
Abstract
Pollen grains represent male gametophytes and are the male partners in sexual reproduction. Following anther dehiscence, pollen grains are exposed into the atmosphere. At the time of shedding, pollen grains contain either 2 cells (a large vegetative cell enclosing the generative cell) or 3 cells (vegetative cell enclosing the two male gametes formed by the division of the generative cell) (Fig. 5.1a, b). The cytology of pollen at the time of shedding (2 or 3 cells) has important correlations with a number of physiological features of the pollen such as viability, storage and in vitro germination. In general, 2-celled pollen show longer viability, store well and germinate on a simple medium when compared to 3-celled pollen. Also the species with 2-celled pollen show gametophytic type of self-incompatibility, whereas those with 3-celled pollen show sporophytic type of self-incompatibility. Mature pollen grains generally contain reserve nutrients in the form of starch (Fig.5.2a) or lipids. The phase of pollen, from their shedding until they land on the stigma, is termed free dispersed phase; it plays a crucial role in plant reproduction as it facilitates gene flow because of their dispersal and transport to other conspecific plants/populations through various pollinating agents.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Pollen grains represent male gametophytes and are the male partners in sexual reproduction. Following anther dehiscence, pollen grains are exposed into the atmosphere. At the time of shedding, pollen grains contain either 2 cells (a large vegetative cell enclosing the generative cell) or 3 cells (vegetative cell enclosing the two male gametes formed by the division of the generative cell) (Fig. 5.1a, b). The cytology of pollen at the time of shedding (2 or 3 cells) has important correlations with a number of physiological features of the pollen such as viability, storage and in vitro germination. In general, 2-celled pollen show longer viability, store well and germinate on a simple medium when compared to 3-celled pollen . Also the species with 2-celled pollen show gametophytic type of self-incompatibility, whereas those with 3-celled pollen show sporophytic type of self-incompatibility. Mature pollen grains generally contain reserve nutrients in the form of starch (Fig.5.2a) or lipids. The phase of pollen, from their shedding until they land on the stigma, is termed free dispersed phase; it plays a crucial role in plant reproduction as it facilitates gene flow because of their dispersal and transport to other conspecific plants/populations through various pollinating agents.

Pollen cytology. (a) Fluorescence micrograph of Feulgen stained pollen grain of oil palm to show intensely stained generative nucleus (gcn) and moderately stained vegetative nucleus (vcn) located close to each other (b) Two intensely stained sperm cells (male gametes) in Dalzelia zeylanica; vegetative nucleus (vcn) is diffuse and thus very lightly stained

Pollen biology. (a) Section of a mature anther to show pollen grains filled with starch grains following PAS staining. (b) Acetocarmine test for pollen fertility. Shrivelled pollen without intense colour (arrows) are sterile pollen. (c) Fluorescein diacetate test for pollen viability. Brightly stained pollen grains (arrow) are viable
Pollen grains resemble seeds in many respects (see Chap. 10); both are desiccated units (their moisture content is generally <20 % when shed), dispersal units and units of gene flow. Similar to the refractory seeds of some species, pollen grains of several species are refractory and are shed with higher moisture content (>30 %); they cannot withstand desiccation. Pollen grains are ubiquitous and present in the air, water and soil. Pollen grains of some anemophilous species travel for long distances in air currents. They have been collected in Arctic and Antarctic regions, over 2,000 km away from the nearest seed plants (Linskens 1995). The amount of pollen produced in some tree species which are wind pollinated is enormous.
Pollen grain wall is not just a passive protective layer but a dynamic system performing various vital functions. It is one of the most complex wall systems found in plants. There are three domains of pollen wall – the surface layer (pollenkitt), the exine made up of sporopollenin and the pectocellulosic intine. Sporopollenin is one of the most resistant organic materials known. It can withstand high temperatures and extreme acidity and alkalinity. All the three domains of the pollen wall contain extracellular components particularly a range of proteins including enzymes. Pollen grain surface, especially in species pollinated by insects, contains, apart from other components, lipids and carotenoids which give a range of colour to pollen of different species. The extracellular components present in the three domains of pollen wall play an important role in pollen germination , pollen tube growth and incompatibility responses (see Shivanna 2003 for details).
Detailed studies on pollen biology are important in understanding reproductive strategies and reproductive success of plant species. Some of the features to be studied under pollen biology are briefly elaborated below.
5.1 Pollen Production
The estimation of pollen production is important to understand many aspects of pollen biology. The amount of pollen produced in each anther/flower varies greatly. In Cannabis sativa, for example, each anther produces >70,000 pollen grains, while some of the cleistogamous flowers in species such as Commelina produce <100 pollen grains per anther. Thus the resource allocated to the formation of pollen is highly variable between species. Pollen production also gives an indication of the type of pollination. Wind- and water-pollinated species generally produce more pollen when compared to animal-pollinated species. The calculation of pollen-to-ovule ratio indicates, to some extent, the possible breeding system of the species (Cruden 1977). The ratio is much higher in cross-pollinated species when compared to self-pollinated species. Pollen grains constitute an important reward for pollinators. Pollen counts are also needed to estimate pollen load on pollinators and the amount of pollen removed by pollinators.
5.2 Pollen Fertility and Viability
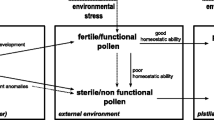
Plants of hybrid origin often show various degrees of pollen sterility. Even adverse environmental conditions such as high or low temperature and water stress induce pollen sterility to different degrees (see Shivanna 2003). Staining pollen grains with acetocarmine is commonly used to score pollen fertility (Fig. 5.2b) (Protocol5.5.6).
Pollen viability refers to the ability of the pollen to complete postpollination events on a compatible, receptive stigma and effect fertilization. Even the pollen samples, which are viable at the time of shedding, lose viability over a period of time; this may vary from less than an hour to several months. Prevailing environmental conditions, particularly temperature and humidity, affect the duration of pollen viability . For successful fertilization and seed set, pollen grains have to be not only fertile but also viable at the time of pollination. Thus, pollen viability is critical for successful fertilization and fruit and seed set (see Shivanna 2003). It is, therefore, important to know the extent of viability of pollen at the time of natural pollination and in pollen samples used for experimental studies. Also, it may be necessary to test the viability of the pollen sample collected from the floral visitor.
A number of methods are available to test pollen viability . The most authentic method to test pollen viability is to use pollen sample to carry out controlled pollinations (see Protocol 9.6.1 for details) and assess the extent of seed set. Alternatively, instead of waiting for seed set, one can study pollen germination and pollen tube growth in manually pollinated pistils to get information on pollen viability . However, manual pollinations and in vivo studies on pollen germination are laborious and time consuming. It also restricts the test period to flowering season of the species. Further, this test tends to be qualitative; even a small proportion of viable pollen can yield full seed set particularly in species with lower number of ovules. Thus, fruit and seed set test cannot be used as a routine test for pollen viability . Many alternative methods (see Protocols 5.5.9, 5.5.10 and 5.5.11) have been used to assess pollen viability (Fig. 5.2c).
5.3 Pollen Vigour
Pollen vigour refers to the speed of germination and the rate of pollen tube growth (Shivanna et al. 1991b; Shivanna 2003). It differs from viability since viable pollen samples may show differences in the vigour. Loss of vigour becomes apparent even before the loss of viability. Loss of vigour is well established in the biology of seeds. Until recently pollen vigour was not taken into consideration in assessing the quality of pollen. Several investigations have shown that aging and many environmental stresses, particularly temperature, humidity and desiccation, affect pollen vigour, and it may affect the ability of such pollen to compete with fresh pollen in effecting fertilization (Shivanna and Cresti 1989; Shivanna et al. 1991a, b; Rao et al. 1992).
When the pollen sample landing on the stigma contains a mixture of fresh and aged pollen (coming from older flowers when their longevity lasts for several days) or self- and cross-pollen, pollen may show variation not only in viability but also in vigour. In such a situation, pollen grains which are more vigorous would disproportionately sire more number of seeds than those with reduced vigour. In some studies concerned with pollination ecology and seed biology, it may be necessary to assess the vigour of the pollen in addition to viability.
5.4 Pollen Morphology
Pollen morphology is important in studies related to taxonomy, phylogeny, aeropalynology and paleobotany. Pollen morphology also plays a key role in several other areas such as archeology and criminology and in testing the purity of honey (Bryant 1990; Shivanna 2003; Warny 2013). As morphological features are specific to each species/genus and also pollen mixtures of each geographic area are different, pollen grains can be used as a tool for identifying the scene of the crime and also in locating the origin of illegally imported drugs, medicines, art objects, food and other products. For effective use of pollen in any of these areas, pollen identification is a basic necessity. The role of pollen morphology in reproductive ecology is limited to describing pollen grains of the focal species and in identifying conspecific and heterospecific pollen load on the floral visitors and on the stigma.
Only a brief description is given here so that the researcher would be familiar with basic aspects of pollen morphology in order to describe and identify the pollen grains of the focal species and also to identify the pollen present on floral visitors. For more details reader may refer to several excellent books available in this field (Erdtman 1969; Nair 1970; Moore and Webb 1978; Faegri and Iversen 1989).
Pollen morphology is generally described in relation to its polarity. Polarity of the pollen grain is established with reference to the arrangement of the microspores in the tetrads. The pore closer to the centre of the tetrad is the proximal pole and the one away from the centre is the distal pole. The hypothetical axis connecting the two poles is the polar axis. The distal half and the proximal half of the pollen is described in relation to an hypothetical line along the equator of the pollen at right angles to the polar axis.
Pollen morphology is largely based on their size, shape, aperture and exine ornamentation (Fig. 5.3a–c). The size and shape of pollen grains vary markedly. Smallest pollen grains (ca 5 μm) are recorded in some members of Boraginaceae, and larger pollen grains are generally seen in members of Cucurbitaceae, Malvaceae and Nyctaginaceae. Perhaps the largest pollen measuring up to 350 μm is recorded in Cymbopetalum odoratissimum (Annonaceae) (Walker 1971). In a majority of the species, pollen size ranges between 15 and 50 μm. Filiform pollen grains measuring up to 5 mm are found in marine angiosperms such as Amphibolis and Zostera (Ducker et al. 1978).

Scanning electron micrographs of pollen grains to show exine ornamentation. (a) Colporate pollen grains (Azadirachta indica). (b) Colporate pollen with reticulate exine ornamentation (Aegle marmelos). (c) Syncolpate (colpae fused to form rings) pollen with striate exine ornamentation (Schleichera oleosa). (d) A compound unit of two pollen grains (dyad) in Zeylanidium olivaceum
The shape of pollen grains is generally described as seen either in the polar view (from the top) or equatorial view (from the side) (see Moore and Webb 1978 for more details). Pollen grains may be spherical, elliptic, triangular, quadrangular, rectangular or rhombic. The shape may vary, to some extent, depending on whether they are desiccated or hydrated. Desiccated pollen grains are frequently subprismatic because of the shrinkage of the exine, while hydrated pollen tends to be spherical or ellipsoidal.
Apertures are distinct regions in the pollen wall through which the pollen tube emerges. The exine is generally thin or even absent in the region of the aperture. Pollen grains are classified on the basis of the shape, number and position of the aperture. They may be in the form of circular pores or elongated boatlike furrows (Fig. 5.3a, b). Pollen grains which bear pores are termed porate and those with furrows as colpate. Colporate pollen grains show a circular pore at the centre of a furrow. In some species, the colpae fuse to form rings or spiral (Fig. 5.3c). The number of apertures is described with a suitable Latin prefixes, mono-, bi-, tri-, tetra-, penta-, hexa- and poly- along with the type of aperture, for example, mono-porate/-colpate/-colporate, bi-porate/-colpate/-colporate and poly-porate/-colpate/-colporate.
Variations in pollen morphology are largely related to the ornamentation of the exine surface (Fig. 5.3b, c). Outer part of the exine, called the sexine, is sculptured, and the inner part, called the nexine, is homogeneous. The sexine is made up of radially directed rods, the baculae/columella; they may stand free or fused with neighbouring ones. When they are free, the pollen grains are called non-tectate. When they are fused to form a roof, such pollen grains are called tectate.
In non-tectate pollen, the head region of the baculae may be club shaped (clavate), pointed (echinate), short and globular (gemmate), or swollen (pilate). When the baculae are fused with neighbouring ones, they form depressed areas (lumina) and elevated wall-like structures (muri); the arrangement of muri and lumina produces different patterns. When the muri form a network, the pollen surface is described as reticulate (Fig. 5.3b), and when they run parallel to each other, as striate (Fig. 5.3c); an intermediate pattern is called rugulate. Instead of rod-like baculae, the sexine may often comprise hemispherical warts (verrucate), tiny flakes (scabrate) or small granules (granulate).
In tectate pollen grains, the baculae are generally referred to as columella and the roof is called the tectum. Pollen grains with partial tectum are described as semi-tectate. Tectate and semi-tectate pollen may bear additional projections on their surface which may be referred to as clavate, echinate, gemmate, pilate, reticulate, regulate, scabrate/granulate, striate or verrucate as in non-tectate grains. To study tectate or non-tectate nature of the pollen, one has to study the sectional view of the pollen wall under compound or electron microscope.
In most of the flowering plants, pollen grains are shed as solitary units (monads). However, there are several families that characteristically release pollen grains in groups (compound pollen). The compound pollen may contain units of two (dyad) (Fig. 5.3d), four (tetrad) or in multiples of four (polyads). In some species such as members of Asclepiadaceae and Orchidaceae, all microspores of a sporangium/anther remain together and are dispersed as massulae or pollinaria. Compound pollen units enhance pollination efficiency; the deposition of a single compound pollen unit may often be sufficient to achieve pollination of all the ovules present in the ovary.
As pointed out earlier, in studies on pollination biology, it is necessary to identify whether pollen grains present on the floral visitor and/or the stigma are conspecific or a mixture of conspecific/heterospecific pollen. A standard method for this is to prepare reference slides of pollen grains (see Protocol 5.5.5) from the focal species and other co-flowering species that are likely to be visited by the floral visitor. Identify pollen preparations collected from the visitor by comparing them with reference slides. Based on the size, shape, details of apertures and exine ornamentation, it is easy to identify the pollen sample (conspecific/heterospecific) carried by the visitor.
In some species lipoidal material on the surface of the pollen may mask its exine ornamentation. The treatment of pollen grains with organic solvents such as hexane, acetone or chloroform would remove the lipoidal material and reveal the surface ornamentation more clearly. The most commonly used technique to study pollen morphology, particularly exine ornamentation, is acetolysis (see Protocol 5.5.4). Acetolysis degrades all other contents of the pollen except the exine, thus revealing the details of apertures and ornamentation more clearly.
5.5 Protocols
5.5.1 Estimation of Pollen Production in Anthers with Limited Number of Pollen Grains
Pollen biologists have been using a number of methods to determine pollen production per anther and per flower. One of the methods used has been through the use of haemocytometre. In recent years, the use of haemocytometre is not very prevalent. One of the reasons is that the amount of pollen suspension used to count pollen is very limited when compared to the total volume. One can also use electronic particle counter, when available, to count pollen grains.
One of the two methods described here counts all the pollen grains produced in an anther; this is ideal when the anthers are small and produce only a limited number of pollen grains. The other method involves the preparation of a pollen suspension in a known volume and counting pollen grains from a sample of the suspension.
5.5.1.1 Special Requirements
-
Acetocarmine or safranin stain
-
Permanent black ink pen with a fine tip
5.5.1.2 Procedure
-
1.
Take a microscope slide, and with the help of a permanent ink pen, draw thin parallel lines about 1 mm apart at right angles to the long axis of the slide. The distance between lines should be such that the two neighbouring lines should be visible under the low magnification of the microscope (10×). The width of the lined area on the slide should be wider than the cover glass you would be placing so that the cover glass will not extend beyond the lined area. Alternately you can gently draw permanent lines on the slide with a glass-etching pen. Such a slide can be used routinely for pollen counting.
-
2.
Take a mature but undehisced anther (about to dehisce) and place it on the other side of the slide (without parallel lines) in a small drop of water/stain (acetocarmine or safranin are suitable) placed at the centre of the lined part of the slide.
-
3.
With the help of needles and/or forceps, crush and squeeze the anther to release all the pollen grains. Remove the debris. Gently lower a cover glass of suitable size in such a way that the liquid does not flow/spread outside the cover slip.
-
4.
Place the slide under a microscope to focus left top corner of the cover glass. Make sure that the lines and the space between the two adjacent lines are covered and in focus under the lower-magnification (10×) objective. Count the pollen grains present between the two lines from the top of the cover glass to the bottom of the cover glass by moving the slide (preferably with mechanical stage) along the parallel lines. Include pollen grains present on the left line while counting. Repeat the counting sequentially covering each parallel line from left to the right of the cover glass so that the whole mounted area is covered for counting. If the mounting medium has spread slightly beyond the boundary of the cover glass, count all the pollen grains present in the mounting medium all around the cover glass. The total number of pollen grains counted from one anther gives the total number of pollen grains produced in an anther.
-
5.
Clean the slide without rubbing the lower surface (marked with parallel lines) and repeat the counting for a number of anthers randomly selected from different flowers and plants. Calculate the average number of pollen grains/anther.
-
6.
Multiply this number with the average number of anthers in a flower to determine the average number of pollen produced in a flower. Present the mean number with SE (standard error)/SD (standard deviation).
5.5.1.3 Modification
When anthers are of different lengths or sizes in a flower, pollen number in each type has to be counted and computed separately.
5.5.2 Estimation of Pollen Production in Anthers with Large Number of Pollen Grains
The procedure described under Protocol 5.5.1 is not suitable for anthers that contain a larger number of pollen grains. As all the pollen grains of an anther cannot be counted, suitable sampling method has to be followed.
5.5.2.1 Special Requirements
-
A glass/plastic vial of suitable size to prepare pollen suspension
-
A glass rod to crush the anther
-
A micropipette (about 100 μl capacity)
-
Sucrose/glycerin solution of suitable density (2–5 %) or a drop of detergent (such as liquid soap) to keep pollen grains in suspension
-
Pasteur pipette or disposable syringe (without needle) of about 2 ml capacity
5.5.2.2 Procedure
-
1.
Suspend one mature but undehisced anther in a suitable amount (1–5 ml depending on the size of the anther and number of pollen grains) of sucrose/glycerin solution or water containing a drop of detergent solution taken in a 5–10 ml vial. Crush the anther with a glass rod to release all the pollen grains from the anther. Mix the suspension. Pollen release and mixing can be enhanced by drawing of the suspension from the vial into a Pasteur pipette or disposable syringe (without needle) and expelling it back to the vial with force repeatedly. Remove visible debris with a pair of fine forceps.
-
2.
While gently shaking the suspension, take (using a micropipette) a suitable volume of the suspension (20–50 μl depending on the concentration of pollen in the suspension so that pollen grains spread sparsely making it convenient to count) and place it on a slide marked with 1 mm wide parallel lines as described in Protocol 5.5.1. The optimization of the amount of liquid used to make pollen suspension and the amount of suspension taken would require some trial runs.
-
3.
Lower a cover glass and count the number of pollen grains as described in the Protocol 5.5.1.
-
4.
Repeat steps 2 and 3 at least for 10 samples of the suspension.
-
5.
Estimate the total number of pollen grains in the suspension by using the following formula:
$$ \frac{\mathrm{Total}\kern0.24em \mathrm{volume}\kern0.24em \mathrm{of}\kern0.24em \mathrm{pollen}\kern0.24em \mathrm{suspension}\times \mathrm{No}.\mathrm{of}\kern0.24em \mathrm{pollen}\kern0.24em \mathrm{grains}\kern0.24em \mathrm{in}\kern0.24em \mathrm{the}\kern0.24em \mathrm{sampled}\kern0.24em \mathrm{suspension}}{\mathrm{Volume}\kern0.24em \mathrm{of}\kern0.24em \mathrm{suspension}\kern0.24em \mathrm{used}\kern0.24em \mathrm{for}\kern0.24em \mathrm{counting}} $$ -
6.
Repeat pollen counts (using the above procedure) for the required number of anthers selected randomly from different flowers of the same plant as well as other plants.
-
7.
Calculate the average number of pollen per anther.
-
8.
Multiply this number with the number of anthers in a flower to determine the average number of pollen per flower.
-
9.
Present the mean number with SE/SD.
When anthers are of different lengths or sizes in a flower, pollen number in each type has to be calculated and computed separately.
5.5.2.3 Modifications
-
1.
Pollen suspension, instead of placing on the slide as a drop, is spread on the slide in the form of a narrow band (the width of which can be covered by low magnification of the microscope). The pollen grains present can be counted by moving the slide from one end of the band to the other.
If the pollen grains are sufficiently large to be clearly seen under a stereomicroscope, the band can be slightly broader and pollen grains can be counted under a stereomicroscope.
-
2.
Kearns and Inouye (1993) have described another simple method. In this one, pollen grains are suspended in a known volume of ethanol and a sample of the suspension is placed on a microscope slide. Addition of a few drops of ethanol onto the slide would enable better spread of pollen. Allow ethanol to evaporate and count the pollen grains under a microscope. Drawing parallel lines with a marker as described in Protocol 5.5.1 would make it convenient to count.
5.5.3 Estimation of Pollen Fertility
5.5.3.1 Special Requirements
-
Acetocarmine stain
5.5.3.2 Procedure
-
1.
Collect pollen from freshly dehisced anthers and place them in a drop of acetocarmine taken on a clean microscope slide.
-
2.
Mix pollen grains with the help of a needle.
-
3.
Lower a cover glass. Warm the slide gently over a spirit lamp/burner and apply gentle pressure over a piece of blotting paper placed on the cover glass to remove excess stain but do not rupture the pollen. Leave the slide for about 5 min for pollen to take up stain. As fertile pollen grains are filled with contents, they take up deep and uniform stain (Fig. 5.2b). Sterile pollen grains are shrivelled/incompletely filled or empty; they do not take up deep, uniform stain.
-
4.
Score pollen grains for sterility/fertility under a microscope using bright illumination. Under each microscope field, count the number of pollen grains with uniform deep colour as fertile and those which are or unstained or partially stained as sterile. Record data using at least ten microscopic fields. Repeat this with a sufficient number of anthers collected from different flowers and plants. Calculate per cent fertility.
5.5.4 Acetolysis of Pollen Grains
This method is used to study exine ornamentation in detail. Acetolysis removes all non-sporopollenin components of the pollen (the contents, intine and components present in and on the pollen wall) leaving only the exine intact.
5.5.4.1 Special Requirements
-
Acetolysis mixture: acetic anhydride and concentrated sulphuric acid (9:1). Add the acid slowly to acetic anhydride in the fume hood; as it results in exothermic reaction, the container gets hot.
-
Low-speed centrifuge.
-
Glacial acetic acid.
5.5.4.2 Procedure
-
1.
Collect sufficient amount of pollen grains (2–4 mg) and suspend them in about 10 ml of acetolysis mixture taken in a test tube.
-
2.
Heat the suspension to boiling in a water bath for 1–2 min. Remove the tube and allow it to cool.
-
3.
Transfer pollen suspension into a centrifuge tube. Remove acetolysis mixture by low-speed centrifugation (about 2,000 rpm).
-
4.
Add about 5 ml of glacial acetic acid to the centrifuge tube. Mix pollen pellet and remove acetic acid by centrifugation.
-
5.
Add about 5 ml of water to centrifuge tube and mix pollen pellet in water.
-
6.
Mount pollen grains on a slide in water or 10 % glycerol and observe under the microscope. They can also be mounted in glycerin jelly as semi-permanent preparations for later observations and also as reference slides (see Protocol 5.5.5).
5.5.4.3 Modification
If acetolysed pollen grains have turned dark, bleach the pollen in chlorine. For this, add 2 ml of glacial acetic acid +2–3 drops of saturated sodium chlorate solution +1–3 drops of concentrated hydrochloric acid. As chlorine is released, gently stir the mixture for about a min. Remove bleaching mixture by centrifugation and suspend pollen grains in water. If necessary, acetolysed pollen grains can be transferred to 70 % ethanol and stored for later studies.
5.5.5 Mounting of Pollen Grains in Glycerin Jelly
For any morphological study such as size, shape, apertures and exine ornamentation, pollen grains have to be mounted in a suitable medium. Although temporary preparations can be made in acetocarmine or safranin, the structure of the pollen grains may not be clear. Glycerin jelly, containing some stain such as basic fuchsin, has been used routinely as a mounting medium by pollen morphologists. Glycerin jelly becomes solid at room temperature, and it has to be melted for mounting. Both fresh and acetolysed pollen can be mounted in glycerin jelly.
5.5.5.1 Special Requirements
-
Glycerin jelly (see Appendix A.3 for details of preparation)
-
Spirit lamp/hot plate
-
Paraffin wax
5.5.5.2 Procedure
-
1.
Take a small amount of pollen sample (to be mounted) on the middle of a clean microscope slide.
-
2.
Scoop a small piece of glycerin jelly by using forceps or a spatula and place the jelly in contact with the pollen sample so that pollen grains adhere to the jelly.
-
3.
Warm the slide gently on a spirit lamp/hot plate until the jelly melts. Do not allow it to boil.
-
4.
Keep the slide under a stereomicroscope and spread the pollen in the melted jelly by gently stirring with a needle. Rewarm the jelly if it solidifies during stirring.
-
5.
Lower a cover glass carefully over the jelly. Allow it to spread into a circle of 4–5 mm and rewarm the slide, if necessary.
-
6.
While the slide is still warm, place the slide inverted (cover slip facing down) with support on both the sides with some solid material so that the cover glass remains hanging without touching anything. This will allow pollen grains to settle near the cover glass so that pollen grains can be observed under higher magnification of the microscope.
-
7.
Observe the preparation or store for later observation.
Glycerin jelly slides remain in good condition for many weeks or even months. They should be stored under dust-free atmosphere.
5.5.5.3 Modification
The slides can be made permanent by sealing the slide with paraffin wax. After step 6, place a small amount of paraffin close to the edge of the cover glass on one side and heat the slide until the wax melts. The wax is drawn under the cover glass and surrounds the glycerin jelly mount. Scrape off the excess wax with a blade or clean the slide with xylene and store in a slide box.
5.5.6 Acetocarmine Squash Preparations to Study Pollen Cytology
The number of cells in the pollen (2 or 3 celled) at the time of shedding is important as it is correlated with a number of physiological responses of the pollen. Pollen cytology can be studied through acetocarmine/Feulgen squash preparation or the use of DNA fluorochromes.
5.5.6.1 Special Requirement
-
Acetocarmine stain
5.5.6.2 Procedure
-
1.
Transfer a small amount of pollen to a drop of acetocarmine taken on a slide.
-
2.
Mix pollen suspension with a needle and lower a cover glass. Warm the slide over a flame but do not boil. Cool the slide. Take out excess stain with a piece of blotting paper.
-
3.
Keep the slide on a flat surface, spread two or three layers of blotting paper pieces on the cover glass and apply pressure with the thumb avoiding any lateral movement of the cover glass. The aim is to rupture the pollen wall and spread the contents evenly. It is desirable to maintain the identity of the contents of individual pollen so that the vegetative nucleus and the generative nucleus/sperm nuclei can be assigned to individual pollen.
-
4.
If the slide is to be observed some time later after the preparation, seal the edges of the cover glass with nail polish or paraffin wax and maintain the slide in a slide box free from dust.
-
5.
Observe the preparation under a high-power/oil immersion objective of the microscope and look for the vegetative nucleus and the generative nucleus/the two sperm nuclei. Generally, the vegetative nucleus takes up faint stain because of its dispersed chromatin, while the generative nucleus/sperm nuclei take deeper stain as they have condensed chromatin. Observe nuclear details in 15–20 pollen grains and record whether the pollen grains are two/three celled.
5.5.7 Feulgen Squash Technique to Study Pollen Cytology
The basis of this technique is hydrolysis of the purine deoxyribose linkages of DNA with warm HCl to open up aldehyde groups on the deoxyribose sugar. The fuchsin reacts with the open aldehyde and produces an insoluble DNA-specific coloured complex.
5.5.7.1 Special Requirements
-
Feulgen stain: Dissolve basic fuchsin (0.5 g) in 100 ml of boiling distilled water, cool to ca 50 °C and filter. Add 10 ml of N-hydrochloric acid and 0.5 g potassium metabisulphite. Shake thoroughly, close the lid tightly and store in dark until use.
-
Table-top centrifuge.
5.5.7.2 Procedure
For sequential changes of pollen (steps 2–7), use low-speed centrifugation of about 2,000 rpm:
-
1.
Fix mature anthers before they dehisce in acetic alcohol or in Carnoy’s fluid (see Appendix A.1) taken in a vial for 2–6 h.
-
2.
Transfer anthers to 70 % ethanol and release pollen by crushing with a glass rod.
-
3.
Hydrolyze pollen in 10 % HCl for about 6 min in a water bath at 60 °C.
-
4.
Rinse 2–3 times in distilled water.
-
5.
Transfer pollen to Feulgen stain for 10–15 min.
-
6.
Remove unbound stain with distilled water.
-
7.
Transfer pollen to 40 % acetic acid.
-
8.
Take a small drop of pollen suspension on a slide and squash pollen sample (follow the procedure of acetocarmine squash, see Protocol 5.5.6).
-
9.
Observe under the microscope. The vegetative and generative/sperm nuclei stain purplish red. The cytoplasm stains green.
5.5.8 DNA Fluorochromes to Study Pollen Cytology
The use of DNA fluorochromes is the most convenient method to study pollen cytology, if a fluorescence microscope is available (Hough et al. 1985). The method is simple, rapid and consistent and can be used to study details of nuclei not only of the mature pollen but also during pollen development, pollen germination and pollen tube growth.
5.5.8.1 Special Requirements
-
Several DNA fluorochromes are available commercially. We have routinely used DAPI (4′,6-diamidino-2-phenylindole) or ethidium bromide (3,8-diamino-5-ethyl-6-phenyl phenanthridinium bromide).
-
DAPI: stock solution – 1 mg/ml dissolved in water. Working solution: 5–50 μg/ml (10 μl of stock solution made up to 2 ml with distilled water = 5 μg/ml working solution).
-
Ethidium bromide: stock solution – 0.1 % in distilled water stored in a fume hood at room temperature. Working solution – 100 μg/ml in distilled water (stock solution diluted 10 times in distilled water).
5.5.8.2 Procedure
-
1.
Take fixed or fresh pollen sample on a microscope slide and add a few drops of working solution of DAPI/ethidium bromide.
-
2.
Lower a cover glass and apply gentle pressure. Sometimes, the reserve material in the pollen may mask the nuclei. For such pollen apply more pressure on the cover glass to rupture the pollen.
-
3.
Observe the preparation under a fluorescence microscope. For DAPI, use UV filter combination; the nuclei show blue fluorescence. For ethidium bromide, use blue filter combination; the nuclei show yellow/orange fluorescence.
-
4.
For later observation, seal the preparation in paraffin/nail polish and store in dark in a refrigerator.
5.5.8.3 Modifications
-
1.
This protocol can be used for cultured pollen grains to study the movement of vegetative nucleus and generative cell into the pollen tube and its division (2-celled pollen ) and the movement of sperm cells in the pollen tube (3-celled pollen). Follow the above procedure for fixed or fresh in vitro germinated pollen tubes after allowing pollen tubes to grow for different lengths.
-
2.
DNA fluorochromes have been used as vital stains (Hough et al. 1985). They can be incorporated into the germination medium, and the details of pollen nuclei can be followed during pollen germination and pollen tube growth by observing pollen suspension taken after different time intervals.
-
3.
For details of other DNA fluorochromes such as Hoechst 33258 and mithramycin, see Shivanna and Rangaswamy (1992).
5.5.9 Tetrazolium Test for Pollen Viability
One of the simple methods used in earlier studies to assess pollen viability has been the tetrazolium test (TTC test ). TTC test is based on the demonstration of the activity of dehydrogenases (an important group of enzymes in the respiratory cycle) in the pollen cytoplasm. When pollen grains are mounted in a solution of colourless soluble tetrazolium salt, dehydrogenases of the pollen, if active, reduce colourless soluble tetrazolium salt to a reddish insoluble substance called formazan. Formazan accumulates in the pollen cytoplasm and gives reddish colour to pollen. 2,3,5-triphenyl tetrazolium chloride and nitroblue tetrazolium are the most commonly used tetrazolium salts.
5.5.9.1 Special Requirements
2,3,5-triphenyltetrazolium chloride (0.2–0.5 % solution prepared in a suitable concentration of sucrose solution to prevent bursting of pollen). TTC solution needs to be stored in a brown bottle as it undergoes photo-oxidation. It can be stored under refrigeration for a couple of weeks.
Simple humidity chamber (see Appendix A.5).
5.5.9.2 Procedure
-
1.
Mount pollen grains in TTC solution and lower a cover glass.
-
2.
Incubate the slide in a humidity chamber in dark (such as table draw) at laboratory temperature for 30–60 min.
-
3.
After the incubation period, observe the slide under the microscope and score pollen grains that have turned red as viable. Oxygen inhibits reduction of TTC; it is better to score pollen grains from the central part of the cover glass. Pollen grains located along the margin of the cover glass may show variable degree of red colouration due to higher oxygen availability.
-
4.
Calculate per cent viable pollen in the sample.
In our experience the results of tetrazolium test are not consistent. It tends to give false-positive results in many pollen samples. Even the heat-treated pollen samples, which fail to respond to germination test, have been reported to give high levels of positive values in TTC test (Heslop-Harrison et al. 1984). Another limitation of this test is that pollen grains often show a gradation in colour development (from very light to deep red) with the result fixing a cut-off point for colour intensity becomes difficult for scoring. In recent years, TTC test is being used very rarely.
5.5.10 In Vitro Germination Test for Pollen Viability
In vitro germination test is based on the ability of the pollen grains to germinate in vitro in a suitable nutrient medium. This is a rapid and reasonably simple test and, in an optimal germination medium, shows correlation with fruit and seed set. In vitro germination test has been a routine test used for pollen viability . This test requires some preliminary studies to standardize the medium that permits optimal germination and the period of incubation. This test cannot be used for those species in which pollen grains cannot be germinated in vitro.
There are a number of methods such as hanging drop cultures, sitting drop cultures, suspension cultures and surface cultures available for in vitro germination. Here, only the sitting-drop culture which is one of the simplest, most convenient and effective methods for in vitro pollen germination is described. For details of other methods, refer to Shivanna and Rangaswamy (1992).
5.5.10.1 Special Requirements
-
Suitable nutrient medium (see Appendix A.4) for composition of various media
-
Humidity chamber (see Appendix A.5) for preparing simple humidity chamber
-
Acetocarmine stain
5.5.10.2 Prehydration of Pollen
Pollen of many species particularly 3-celled pollen require prehydration/controlled hydration/vapour-phase hydration to give consistent results. This is carried out by exposing pollen grains to high humidity for about 30 min. One of the easier methods of prehydration is to spread the pollen sample on a dry slide and keep the slide in a humidity chamber for 30 min.
The plasma membrane in desiccated pollen is not in a functional state. When they are cultured directly in the liquid medium, they leak soluble metabolites into the surrounding medium. This imbibitional leakage seems to be the major limitation for achieving optimal germination. Prehydration of pollen by exposing them to high humidity permits gradual hydration which is suitable for the restoration of membrane integrity (see Shivanna 2003). As prehydration does not have any negative effects, we routinely expose pollen sample to prehydration before culturing.
5.5.10.3 Procedure
-
1.
Collect pollen sample to be tested for viability and spread them as a thin layer on a dry microscope slide.
-
2.
Keep the slide in a humidity chamber for prehydration for 30–60 min.
-
3.
Collect suitable amount of prehydrated pollen with a clean needle or forceps from the slide maintained in the humidity chamber and disperse them in a drop (about 20–30 μl) of standardized germination medium taken on a microscope slide. As the pollen grains tend to clump and settle at the periphery of the drop, try to make homogeneous distribution of pollen in the drop with the help of a clean and dry needle. Pollen density is also important; too dense or too dilute suspension would not yield optimal results. A good sitting drop culture should appear neither turbid nor transparent. Take minimum time to prepare sitting drop culture; too much of preparation time results in evaporation of the medium and consequent changes in the concentration of its constituents. Transfer pollen cultures into a humidity chamber.
-
4.
Maintain the pollen cultures in the humidity chamber for optimal period on the basis of your preliminary studies (1–4 h depending on the species). For each sample, an enough number of replicates have to be cultured (4–6 sitting drop cultures).
-
5.
At the end of the incubation period, add a small drop of a fixative (FAA/acetic alcohol) or a drop of acetocarmine, remix pollen suspension as uniformly as possible and lower a cover glass.
-
6.
Score the cultures to calculate per cent germination. Pollen grain is considered germinated when the length of its tube is more than the diameter of the pollen grain. For each culture, it would be desirable to score pollen from 6 to 10 microscope fields, selected randomly. Enter the scores of each microscope field as shown in the sample Table 5.1. To eliminate the probability of scoring the same sample of pollen grains more than once, it would be desirable to move the preparation under the microscope in consecutive rows and score 2–3 arbitrary fields in each row. From each microscope field, count the total number of pollen grains and the number of pollen grains germinated.
Table 5.1 Arbitrary scores for calculating pollen germination and pollen tube growth in pollen cultures raised to test pollen viability* -
7.
Calculate per cent pollen germination as shown in sample Table 5.1.
5.5.11 Fluorescein Diacetate Test for Pollen Viability
Fluorescein diacetate (FDA) test is also referred to as fluorochromatic reaction (FCR) test. The test was introduced by Heslop-Harrison and Heslop-Harrison (1970). Since then it has become one of the standard tests to assess pollen viability . It is simple and can be performed within a few minutes. The only limitation is that it requires a fluorescence microscope. In this test, pollen grains are mounted in FDA solution. The nonpolar and nonfluorescent FDA readily enters pollen cytoplasm as the plasma membrane does not act as a barrier. Cytoplasmic esterases, if active, hydrolyze FDA and release fluorescein, which is polar and fluorescent. Unlike FDA, fluorescein passes through the intact plasma membrane sparingly and therefore accumulates in the cytoplasm of viable grains and gives a bright green or yellowish green fluorescence under the fluorescence microscope (Fig. 5.2c). If the plasma membrane of the pollen is not intact, fluorescein comes out readily through the membrane into the mounting medium to give a dull fluorescence of the mounting medium; such pollen grains do not show bright fluorescence. Similarly if pollen grains lack active esterases, they do not produce fluorescein and thus do not fluoresce. FDA test, therefore, assesses two properties of the pollen grain: the integrity of the plasma membrane and the activity of esterases. The FDA test has been found satisfactory for pollen of a range of species and correlates with in vitro germination test (Shivanna and Heslop-Harrison 1981; Heslop-Harrison et al. 1984; Shivanna et al. 1991a, b). We have been using FDA test routinely to assess the pollen viability (Kuriakose et al. 2009; Sharma et al. 2010).
5.5.11.1 Special Requirements
-
Fluorescein diacetate (FDA): stock solution – since FDA does not dissolve in water, stock solution has to be prepared in acetone (2 mg/ml). Stock solution can be stored in a refrigerator for months.
-
Sucrose solution of suitable concentration to prevent bursting of pollen grains. 8–10 % works well for most of the pollen systems.
-
Humidity chamber (Appendix A.5)
5.5.11.2 Procedure
-
1.
Prepare working solution of FDA by taking 2–5 ml of sucrose solution/germination medium in a glass vial and adding drops of FDA stock solution with a pipette until the solution shows persistent turbidity. The working solution should be used within 30 min of its preparation; otherwise most of the FDA would precipitate.
-
2.
Take a drop of working solution on a microscope slide and suspend sufficient amount of pollen grains in the drop and mix the suspension with a needle and lower a cover glass.
-
3.
Incubate the preparation in a humidity chamber to prevent evaporation of the solution for 3–5 min. The incubation time may vary depending on the species. Pollen of some species show bright fluorescence soon after making the preparation.
-
4.
After the incubation period, observe the preparation under the fluorescence microscope using blue filter combination.
-
5.
Score pollen grains that fluoresce brightly as viable by following the procedure described under Protocol 5.5.10. Scoring has to be done within a few minutes of preparation. If they are left for longer period, the fluorescence of pollen becomes dull as the fluorescein continues to move from pollen cytoplasm into the medium although at a slower rate and the background also starts showing dull fluorescence.
5.5.11.3 Modification
Pollen grains of many species may require prehydration for 30–60 min to provide better conditions for restoration of membrane integrity to enhance pollen response to the FDA test. In such species, it would be better to prehydrate pollen samples (see Protocol 5.5.10 for details) before testing for viability.
5.5.12 In Vitro Germination Test for Pollen Vigour
One of the simple methods to assess the vigour of pollen samples is to use pollen samples for controlled pollinations. One set of pistils is pollinated with fresh pollen which are expected to show maximum viability and vigour and another set with pollen sample to be tested for vigour. Some pistils from each set are fixed at intervals and used to study pollen germination and pollen tube growth using aniline blue test (for details see Protocol 8.5.1) and record the extent of tube growth in terms of mm/cm (depending on the length of the pistil ) in a unit time. Less vigorous pollen show slower growth through the pistil. For example, pistils pollinated with fresh pollen may show pollen tubes in the middle part of the style at a given time, whereas those pollinated with test pollen may show pollen tubes in the upper part of the style. This procedure again requires too much of time and also a fluorescence microscope for clear documentation of pollen tube growth in the pistil.
In vitro germination assay can also be used to assess pollen vigour. In the protocol to assess pollen viability , per cent pollen germination is scored at one time without consideration of the time factor; the cultures are scored after maintaining them for a much longer period than that required for germination. To assess pollen vigour, however, germination and tube length in fresh pollen samples and test pollen samples are scored at regular intervals after culture and the responses are compared. The results on pollen germination and tube length are presented in the form of a graph. Pollen grains with reduced vigour take longer time when compared to control (fresh pollen) to attain maximum germinability (Shivanna and Cresti 1989). Similarly, pollen tube length remains significantly shorter at any given time in pollen samples with reduced vigour.
5.5.12.1 Special Requirements
-
As in Protocol 5.5.10 on in vitro pollen germination test
5.5.12.2 Procedure
-
1.
Raise sitting drop cultures of fresh pollen and test pollen as described in Protocol 5.5.10. As the same culture cannot be used for scoring at regular intervals, raise more number of cultures for each pollen sample depending on the scoring time intervals. Scoring intervals depend on the time taken for germination and the rate of pollen tube growth. Record the time of raising each culture.
-
2.
Incubate the cultures in the humidity chamber.
-
3.
After each time interval (for many species, 30-min intervals are satisfactory, and others may require 1-h intervals), take two cultures from each pollen sample, add a drop of fixative or acetocarmine, mix the pollen suspension and lower a cover glass. Keep the slide under humidity chamber to prevent drying if they are not scored immediately.
-
4.
Repeat the procedure at selected time intervals.
-
5.
Score all the cultures (2 from fresh pollen and 2 from test pollen, at each selected time interval) for pollen germination (following the procedure given under Protocol 5.5.10 and Table 5.1). Measure pollen tube length from each field used for scoring germination as follows:
-
(a)
Adjust the intensity of microscope illumination with the help of diaphragm so that the boundary of each grain and tube are clearly visible. From each microscope field used for germination score, measure the length of 10 pollen tubes selected randomly (in ocular units) by using an ocular micrometre. Omit curved pollen tubes which cannot be measured accurately. If the number of pollen tubes in each microscope field is <10, measure all the pollen tubes in the field. If the number of tubes is >10, measure 10 randomly selected pollen tubes.
-
(b)
Enter both germination and tube length data in a tabular form (see the sample Table 5.1). Repeat the scoring for all the cultures. Calculate mean pollen germination and pollen tube length for each pollen sample at each time interval and present the same with SE/SD. If necessary, ocular units can be converted into micrometres by calibrating the microscope with the use of stage micrometre (see Table 5.1 for details).
-
(a)
-
6.
Present the results for germination and tube length in the form of a graph over time for each reading.
5.5.12.3 Modification
The semi-vivo method can also be used to test pollen vigour. The details are described in Protocol 8.5.3.
References
Bryant VM Jr (1990) Pollen: nature’s fingerprints of plants. Encyclopedia Britannica 93:111
Cruden RW (1977) Pollen-ovule ratios: a conservative indicator of breeding systems in flowering plants. Evolution 31:32–46
Ducker SC, Pettitt J, Knox RB (1978) Biology of Australian seagrasses: pollen development and submarine pollination in Amphibolis antarctica and Thalassodendron ciliatum (Cymodoceaceae). Aust J Bot 26:265–285
Erdtman G (1969) Handbook of palynology. Hafner, New York
Fægri K, Iversen J (1989) Textbook of pollen analysis, 4th edn. Wiley, Chichester
Heslop-Harrison J, Heslop-Harrison Y (1970) Evaluation of pollen viability by enzymatically induced fluorescence; intracellular hydrolysis of fluorescein diacetate. Stain Technol 45:115–120
Heslop-Harrison J, Heslop-Harrison Y, Shivanna KR (1984) The evaluation of pollen quality and a further appraisal of the fluorochromatic (FCR) test procedure. Theor Appl Genet 67:367–375
Hough T, Bernhardt P, Knox RB, Williams EG (1985) Application of fluorochromes to pollen biology. II. The DNA probes ethidium bromide and Hoechst 33258 in conjunction with callose-specific aniline blue fluorochrome. Stain Tech 60:155–162
Kearns A, Inouye DW (1993) Techniques for pollination biologists. University Press Colorado, Niwot
Kuriakose G, Sinu PA, Shivanna KR (2009) Domestication of cardamom (Elettaria cardamomum) in Western Ghats, India: divergence in productive traits and a shift in major pollinators. Ann Bot 103:727–733
Linskens HF (1995) Pollen deposition in mosses from north-west Greenland. Proc Kon Ned Akad Westensch 98:45–53
Moore PD, Webb JA (1978) An illustrated guide to pollen analysis. Hodder & Stoughton, London
Nair PKK (1970) Pollen morphology of angiosperms. Scholar Publishing House, Lucknow
Rao GU, Jain A, Shivanna KR (1992) Effects of high temperature stress on Brassica pollen: viability, germination and ability to set fruits and seeds. Ann Bot 69:193–198
Sharma MV, Shaanker RU, Vasudeva R, Shivanna KR (2010) Functional dioecy in Nothapodytes nimmoniana, a distylous species in the Western Ghats. Curr Sci 99:1444–1449
Shivanna KR (2003) Pollen biology and biotechnology. Science Publishers, Inc., Enfield/Plymouth
Shivanna KR, Cresti M (1989) Effects of high humidity and temperature stress on pollen membrane and pollen vigour. Sex Plant Repro 2:137–141
Shivanna KR, Heslop-Harrison J (1981) Membrane state and pollen viability. Ann Bot 47:759–770
Shivanna KR, Rangaswamy NS (1992) Pollen biology: a laboratory manual. Springer, Berlin/Heidelberg
Shivanna KR, Linskens HF, Cresti M (1991a) Pollen viability and pollen vigour. Theor Appl Genet 81:38–42
Shivanna KR, Linskens HF, Cresti M (1991b) Responses of tobacco pollen to high humidity and heat stress: germination in vitro and in vivo. Sex Plant Reprod 4:104–109
Walker JW (1971) Pollen morphology, phytogeography, and phylogeny of the Annonaceae. Contrib Gray Herb 202:1–131
Warny S (2013) Museums’ role: pollen and forensic science. Science 339:1149
Author information
Authors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer India
About this chapter
Cite this chapter
Shivanna, K.R., Tandon, R. (2014). Pollen Biology. In: Reproductive Ecology of Flowering Plants: A Manual. Springer, New Delhi. https://doi.org/10.1007/978-81-322-2003-9_5
Download citation
DOI: https://doi.org/10.1007/978-81-322-2003-9_5
Published:
Publisher Name: Springer, New Delhi
Print ISBN: 978-81-322-2002-2
Online ISBN: 978-81-322-2003-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)