
Abstract
Amyloidoses are characterised by the deposition of insoluble protein that occurs in the extracellular compartment of various tissues. One form of amyloidosis is caused by transthyretin (TTR) misfolding and deposition in target tissues. It is clear that many amyloidoses share common features of fibrillogenesis and toxicity. This chapter examines the mechanisms of TTR aggregation with a view to understanding the possible therapeutic interventions in amyloid disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The amyloidoses constitute a disparate group of diseases characterised by the aggregation and extracellular deposition of insoluble protein (amyloid) in a variety of tissues. Accumulation of amyloid can lead to dysfunction of the affected organs. The underlying pathophysiology of the amyloidoses is still not completely understood and a large body of evidence now exists implicating amyloid formation in the pathogenesis of diseases such as Alzheimer’s disease , prion diseases and the British and Danish dementias. There is increasing evidence that the mechanisms of pathogenesis of amyloidoses are similar and that it is the conformation of the amyloid, rather than the specific amino-acid sequence that is the key determinant of disease progression and toxicity (Bucciantini et al. 2002). A striking, but common feature of all amyloids is their high b-sheet content and their fibrillar ultrastructure. These observations have strengthened the notion that amyloidogenic proteins cause neurotoxicity by a similar mechanism. Therefore, a more complete understanding of the mechanisms of one amyloidosis may provide insights into the biology of other diseases.
The familial amyloidotic polyneuropathies (FAPs) are a group of diseases characterised by deposition of amyloid in the peripheral nervous system . One FAP is transthyretin (TTR)-related amyloidosis. TTR FAP is a rare autosomal dominant disease characterised by the deposition of TTR in nerves that can cause peripheral pain , paresthesia and muscular weakness leading ultimately to cardiac and renal system dysfunction (Ando et al. 2005a). The underlying molecular mechanisms responsible for TTR amyloidosis and the subsequent pathophysiological responses in tissues are yet to be fully understood. This chapter will review our current understanding of the mechanisms of TTR aggregation and toxicity in vitro . It also explores how this knowledge may inform the process of designing therapeutic agents to treat FAP . The implication of these studies on FAP and other important neurodegenerative diseases of the central nervous system such as Alzheimer’s disease will also be discussed.
2 Transthyretin in Health and Disease
2.1 Structure and Function of TTR
TTR is a thyroid hormone transport protein that is highly conserved in sequence similarity and biological function (Blake and Oatley 1977). The primary structure is highly conserved throughout eutherian, marsupial, bird, reptile and fish species (Fung et al. 1988). TTR possesses a conserved structural motif of four identical monomeric subunits arranged to form a central channel, or binding domain. In humans, TTR is a relatively abundant protein that is primarily produced in the liver, and to a lesser extent in extra-hepatic tissues such as the choroid plexus. TTR is also involved in the transportation of retinol by forming a complex with a smaller, retinol- binding protein (Hagen and Elliott 1973).
The crystal structure of human TTR has been determined (Blake et al. 1971), and shown to be a 55 kDa tetramer with four identical 127 amino-acid subunits exhibiting an unusually high b-sheet content. Almost 45 % of the residues in a TTR monomer are organised into eight b-strands, identified as A-H, connected by loops, or short helix, as occurs between strands E and F (Fig. 9.1a). Strands CBEF are oriented orthogonally to strands DAGH, forming a prominent b-barrel (Fig. 9.1b). Extensive hydrogen bonding at the contact region between two monomers results in strong dimeric interactions (Blake et al. 1974). The native tetrameric structure of TTR is formed by two TTR dimers that bind through relatively weak hydrophobic and hydrophilic interactions between the AB loop of one monomer and the H strand of the two primed monomers, creating a 50 Å central channel that contains two thyroid hormone binding sites. The strength and extent of the monomeric interactions suggest that the dimers, rather than monomers or tetramers are the basic unit of TTR structure.

Schematic representation of thransthyretin monomer and tetramer structures. a TTR monomer with inner b-sheet (DAGH) shown in blue and the outer sheet (CBEF) shown by yellow shading. b Predicted structure of TTR tetramer complexed with 3¢,5¢-dibromo-2¢,4,4¢,6-tetrahydroxyaurone, a flavone derivative. The diagram shows four inner, monomer b-sheets (shaded in magenta and blue) forming a prominent b-barrel. The diagrams were constructed using MacPyMOL (Schrodinger 2010) using coordinates from file PDB ID:1THC (Ciszak et al. 1992)
2.2 Transthyretin Amyloidosis and FAP
TTR amyloidosis is a relatively common inherited amyloid disease . However, the amyloidosis also exists as a sporadic, but asymptomatic disease in the elderly known as senile systemic amyloidosis (SSA) . In SSA, deposits of wild-type TTR accumulate in cardiac tissue and only infrequently cause complications such as cardiomegaly and congestive cardiac failure (Westermark et al. 1990). FAP is associated with systemic extracellular amyloid deposition in the peripheral nervous system . It is now established that the inherited forms of TTR amyloidosis are associated with over 100 mutations in the TTR gene with affected individuals or single families having a single mutation. Significantly, a single mutation can exhibit a wide variety of clinical presentations, age of onset and organ involvement suggesting a complicated picture of amyloid pathogenesis. FAP was originally described in a small group of patients in Portugal (Andrade 1952b). The disease was initially described as a peripheral neuropathy with some involvement of the renal and cardiac systems. The Portuguese kindred has since been extensively studied (Ando 2005; Ando et al. 2005b; Lashuel et al. 1998; Quintas et al. 1997a, 1999) and the typical presentation of this FAP involves small diameter fiber loss in the lower extremities, coupled with loss of temperature and touch sensation. Pain is common with a distal to proximal neuropathic presentation. Parathesias, lower limb motor impairment and generalised autonomic symptoms of constipation or diarrhoea can occur with further involvement of cardiac, renal and gastrointestinal systems and invariably, death of affected individuals.
Histopathological studies of TTR FAP have demonstrated that axonal degeneration and neuronal loss are associated with extensive endoneurial amyloid (Sousa 2003). Biopsy and autopsy material of patients with FAP show amyloid deposition in nerve trunks, plexuses and sensory and autonomic ganglia (Coimbra and Andrade 1971). Amyloid deposits are mainly present in the endoneurium, usually accompanied by destruction of the myelin sheath, degeneration of nerve fibers and neuronal loss (Takahashi et al. 1997). Amyloid deposits have also been detected in the choroid plexus, cardiovascular system and kidneys. The oculoleptomeningeal form of FAP is characterised by severe, diffuse amyloidosis of the leptomeninges and subarachnoid vessels associated with patchy fibrosis, obliteration of the subarachnoid space and widespread neuronal loss (Herrick et al. 1996; Uitti et al. 1988).
The most common (and extensively studied) variant, Val30Met (V30M) occurs in Portuguese, Swedish and Japanese kindreds, with age of presentation and severity of disease symptoms varying considerably. Similar variability in phenotype occurs in other less frequent mutations such as Leu58His, Thr60Ala, Ser77Tyr, Ile84Ser and Val22Ile and the rare, but highly aggressive Leu55Pro (L55P) mutation which will be referred to extensively in this chapter.
3 Transthyretin Aggregation in vitro
The presence of amyloid in diseased tissue was originally documented in the mid 19th century (reviewed by Cohen and Calkins 1959) . Advances in microscopy and histochemical staining techniques in the early part of the 20th century enabled researchers to apply the criteria of congophilic staining and birefringence under polarized light to positively identify amyloid deposits from biopsy and post-mortem tissue. Electron microscopic studies have since confirmed that amyloid deposits from diverse proteins all exhibit a similar, fibrillar structure . Generally, amyloid deposits are composed of rigid, unbranched fibrils ranging in width from 60–130 Å and 0.1–16 nm in length (Cohen and Calkins 1959; Sipe and Cohen 2000). These observations have formed the widely held view that the amyloidoses are a clinically diverse group of diseases that all involve proteins which have a common propensity to aggregate and produce insoluble fibrils. Proteins with little or no sequence similarity such as TTR, the Ab protein of Alzheimer’s disease, diabetes-associated islet amyloid protein (IAPP) and the prion proteins (PrP) can aggregate to form similar insoluble fibrillar deposits in vivo (Bucciantini et al. 2002; Dobson 2003; Stefani and Dobson 2003; Vendruscolo et al. 2003). Significantly, X-ray diffraction studies of protein fibrils has revealed a common structural theme of ordered secondary structure, with the peptide backbone of b-pleated sheets oriented perpendicular to the fibril axis (Jaroniec et al. 2004).
While many studies have examined the fibrillogenesis of TTR and other amyloidogenic proteins in vitro, there is little, if any, evidence correlating the aggregated species with cellular toxicity . Identification of toxic amyloidogenic aggregates would greatly accelerate the search for an effective treatment for a fatal disease such as FAP . Several studies have shown that mutant TTR aggregates to form high molecular weight oligomers more readily than wild-type TTR, and that further aggregation leads to the formation of amyloid fibrils (Kayed et al. 2003; Reixach et al. 2004). There is a good correlation between the rate of aggregation of TTR in vitro and the extent or severity of the disease phenotype (Hurshman et al. 2008; Lashuel et al. 1999; Quintas et al. 1997a). For example, the rare L55P mutation produces a significantly more aggressive amyloidosis than the more common V30M mutation, and in vitro studies have shown that L55P aggregates more much readily than V30M (Quintas et al. 1997a; Lashuel et al. 1998, 1999; Hammarström et al. 2002b; Hou et al. 2005; Pokrzywa et al. 2007; Hurshman et al. 2008). There is considerable scope therefore, to justify the use of TTR mutant proteins to study the pathogenesis and possible treatment of TTR FAP.
3.1 Mechanisms of TTR Aggregation
The mechanism by which TTR forms fibrils is not well understood. While it is generally accepted that some form of amyloidogenic intermediate species is important to initiate fibrillogenesis, the precise nature and sequence of molecular events that drives TTR association into fibrils are yet to be elucidated. Furthermore, it is not clear at what stage during fibrillogenesis that toxic species are formed. Over the last decade various groups have elucidated the crystal structure of over 20 mutant TTR variants. While these studies have shown structural differences that could be explained in terms of fibrillar structures, there is no apparent mechanism or conformational change that would explain fibril formation by all mutant forms of TTR (Hörnberg et al. 2000). Early attempts to understand TTR fibrillogenesis employed methods designed to solubilise mature fibrils and to determine their species composition (Costa et al. 1978), however the identity of the building blocks of TTR fibrils has remained elusive.
More recently, several studies have suggested mutations of wild-type TTR result in the destabilisation and dissociation of TTR tetramers into unfolded monomers and dimers which undergo further partial refolding, forming amyloidogenic intermediates (Cardoso et al. 2007; Colón et al. 1996; Lai et al. 1996; Lashuel et al. 1998, 1999). Significantly, there is a strong correlation between the thermodynamic stability of TTR variants and their propensity to form unfolded, soluble aggregates (Quintas et al. 2001; Shnyrov et al. 2000). The V30M mutation is the most frequently occurring variant and an examination of its crystal structure suggests the mutant substitution results in a conformational change in strand A, exposing Cys10, and rendering the thiol group more exposed (Terry et al. 1993). The formation of fibrils resulting from the association of TTR through disulphide bridges has been suggested (Thylén et al. 1993), however the existence of an amyloidogenic Cys10Arg(C10 A) variant would suggest that such a mechanism is not a significant factor in V30M fibrillogenesis. A “hot spot” for amyloidogenic mutations occurs in the region between residues 45 and 58. This region contains the C strand, C-D loop, and D strand which are located at the edge of each dimer (Serpell et al. 1995). The structure of the highly amyloidogenic and clinically aggressive L55P variant crystallizes in a different space to that of wild-type and other variant forms of TTR (Quintas et al. 1997b). Analysis of the structure suggests that strands C and D are disrupted, altering the hydrogen bonds between the AB loop of one dimer and strand H of the other dimer (Sebastião et al. 1998), in an area that defines weak, native dimer-dimer interactions . These observations suggest a significant destabilisation of the L55P tetramer with the formation of, as yet, undefined intermediate species that subsequently aggregate further to form fibrils. Whether these destabilised intermediates serve a dual purpose, namely, seeds for further polymerisation and aggregation or soluble, toxic oligomeric species responsible for the pathogenesis of FAP , has yet to be determined. Since the L55P variant is clinically aggressive it has been the focus of much of our own work investigating the mechanisms responsible for the toxicity of L55P in sensory neurons (Gasperini et al. 2011) .
3.2 Monitoring TTR Aggregation in vitro
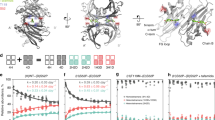
We have compared the aggregation properties of the L55P variant with wild-type TTR protein using atomic force microscopy (AFM) and dynamic light scattering (DLS) techniques. Freshly prepared L55P and wild-type TTR in physiological buffers contains predominantly globular or amorphous particles ranging in apparent size from 10–50 nm in diameter (Fig. 9.2a, 9.2b). The smallest particles observed by AFM are approximately 10 nm in diameter and are probably tetramers. Significantly, using quantitative image analysis of particle cross-sectional area (Fig. 9.2c), the average AFM particle sizes of L55P are greater than those of wild-type preparations confirming that L55P is highly unstable at physiological pH and ionic strength (Quintas et al. 1997b).

Aggregation of transthyretin in vitro. AFM images of freshly prepared a Wild-type and b L55P transthyretin. Arrows denote large, oligomeric aggregates of L55P TTR (scale bar is 100 nm). c When particle cross-sectional areas from the AFM are quantitated, L55P (red) contain a higher proportion of large particles than wild-type preparation (green). d Using a dynamic light scattering technique, L55P (red) contains a population of large, soluble oligomeric aggregates ranging in size from 100–300 nm in diameter. e Qualitatively, when L55P is monitored by DLS at 37 °C over 36 h, these oligomeric species decrease in average size and appear to form much larger (1,000 nm) aggregates, probably protofibrils
Are these larger aggregates amyloidogenic intermediates or toxic oligomers? To answer this question, we have examined freshly prepared solutions of L55P more closely by measuring aggregate size distributions in real time by DLS (Fig. 9.3d). This technique allows the measurement of size distribution (as measured by hydrodynamic radius) of protein preparations in real time. Using this technique, freshly prepared solutions of L55P at neutral pH show the presence of large molecular weight, pre-fibrillar aggregates in addition to the natively folded tetramer. Significantly monomers are not observed by DLS. However, the existence of small amounts of monomeric material cannot be excluded using this technique. Assuming a globular structure and a specific density of 1.39 g/cm3, which would be typical for many proteins, these intermediate aggregates would correspond to a molecular mass of 1810 ± 160 kDa. These molecular species would therefore be predicted to contain approximately 130 TTR monomers. Interestingly, these intermediate species increase in size over a period of 12–36 h, during which a concomitant reduction in abundance of native tetramer occurs (Fig. 9.3e). After 36–48 h, oligomeric species are possibly converted into much larger, probably protofibril aggregates, in a nucleation-dependent manner . These observations are consistent, in part, with other studies (Hammarström et al. 2002a; Hurshman et al. 2008; Lai et al. 1996; Lashuel et al. 1998, 1999) that have demonstrated L55P dissociation into monomeric intermediates at neutral pH to form protofibrils 60–65 Å in diameter and approximately 50 nm in length. Another possible explanation for these observations would include the polymerization of natively folded, but conformationally distorted tertramers via a novel “b-slip” mechanism (Eneqvist et al. 2000). This mechanism is not dependent on the formation of unfolded monomers as aggregation intermediates. A triple TTR variant, G53S/E54D/L55S has been generated to target the proposed amyloidogenic “hot-spot” discussed previously. This mutation induces a shift in strand B creating significantly extended C-D loops and shortened D-E loops. This “b-slip” creates new interactions at a potential amyloid packing site, in which distorted but intact tetramers are the basic building blocks for TTR amyloidogenesis . Such a mechanism would be consistent with our experimental findings.

Effect of glycosoaminoglycans on TTR aggregation. a L55P TTR was incubated either without (open circles) or with heparin (cross), ChSA (triangles), ChSB (squares) or ChSC (inverted triangles) for 48 h and particle diameter measured using DLS. Heparin, ChSA and ChSB accelerated the increase TTR aggregation over time, while ChSC inhibited TTR aggregation. b L55P TTR was incubated either without (circles) or with heparin (triangles), 5 kDa heparin (crosses), 3–4 kDa heparin (squares) or heparin disaccharide (inverted triangles) for 48 h, and particle diameter measured using DLS. All heparin derivatives stimulated L55P TTR aggregation
It is apparent that while DLS may lack the required sensitivity to detect monomeric TTR in appreciable quantities, the ability of DLS to monitor the self-assembly of products of tetramer disassociation into pre-fibrillar aggregates provides a valuable insight into TTR fibrillogenesis. The concept of partially unfolded monomers as quantal assemblies of intermediate aggregates in vitro has gained momentum in the field. Our current understanding of TTR fibrillogenesis would suggest that irrespective of the actual nature of the amyloidogenic intermediate(s), the in-vitro assembly of fibrils is probably a nucleation-dependent process . The precise mechanistic explanation for these observations remains unclear.
However, many crucial aspects of TTR fibrillogenesis remain unresolved. In particular, how does this complex picture of denaturation, refolding, aggregation and fibril formation at varying degrees of acidity and ionic strength drive the pathogenesis of TTR toxicity in vivo? In the context of an extracellular amyloidosis, is TTR fibrillogenesis a determinant of the severity of FAP? And finally, will a more complete understanding of TTR fibrillogenesis ultimately lead to specific, targeted therapeutic agents for FAP?
Using electron microscopy and image analysis techniques, Inoue et al. (1998) investigated the ultrastructure of mature and immature TTR fibrils in sural nerve biopsies of V30M FAP patients. Detailed analysis of averaged EM images showed mature TTR fibrils composed of a complex linear arrangement of TTR protofibrils, heparan sulphate (HSPG) and chondroitin sulphate proteoglycans (CSPG) . While these studies have not been replicated, they suggest a mechanism for the regional and tissue-specific differences seen in amyloid deposits from different TTR variant FAP’s.
Our group has recently investigated the effects of related molecules, the glycosoaminoglycans (GAGs) , on TTR aggregation in-vitro. Incubation of L55P TTR with heparin , chondroitin sulphate A (ChSA) or chondroitin sulphate B (ChSB) resulted in an acceleration of aggregation while incubation with chondroitin sulphate C (ChSC) resulted in inhibition of formation of soluble oligomeric species (Fig. 9.3a) . Since inhibiting aggregation of proteins into toxic oligomers or fibrils would be a promising target for therapeutic intervention in FAP , the ability of small heparinoid molecules to influence aggregation of L55P was also examined. Three low molecular weight mucosal heparin fragments, a heparin disaccharide (MH di), a 3–4 kDa heparin fragment (MH 3–4 kDa) and a 5 kDa heparin fragment (MH 5 kDa) were tested. All the heparin derivatives tested stimulated aggregation of L55P, with the magnitude of aggregation dependent on the size of the fragment tested (Fig. 9.3b). Significantly, the differing effects of GAGs on TTR aggregation may have implications for our understanding of the pathogenesis of FAP and potential treatment strategies. There is evidence that GAGs can influence nucleation of other amyloidogenic proteins such as Ab (McLaurin et al. 1999) and a-synuclein (Liu et al. 2005), suggesting that effects on nucleation are a common theme behind the effect of GAGs on aggregation of amyloidogenic proteins. If GAGs function through modification of nucleation-dependent aggregation processes they could be used as anti-amyloidogenic agents in FAP .
4 Mechanisms of Transthyretin Neurotoxicity in FAP
Understanding the mechanism of transthyretin toxicity may provide useful insights into the mechanism of amyloid toxicity in other significant amyloidoses such as Alzheimer’s disease . TTR has been shown to be toxic to cells in culture (Hou et al. 2007; Sousa et al. 2001a, 2001b). However, the complete biochemical mechanisms responsible for cellular toxicity and death are still unclear. There is good experimental evidence that misfolded proteins exert their toxic effects by interacting with cells by binding directly to lipid-rich areas of the cell membrane (Hou et al. 2005; Subasinghe et al. 2003). It has also been suggested that TTR-induced toxicity may be mediated by the receptor for advanced glycation end-products (RAGE ) and that activation of RAGE leads to endoplasmic reticulum stress, activation of ERK1/2 and caspase-dependent apoptosis (Monteiro et al. 2006).
There is evidence suggesting the toxicity of amyloid is caused by an increase in cation permeability of neuronal cell membranes resulting in chronic cytosolic calcium dysregulation and subsequent cell death or dysfunction (Arispe et al. 1996; Koopmans et al. 1992; Mattson et al. 1992; Moe and Sprague 1992; Saito et al. 1993). However, the mechanism by which amyloid proteins induce calcium entry in cells is poorly understood. Some studies have suggested protofibrillar aggregates of interacting amyloidogenic proteins might embed in the membrane, leading to the formation of pore-like structures with consequent aberrant ion conductance (Tsigelny et al. 2008). Previously, we have shown TTR-induced calcium permeability in cell lines is primarily mediated by voltage-gated calcium channels (VGCC) , with a small proportion (~20 %) of the calcium influx through voltage-independent channels (Hou et al. 2007). Using embryonic sensory neurons from rodent dorsal root ganglia (DRG) , we have demonstrated a novel mechanism of calcium entry requiring the coordinated activation of Nav1.8 voltage-gated sodium channels and transient receptor potential (TRP) M8 channels (Gasperini et al. 2011). The precise nature of the TTR aggregates that mediate this effect on sensory neurons is yet to be elucidated. One possibility is that TTR binds directly to cationic channels causing a conformational change leading to channel gating. However, there is no direct evidence for this. Another possibility is that TTR binds to lipid membrane components (Hou et al. 2005), thus altering cell membrane fluidity and triggering the opening of TRPM8 in DRG sensory neurons (Gasperini et al. 2011). TRPM8 channels have been described as the prototypic thermosensitive ion channel (McKemy et al. 2002; Peier et al. 2002), and are a prominent receptor subtype on small diameter (C and Ad fibre) sensory neurons (Staaf et al. 2010). Since FAP exhibits a variety of clinical manifestations, including progressive parasthesias involving the lower limbs and affecting thermosensation and nociception (Andrade 1952a), it is possible that TTR toxicity mediated through TRPM8 channels is a key molecular correlate of FAP pathogenesis.
5 Summary
At the present time, amyloidoses carry a significant economic burden to the health of our ageing society. It is generally accepted that this burden will continue to increase and overtake many other degenerative diseases. Our current understanding of the mechanisms responsible for aggregation and fibrillogenesis of many amyloidogenic proteins remains unclear. The design and delivery of effective therapeutic agents to inhibit or dissociate protein aggregates in vivo will require a complete understanding of how amyloid is deposited in target tissues and more importantly, how these aggregates mediate their toxic effects on cells. It is clear that what is learnt from studying TTR fibrillogenesis may have considerable application to other amyloidoses. In conclusion, TTR aggregation and its effects on a variety of cell types in vitro provides many testable hypotheses to study the pathogenesis of important diseases such as AD, prion diseases and the British and Danish dementias.
Abbreviations
- TTR:
-
Transthyretin
- AFM:
-
Atomic force microscopy
- DLS:
-
Dynamic light scattering
- FAP:
-
Familial amyloidotic polyneuropathy
- Ab:
-
Amyloid beta protein
- Å:
-
Angstrom
- nm:
-
Nanometer
- kDa:
-
Kilodalton
- SSA:
-
Senile systemic amyloidosis
- IAPP:
-
Diabetes-associated islet amyloid polypeptide
- PrP:
-
Prion protein
- HSPG:
-
Heparan sulphate proteoglycan
- CSPG:
-
Chondroitin sulphate proteoglycan
- GAG:
-
Glycosoaminoglycan
- VGCC:
-
Voltage-gated calcium channel
- TRPM8:
-
Transient receptor potential (melastatin) channel
References
Ando Y (2005) Liver transplantation and new therapeutic approaches for familial amyloidotic polyneuropathy (FAP). Med Mol Morphol 38(3):142–154
Ando Y, Nakamura M, Araki S (2005a) Transthyretin-related familial amyloidotic polyneuropathy. Arch Neuol 62(7):1057–1062
Ando Y, Nakamura M, Araki S (2005b) Transthyretin-related familial amyloidotic polyneuropathy. Arch Neurol 62(7):1057–1062
Andrade C (1952a) A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 75(3):408–427
Andrade C (1952b) A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 75(3):408–427
Arispe N, Pollard HB, Rojas E (1996) Zn2+ interaction with Alzheimer amyloid beta protein calcium channels. Proc Natl Acad Sci USA 93(4):1710–1715
Blake CC, Swan ID, Rerat C, Berthou J, Laurent A, Rerat B (1971) An x-ray study of the subunit structure of prealbumin. J Mol Biol 61(1):217–224
Blake CC, Geisow MJ, Swan ID, Rerat C, Rerat B (1974) Structure of human plasma prealbumin at 2–5 A resolution. A preliminary report on the polypeptide chain conformation, quaternary structure and thyroxine binding. J Mol Biol 88(1):1–12
Blake CC, Oatley SJ (1977) Protein-DNA and protein-hormone interactions in prealbumin: a model of the thyroid hormone nuclear receptor? Nature 268(5616):115–120
Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416(6880):507–511
Cardoso I, Almeida MR, Ferreira N, Arsequell G, Valencia G, Saraiva MJ (2007) Comparative in vitro and ex vivo activities of selected inhibitors of transthyretin aggregation: relevance in drug design. Biochem J 408(1):131
Ciszak E, Cody V, Luft JR (1992) Crystal structure determination at 2.3-Å resolution of human transthyretin—3¢,5¢-dibromo-2¢,4,4¢,6-tetrahydroxyaurone complex. Proc Natl Acad Sci U S A 89(14):6644–6648
Cohen AS, Calkins E (1959) Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature 183(4669):1202–1203
Coimbra A, Andrade C (1971) Familial amyloid polyneuropathy: an electron microscope study of the peripheral nerve in five cases. I. Interstitial changes. Brain 94(2):199–206
Colón W, Lai Z, McCutchen SL, Miroy GJ, Strang C, Kelly JW (1996) FAP mutations destabilize transthyretin facilitating conformational changes required for amyloid formation. Ciba Found Symp 199:228–238; (discussion 239–242)
Costa PP, Figueira AS, Bravo FR (1978) Amyloid fibril protein related to prealbumin in familial amyloidotic polyneuropathy. Proc Natl Acad Sci U S A 75(9):4499–4503
Dobson CM (2003) Protein folding and misfolding. Nature 426(6968):884–890
Eneqvist T, Andersson K, Olofsson A, Lundgren E, Sauer-Eriksson AE (2000) The beta-slip: a novel concept in transthyretin amyloidosis. Mol Cell 6(5):1207–1218
Fung WP, Thomas T, Dickson PW, Aldred AR, Milland J, Dziadek M, Power B, Hudson P, Schreiber G (1988) Structure and expression of the rat transthyretin (prealbumin) gene. J Biol Chem 263(1):480–488
Gasperini RJ, Hou X, Parkington H, Coleman H, Klaver DW, Vincent AJ, Foa LC, Small DH (2011) TRPM8 and Nav1.8 sodium channels are required for transthyretin-induced calcium influx in growth cones of small-diameter TrkA-positive sensory neurons. Mol Neurodegener 6(1):19
Hagen GA, Elliott WJ (1973) Transport of thyroid hormones in serum and cerebrospinal fluid. J Clin Endocrinol Metab 37(3):415–422
Hammarström P, Jiang X, Hurshman AR, Powers ET, Kelly JW (2002a) Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci U S A 99 Suppl 4:16427–16432
Hammarström P, Jiang X, Hurshman AR, Powers ET, Kelly JW (2002b) Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci U S A 99 Suppl 4:16427–16432
Herrick MK, DeBruyne K, Horoupian DS, Skare J, Vanefsky MA, Ong T (1996) Massive leptomeningeal amyloidosis associated with a Val30Met transthyretin gene. Neurology 47(4):988–992
Hörnberg A, Eneqvist T, Olofsson A, Lundgren E, Sauer-Eriksson AE (2000) A comparative analysis of 23 structures of the amyloidogenic protein transthyretin. J Mol Biol 302(3):649–669
Hou X, Richardson SJ, Aguilar M-I, Small DH (2005) Binding of amyloidogenic transthyretin to the plasma membrane alters membrane fluidity and induces neurotoxicity. Biochemistry 44(34):11618–11627
Hou X, Parkington HC, Coleman HA, Mechler A, Martin LL, Aguilar M-I, Small DH (2007) Transthyretin oligomers induce calcium influx via voltage-gated calcium channels. J Neurochem 100(2):446–457
Hurshman B, Amy R, Powers ET, Kelly JW (2008) Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: the relationship between stability and amyloidosis. Biochemistry 47(26):6969–6984
Inoue S, Kuroiwa M, Saraiva MJ, Guimarães A, Kisilevsky R (1998) Ultrastructure of familial amyloid polyneuropathy amyloid fibrils: examination with high-resolution electron microscopy. J Struct Biol 124(1):1–12
Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM, Griffin RG (2004) High-resolution molecular structure of a peptide in an amyloid fibril determined by magic angle spinning NMR spectroscopy. Proc Natl Acad Sci U S A 101(3):711–716
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300(5618):486–489
Koopmans SJ, Radder JK, Krans HM, Barge RM (1992) Biological action of pancreatic amylin: relationship with glucose metabolism, diabetes, obesity and calcium metabolism. Neth J Med 41(1–2):82–90
Lai Z, Colón W, Kelly JW (1996) The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry 35(20):6470–6482
Lashuel HA, Lai Z, Kelly JW (1998) Characterization of the transthyretin acid denaturation pathways by analytical ultracentrifugation: implications for wild-type, V30M, and L55P amyloid fibril formation. Biochemistry 37(51):17851–17864
Lashuel HA, Wurth C, Woo L, Kelly JW (1999) The most pathogenic transthyretin variant, L55P, forms amyloid fibrils under acidic conditions and protofilaments under physiological conditions. Biochemistry 38(41):13560–13573
Liu I-H, Uversky VN, Munishkina LA, Fink AL, Halfter W, Cole GJ (2005) Agrin binds alpha-synuclein and modulates alpha-synuclein fibrillation. Glycobiology 15(12):1320–1331
Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE (1992) beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 12(2):376–389
McKemy DD, Neuhausser WM, Julius D (2002) Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416(6876):52–58
McLaurin J, Franklin T, Zhang X, Deng J, Fraser PE (1999) Interactions of Alzheimer amyloid-beta peptides with glycosaminoglycans effects on fibril nucleation and growth. Eur J Biochem 266(3):1101–1110
Moe SM, Sprague SM (1992) Beta 2-microglobulin induces calcium efflux from cultured neonatal mouse calvariae. Am J Physiol 263 (3 Pt 2):F540–545
Monteiro FA, Sousam MM, Cardoso I, do Amaral JB, Guimarães A, Saraiva MJ (2006) Activation of ERK1/2 MAP kinases in familial amyloidotic polyneuropathy. J Neurochem 97(1):151–161
Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, Earley TJ, Dragoni I, McIntyre P, Bevan S, Patapoutian A (2002) A TRP channel that senses cold stimuli and menthol. Cell 108(5):705–715
Pokrzywa M, Dacklin I, Hultmark D, Lundgren E (2007) Misfolded transthyretin causes behavioral changes in a Drosophila model for transthyretin-associated amyloidosis. Eur J Neurosci 26(4):913–924
Quintas A, Saraiva MJ, Brito RM (1997a) The amyloidogenic potential of transthyretin variants correlates with their tendency to aggregate in solution. FEBS Lett 418(3):297–300
Quintas A, Saraiva MJ, Brito RM (1997b) The amyloidogenic potential of transthyretin variants correlates with their tendency to aggregate in solution. FEBS Lett 418(3):297–300
Quintas, A, Saraivam MJ, Brito RM (1999) The tetrameric protein transthyretin dissociates to a non-native monomer in solution. A novel model for amyloidogenesis. J Biol Chem 274(46):32943–32949
Quintas A, Vaz DC, Cardoso I, Saraiva MJ, Brito RM (2001) Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J Biol Chem 276(29):27207–27213
Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN (2004) Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci U S A 101(9):2817–2822
Saito K, Elcem JS, Hamos JE, Nixon RA (1993) Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci USA 90(7):2628–2632
Schrodinger LLC (2010) The AxPyMOL Molecular Graphics Plugin for Microsoft Powerpoint, Version 1.0
Sebastião MP, Saraiva MJ, Damas AM (1998) The crystal structure of amyloidogenic Leu55 → Pro transthyretin variant reveals a possible pathway for transthyretin polymerization into amyloid fibrils. J Biol Chem 273(38):24715–24722
Serpell LC, Sunde M, Fraser PE, Luther PK, Morris EP, Sangren O, Lundgren E, Blake CC (1995) Examination of the structure of the transthyretin amyloid fibril by image reconstruction from electron micrographs. J Mol Biol 254(2):113–118
Shnyrov VL, Villar E, Zhadan GG, Sanchez-Ruiz JM, Quintas A, Saraiva MJ, Brito RM (2000) Comparative calorimetric study of non-amyloidogenic and amyloidogenic variants of the homotetrameric protein transthyretin. Biophys Chem 88(1–3):61–67
Sipe JD, Cohen AS (2000) Review: History of the Amyloid Fibril. J Struct Biol 130 (2–3):88–98
Sousa MM, Cardoso I, Fernandes R, Guimarães A, Saraiva MJ (2001a) Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: evidence for toxicity of nonfibrillar aggregates. Am J Pathol 159(6):1993–2000
Sousa MM, Du Yan S, Fernandes R, Guimarães A, Stern D, Saraiva MJ (2001b) Familial amyloid polyneuropathy: receptor for advanced glycation end products-dependent triggering of neuronal inflammatory and apoptotic pathways. J Neurosci 21(19):7576–7586
Sousa M (2003) Neurodegeneration in familial amyloid polyneuropathy: from pathology to molecular signaling. Prog Neurobiol 71(5):385–400
Staaf S, Franck MCM, Marmigère F, Mattsson JP, Ernfors P (2010) Dynamic expression of the TRPM subgroup of ion channels in developing mouse sensory neurons. Gene Expr Patterns 10(1):65–74
Stefani M, Dobson CM (2003) Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med 81(11):678–99
Subasinghe S, Barrow CJ, Aguilar MI, Small DH (2003) Cholesterol is necessary both for the toxic effect of Abeta peptides on vascular smooth muscle cells and for Abeta binding to vascular smooth muscle cell membranes. J Neurochem 84(3):471–479
Takahashi K, Sakashita N, Ando Y, Suga M, Ando M (1997) Late onset type I familial amyloidotic polyneuropathy: presentation of three autopsy cases in comparison with 19 autopsy cases of the ordinary type. Pathol Int 47(6):353–359
Terry CJ, Damas AM, Oliveira P, Saraiva MJ, Alves IL, Costa PP, Matias PM, Sakaki Y, Blake CC (1993) Structure of Met30 variant of transthyretin and its amyloidogenic implications. EMBO J 12(2):735–741
Thylén C, Wahlqvist J, Haettner E, Sandgren O, Holmgren G, Lundgren E (1993) Modifications of transthyretin in amyloid fibrils: analysis of amyloid from homozygous and heterozygous individuals with the Met30 mutation. EMBO J 12(2):743–748
Tsigelny IF, Crews L, Desplats P, Shaked GM, Sharikov Y, Mizuno H, Spencer B, Rockenstein E, Trejo M, Platoshyn O, Yuan JXJ, Masliah E (2008) Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS ONE 3(9):e3135
Uitti RJ, Donat JR, Rozdilsky B, Schneider RJ, Koeppen AH (1988) Familial oculoleptomeningeal amyloidosis. Report of a new family with unusual features. Arch Neurol 45(10):1118–1122
Vendruscolo M, Zurdo J, MacPhee CE, Dobson CM (2003) Protein folding and misfolding: a paradigm of self-assembly and regulation in complex biological systems. Philos Trans Ser A Math Phys Eng Sci 361(1807):1205–1222
Westermark P, Sletten K, Johansson B, Cornwell GG (1990) Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci U S A 87(7):2843–2845
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Gasperini, R., Klaver, D., Hou, X., Aguilar, MI., Small, D. (2012). Mechanisms of Transthyretin Aggregation and Toxicity. In: Harris, J. (eds) Protein Aggregation and Fibrillogenesis in Cerebral and Systemic Amyloid Disease. Subcellular Biochemistry, vol 65. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-5416-4_9
Download citation
DOI: https://doi.org/10.1007/978-94-007-5416-4_9
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-5415-7
Online ISBN: 978-94-007-5416-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)