mislocalisation of TJ proteins could initiate aberrant signalling cascades that culminate in abnormal differentiation, proliferation, migration, and invasion. However, given the multi-functional roles of TJ proteins in several biological processes, it remains unlikely that a simple paradigm of either TJ protein loss or TJ protein gain will explain the complexities of different cancers.
In this review, we will firstly outline the processes that influence tight junction structure and function at a physiological level; then focus on emerging data describing mechanisms whereby alterations in TJ proteins may facilitate processes critical for tumor formation and progression. Given the huge expansion in the TJ literature over the last number of years, it has not been possible to give an exhaustive overview of all the relevant literature. We therefore chose to focus on some selected aspects, and apologise to those authors whose work we did not have space to include.
1.2 Physiological Processes Which Involve Tight Junction Proteins
The strategic location of TJs at the apical-most interface of the lateral intercellular membrane of polarized epithelia or endothelia facilitates their dynamic regulation by both extracellular and intracellular factors during a variety of physiological processes. Some of these will be summarized in the upcoming paragraphs.
1.2.1 Establishment of Cell Adhesion and Polarity
Tight junction proteins are critically important for the establishment of cellular adhesion and cell polarity through interactions with polarity complex proteins and RhoGTPases. For example, TJ barrier function is strongly regulated through homotypic binding of claudin proteins on adjacent cells, which control permeability through the formation of aqueous pores (Van Itallie and Anderson 2006). However, formation of apico-basal polarity requires the coordinate spatial regulation of the Par, Crumbs, and Scribble core polarity complexes.
Nascent cell adhesions are enriched in the TJ proteins JAM-A and ZO1, and also the adherens junction protein E-cadherin (which activates Rac1 and suppresses RhoA to facilitate junction formation (Nakagawa et al. 2001)). Upon initiation of cell polarity, Par3 is transported to the apical cortex (Harris and Peifer 2004) where its association with TJ proteins such as JAM-A (Itoh et al. 2001) and PTEN (Feng et al. 2008) facilitates Par3-TIAM1 interactions which mediates stabilization and maturation of junctions (Chen and Macara 2005). Subsequent binding of the small GTPase Cdc42 to Par6 provides the stimulus for recruitment of atypical PKC (aPKC) to the apical surface, which serves to maintain apical domain integrity (Martin-Belmonte et al. 2007) and to recruit the Crumbs complex to the apical cortex via an interaction with PALS1 (Hurd et al. 2003). The basolateral-associated Scribble complex is formed through co-localization of Scribble (Albertson et al. 2004) and Dlg (Dow et al. 2003) proteins at the basolateral cortex; with recent data suggesting that Scribble association with ZO1 may be critical for TJ assembly (Ivanov et al. 2010). This data indicate that the interplay between TJ and polarity proteins is crucial for both maturation of apical junctions and the formation of apico-basal polarity.
1.2.2 Cell-Matrix Signaling
TJ proteins can also influence cell-matrix interactions, transmitting signals to and from the microenvironment to control cell polarity and processes such as migration and invasion. For example, JAM-A-deficient neutrophils show impaired activation of the small GTPase Rap1A (Cera et al. 2009), which is known to promote β1-integrin activation in a Talin-dependent manner (Boettner and Van Aelst 2009). JAM-A knockdown or inhibition using blocking antibodies has also been shown to reduce Rap1-GTPase activity and to decrease cell migration in colonic and breast epithelial cells (McSherry et al. 2011; Severson et al. 2009).
Correct cell-matrix interactions are also critically important for developmental processes involving the generation of multi-dimensional glandular structures such as acini or organoids. For example, activity of the extracellular matrix (ECM)- degrading protein, matrix metalloproteinase MT1-MMP, has been shown to be crucial for normal branching during mammary gland development (Mori et al. 2009). Interestingly, the TJ protein ZO1 has recently been shown to regulate MT1-MMP expression in breast cell lines, suggesting that TJs may participate in modulating cell-matrix interactions during normal morphogenesis (Polette et al. 2005, 2007).
Bidirectionality in the signalling cascades between cell-cell and cell-matrix complexes is also evident, as typified by functional interactions between the cell-matrix protein CD44 and TJs. CD44 binds ECM components such as hyaluronic acid, collagen, fibronectin, laminin, and chondroitin sulfate (Naor et al. 1997). Recent studies have demonstrated that CD44 can regulate TJ assembly and barrier function in keratinocyte epithelial cells (Kirschner et al. 2011). Specifically, CD44 knockout mice exhibited alterations in expression and/or localization of TJ proteins including Claudins-1 and -4, ZO1, and Par3; and a reduction in Rac1 activity culminating in a loss of cell polarity and decreased epidermal barrier function.
1.2.3 Alterations in Ion Transport
A number of epithelial ion channels have emerged as important regulators of TJ function, of which the sodium potassium ATPase (Na+ K+ ATPase) is the best studied. This heterodimeric protein exports three sodium ions and imports two of potassium against their concentration gradients in an ATP-dependent reaction to maintain transmembrane ion concentrations (Lingrel and Kuntzweiler 1994; Kaplan 2002; Malik et al. 1996; Sweadner 1989). This serves to maintain transmembrane potential, driving multiple transport mechanisms and controlling cell volume and osmolality.
A functional Na+ K+ ATPase plays important roles in the assembly of TJs, establishment of cell polarity and regulation of paracellular permeability. Studies in various epithelial cell types by Rajasekeran et al. (Rajasekaran et al. 2003, 2007, 2008) have shown reversible inhibition of TJ formation upon inhibition of Na+ K+ ATPase, either by K+ depletion or treatment with the inhibitor ouabain, in a sodium-dependent fashion. In addition, expression of Na+ K+ ATPase subunits and E-cadherin, in addition to adequate Na+ K+ ATPase pump function, have been shown to be necessary for TJ formation and normal epithelial polarization (Rajasekaran et al. 2003, 2007). Accordingly, it has been hypothesised that Na+ K+ ATPase and E-cadherin function synergistically in assembling TJs (Rajasekaran et al. 2008).
Interestingly, while treatment with high concentrations of ouabain that inhibit Na+ K+ ATPase pump function increase permeability and decrease transepithelial resistance (Rajasekaran et al. 2003; Contreras et al. 1999); treatment with nanomolar ouabain concentrations that do not affect pump function actually decrease TJ permeability to both ions and non-ionic molecules (Larre et al. 2010). The latter has been attributed in part to alterations in the expression of claudins -1, -2 and -4 (Larre et al. 2010). It is intriguing to speculate that such profoundly opposing effects on TJ function may in fact be subject to physiological regulation by endogenous hormone-like molecules in addition to exogenous drugs, with reports that an endogenous form of ouabain is synthesized and stored in the mammalian adrenal cortex and hypothalamus (Schoner and Scheiner-Bobis 2007).
Other ion channels which have been implicated in regulatory control of TJ functions include the Na+/glucose co-transporter SGLT-1. Glucose uptake by apically-expressed SGLT-1 in the intestinal brush border has been shown to induce a drop in transepithelial electrical resistance and to increase in the paracellular uptake of small nutrients in vitro (Turner et al. 1997) and in vivo in both rats (Pappenheimer and Reiss 1987) and humans (Turner et al. 2000). This has been associated with TJ strand disruption (Madara and Pappenheimer 1987), dissociation of ZO1 from tight junctions (Atisook and Madara 1991) and phosphorylation of myosin regulatory light chain at the epithelial perijunctional ring (Turner et al. 1997).
Energy-dependent ion channels are not the only ones to have been functionally linked to TJs, with the passive transport chloride transporter ClC-2 also known to localise at TJs in intestinal epithelia. Activation of this channel reportedly stimulates an increase in transepithelial electrical resistance and a concomitant reduction in paracellular permeability (Moeser et al. 2004). Furthermore expression of the chloride channel CFTR has been shown to increase transepithelial resistance (LeSimple et al. 2010); while CLIC-4 co-localises with ZO1 in apical regions of epithelial cells, suggesting a possible but unproven role in regulation of TJs (Berryman and Goldenring 2003). Finally, the transmembrane water channels termed aquaporins are also thought to regulate TJs, with aquaporin-5 in particular being shown to modulate epithelial paracellular permeability (Turner et al. 1997; Murakami et al. 2006).
1.2.4 Hormone Effects
Several hormones from the steroid receptor family and otherwise have been shown to regulate TJs, consistent with the physiological need to actively modulate tissue permeability or other important functions of TJs at different stages of development or hormonal cycles.
Among the most prominent, estrogen has been demonstrated to profoundly affect the TJs of sex hormone-sensitive epithelia ranging from reproductive tissue to the intestinal tract. In cervico-vaginal epithelium, oestrogen can reportedly decrease the resistance of both epithelial TJs and the lateral intercellular space via matrix metalloproteinase 7-induced modulation of occludin, with the net effect of increasing epithelial permeability (Gorodeski 2005, 2007).
Oestrogen receptor-β (ERβ) is expressed in intestinal epithelial cells, where it appears to regulate paracellular permeability in a manner not strictly dependent on the oestrus cycle. In fact both male and female rats that under-express ERβ exhibit greater epithelial permeability and susceptibility to barrier-disruptive injury than their wild type female counterparts (Wada-Hiraike et al. 2006; Looijer-van Langen et al. 2011). Female rats under-expressing ERβ also show ultrastructural evidence of altered TJ and desmosomal morphology (Wada-Hiraike et al. 2006).
Hormonal regulation of mammary epithelial permeability during pregnancy and lactation occurs via not just the complex effects of oestrogen, but rather its interplay with other hormones such as progesterone, glucocorticoids, prolactin and serotonin (5-HT). During pregnancy the mammary gland reaches the expanded alveolar stage of development, however milk synthesis cannot begin until after parturition in conjunction with prolactin and glucocorticoid secretion which dynamically regulate TJ opening to facilitate the delivery of milk proteins during breastfeeding. (Thompson 1996; Zettl et al. 1992; Stelwagen et al. 1999).
The neurotransmitter serotonin (5-HT) also appears to regulate epithelial homeostasis in several organ systems including the mammary gland, where it is locally synthesized (Matsuda et al. 2004). 5-HT regulates the lactation to involution switch, and exhibits biphasic effects on tight junctions in vitro; increasing transepithelial resistance at low concentrations and early time points via protein kinase A while disrupting TJs via p38 MAP kinase signalling following sustained exposure to higher concentrations (Pai and Horseman 2008, 2011).
1.2.5 Pro-inflammatory Cytokines
Prototypic pro-inflammatory cytokines including interleukins-1, -6, -17, -18 (IL-1, -6, -17, -18), tumour necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) are secreted from multiple cellular sources under physiological and pathophysiological circumstances. Among their pleiotrophic effects include profound remodelling of TJs, which often induces endothelial or epithelial barrier disruption and perpetuates inflammation (for a recent review see (Capaldo and Nusrat 2009)). The near-ubiquitous expression of cytokine receptors has fuelled reports of cytokine-induced TJ disruption in most epithelial and endothelial barriers, yet despite functional overlap between different cytokines there is no unifying paradigm of specific alterations which are essential for barrier dysfunction.
IL-1 has been reported to induce a variable combination of reduced transepithelial resistance or increased paracellular permeability in conjunction with occludin and ZO-1 degradation/redistribution in epithelial cells from the intestine (Al-Sadi and Ma 2007), thyroid (Nilsson et al. 1998) and cornea (Kimura et al. 2009) in addition to models of the blood-brain barrier (Bolton et al. 1998), blood-retinal barrier (Bamforth et al. 1997) and blood-testis barrier (Lie et al. 2011). The barrier-disruptive mechanisms associated with IL-1 exposure in both intestinal (Al-Sadi and Ma 2007; Al-Sadi et al. 2008, 2010) and corneal (Kimura et al. 2009) epithelial cells as well as an in vitro model of the blood-brain barrier (Afonso et al. 2007) have been ascribed to canonical NFκB signalling via upstream activators such as MEKK and downstream effectors including myosin light chain kinase.
In addition to phenocopying several noted effects of IL-1 on barrier function and occludin/ZO-1 distribution, TNF-α has been observed to reduce the structural complexity (Schmitz et al. 1999) of claudin-containing TJ strands (Furuse et al. 1998). Also in common with IL-1 signalling mechanisms, TNF-dependent reductions in barrier function have been linked to activation of NFκB in retinal endothelial cells (Aveleira et al. 2010) and corneal epithelial cells (Kimura et al. 2008). Barrier disruption downstream of TNF-α signalling in intestinal epithelial models has alternately been proposed to reflect expressional enhancement of specific micro-RNAs targeting occludin for degradation (Ye et al. 2011) or enhanced removal of occludin from tight junctions via caveolar-mediated endocytosis (Marchiando et al. 2010b).
Observations of IFN-γ-induced epithelial (Madara and Stafford 1989; Youakim and Ahdieh 1999; Adams et al. 1993) or microvasculature endothelial (Oshima et al. 2001) barrier disruption in conjunction with degradation/mislocalization of ZO/occludin proteins might seem to mirror the cellular mechanisms discussed above in response to IL-1 or TNF-α exposure. However several lines of evidence suggest not only overlapping mechanisms but also unique ones whereby IFN-γ disrupts barrier function. One contends that the barrier-disruptive effects of IFN-γ in intestinal epithelia involve PI3-kinase/NFκB cross-talk (Boivin et al. 2009); another that macropinocytotic internalisation of occludin is responsible for induced deficits (Bruewer et al. 2005), while yet another possibility is that IFN-induced protease activation cleaves supporting TJ proteins such as claudin-2 (Willemsen et al. 2005). Reported alterations in the lipid composition of membrane microdomains following IFN/TNF co-treatment (Li et al. 2008) also offer a novel explanation for putative sub-membranous displacement of occludin and ZO-1 from tight junctions. Accordingly, although synergism between IFN-γ and TNF-α has been reported in many instances (Rodriguez et al. 1995; Wang et al. 2006; Bruewer et al. 2003), it is interesting to note that this can be dissociated from the pro-apoptotic effects of some pro-inflammatory cytokines (Bruewer et al. 2003).
1.2.6 Hypoxia
The adaptive response to reduced oxygen tension, termed hypoxia, also plays an important role in influencing TJ structure and function in physiological and pathophysiological settings. Physiological differences in vascular perfusion between tissues dictate that some body compartments exist in normoxic states (e.g. lung alveoli) while others are relatively hypoxic (e.g. colon). In pathophysiological settings, hypoxia reportedly activates Notch signalling (Chen et al. 2010); which in turn has been implicated in reducing gene expression of TJ components such as occludin and ZO-1 during EMT in airway epithelial cells (Aoyagi-Ikeda et al. 2011). While this implies that Notch activation secondary to hypoxia exerts a negative effect on lung epithelial barrier function, it is interesting to note that inactivation of Notch signalling may have a similar net effect in intestinal epithelia (Dahan et al. 2011). Whether this reflects innate differences in sensitivity to hypoxic signalling in tissues with disparate basal oxygen tensions, or merely illustrates the importance of carefully balancing Notch activity levels for barrier function homeostasis in any epithelial tissue is not yet clear.
What is clear is that regulation of epithelial barrier function by hypoxic signalling is complex and multi-factorial. Temporal activation of the transcription factors Slug and Snail during hypoxia (Kurrey et al. 2005) can also trigger junctional disassembly via repression of occludin, ZO-1 and claudin-1 expression (Martinez-Estrada et al. 2006; Ohkubo and Ozawa 2004; Wang et al. 2007). Similarly, reduced expression of occludin and claudin-1 have been demonstrated both in vitro and in vivo in renal epithelial cells deficient in the tumour suppressor gene von Hippel-Lindau (VHL) (Harten et al. 2009). Consequently, VHL inactivation has been associated with loss of barrier function and structural disorder of the renal epithelial phenotype (Calzada et al. 2006). Since a major function of the VHL protein product is to promote proteasomal degradation of hypoxia inducible factors (HIFs) (Maxwell et al. 1999), much interest has focussed on the potential role of HIFs in regulating TJs in various tissues. Recent evidence has suggested that HIF-1α antagonism can temper occludin/ZO-1 redistribution and the associated defects in intestinal epithelial barrier function induced by pro-inflammatory cytokines (Liu et al. 2011). A similar regulatory role has been noted in endothelial tight junctions, with loss of HIF-1α promoting TJ re-sealing in brain microvascular endothelial sheets compromised by prior exposure to either high glucose levels (Yan et al. 2012) or hypoxia-reoxygenation injury (Yeh et al. 2007). Taken together with the links between enhanced HIF activity and tumour progression or metastasis (Liao et al. 2007), the importance of hypoxia as an upstream regulator of tight junctions and barrier function cannot be underestimated.
As described above, TJ proteins are important for the maintenance of cell polarity and for hormonal and cytokine regulation of cellular homeostasis among a myriad of associated functions. Disruptions in cell polarity and tissue architecture are hallmarks of de-differentiation and early features of malignancy (Molitoris and Nelson 1990). In addition, several TJ-associated proteins have recently been shown to be targeted by oncogenes such as ERBB2 (Aranda et al. 2006) and MYC (Zhan et al. 2008) to facilitate malignant transformation. Furthermore, TJ proteins including Scribble (Javier 2008) and ZO2 (Glaunsinger et al. 2001) have been shown to be targeted by oncogenic viruses such as the human papilloma virus. Collectively, these studies provide strong evidence that TJ proteins may indeed act as key regulators of cancer initiation and progression. This will next be addressed.
1.3 Pathophysiological Processes in Cancer Influenced by Tight Junctions
1.3.1 Regulation of Cell Proliferation and Apoptosis
Tumour formation requires the acquisition of alterations that facilitate sustained proliferative capacity, whilst resisting cellular senescence and apoptotic cell death (Hanahan and Weinberg 2011). Several studies have described how alterations in several TJ-associated proteins may upset the delicate balance of growth and death signalling to result in malignant transformation. As noted earlier, however, the complex and tissue-specific regulation of TJ function in various endothelial and epithelial cells makes it unlikely that a single paradigm of simple expressional upregulation or downregulation will emerge to explain the many functional events associated with tumour initiation and progression.
Regardless, members of the largest family of integral membrane TJ proteins, the claudins, are frequently dysregulated in many cancers and appear to have a central role in determining cell fate (Escudero-Esparza et al. 2011). With respect to tumour initiation, Claudin-6 downregulation has been shown to result in increased resistance to apoptosis in vitro (Osanai et al. 2007). Claudin-1 expression, though increased in senescent cells (Swisshelm et al. 1999), has been reported to be decreased throughout several tumour types (Martin et al. 2011). Similarly downregulation of occludin has been correlated with dedifferentiation and progression of several cancers including endometrial (Tobioka et al. 2004a) and lung (Tobioka et al. 2004b). These effects may be due to occludin-mediated regulation of apoptosis, as occludin loss results in decreased expression of pro-apoptotic proteins including Bax (Osanai et al. 2006).
Alterations in proteins of the junctional adhesion molecule (JAM) family have also been shown in breast and renal cancers (McSherry et al. 2011; Gutwein et al. 2009) and melanoma (Langer et al. 2011), where many have demonstrated prognostic value in determining levels of JAM-A expression in patient cohorts. Indeed aberrant expression of microRNA mir145 may be mechanistically responsible for observed overexpression of JAM-A in breast cancer patient tissues which correlate strongly with poor survival outcomes (Gotte et al. 2010; McSherry et al. 2009). Although generally accepted as primarily functioning in adhesive and barrier roles at the TJ, compelling data have recently emerged regarding a role for JAM-A in both apoptosis and proliferation control. Colonic epithelial cells of JAM-A-deficient mice show enhanced crypt proliferation as measured by Ki67 staining (Laukoetter et al. 2007). Specifically, JAM-A appears to control cell proliferation through inhibition of Akt-dependent β-catenin activation (Nava et al. 2011); with Akt inhibition reversing crypt proliferation in JAM-A-deficient mice. Somewhat conversely in the context of cancer, JAM-A deficient mice display significantly reduced tumor growth in a pancreatic tumor model, likely due to decreased angiogenesis and increased immune responses (Murakami et al. 2010). Furthermore, in a breast cancer mouse model, JAM-A deficient mice show significantly decreased tumour growth; with tumour cells displaying increased rates of apoptosis in vivo and in vitro (Murakami et al. 2011). Together these studies suggest that, in contrast to loss of TJ proteins such as claudins and occludin, upregulation of JAM-A may in fact facilitate increased tumor growth and survival by promoting signalling events which protect cells from apoptosis.
As mentioned, JAM-A associates with the peripheral TJ protein Par3 during junctional maturation and establishment of cell polarity (Ebnet et al. 2001). In a mouse model of mammary morphogenesis, Par3 depletion in mammary progenitor cells disrupted mammary development, resulting in ductal hyperplasia. Re-expression of full length Par3 (but not truncated Par3) rescued this defect, demonstrating that Par3/aPKC interaction is essential for normal breast morphogenesis (McCaffrey and Macara 2009). The interaction of another TJ-associated protein and Par polarity complex member, Par 6, with aPKC has also been shown to be required for ErbB2 oncogene-driven evasion of apoptosis and disruption of breast cellular morphogenesis in vitro (Aranda et al. 2006).
Interestingly, association of ZO-1 with the transcription factor ZONAB can directly promote expression of the ErbB2 oncogene (Balda and Matter 2000). Furthermore, ZONAB is a critical determinant of cell cycle progress through effects on cyclin D1 and cdk4 (Balda et al. 2003). Similarly ZO2 can control cell proliferation through sequestration of transcription factors such as Jun and Fos at the TJ in a density-dependent manner (Huerta et al. 2007). Finally, interactions between ZO1 and the polarity complex member Scribble play an important role in normal regulation of cell adhesion (Ivanov et al. 2010). Interestingly, correct localization and expression of Scribble mediates pro-apoptotic signalling critical for both normal mammary gland morphogenesis and resisting MYC–induced transformation (Zhan et al. 2008).
In summary, data suggests that TJ proteins may be critical determinants of cancer initiation through effects on oncogene expression and imbalances in cell proliferative and apoptotic signaling.
1.3.2 Migration and Invasion
Although uncontrolled growth is a fundamental requirement during transformation, cancer cells must acquire both migratory and invasive capabilities in order to successfully disseminate from a primary tumour before seeding metastases at distant sites. Generally, cell migration consists of three main steps: the activation of Rho GTPases extend cell protrusions (through assembly of focal contacts with extracellular matrix proteins), the cell is dragged forward (through myosin II-mediated cell contraction), and finally cell adhesions are disassembled at the rear of the cell. This cyclical process (similar to regulation of apico-basal polarity and establishment of cell adhesion) requires crosstalk between junctional proteins, core polarity regulators (Etienne-Manneville and Hall 2001; Huo et al. 2011), and Rho family GTPases (Etienne-Manneville 2008; Iden and Collard 2008). Malignant cells can hijack these pro-migratory pathways and several TJ associated proteins have been implicated as having a causal role in cancer progression.
Loss of claudin-7 expression has been correlated with increased migration and invasion in lung (Lu et al. 2011), colorectal (Oshima et al. 2008) and oesophageal cancer (Lioni et al. 2007). Specifically, claudin-7 loss or mis-localisation in oesophageal cancer can lead to decreased E-cadherin expression and increased three-dimensional invasion in vitro (Lioni et al. 2007). Furthermore, re-expression of claudin-7 in claudin-7 deficient lung cancer cells resulted in decreased hepatocyte growth factor-mediated in vitro migration and invasion, and decreased in vivo tumour growth via regulation of ERK/MAPK signalling (Lu et al. 2011). Several other claudins have been implicated in regulating invasion through effects on matrix degrading enzymes from the matrix metalloproteinase (MMP) family. Claudin-1 expression in liver cancer cells promotes increased MMP2 activity and migration and invasion through activation of a c-Abl-PKCdelta signaling pathway (Yoon et al. 2010). Conversely, claudin-6 loss has been demonstrated to increase MMP activity and promote invasion of breast cancer cells (Osanai et al. 2007).
JAMs also have established roles in promoting normal leukocyte (Ostermann et al. 2002) and neutrophil migration (Cera et al. 2009), with JAM-A loss in endothelial cells functioning to decrease motility (Bazzoni et al. 2005). With respect to cancer, the majority of studies suggest that JAM proteins signal to increase cancer cell migration and invasion. JAM-A overexpression is associated with increased breast cancer metastasis (McSherry et al. 2009); potentially due to downstream regulation of the migratory protein β1-integrin through AF-6 and Rap1 GTPase adaptor proteins (McSherry et al. 2011; Severson et al. 2009). Furthermore, JAM-C is required for melanoma cell transendothelial migration in vitro (Ghislin et al. 2011); with its increased expression linked to melanoma invasion and metastasis in vivo (Fuse et al. 2007).
JAM proteins interact with several TJ adaptor proteins including AF6 and ZO proteins (Schneeberger and Lynch 2004). Fusion of AF6 and MLL represents the most common alteration in mixed lineage leukemia (MLL), where the Ras association-1 domain of AF6 likely activates the oncogenic potential of the MLL-AF6 protein (Liedtke et al. 2010). Recently, loss of AF6 in breast cancer has also been linked with poor prognosis (Letessier et al. 2007). Further work has demonstrated that AF6 loss dramatically increased heregulin-induced in vitro migration and invasion through activation of RAS/MAPK and Src kinase pathways; as well as significantly increased tumour growth and metastasis in an SKBR3 orthotopic mouse model (Fournier et al. 2011).
Interestingly, ZO1 has been shown to regulate the expression of the matrix metalloproteinase MT1-MMP, with knockdown of ZO1 in breast cancer cell lines reducing MT1-MMP expression and three-dimensional in vitro invasion (Polette et al. 2005). Recently, the TGF-β/Smad pathway (known to target the Par polarity complex (Viloria-Petit et al. 2009)) was demonstrated to induce breast cancer cell invasion through up-regulation of MMPs -2 and -9, reinforcing a potential link between matrix degradation and TJ-associated proteins (Wiercinska et al. 2011).
Furthermore, interactions between the Par complex members Par6 and aPKC lead to Rac GTPase activation in non-small cell lung cancer (NSCLC) cells, which drives anchorage-independent growth and invasion through activation of MMP10 (Frederick et al. 2008). The evidence for an involvement of Par3 in cancer cell migration has also been strengthened by studies demonstrating that Par-3 engages in the spatial regulation of Rac activity. Par3 directly interacts with Tiam1, a Rac1-specific guanine nucleotide exchange factor, to form a complex with aPKC-PAR-6-Cdc42, leading to Rac1 activation (Chen and Macara 2005). Recently, Par3 has been suggested to also be important in regulating squamous cell carcinoma collective cell migration. Recruitment of Par3 by DDR1 reduced actinomyosin contractile activity at cell-cell contacts and antagonized ROCK activity to Rac activation, thus keeping migrating cells clustered together and promoting more efficient collective migration (Hidalgo-Carcedo 2011).
Finally loss or mislocalisation of the ZO1 interacting protein, Scribble, increases migration and invasion of breast cancer cell lines (Zhan et al. 2008; Vaira et al. 2012), and cooperation of Scribble with the Ras oncogene increases MEK-ERK-dependent matrix invasion in a 3D breast acinar morphogenesis model (Dow et al. 2008).
Together, the above studies underline the importance of TJ proteins in mediating pro-migratory and pro-invasive signals and also suggest that targeting these proteins in cancer may be of therapeutic value.
1.3.3 Cell Fate and Differentiation
Recent work has provided evidence that several TJ proteins may regulate cell fate and differentiation during normal development (Balda and Matter 2009; Koch and Nusrat 2009). Expression levels of Claudin-4, ZO1 and ZO2 regulate murine stem cell commitment to hematopoetic or endothelial cell lineages (Stankovich et al. 2011). In addition, JAM proteins have been shown to be required for maintenance of hematopoietic stem cells in bone marrow (Arcangeli et al. 2011), spermatid differentiation (Gliki et al. 2004) and dendritic cell differentiation (Ogasawara et al. 2009). Furthermore, as mentioned above, TJ proteins interact with polarity complexes such as the Scribble and Par complexes to influence cell fate through processes including EMT, which allows cancer cells to alter their cell morphology and acquire pro-invasive phenotypes that might facilitate their migration to optimally-supportive growth niches (Viloria-Petit et al. 2009; Dow et al. 2008; Ozdamar et al. 2005).
The claudin-low aggressive breast cancer subtype is characterized by near absence of luminal differentiation markers, and increased expression of EMT and cancer stem cell-like markers (Prat et al. 2010). Indeed, gene expression signatures derived from normal human breast cells undergoing EMT in response to snail/slug activation or TGFβ treatment were recently shown to closely resemble those derived from claudin-low breast cancer tissues (Taube et al. 2010). Poor prognosis claudin-low tumour cells could undergo EMT through changes in several Zeb1 transcription factor–regulated genes. Zeb1 expression, through its repression of junctional proteins, may therefore also have a causal role in cancer types including breast (Aigner et al. 2007) and colorectal (Spaderna et al. 2008). Downregulation of Mir200c in breast cancer cells prevents expression of Zeb1, and reduces cancer cell migration (Cochrane et al. 2010). Furthermore, knockdown of Zeb1 in MDA-MB-231 breast cancer cells promotes EMT reversion whereby induced re-expression of the TJ proteins JAM-A, Occludin, Crumbs and PATJ partially re-establishes cell polarity and epithelial morphology, and significantly decreases cancer cell migration. Encouragingly, Zeb1 knockdown in a mouse model of metastatic colorectal cancer resulted in complete suppression of liver metastasis (Spaderna et al. 2008), suggesting that targeting Zeb1 may be a valuable therapeutic modality.
The TJ peripheral protein Par6 has been demonstrated to be required for TGFβ-induced EMT in breast epithelial cells (Viloria-Petit et al. 2009). Specifically, TGFβ-dependent phosphorylation of Par6 mediated recruitment of Smurf I (an E3 ubiquitin ligase) to promote degradation of RhoA and dissolution of the TJs, a crucial step in EMT (Ozdamar et al. 2005). In addition, TGFβ-Par6 signalling led to a loss of cell polarity and induced local invasion of MMECs in vitro and in vivo (Viloria-Petit et al. 2009).
ZO1 and its associated transcription factor ZONAB have also been implicated in the regulation of epithelial homeostasis and differentiation (Balda et al. 2003; Georgiadis et al. 2010; Sourisseau et al. 2006), through downstream regulation of cell cycle genes such as cyclin D1 and PCNA. Overexpression of ZONAB or knockdown of ZO-1 in mouse epithelial cells resulted in increased proliferation, and induced EMT-like morphological and protein expression changes that disrupted normal epithelial differentiation. This suggests that ZO1 loss, as seen in several cancers (Hoover et al. 1998; Kaihara et al. 2003), may phenocopy ZONAB over-expression in vitro thus altering cell differentiation through the induction of EMT.
Finally, several recent studies have suggested novel roles for TJ-associated proteins in controlling cellular homeostasis through regulation of spindle orientation and cell division. As mentioned above, transplantation of Par3-depleted stem cells into murine mammary fat pads resulted in disrupted ductal morphogenesis (McCaffrey and Macara 2009). Interestingly however, an expansion in the luminal progenitor cell population and reduction in myoepithelial cell population was evident in Par3-depleted mammary glands. This suggests a role for Par3 and its TJ binding partners in the regulation of progenitor differentiation and epithelial morphogenesis in vivo.
Recent studies in MDCK renal epithelial cells have shown that Par3 knockdown disrupts aPKC association with the apical cortex, and causes spindle misorientation leading to the appearance of multiple lumens in 3D cysts (Hao et al. 2010). Similarly, depletion or inhibition of Par6B or aPKC induces misorientation of the mitotic spindle to drive formation of aberrant Caco-2 intestinal epithelial cysts, with cell survival and apical positioning dependent upon aPKC expression levels (Durgan et al. 2011). Together these results suggest that TJ-associated proteins may have a role in spindle orientation and cell differentiation in vivo, and that their alteration may facilitate tumour formation by affecting the spatial regulation of cell division.
1.3.4 Metastasis and the Blood-Brain Barrier
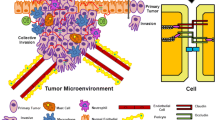
In addition to their regulatory roles in cell fate and differentiation, the functional integrity of TJs also play an intrinsic part in preventing cancer metastasis. In order for metastasis to occur, invading cells must first detach from the primary tumour and invade into the bloodstream. At the site of metastasis, the tumour cells must extravasate. This is similar to leukocyte extravasation and consists of three steps; firstly loose attachment and rolling on the endothelial surface, secondly tight attachment to the endothelium, and thirdly diapedesis or transmigration through the endothelium, either by the transcellular or paracellular route. While leukocytes and tumour cells share many similarities during the first two steps, the third step of diapedesis differs in that tumour cell migration irreversibly alters endothelial morphology (Heyder et al. 2002). This in turn induces endothelial cell retraction and in some cases apoptosis, possibly due to loss of cell-cell contacts (Brandt et al. 2005; Uchide et al. 2007; Kebers et al. 1998) via molecules including N-cadherin (Qi et al. 2005; Strell et al. 2007). The net effect of this destructive form of transmigration is the formation of gaps in the endothelial barrier, which can allow permissive access of tumour cells to the circulation and facilitate the early steps of metastasis.
A unique and highly-specialised form of the endothelium that poses a significant barrier to metastasis is the blood brain barrier (BBB), a complex structure consisting of nonfenestrated brain microvascular endothelial cells (BMECs) held together by abundant TJs and adherens junctions [reviewed in (Arshad et al. 2010; Abbott et al. 2010)]. Relative to other endothelial cells, BMECs exhibit higher transepithelial resistances and lower solute permeability, while TJs are structurally more complex and restrictive to diffusion of polar solutes via the paracellular pathway. The basement membrane is also thicker, and a layer of underlying astrocytes adds an extra regulatory component that restricts flow across the barrier. Collectively, the layers that compose the BBB represent a formidable challenge to the cancer cells which must breach it in order to form brain metastases. Since the brain lacks lymphatic drainage, brain metastasis occurs solely via the haematogenous route. It often carries a dire prognosis due to limited available treatment modalities, since the BBB is as impermeable to most drugs as it is to cells. Thus in recent years much interest has focused upon the regulation of BBB TJs, both to prevent cross-trafficking of tumour cells and also to understand mechanisms of selectively enhancing permeability to facilitate chemotherapeutic drug delivery. In particular, claudins have emerged as promising targets since losses in claudins-3 and -5 have been associated with increased leakiness of the BBB (Wolburg et al. 2003; Nitta et al. 2003).
With regard to specific types of cancers, melanoma displays the highest propensity of all primary tumours to metastasize to brain (Denkins et al. 2004). Melanoma cells have been shown in vitro to degrade brain endothelial TJs, resulting in decreased transepithelial resistance and decreased expression of claudin-5, ZO-1 and occludin (Fazakas et al. 2011). While the mechanisms of such events remain incompletely understood, TJ protein degradation may be facilitated by the fact that melanoma cells express several proteases, including matrix metalloproteinases (Hofmann et al. 2000), urokinase type plasminogen activator (Artym et al. 2002), seprase (Piñeiro-Sánchez et al. 1997) and serine proteinases (Fazakas et al. 2011).
Similarly, breast cancer displays a high propensity for brain metastasis, with a prevalence of approximately 30% at autopsy (Tsukada et al. 1983; Cho and Choi 1980). Risk factors include young age, grade and stage, oestrogen receptor negativity and Her2/neu overexpression (Pestalozzi et al. 2006; Hicks et al. 2006). While many soluble and cell-fixed factors are potentially involved in the transit of breast cancer cells across the BBB, one intriguing pathway that has recently emerged involves the chemokine stromal cell derived factor-1α (SDF-1α) and its receptor CXCR4. SDF-1α is secreted by several organs including the central nervous system, and SDF-1α treatment has been shown to increase the permeability of BMEC monolayers to breast tumour cell invasion by activating the PI-3K/AKT signalling pathway and causing endothelial cell retraction (Lee et al. 2004). Interestingly CXCR4 is expressed on breast cancer cells and is sensitive to upregulation by the oncogene Her2/Neu, which is associated with aggressive and highly-metastatic forms of breast cancer (Li et al. 2004). As with melanoma metastasis, however, it is clear that many other pathways could also govern transit of breast cancer cells across the BBB. Some of these mechanisms are likely to be convergent, for example the activity of degradative enzymes such as MMP-2 and -3 has been shown to be increased in in vivo breast cancer models (Mendes et al. 2005; Tester et al. 2004), while that of MMP-1 and -9 has been shown to be increased in in vitro settings of breast cancer (Stark et al. 2007). Given the links between MMPs and TJs alluded to earlier in this article, cross-regulation in the context of enhancing BBB permeability could therefore have serious implications for the development of brain metastases.
1.3.5 Angiogenesis
Metastasis is a complex and multi-step process which requires many forms of sophisticated functional adaptation in addition to the relatively simple requirement for mechanical movement of cells across biological barriers. One biological process which exerts a key influence on the ability of metastasized tumours (and indeed primary tumours) to survive is the generation of a vascular supply to nourish the growing tumour, termed tumour angiogenesis. Several TJ proteins have been implicated in both physiological and pathological angiogenesis. Junction adhesional molecules, and in particular JAM-A, are known to be important regulators of angiogenesis. JAM-A is expressed in the early vasculature of the developing mouse embryo (Parris et al. 2005), and is a vital component of basic fibroblast growth factor (bFGF)- induced angiogenesis. In the latter context it forms an inhibitory complex with αvβ3 integrin, which disassembles in response to bFGF signalling. JAM-A then facilitates MAP kinase activation, which in turn induces endothelial tube formation and angiogenesis (Naik et al. 2003; Naik and Naik 2006). Accordingly, transient knockdown of JAM-A has been shown to prevent bFGF-induced endothelial cell migration in an ECM substrate-specific fashion (Naik et al. 2003). Similarly, bFGF cannot induce angiogenesis in JAM-A deficient mice (Cooke et al. 2006), and pancreatic islet cell carcinomas grown in JAM-A null mice have been shown to exhibit a small decrease in angiogenesis compared to JAM-A-expressing mice (Murakami et al. 2010).
Other JAM proteins also appear to regulate angiogenesis in ways that could be relevant to tumour angiogenesis, or the pharmacological antagonism thereof. Soluble JAM-C levels have been shown to be increased in the serum of patients with rheumatoid arthritis, and treatment with exogenous JAM-C has the potential to induce angiogenesis in vitro (Rabquer et al. 2010). Furthermore JAM-C blockade has been shown to reduce angiogenesis by 50% in a mouse model of hypoxia-induced retinopathy (Orlova et al. 2006). Others have reported that functional antagonism of JAM-C with a monoclonal antibody can inhibit angiogenesis both in vitro and in vivo (Rabquer et al. 2010; Lamagna et al. 2005). Conversely, overexpression of JAM-B in a mouse model of Downs syndrome has been shown to inhibit the angiogenic response to vascular endothelial growth factor (VEGF) (Reynolds et al. 2010).
Other TJ proteins such as the claudins also play a complex role in angiogenesis. Claudin-5 has been shown to reduce endothelial cell motility via N-WASP and ROCK signalling cascades (Escudero-Esparza et al. 2011). Claudin-4-expressing ovarian epithelial cells reportedly feature upregulation of several genes encoding pro-angiogenic cytokines, and can induce angiogenesis both in vitro and in in vivo mouse models (Li et al. 2009). Claudins -1, -2 and -5 are expressed in normal murine retinal vessel development; while claudins -2 and -5 are overexpressed in vessels in an oxygen-induced retinopathy model (Luo et al. 2011).
Similarly expression of occludin can be altered by a number of angiogenic factors. Increased occludin expression has been linked with the secretion of angiopoetin-1 from pericytes (Hori et al. 2004), while decreased occludin expression in conjunction with increased paracellular permeability has been noted in retinal endothelial cells treated with vascular endothelial growth factor (VEGF) (Antonetti et al. 1998; Behzadian et al. 2003).
Taken together, the above points illustrate a complex and dynamic relationship between TJ proteins and angiogenic cascades. We believe this shows much potential for interrogation to better understand not only the mechanisms of tumour angiogenesis, but also to drive forward the design of new TJ-based therapeutics aimed at interfering with this process.
1.4 Conclusion
This review has attempted to summarise the molecular aspects of TJs regarding their regulation by normal physiological processes and their contributions to pathophysiological behaviours characteristic of cancers. What has emerged is that TJs are intrinsic downstream components of a number of important cascades regulating physiological processes as diverse as polarity, ion transport and responsiveness to paracrine or endocrine factors. Perhaps more importantly, it has also illustrated that while TJs may act as upstream regulators of functional behaviours intrinsically associated with cancer, there is no universal paradigm whereby simple loss or gain of TJ proteins drives processes like cell proliferation, migration or angiogenesis. Instead this review suggests that complex spatial and temporal regulation of TJ signalling must be elucidated on an individual protein basis, but may bear fruit in the design of future drugs to target tumourigenic behaviour.
References
Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37:13–25
Adams RB, Planchon SM, Roche JK (1993) IFN-gamma modulation of epithelial barrier function. Time course, reversibility, and site of cytokine binding. J Immunol 150:2356–2363
Afonso PV, Ozden S, Prevost MC, Schmitt C, Seilhean D, Weksler B, Couraud PO, Gessain A, Romero IA, Ceccaldi PE (2007) Human blood-brain barrier disruption by retroviral-infected lymphocytes: role of myosin light chain kinase in endothelial tight-junction disorganization. J Immunol 179:2576–2583
Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist P et al (2007) The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26:6979–6988
Albertson R, Chabu C, Sheehan A, Doe CQ (2004) Scribble protein domain mapping reveals a multistep localization mechanism and domains necessary for establishing cortical polarity. J Cell Sci 117:6061–6070
Al-Sadi RM, Ma TY (2007) IL-1beta causes an increase in intestinal epithelial tight junction permeability. J Immunol 178:4641–4649
Al-Sadi R, Ye D, Dokladny K, Ma TY (2008) Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction permeability. J Immunol 180:5653–5661
Al-Sadi R, Ye D, Said HM, Ma TY (2010) IL-1beta-induced increase in intestinal epithelial tight junction permeability is mediated by MEKK-1 activation of canonical NF-kappaB pathway. Am J Pathol 177:2310–2322
Antonetti DA, Barber AJ, Khin S, Lieth E, Tarbell JM, Gardner TW (1998) Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: vascular endothelial growth factor decreases occludin in retinal endothelial cells. Penn State Retina Research Group. Diabetes 47:1953–1959
Aoyagi-Ikeda K, Maeno T, Matsui H, Ueno M, Hara K, Aoki Y, Aoki F, Shimizu T, Doi H, Kawai-Kowase K et al (2011) Notch induces myofibroblast differentiation of alveolar epithelial cells via transforming growth factor-{beta}-Smad3 pathway. Am J Respir Cell Mol Biol 45:136–144
Aranda V, Haire T, Nolan ME, Calarco JP, Rosenberg AZ, Fawcett JP, Pawson T, Muthuswamy SK (2006) Par6-aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nat Cell Biol 8:1235–1245
Arcangeli ML, Frontera V, Bardin F, Obrados E, Adams S, Chabannon C, Schiff C, Mancini SJ, Adams RH, Aurrand-Lions M (2011) JAM-B regulates maintenance of hematopoietic stem cells in the bone marrow. Blood 118:4609–4619
Arshad F, Wang L, Sy C, Avraham S, Avraham HK (2010) Blood-brain barrier integrity and breast cancer metastasis to the brain. Pathol Res Int 2011:920509
Artym VV, Kindzelskii AL, Chen WT, Petty HR (2002) Molecular proximity of seprase and the urokinase-type plasminogen activator receptor on malignant melanoma cell membranes: dependence on beta1 integrins and the cytoskeleton. Carcinogenesis 23:1593–1601
Atisook K, Madara JL (1991) An oligopeptide permeates intestinal tight junctions at glucose-elicited dilatations. Implications for oligopeptide absorption. Gastroenterology 100:719–724
Aveleira CA, Lin CM, Abcouwer SF, Ambrosio AF, Antonetti DA (2010) TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes 59:2872–2882
Balda MS, Matter K (2000) The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J 19:2024–2033
Balda MS, Matter K (2009) Tight junctions and the regulation of gene expression. Biochim Biophys Acta 1788:761–767
Balda MS, Garrett MD, Matter K (2003) The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J Cell Biol 160:423–432
Bamforth SD, Lightman SL, Greenwood J (1997) Ultrastructural analysis of interleukin-1 beta-induced leukocyte recruitment to the rat retina. Invest Ophthalmol Vis Sci 38:25–35
Bazzoni G, Tonetti P, Manzi L, Cera MR, Balconi G, Dejana E (2005) Expression of junctional adhesion molecule-A prevents spontaneous and random motility. J Cell Sci 118:623–632
Behzadian MA, Windsor LJ, Ghaly N, Liou G, Tsai NT, Caldwell RB (2003) VEGF-induced paracellular permeability in cultured endothelial cells involves urokinase and its receptor. FASEB J 17:752–754
Berryman MA, Goldenring JR (2003) CLIC4 is enriched at cell-cell junctions and colocalizes with AKAP350 at the centrosome and midbody of cultured mammalian cells. Cell Motil Cytoskeleton 56:159–172
Boettner B, Van Aelst L (2009) Control of cell adhesion dynamics by Rap1 signaling. Curr Opin Cell Biol 21:684–693
Boivin MA, Roy PK, Bradley A, Kennedy JC, Rihani T, Ma TY (2009) Mechanism of interferon-gamma-induced increase in T84 intestinal epithelial tight junction. J Interferon Cytokine Res 29:45–54
Bolton SJ, Anthony DC, Perry VH (1998) Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience 86:1245–1257
Brandt B, Heyder C, Gloria-Maercker E, Hatzmann W, Rötger A, Kemming D, Zänker KS, Entschladen F, Dittmar T (2005) 3D-extravasation model – selection of highly motile and metastatic cancer cells. Semin Cancer Biol 15:387–395
Brennan K, Offiah G, McSherry EA, Hopkins AM (2010) Tight junctions: a barrier to the initiation and progression of breast cancer? J Biomed Biotechnol 2010:460607
Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, Nusrat A (2003) Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol 171:6164–6172
Bruewer M, Utech M, Ivanov AI, Hopkins AM, Parkos CA, Nusrat A (2005) Interferon-gamma induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J 19:923–933
Calzada MJ, Esteban MA, Feijoo-Cuaresma M, Castellanos MC, Naranjo-Suarez S, Temes E, Mendez F, Yanez-Mo M, Ohh M, Landazuri MO (2006) von Hippel-Lindau tumor suppressor protein regulates the assembly of intercellular junctions in renal cancer cells through hypoxia-inducible factor-independent mechanisms. Cancer Res 66:1553–1560
Capaldo CT, Nusrat A (2009) Cytokine regulation of tight junctions. Biochim Biophys Acta 1788:864–871
Cera MR, Fabbri M, Molendini C, Corada M, Orsenigo F, Rehberg M, Reichel CA, Krombach F, Pardi R, Dejana E (2009) JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J Cell Sci 122:268–277
Chen X, Macara IG (2005) Par-3 controls tight junction assembly through the Rac exchange factor Tiam1. Nat Cell Biol 7:262–269
Chen J, Imanaka N, Chen J, Griffin JD (2010) Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. Br J Cancer 102:351–360
Cho SY, Choi HY (1980) Causes of death and metastatic patterns in patients with mammary cancer. Ten-year autopsy study. Am J Clin Pathol 73:232–234
Cochrane DR, Howe EN, Spoelstra NS, Richer JK (2010) Loss of miR-200c: a marker of aggressiveness and chemoresistance in female reproductive cancers. J Oncol 2010:821717
Contreras RG, Shoshani L, Flores-Maldonado C, Lázaro A, Cereijido M (1999) Relationship between Na(+), K(+)-ATPase and cell attachment. J Cell Sci 112:4223–4232
Cooke VG, Naik MU, Naik UP (2006) Fibroblast growth factor-2 failed to induce angiogenesis in junctional adhesion molecule-A-deficient mice. Arterioscler Thromb Vasc Biol 26:2005–2011
Dahan S, Rabinowitz KM, Martin AP, Berin MC, Unkeless JC, Mayer L (2011) Notch-1 signaling regulates intestinal epithelial barrier function, through interaction with CD4+ T cells, in mice and humans. Gastroenterology 140:550–559
Denkins Y, Reiland J, Roy M, Sinnappah-Kang ND, Galjour J, Murry BP, Blust J, Aucoin R, Marchetti D (2004) Brain metastases in melanoma: roles of neurotrophins. Neuro Oncol 6:154–165
Dow LE, Brumby AM, Muratore R, Coombe ML, Sedelies KA, Trapani JA, Russell SM, Richardson HE, Humbert PO (2003) hScrib is a functional homologue of the Drosophila tumour suppressor Scribble. Oncogene 22:9225–9230
Dow LE, Elsum IA, King CL, Kinross KM, Richardson HE, Humbert PO (2008) Loss of human Scribble cooperates with H-Ras to promote cell invasion through deregulation of MAPK signalling. Oncogene 27:5988–6001
Durgan J, Kaji N, Jin D, Hall A (2011) Par6B and atypical PKC regulate mitotic spindle orientation during epithelial morphogenesis. J Biol Chem 286:12461–12474
Ebnet K, Suzuki A, Horikoshi Y, Hirose T, Meyer Zu Brickwedde MK, Ohno S, Vestweber D (2001) The cell polarity protein ASIP/PAR-3 directly associates with junctional adhesion molecule (JAM). EMBO J 20:3738–3748
Escudero-Esparza A, Jiang WG, Martin TA (2011) Claudin-5 participates in the regulation of endothelial cell motility. Mol Cell Biochem 362(1–2):71–85
Etienne-Manneville S (2008) Polarity proteins in migration and invasion. Oncogene 27:6970–6980
Etienne-Manneville S, Hall A (2001) Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell 106:489–498
Fazakas C, Wilhelm I, Nagyoszi P, Farkas AE, Haskó J, Molnár J, Bauer H, Bauer HC, Ayaydin F, Dung NT, Siklós L, Krizbai IA (2011) Transmigration of melanoma cells through the blood-brain barrier: role of endothelial tight junctions and melanoma-released serine proteases. PLoS One 6:e20758
Feng W, Wu H, Chan LN, Zhang M (2008) Par-3-mediated junctional localization of the lipid phosphatase PTEN is required for cell polarity establishment. J Biol Chem 283:23440–23449
Fournier G, Cabaud O, Josselin E, Chaix A, Adelaide J, Isnardon D, Restouin A, Castellano R, Dubreuil P, Chaffanet M et al (2011) Loss of AF6/afadin, a marker of poor outcome in breast cancer, induces cell migration, invasiveness and tumor growth. Oncogene 30:3862–3874
Frederick LA, Matthews JA, Jamieson L, Justilien V, Thompson EA, Radisky DC, Fields AP (2008) Matrix metalloproteinase-10 is a critical effector of protein kinase Ciota-Par6alpha-mediated lung cancer. Oncogene 27:4841–4853
Furuse M, Sasaki H, Fujimoto K, Tsukita S (1998) A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol 143:391–401
Fuse C, Ishida Y, Hikita T, Asai T, Oku N (2007) Junctional adhesion molecule-C promotes metastatic potential of HT1080 human fibrosarcoma. J Biol Chem 282:8276–8283
Georgiadis A, Tschernutter M, Bainbridge JW, Balaggan KS, Mowat F, West EL, Munro PM, Thrasher AJ, Matter K, Balda MS et al (2010) The tight junction associated signalling proteins ZO-1 and ZONAB regulate retinal pigment epithelium homeostasis in mice. PloS one 5:e15730
Ghislin S, Obino D, Middendorp S, Boggetto N, Alcaide-Loridan C, Deshayes F (2011) Junctional adhesion molecules are required for melanoma cell lines transendothelial migration in vitro. Pigment Cell Melanoma Res 24:504–511
Glaunsinger BA, Weiss RS, Lee SS, Javier R (2001) Link of the unique oncogenic properties of adenovirus type 9 E4-ORF1 to a select interaction with the candidate tumor suppressor protein ZO-2. EMBO J 20:5578–5586
Gliki G, Ebnet K, Aurrand-Lions M, Imhof BA, Adams RH (2004) Spermatid differentiation requires the assembly of a cell polarity complex downstream of junctional adhesion molecule-C. Nature 431:320–324
Gorodeski GI (2005) Aging and estrogen effects on transcervical-transvaginal epithelial permeability. J Clin Endocrinol Metab 90:345–351
Gorodeski GI (2007) Estrogen decrease in tight junctional resistance involves matrix-metalloproteinase-7-mediated remodeling of occludin. Endocrinology 148:218–231
Gotte M, Mohr C, Koo CY, Stock C, Vaske AK, Viola M, Ibrahim SA, Peddibhotla S, Teng YH, Low JY et al (2010) miR-145-dependent targeting of junctional adhesion molecule A and modulation of fascin expression are associated with reduced breast cancer cell motility and invasiveness. Oncogene 29:6569–6580
Gutwein P, Schramme A, Voss B, Abdel-Bakky MS, Doberstein K, Ludwig A, Altevogt P, Hansmann ML, Moch H, Kristiansen G et al (2009) Downregulation of junctional adhesion molecule-A is involved in the progression of clear cell renal cell carcinoma. Biochem Biophys Res Commun 380:387–391
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hao Y, Du Q, Chen X, Zheng Z, Balsbaugh JL, Maitra S, Shabanowitz J, Hunt DF, Macara IG (2010) Par3 controls epithelial spindle orientation by aPKC-mediated phosphorylation of apical pins. Curr Biol 20:1809–1818
Harris TJ, Peifer M (2004) Adherens junction-dependent and -independent steps in the establishment of epithelial cell polarity in Drosophila. J Cell Biol 167:135–147
Harten SK, Shukla D, Barod R, Hergovich A, Balda MS, Matter K, Esteban MA, Maxwell PH (2009) Regulation of renal epithelial tight junctions by the von Hippel-Lindau tumor suppressor gene involves occludin and claudin 1 and is independent of E-cadherin. Mol Biol Cell 20:1089–1101
Heyder C, Gloria-Maercker E, Entschladen F, Hatzmann W, Niggemann B, Zänker KS, Dittmar T (2002) Realtime visualization of tumor cell/endothelial cell interactions during transmigration across the endothelial barrier. J Cancer Res Clin Oncol 128:533–538
Hicks DG, Short SM, Prescott NL, Tarr SM, Coleman KA, Yoder BJ, Crowe JP, Choueiri TK, Dawson AE, Budd GT, Tubbs RR, Casey G, Weil RJ (2006) Breast cancers with brain metastases are more likely to be estrogen receptor negative, express the basal cytokeratin CK5/6, and overexpress HER2 or EGFR. Am J Surg Pathol 30:1097–1104
Hidalgo-Carcedo C, Hooper S, Chaudhry SI, Williamson P, Harrington K, Leitinger B, Sahai E (2011) E Collective cell migration requires suppression of actomyosin at cell-cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat Cell Biol 13:49–58
Hofmann UB, Westphal JR, Zendman AJ, Becker JC, Ruiter DJ, van Muijen GN (2000) Expression and activation of matrix metalloproteinase-2 (MMP-2) and its co-localization with membrane-type 1 matrix metalloproteinase (MT1-MMP) correlate with melanoma progression. J Pathol 191:245–256
Hollander D (1988) Crohn’s disease–a permeability disorder of the tight junction? Gut 29:1621–1624
Hoover KB, Liao SY, Bryant PJ (1998) Loss of the tight junction MAGUK ZO-1 in breast cancer: relationship to glandular differentiation and loss of heterozygosity. Am J Pathol 153:1767–1773
Hori S, Ohtsuki S, Hosoya K, Nakashima E, Terasaki T (2004) A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J Neurochem 89:503–513
Huerta M, Munoz R, Tapia R, Soto-Reyes E, Ramirez L, Recillas-Targa F, Gonzalez-Mariscal L, Lopez-Bayghen E (2007) Cyclin D1 is transcriptionally down-regulated by ZO-2 via an E box and the transcription factor c-Myc. Mol Biol Cell 18:4826–4836
Huo L, Wen W, Wang R, Kam C, Xia J, Feng W, Zhang M (2011) Cdc42-dependent formation of the ZO-1/MRCKbeta complex at the leading edge controls cell migration. EMBO J 30:665–678
Hurd TW, Gao L, Roh MH, Macara IG, Margolis B (2003) Direct interaction of two polarity complexes implicated in epithelial tight junction assembly. Nat Cell Biol 5:137–142
Iden S, Collard JG (2008) Crosstalk between small GTPases and polarity proteins in cell polarization. Nat Rev Mol Cell Biol 9:846–859
Itoh M, Sasaki H, Furuse M, Ozaki H, Kita T, Tsukita S (2001) Junctional adhesion molecule (JAM) binds to PAR-3: a possible mechanism for the recruitment of PAR-3 to tight junctions. J Cell Biol 154:491–497
Ivanov AI, Young C, Den Beste K, Capaldo CT, Humbert PO, Brennwald P, Parkos CA, Nusrat A (2010) Tumor suppressor scribble regulates assembly of tight junctions in the intestinal epithelium. Am J Pathol 176:134–145
Javier RT (2008) Cell polarity proteins: common targets for tumorigenic human viruses. Oncogene 27:7031–7046
Kaihara T, Kusaka T, Nishi M, Kawamata H, Imura J, Kitajima K, Itoh-Minami R, Aoyama N, Kasuga M, Oda Y et al (2003) Dedifferentiation and decreased expression of adhesion molecules, E-cadherin and ZO-1, in colorectal cancer are closely related to liver metastasis. J Exp Clin Cancer Res 22:117–123
Kaplan JH (2002) Biochemistry of Na, K-ATPase. Annu Rev Biochem 71:511–535
Kebers F, Lewalle JM, Desreux J, Munaut C, Devy L, Foidart JM, Noël A (1998) Induction of endothelial cell apoptosis by solid tumor cells. Exp Cell Res 240:197–205
Kimura K, Teranishi S, Fukuda K, Kawamoto K, Nishida T (2008) Delayed disruption of barrier function in cultured human corneal epithelial cells induced by tumor necrosis factor-alpha in a manner dependent on NF-kappaB. Invest Ophthalmol Vis Sci 49:565–571
Kimura K, Teranishi S, Nishida T (2009) Interleukin-1beta-induced disruption of barrier function in cultured human corneal epithelial cells. Invest Ophthalmol Vis Sci 50:597–603
Kirschner N, Haftek M, Niessen CM, Behne MJ, Furuse M, Moll I, Brandner JM (2011) CD44 regulates tight-junction assembly and barrier function. J Invest Dermatol 131:932–943
Koch S, Nusrat A (2009) Dynamic regulation of epithelial cell fate and barrier function by intercellular junctions. Ann N Y Acad Sci 1165:220–227
Kurrey NK, Amit K, Bapat SA (2005) Snail and Slug are major determinants of ovarian cancer invasiveness at the transcription level. Gynecol Oncol 97:155–165
Lamagna C, Hodivala-Dilke KM, Imhof BA, Aurrand-Lions M (2005) Antibody against junctional adhesion molecule-C inhibits angiogenesis and tumor growth. Cancer Res 65:5703–5710
Langer HF, Orlova VV, Xie C, Kaul S, Schneider D, Lonsdorf AS, Fahrleitner M, Choi EY, Dutoit V, Pellegrini M et al (2011) A novel function of junctional adhesion molecule-C in mediating melanoma cell metastasis. Cancer Res 71:4096–4105
Larre I, Lazaro A, Contreras RG, Balda MS, Matter K, Flores-Maldonado C, Ponce A, Flores-Benitez D, Rincon-Heredia R, Padilla-Benavides T, Castillo A, Shoshani L, Cereijido M (2010) Ouabain modulates epithelial cell tight junction. Proc Natl Acad Sci U S A 107:11387–11392
Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA et al (2007) JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med 204:3067–3076
Lee BC, Lee TH, Avraham S, Avraham HK (2004) Involvement of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1alpha in breast cancer cell migration through human brain microvascular endothelial cells. Mol Cancer Res 2:327–338
LeSimple P, Liao J, Robert R, Gruenert DC, Hanrahan JW (2010) Cystic fibrosis transmembrane conductance regulator trafficking modulates the barrier function of airway epithelial cell monolayers. J Physiol 588:1195–1209
Letessier A, Garrido-Urbani S, Ginestier C, Fournier G, Esterni B, Monville F, Adelaide J, Geneix J, Xerri L, Dubreuil P et al (2007) Correlated break at PARK2/FRA6E and loss of AF-6/Afadin protein expression are associated with poor outcome in breast cancer. Oncogene 26:298–307
Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, Zhou X, Xia W, Hortobagyi GN, Yu D, Hung MC (2004) Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell 6:459–469
Li Q, Zhang Q, Wang M, Zhao S, Ma J, Luo N, Li N, Li Y, Xu G, Li J (2008) Interferon-gamma and tumor necrosis factor-alpha disrupt epithelial barrier function by altering lipid composition in membrane microdomains of tight junction. Clin Immunol 126:67–80, Orlando, FL
Li J, Chigurupati S, Agarwal R, Mughal MR, Mattson MP, Becker KG, Wood WH 3rd, Zhang Y, Morin PJ (2009) Possible angiogenic roles for claudin-4 in ovarian cancer. Cancer Biol Ther 8:1806–1814
Liao D, Corle C, Seagroves TN, Johnson RS (2007) Hypoxia-inducible factor-1alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res 67:563–572
Lie PP, Cheng CY, Mruk DD (2011) Interleukin-1alpha is a regulator of the blood-testis barrier. FASEB J 25:1244–1253
Liedtke M, Ayton PM, Somervaille TC, Smith KS, Cleary ML (2010) Self-association mediated by the Ras association 1 domain of AF6 activates the oncogenic potential of MLL-AF6. Blood 116:63–70
Lingrel JB, Kuntzweiler T (1994) Na+, K(+)-ATPase. J Biol Chem 5:19659–19662
Lioni M, Brafford P, Andl C, Rustgi A, El-Deiry W, Herlyn M, Smalley KS (2007) Dysregulation of claudin-7 leads to loss of E-cadherin expression and the increased invasion of esophageal squamous cell carcinoma cells. Am J Pathol 170:709–721
Liu H, Li M, Wang P, Wang F (2011) Blockade of hypoxia-inducible factor-1alpha by YC-1 attenuates interferon-gamma and tumor necrosis factor-alpha-induced intestinal epithelial barrier dysfunction. Cytokine 56:581–588
Looijer-van Langen M, Hotte N, Dieleman LA, Albert E, Mulder C, Madsen KL (2011) Estrogen receptor-β signaling modulates epithelial barrier function. Am J Physiol Gastrointest Liver Physiol 300:G621–G626
Lu Z, Ding L, Hong H, Hoggard J, Lu Q, Chen YH (2011) Claudin-7 inhibits human lung cancer cell migration and invasion through ERK/MAPK signaling pathway. Exp Cell Res 317:1935–1946
Luo Y, Xiao W, Zhu X, Mao Y, Liu X, Chen X, Huang J, Tang S, Rizzolo LJ (2011) Differential expression of claudins in retinas during normal development and the angiogenesis of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci 52:7556–7564
Madara JL, Pappenheimer JR (1987) Structural basis for physiological regulation of paracellular pathways in intestinal epithelia. J Membr Biol 100:149–164
Madara JL, Stafford J (1989) Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Invest 83:724–727
Malik N, Canfield VA, Beckers MC, Gros P, Levenson R (1996) Identification of the mammalian Na, K-ATPase 3 subunit. J Biol Chem 13:22754–22758
Marchiando AM, Graham WV, Turner JR (2010a) Epithelial barriers in homeostasis and disease. Annu Rev Pathol 5:119–144
Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR 2nd, Raleigh DR, Guan Y, Watson AJ, Montrose MH et al (2010b) Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol 189:111–126
Martin TA, Mason MD, Jiang WG (2011) Tight junctions in cancer metastasis. Front Biosci 16:898–936
Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, Mostov K (2007) PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell 128:383–397
Martinez-Estrada OM, Culleres A, Soriano FX, Peinado H, Bolos V, Martinez FO, Reina M, Cano A, Fabre M, Vilaro S (2006) The transcription factors Slug and Snail act as repressors of Claudin-1 expression in epithelial cells. Biochem J 394:449–457
Matsuda M, Imaoka T, Vomachka AJ, Gudelsky GA, Hou Z, Mistry M, Bailey JP, Nieport KM, Walther DJ, Bader M, Horseman ND (2004) Serotonin regulates mammary gland development via an autocrine-paracrine loop. Dev Cell 6:193–203
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399:271–275
McCaffrey LM, Macara IG (2009) The Par3/aPKC interaction is essential for end bud remodeling and progenitor differentiation during mammary gland morphogenesis. Genes Dev 23:1450–1460
McSherry EA, McGee SF, Jirstrom K, Doyle EM, Brennan DJ, Landberg G, Dervan PA, Hopkins AM, Gallagher WM (2009) JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int J Cancer 125:1343–1351
McSherry EA, Brennan K, Hudson L, Hill AD, Hopkins AM (2011) Breast cancer cell migration is regulated through junctional adhesion molecule-A-mediated activation of Rap1 GTPase. Breast Cancer Res 13:R31
Mendes O, Kim HT, Stoica G (2005) Expression of MMP2, MMP9 and MMP3 in breast cancer brain metastasis in a rat model. Clin Exp Metastasis 22:237–246
Moeser AJ, Nighot PK, Engelke KJ, Ueno R, Blikslager AT (2004) Recovery of mucosal barrier function in ischemic porcine ileum and colon is stimulated by a novel agonist of the ClC-2 chloride channel, lubiprostone. Am J Physiol Gastrointest Liver Physiol 292:G647–G656
Molitoris BA, Nelson WJ (1990) Alterations in the establishment and maintenance of epithelial cell polarity as a basis for disease processes. J Clin Invest 85:3–9
Mori H, Gjorevski N, Inman JL, Bissell MJ, Nelson CM (2009) Self-organization of engineered epithelial tubules by differential cellular motility. Proc Natl Acad Sci USA 106:14890–14895
Murakami M, Murdiastuti K, Hosoi K, Hill AE (2006) AQP and the control of fluid transport in a salivary gland. J Membr Biol 210:91–103
Murakami M, Francavilla C, Torselli I, Corada M, Maddaluno L, Sica A, Matteoli G, Iliev ID, Mantovani A, Rescigno M et al (2010) Inactivation of junctional adhesion molecule-A enhances antitumoral immune response by promoting dendritic cell and T lymphocyte infiltration. Cancer Res 70:1759–1765
Murakami M, Giampietro C, Giannotta M, Corada M, Torselli I, Orsenigo F, Cocito A, D’Ario G, Mazzarol G, Confalonieri S et al (2011) Abrogation of junctional adhesion molecule-A expression induces cell apoptosis and reduces breast cancer progression. PloS one 6:e21242
Naik MU, Naik UP (2006) Junctional adhesion molecule-A-induced endothelial cell migration on vitronectin is integrin alpha v beta 3 specific. J Cell Sci 119:490–499
Naik MU, Vuppalanchi D, Naik UP (2003) Essential role of junctional adhesion molecule-1 in basic fibroblast growth factor-induced endothelial cell migration. Arterioscler Thromb Vasc Biol 23:2165–2171
Nakagawa M, Fukata M, Yamaga M, Itoh N, Kaibuchi K (2001) Recruitment and activation of Rac1 by the formation of E-cadherin-mediated cell-cell adhesion sites. J Cell Sci 114:1829–1838
Naor D, Sionov RV, Ish-Shalom D (1997) CD44: structure, function, and association with the malignant process. Adv Cancer Res 71:241–319
Nava P, Capaldo CT, Koch S, Kolegraff K, Rankin CR, Farkas AE, Feasel ME, Li L, Addis C, Parkos CA et al (2011) JAM-A regulates epithelial proliferation through Akt/beta-catenin signalling. EMBO Rep 12:314–320
Nilsson M, Husmark J, Bjorkman U, Ericson LE (1998) Cytokines and thyroid epithelial integrity: interleukin-1alpha induces dissociation of the junctional complex and paracellular leakage in filter-cultured human thyrocytes. J Clin Endocrinol Metab 83:945–952
Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S (2003) Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol 161:653–660
Ogasawara N, Kojima T, Go M, Fuchimoto J, Kamekura R, Koizumi J, Ohkuni T, Masaki T, Murata M, Tanaka S et al (2009) Induction of JAM-A during differentiation of human THP-1 dendritic cells. Biochem Biophys Res Commun 389:543–549
Ohkubo T, Ozawa M (2004) The transcription factor Snail downregulates the tight junction components independently of E-cadherin downregulation. J Cell Sci 117:1675–1685
Orlova VV, Economopoulou M, Lupu F, Santoso S, Chavakis T (2006) Junctional adhesion molecule-C regulates vascular endothelial permeability by modulating VE-cadherin-mediated cell-cell contacts. J Exp Med 203:2703–2714
Osanai M, Murata M, Nishikiori N, Chiba H, Kojima T, Sawada N (2006) Epigenetic silencing of occludin promotes tumorigenic and metastatic properties of cancer cells via modulations of unique sets of apoptosis-associated genes. Cancer Res 66:9125–9133
Osanai M, Murata M, Chiba H, Kojima T, Sawada N (2007) Epigenetic silencing of claudin-6 promotes anchorage-independent growth of breast carcinoma cells. Cancer Sci 98:1557–1562
Oshima T, Laroux FS, Coe LL, Morise Z, Kawachi S, Bauer P, Grisham MB, Specian RD, Carter P, Jennings S et al (2001) Interferon-gamma and interleukin-10 reciprocally regulate endothelial junction integrity and barrier function. Microvasc Res 61:130–143
Oshima T, Kunisaki C, Yoshihara K, Yamada R, Yamamoto N, Sato T, Makino H, Yamagishi S, Nagano Y, Fujii S et al (2008) Reduced expression of the claudin-7 gene correlates with venous invasion and liver metastasis in colorectal cancer. Oncol Rep 19:953–959
Ostermann G, Weber KS, Zernecke A, Schroder A, Weber C (2002) JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol 3:151–158
Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL (2005) Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science 307:1603–1609
Pai VP, Horseman ND (2008) Biphasic regulation of mammary epithelial resistance by serotonin through activation of multiple pathways. J Biol Chem 283:30901–30910
Pai VP, Horseman ND (2011) Multiple cellular responses to serotonin contribute to epithelial homeostasis. PLoS One 6:e17028
Pappenheimer JR, Reiss KZ (1987) Contribution of solvent drag through intercellular junctions to absorption of nutrients by the small intestine of the rat. J Membr Biol 100:123–136
Parris JJ, Cooke VG, Skarnes WC, Duncan MK, Naik UP (2005) JAM-A expression during embryonic development. Dev Dyn 233:1517–1524
Pestalozzi BC, Zahrieh D, Price KN, Holmberg SB, Lindtner J, Collins J, Crivellari D, Fey MF, Murray E, Pagani O, Simoncini E, Castiglione-Gertsch M, Gelber RD, Coates AS, Goldhirsch A, International Breast Cancer Study Group (IBCSG) (2006) Identifying breast cancer patients at risk for Central Nervous System (CNS) metastases in trials of the International Breast Cancer Study Group (IBCSG). Ann Oncol 17:935–944
Piñeiro-Sánchez ML, Goldstein LA, Dodt J, Howard L, Yeh Y, Tran H, Argraves WS, Chen WT (1997) Identification of the 170-kDa melanoma membrane-bound gelatinase (seprase) as a serine integral membrane protease. J Biol Chem 272:7595–7601
Polette M, Gilles C, Nawrocki-Raby B, Lohi J, Hunziker W, Foidart JM, Birembaut P (2005) Membrane-type 1 matrix metalloproteinase expression is regulated by zonula occludens-1 in human breast cancer cells. Cancer Res 65:7691–7698
Polette M, Mestdagt M, Bindels S, Nawrocki-Raby B, Hunziker W, Foidart JM, Birembaut P, Gilles C (2007) Beta-catenin and ZO-1: shuttle molecules involved in tumor invasion-associated epithelial-mesenchymal transition processes. Cells Tissues Organs 185:61–65
Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM (2010) Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res 12:R68
Qi J, Chen N, Wang J, Siu CH (2005) Transendothelial migration of melanoma cells involves N-cadherin-mediated adhesion and activation of the beta-catenin signaling pathway. Mol Biol Cell 16:4386–4397
Rabquer BJ, Amin MA, Teegala N, Shaheen MK, Tsou PS, Ruth JH, Lesch CA, Imhof BA, Koch AE (2010) Junctional adhesion molecule-C is a soluble mediator of angiogenesis. J Immunol 185:1777–1785
Rajasekaran SA, Hu J, Gopal J, Gallemore R, Ryazantsev S, Bok D, Rajasekaran AK (2003) Na, K-ATPase inhibition alters tight junction structure and permeability in human retinal pigment epithelial cells. Am J Physiol Cell Physiol 284:C1497–C1507
Rajasekaran SA, Barwe SP, Gopal J, Ryazantsev S, Schneeberger EE, Rajasekaran AK (2007) Na-K-ATPase regulates tight junction permeability through occludin phosphorylation in pancreatic epithelial cells. Am J Physiol Gastrointest Liver Physiol 292:G124–G133
Rajasekaran SA, Beyenbach KW, Rajasekaran AK (2008) Interactions of tight junctions with membrane channels and transporters. Biochim Biophys Acta 1778:757–769
Reynolds LE, Watson AR, Baker M, Jones TA, D’Amico G, Robinson SD, Joffre C, Garrido-Urbani S, Rodriguez-Manzaneque JC, Martino-Echarri E, Aurrand-Lions M, Sheer D, Dagna-Bricarelli F, Nizetic D, McCabe CJ, Turnell AS, Kermorgant S, Imhof BA, Adams R, Fisher EM, Tybulewicz VL, Hart IR, Hodivala-Dilke KM (2010) Tumour angiogenesis is reduced in the Tc1 mouse model of Down’s syndrome. Nature 465:813–817
Rodriguez P, Heyman M, Candalh C, Blaton MA, Bouchaud C (1995) Tumour necrosis factor-alpha induces morphological and functional alterations of intestinal HT29 cl.19A cell monolayers. Cytokine 7:441–448
Schmitz H, Fromm M, Bentzel CJ, Scholz P, Detjen K, Mankertz J, Bode H, Epple HJ, Riecken EO, Schulzke JD (1999) Tumor necrosis factor-alpha (TNFalpha) regulates the epithelial barrier in the human intestinal cell line HT-29/B6. J Cell Sci 112(Pt 1):137–146
Schneeberger EE, Lynch RD (2004) The tight junction: a multifunctional complex. Am J Physiol 286:C1213–C1228
Schoner W, Scheiner-Bobis G (2007) Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am J Cardiovasc Drugs 7:173–189
Schulzke JD, Ploeger S, Amasheh M, Fromm A, Zeissig S, Troeger H, Richter J, Bojarski C, Schumann M, Fromm M (2009) Epithelial tight junctions in intestinal inflammation. Ann N Y Acad Sci 1165:294–300
Severson EA, Lee WY, Capaldo CT, Nusrat A, Parkos CA (2009) Junctional adhesion molecule A interacts with Afadin and PDZ-GEF2 to activate Rap1A, regulate beta1 integrin levels, and enhance cell migration. Mol Biol Cell 20:1916–1925
Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, Casari G, Bettinelli A, Colussi G, Rodriguez-Soriano J et al (1999) Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 285:103–106
Sourisseau T, Georgiadis A, Tsapara A, Ali RR, Pestell R, Matter K, Balda MS (2006) Regulation of PCNA and cyclin D1 expression and epithelial morphogenesis by the ZO-1-regulated transcription factor ZONAB/DbpA. Mol Cell Biol 26:2387–2398
Spaderna S, Schmalhofer O, Wahlbuhl M, Dimmler A, Bauer K, Sultan A, Hlubek F, Jung A, Strand D, Eger A et al (2008) The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res 68:537–544
Stankovich BL, Aguayo E, Barragan F, Sharma A, Pallavicini MG (2011) Differential adhesion molecule expression during murine embryonic stem cell commitment to the hematopoietic and endothelial lineages. PloS one 6:e23810
Stark AM, Anuszkiewicz B, Mentlein R, Yoneda T, Mehdorn HM, Held-Feindt J (2007) Differential expression of matrix metalloproteinases in brain- and bone-seeking clones of metastatic MDA-MB-231 breast cancer cells. J Neurooncol 81:39–48
Stelwagen K, McFadden HA, Demmer J (1999) Prolactin, alone or in combination with glucocorticoids, enhances tight junction formation and expression of the tight junction protein occludin in mammary cells. Mol Cell Endocrinol 156:55–61
Strell C, Lang K, Niggemann B, Zaenker KS, Entschladen F (2007) Surface molecules regulating rolling and adhesion to endothelium of neutrophil granulocytes and MDA-MB-468 breast carcinoma cells and their interaction. Cell Mol Life Sci 64:3306–3316
Sweadner KJ (1989) Isozymes of the Na+/K + −ATPase. Biochim Biophys Acta 988:185–220
Swisshelm K, Machl A, Planitzer S, Robertson R, Kubbies M, Hosier S (1999) SEMP1, a senescence-associated cDNA isolated from human mammary epithelial cells, is a member of an epithelial membrane protein superfamily. Gene 226:285–295
Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW et al (2010) Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A 107:15449–15454
Tester AM, Waltham M, Oh SJ, Bae SN, Bills MM, Walker EC, Kern FG, Stetler-Stevenson WG, Lippman ME, Thompson EW (2004) Pro-matrix metalloproteinase-2 transfection increases orthotopic primary growth and experimental metastasis of MDA-MB-231 human breast cancer cells in nude mice. Cancer Res 64:652–658
Thompson GE (1996) Cortisol and regulation of tight junctions in the mammary gland of the late-pregnant goat. J Dairy Res 63:305–308
Tobioka H, Isomura H, Kokai Y, Tokunaga Y, Yamaguchi J, Sawada N (2004a) Occludin expression decreases with the progression of human endometrial carcinoma. Hum Pathol 35:159–164
Tobioka H, Tokunaga Y, Isomura H, Kokai Y, Yamaguchi J, Sawada N (2004b) Expression of occludin, a tight-junction-associated protein, in human lung carcinomas. Virchows Arch 445:472–476
Tsukada Y, Fouad A, Pickren JW, Lane WW (1983) Central nervous system metastasis from breast carcinoma. Autopsy study. Cancer 52:2359–2354
Tsukita S, Furuse M, Itoh M (2001) Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol 2:285–293
Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL (1997) Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol 273:C1378–C1385
Turner JR, Cohen DE, Mrsny RJ, Madara JL (2000) Noninvasive in vivo analysis of human small intestinal paracellular absorption: regulation by Na + −glucose cotransport. Dig Dis Sci 45:2122–2126
Uchide K, Sakon M, Ariyoshi H, Nakamori S, Tokunaga M, Monden M (2007) Cancer cells cause vascular endothelial cell (vEC) retraction via 12(S)HETE secretion; the possible role of cancer cell derived microparticle. Ann Surg Oncol 14:862–868
Vaira V, Faversani A, Dohi T, Montorsi M, Augello C, Gatti S, Coggi G, Altieri DC, Bosari S (2012) miR-296 regulation of a cell polarity-cell plasticity module controls tumor progression. Oncogene 31:27
Van Itallie CM, Anderson JM (2006) Claudins and epithelial paracellular transport. Annu Rev Physiol 68:403–429
Viloria-Petit AM, David L, Jia JY, Erdemir T, Bane AL, Pinnaduwage D, Roncari L, Narimatsu M, Bose R, Moffat J et al (2009) A role for the TGFbeta-Par6 polarity pathway in breast cancer progression. Proc Natl Acad Sci U S A 106:14028–14033
Wada-Hiraike O, Imamov O, Hiraike H, Hultenby K, Schwend T, Omoto Y, Warner M, Gustafsson JA (2006) Role of estrogen receptor beta in colonic epithelium. Proc Natl Acad Sci USA 103:2959–2964
Wang F, Schwarz BT, Graham WV, Wang Y, Su L, Clayburgh DR, Abraham C, Turner JR (2006) IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology 131:1153–1163
Wang Z, Wade P, Mandell KJ, Akyildiz A, Parkos CA, Mrsny RJ, Nusrat A (2007) Raf 1 represses expression of the tight junction protein occludin via activation of the zinc-finger transcription factor slug. Oncogene 26:1222–1230
Wiercinska E, Naber HP, Pardali E, van der Pluijm G, van Dam H, ten Dijke P (2011) The TGF-beta/Smad pathway induces breast cancer cell invasion through the up-regulation of matrix metalloproteinase 2 and 9 in a spheroid invasion model system. Breast Cancer Res Treat 128:657–666
Willemsen LE, Hoetjes JP, van Deventer SJ, van Tol EA (2005) Abrogation of IFN-gamma mediated epithelial barrier disruption by serine protease inhibition. Clin Exp Immunol 142:275–284
Wolburg H, Wolburg-Buchholz K, Kraus J, Rascher-Eggstein G, Liebner S, Hamm S, Duffner F, Grote EH, Risau W, Engelhardt B (2003) Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol 105:586–592
Yan J, Zhang Z, Shi H (2012) HIF-1 is involved in high glucose-induced paracellular permeability of brain endothelial cells. Cell Mol Life Sci 69:115
Ye D, Guo S, Al-Sadi R, Ma TY (2011) MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology 141:1323–1333
Yeh WL, Lu DY, Lin CJ, Liou HC, Fu WM (2007) Inhibition of hypoxia-induced increase of blood-brain barrier permeability by YC-1 through the antagonism of HIF-1alpha accumulation and VEGF expression. Mol Pharmacol 72:440–449
Yoon CH, Kim MJ, Park MJ, Park IC, Hwang SG, An S, Choi YH, Yoon G, Lee SJ (2010) Claudin-1 acts through c-Abl-protein kinase Cdelta (PKCdelta) signaling and has a causal role in the acquisition of invasive capacity in human liver cells. J Biol Chem 285:226–233
Youakim A, Ahdieh M (1999) Interferon-gamma decreases barrier function in T84 cells by reducing ZO-1 levels and disrupting apical actin. Am J Physiol 276:G1279–G1288
Zettl KS, Sjaastad MD, Riskin PM, Parry G, Machen TE, Firestone GL (1992) Glucocorticoid-induced formation of tight junctions in mouse mammary epithelial cells in vitro. Proc Natl Acad Sci U S A 89:9069–9073
Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB, Allred C, Muthuswamy SK (2008) Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 135:865–878
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
McSherry, E.A., Owens, M.B., Hopkins, A.M. (2013). The Molecular Aspects of Tight Junctions. In: Martin, T., Jiang, W. (eds) Tight Junctions in Cancer Metastasis. Cancer Metastasis - Biology and Treatment, vol 19. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6028-8_1
Download citation
DOI: https://doi.org/10.1007/978-94-007-6028-8_1
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6027-1
Online ISBN: 978-94-007-6028-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)