
Abstract
Recombination is a key molecular process of chromosome reshuffling that takes place producing the gametes during sexual reproduction. The frequency of recombination varies greatly along the chromosomes, and its variation along the chromosome can be quantified through the measure of linkage disequilibrium (LD). Thus, recombination, through the measure of linkage disequilibrium, is at the base of all progress in mapping traits in the genome, is critical for GWAS studies, and has therefore fostered advancements in medical genetics. A major limitation that hampers a more extended use of LD is due to the fact that most current experimental technologies do not solve the phasing, along with a low accuracy of computational phasing algorithms.
Besides LD, recombination events by themselves, that is, the presence or absence of specific recombination events, can be used as genetic markers to study human population diversity as well as the dynamics of the recombination process. Differences in the recombination rates are found not only along the genome but also between populations and individuals. When LD has been applied to reconstructing the demographic history of modern human populations, the complementation of genetic diversity measures with LD has proved to be critical in solving the problem of defining ancestral populations. Despite the wealth of information contained in the recombination footprint, the full use of recombination and linkage disequilibrium data in population genetics is still in its infancy, and many more possibilities are waiting to be uncovered.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
1 Recombination and Its Distribution Along the Genome, Linkage Disequilibrium
Sexual reproduction requires diploid number of chromosomes, which reduces to haploid by meiosis to generate the gametes (eggs or spermatozoa). During this process, homologous chromosomes align and switch over segments. As a consequence, the haploid content of a gamete does not have any full chromosomes from the maternal or paternal genome, so each gamete is made up of pieces or a complex mosaic of their parental chromosomes.
Recombination happens all over the genome except in a few specific locations: the mitochondrial DNA (mtDNA) and most of the Y chromosomes (the non-recombinant part or NRY). Thus, genes will only be transmitted in a group if recombination has not occurred between them. From classical genetics principles, it is possible to calculate the number of cases in which two genes (or any genetic fragment or locus) which are neighbors on a chromosome were set apart by recombination while producing gametes. This ascertains a distance between the two genes (or two loci) defined as the percentage of cases in which recombination has occurred between them. This is the base of genetic distances , measured in centimorgans (one centimorgan means that 1% of the gametes will have a recombination event between the two specific points for which the distance is measured). A general average for the genome is that 1% of recombination events happen at a distance of one megabase (1 cM = 1 MB).
The frequency of recombination varies greatly along the chromosomes featuring regions with high recombination, while others are depleted or even totally absent (Paigen and Petkov 2010). The short regions (1–2 Kb) where most of the recombinations happen are called recombination hotspots and have a very strong concentration of recombination events. One of the most interesting advances in genomics in the last few years has been the discovery of the molecular bases for the formation of recombination hotspots in the genome and their complex evolutionary dynamics (Baudat et al. 2013).
Differences in the recombination landscape are found not only along the genome or a chromosome but also between sexes (women recombine more than men), between populations (slight differences among human populations have been observed, with some hotspots being population specific), and among species (the recombination map of humans and chimpanzees differs greatly). Kong et al. (2010) were able to construct the first recombination map based on directly observed recombinations with a resolution that is effective down to 10 kilobases (Kb) from the observation of thousands of parent-offspring pairs. They were able to pinpoint differences among sexes, populations, and even individuals. Thus, recombination is a fast-evolving trait with a birth and death process of recombination hotspots.
The existence of recombination breaks indicates that the genome content along a single arm of a chromosome (or along the DNA that constitutes it) may be different from one to another. This is why we talk about haplotypes to refer to the genetic content along a single DNA molecule; if several variants (or alleles) for the consecutive genome regions exist, the specific combination in each chromosome constitutes a haplotype. With n number of variants in a given fragment of a chromosome, the number of possible haplotypes is 2n−1. In fact, given the information of diploid variants (like a DNA sequence), it may be impossible to know the phase in which the variants are found along the two chromosomes, but we explore below methods to solve it, based on the information in pedigrees or using statistical methods.
Given the irregular pattern of recombination endowed with strong differences among genome regions, we can wonder to which extent the information at a specific genome position is independent of the information at a position nearby: if we have two close genomic positions, the genomic information of the two may be independent if it is a recombination hotspot between them, or they may be fully correlated if there is no recombination, and the information in one tells us what the state is of the other. It is similar to the concept of pants and jackets—they can be separate objects, yet linked completely like in the case of a suit. This idea is unified by a concept: linkage disequilibrium (LD) , which tells the degree of interdependence between two positions in the genome, from independence (total lack of LD) to full dependence (total LD) with all possible states in between. It is a statistical concept and the most usual measures vary from 0 (no LD) to 1 (total LD).
When looking at the LD pattern in a given genome region (Fig. 6.1), information for a population of individuals is needed, because LD is a population-level statistic. Linkage disequilibrium is at the base of all progress in mapping traits in the genome. The advancement of medical genetics owes most of its achievements to the existence of LD, as it has aided in the mapping of traits in the genome to the linkage between the gene of interest and a specific genetic marker, usually a SNP (single nucleotide polymorphism). All the GWAS (genome-wide association studies) that analyze hundreds of thousands or millions of SNPs in a group of affected people and unaffected controls will be able to find the genetic bases of the trait if LD is present between one (or several) of the SNPs and the causative variant(s) in the genome.

Structure of linkage disequilibrium in a region in the human genome. Red squares indicate high linkage disequilibrium between the two SNPs. It is possible to see long stretches of strong linkage disequilibrium, which usually are flanked by recombination hotspots that are the cause of breakage of linkage disequilibrium blocks. Data for European (Catalans) and African (Mandenka) populations. African populations show much less linkage disequilibrium than in non-African
The haplotype is of interest in many areas of genome studies, as variants in the genome are not independent among them. But, most of the present technologies give information of both chromosomes together; in fact many technologies allow working on a single DNA strand, but difficulties remain in separating chromosomes. Some basic techniques based on long-range PCR or on the separation of single chromosomes may be used to solve the issue, but both are difficult and with limitations. The technology of second-generation sequencing does not solve the issue, but third-generation technology will be able to sequence long single DNA molecules with long reads and haplotype information.
Computationally, there are interesting statistical methods to solve the phasing of a set of linked variants. These methods use coalescent theory to reconstruct the phase of several variants, and their power is strongly dependent on the amount of LD: the more LD, the better the reconstruction will be. One important consideration is that the accuracy of the phasing of genotyped data depends on LD, and thus no algorithm can make accurate phasing estimations in the absence of LD . The low accuracy of these phasing algorithms, especially in populations with very low linkage disequilibrium, is often not adequately taken into account, like in the data provided in the 1000 Genomes project.
The detection of specific recombination events in a set of sequences allows the study of “junctions” in the genome (as in the classical approach by Fisher) as genetic markers and opens the door to the study of recent population events (Melé et al. 2010). In fact, at the beginning of modern genetics, Ronald Fisher postulated that beyond the diversity given by the allelic state of genetic variation (a change of one nucleotide in the case of a SNP), the way in which the successive pieces of DNA had been assembled by recombinations could be used as genetic characteristics of a sequence. Nonetheless, this approach has not been perused due to the difficulties of recognizing these junctions. To test this hypothesis, it is necessary to reconstruct the ARG (ancestral recombination graph), which is a graph that tries to reconstruct the gene genealogy of a given amount of linked variants not only by mutation but also by recombination.
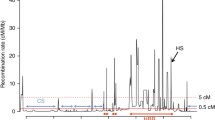
The inference of past recombination events and the reconstruction of the ARG have been addressed, for example, through the IRIS software (Melé et al. 2010). This software uses the patterns of adjacent SNPs created by linkage disequilibrium to infer past recombination events by means of a combinatory as well as statistical algorithm based on pattern-switch recognition. Through this and other methods, it is possible to infer recombination events with the specific position and the parental haplotypes . Recombination events inferred though these methods strongly correlate with recombination rates inferred through methods based on linkage disequilibrium and on sperm typing (Fig. 6.2). The presence or absence of a specific recombination event can be taken as a genetic marker, opening the way to consider the whole set of recombinations in a chromosome as a set of consecutive markers, which have been named “recotypes” that can be analyzed with the same toolkit available for SNPs and haplotypes. The utilization of recombination events as genetic markers is not only useful for human population genetics but also for achieving a deeper understanding of how recombination shapes genomes. Figure 6.3 shows the coalescence with recombination of the extant genetic variation for Africa (in blue) and Out of Africa (Europe and East Asia). The small gray spheres represent coalescent nodes and blue spheres indicate recombinant nodes. Red spheres are the recombinant nodes that have been inferred. In all, it is a quite complete reconstruction of the history of a genome fragment, including its demographic history.

Recombination rates obtained by three different methods: direct observation by sperm typing (top), inferred through linkage disequilibrium using the program LDhat (center), and reconstructing the ancestral recombination graph and, from it, counting the recombinations that have taken place (bottom). Produced for a region of chromosome 1, near MS32 minisatellite, with 365 SNPs and using 120 sequences from HapMap CEU population. A very strong correlation is observed among the methods

A representation of the evolutionary diversification of the human genome since the origin of modern humans. The Out of Africa is clear and both mutations and recombinations have been considered
2 Linkage Disequilibrium and a Population View
We have seen that LD is a population concept that enriches the most used concepts of genome variation. In fact, haplotypes contain more information than SNPs; nonetheless, their use is hampered by the difficulties of defining their length in terms of the number of variant positions they encompass.
Much effort has been concentrated on describing genome variation among human populations based on single nucleotide variants, but very few studies are based on the information that the whole process of recombination can provide. A single genetic distance could be much more informative than just the independent information of each SNP. It is interesting to mention a very efficient study in the analysis of a worldwide survey of high-density SNPs (Jakobsson et al. 2008), in which the haplotype information proved to be more reliable than the independent information given by SNPs. This approach should be further developed and used in population genetics studies.
It is becoming clearer that recombination rates vary among populations, and in fine detail differences (like the presence/absence of recombination hotspots in specific populations) may become a powerful tool for population genetics. As an example, Fig. 6.4, with data extracted from Li et al. (2008), showed that the recombination landscapes vary among populations and continental groups: some hotspots are population specific and others follow a continental pattern, in agreement with the pace of the expansion of modern humans. The wealth of information contained in the recombination footprint in the genome has hardly been used in human population genetics and deserves further attention.

Heatmap showing recombination rates , including recombination hotspots, observed for 258 SNPs of chromosome 22 for 28 human populations, grouped according to their geographical region (AME, Americas; EASIA, Eastern Asia; CSASIA, Central and South Asia; EUR, Europeans; MENA, Middle East and North Africa; SSAFR, sub-Saharan Africa). The 258 SNPs of chromosome 22 are reported on the X axis. In color, for each population, the value of the recombination estimate (4N e r/kb) (corrected for effective population size) for that SNP is shown by a gradient from blue (low recombination values) to green (high recombination values). This figure shows that recombination rates vary not only along the genome (with clear hotspots) but also among continental groups and, to a lesser extent, among populations. Some recombination hotspots are strong in some populations and do not exist in others. This plot indicates the complexity of the distribution of recombination rates and subsequently of linkage disequilibrium and haplotype distribution which are key factors for interpreting the genetic diversity data in rich and robust terms of population history
Differences in recombination rates among human populations provide a useful temporal framework to analyze the evolution of the recombination landscape, which is recent enough to capture fast evolutionary changes. The basal branches of the genetic diversification of human populations happened some 150,000 years ago, a much shorter time than the split between humans and chimpanzees (around 6 million years). The comparison of the recombination patterns among human populations provides a means to verify whether recombination landscapes evolve over time. To address this issue, Laayouni et al. (2011) analyzed whether differences in recombination rates among human populations are correlated with their genetic differences computed as genetic distances . Results show a positive and strong correlation of 0.894 (p < 0.0001) indicating that differences in recombination rates among populations increase with their genetic distance. It is a clear indicator of the pace of evolution of recombination, which in longer time periods (like that of separation of humans and chimpanzees) would give a totally different recombination landscape along the genome.
Recombination rate appears to be a rapidly changing parameter, indicating that the underlying factors shaping the likelihood of a recombination event, such as DNA sequences controlling recombination rate variation, also change. This shows that recombination is not a fixed feature of the genome of a species but a phenotype with broad genetic variation.
3 The Role of Linkage Disequilibrium in Reconstructing Our Past and the Origins of Modern Humans
Traditionally, most inferences in human population genetics have been based on the non-recombining mtDNA and NRY, while the action of recombination in autosomal and X-chromosome data has been considered a hurdle. However, it is possible to study human population diversity using recombination events as genetic markers. In fact, the fragmentation produced by recombinations in the genome produces a mosaic structure of haplotypes rich in information that has yet to be fully used. This analysis focuses on reconstructing the recombination events, using haplotypes instead of SNPs to encompass the genetic diversity, and the direct use of recombination rates and measures of LD .
A paramount accomplishment of genetics has been to provide an incredible source of data from which the demographic history of modern human populations can be deeply investigated. A landmark achievement in this field has been the discovery of the African origin of modern humankind, which was followed by the “Out of Africa” migration, leading to the spread of modern human populations over the whole world. A major piece of evidence of the African origin is that the genetic variability of all non-African populations is reduced and comprised of African populations, seen clearly in mtDNA (Behar et al. 2008) and Y-chromosome (Underhill and Kivisild 2007) studies. Reconstructing the demographic history of African populations before the migration, however, remains a difficult task. Indeed genetic studies of African populations, carried out with different methods (uniparental markers (mtDNA and Y chromosome), microsatellites, SNP genotyping arrays, or whole genome sequences), have revealed a high level of complexity in the structure of African populations, suggesting a complicated intertwining of human groups, some of which have been recognized in their present form, thanks to distinctive forms of leaving and languages. In fact, the complex history of human groups before the “Out of Africa” migration (Tishkoff et al. 2009; Sikora et al. 2011) makes it arduous (or even nearly impossible) to ultimately identify the ancestral genetic pool of modern humans and its region of origin. Genetic data analyzed so far have generally supported the hypothesis that the most ancient population of modern humans originated in eastern Africa, mainly because populations outside of Africa carry a subset of the genetic diversity found in eastern Africa, along with tentative support from the fossil record. The issue of finding the ancestral genetic pool of modern humans and thus localizing their place of origin within the African continent has been readdressed (Henn et al. 2011) and postulates an origin of modern humans in South Africa, with a shared common ancestry of all hunter-gatherer populations distinct from agriculturalists.
Since the pioneering work of Cavalli-Sforza, genetic data have provided formidable evidence to elucidate relationships between extant populations and reconstructed past events of their ancestors, making it possible to infer many aspects of ancient population history such as expansion and migration events, admixture, gene flow, and dramatic changes in population size. Among the tools that have been used, recombination has an interesting role. The key question is to which extent genetic footprints of past historical and demographic events allow us to disentangle the “source” population from those stemming from it at different times during the history of the species. In situations where the evolutionary process is not simply a linear dynamic process of growth, dispersion, and fusion and fission in which the initial characteristics would be much conserved, a founder situation in genetic terms has to reshape the original characteristics, a kind of “zero point” of the process. This can only be created by an offshoot of a small number of individuals and would have two main characteristics: reduction of genetic diversity and creation of linkage disequilibrium. Thus, we should look for, as a proxy for ancestral human populations, those groups having more genetic diversity and less LD. High levels of genetic diversity for a given population may be simply found as a high number of nucleotide variants or a higher heterozygosity for a genome region, though it is something difficult to assess if done through the genotyping of already known variant positions or SNPs and not through retrieving all the variants via DNA sequencing. In the case of genotypes, instead of using direct measures of gene diversity, it is more useful to use the information of genetic distances among populations, as those populations less-derived (and thus, closer to the ancestral one) will have higher differences (measured by the standard Fst statistic, easily understood if the differentiation process is mainly produced by drift) than the rest. In this case it is assumed that the amount of variation in the pre-existing African populations is higher than that produced in non-Africans due to the differentiation process of time and drift, and the data for worldwide surveys confirms this.
But the use of LD enhances our power to understand the past. No doubt the LD differences between Africans and non-Africans (descendants of the Out of Africa ) are dramatic, with strong LD landscape outside Africa and low levels in South Saharan Africa. The African analysis done by Henn et al. (2011) makes an interesting use of LD data: low values of LD reflect higher effective population sizes and, therefore, denote a more ancient population because it is closer to the equilibrium than any derived population. By regressing LD estimates for the different African populations with geographical coordinates, they are able to localize a hypothetical point of origin of modern humans as the region for which this correlation is highest; and this region has been identified as South Africa.
There have been other attempts to use recombination to reconstruct the human past. As the number of past recombination events in a population sample is a function of its effective population size (Ne), it has been possible to detect specific past recombination events in Old World populations to infer their Ne. Results show that sub-Saharan African populations have an Ne that is ~4 times greater than those of non-African populations, and outside of Africa, the South Asian populations had the largest Ne. It has also been possible to use this information to reconstruct the “Out of Africa” route into Eurasia. Observing how the patterns of recombination diversity of the Eurasian populations correlates with distance from Africa, which shows a clearly significant correlation with that distance measured along a path crossing South Arabia, while no such correlation is found through the Sinai route (usually taken as the route of the Out of Africa), suggesting that anatomically modern humans first left Africa through the Bab-el-Mandeb Strait rather than through present Egypt (Melé et al. 2012).
The full use of recombination and linkage disequilibrium data in population genetics is still in its infancy, and many more possibilities are waiting to be uncovered and fully used. Its full development requires good algorithms for detection of recombination points, a close approach to the ancestral recombination graph , and a high-resolution description of the genome, something that is being achieved with the third generation of sequencing technologies that allow obtaining long DNA sequences (and haploid sequences ) of single molecules. Thus, not only the genetic variation is of interest but also how that variation is organized along the chromosomes, the departure point of making diploid individuals and population genome pools.
References
Baudat F, Imai Y, de Massy B (2013) Meiotic recombination in mammals: localization and regulation. Nat Rev Genet 14(11):794–806
Behar DM, Villems R, Soodyall H, Blue-Smith J, Pereira L, Metspalu E, Scozzari R, Makkan H, Tzur S, Comas D, Bertranpetit J, Quintana-Murci L, Tyler-Smith C, Wells RS, Rosset S, Genographic Consortium (2008) The dawn of human matrilineal diversity. Am J Hum Genet 82(5):1130–1140
Henn BM, Gignoux CR, Jobin M, Granka JM, Macpherson JM, Kidd JM, Rodríguez-Botigué L, Ramachandran S, Hon L, Brisbin A, Lin AA, Underhill PA, Comas D, Kidd KK, Norman PJ, Parham P, Bustamante CD, Mountain JL, Feldman MW (2011) Hunter-gatherer genomic diversity suggests a Southern African origin for modern humans. Proc Natl Acad Sci U S A 108(13):5154–5162
Jakobsson M et al (2008) Genotype, haplotype and copy-number variation in worldwide human populations. Nature 451(7181):998–1003
Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, Jonasdottir A, Walters GB, Jonasdottir A, Gylfason A, Kristinsson KT, Gudjonsson SA, Frigge ML, Helgason A, Thorsteinsdottir U, Stefansson K (2010) Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467(7319):1099–1103
Laayouni H, Montanucci L, Sikora M, Melé M, Dall’Olio GM, Lorente-Galdos B, McGee KM, Graffelman J, Awadalla P, Bosch E, Comas D, Navarro A, Calafell F, Casals F, Bertranpetit J (2011) Similarity in recombination rate estimates highly correlates with genetic differentiation in humans. PlosOne 6(3):e17913
Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, Ramachandran S, Cann HM, Barsh GS, Feldman M, Cavalli-Sforza LL, Myers RM (2008) Worldwide human relationships inferred from genome-wide patterns of variation. Science 319:1100–1104
Melé M, Javed A, Pybus M, Calafell F, Parida L, Bertranpetit J, Genographic Consortium (2010) A new method to reconstruct recombination events at a genomic scale. PLoS Comput Biol 6(11):e1001010
Melé M, Javed A, Pybus M, Zalloua P, Haber M, Comas D, Netea MG, Balanovsky O, Balanovska E, Jin L, Yang Y, Pitchappan RM, Arunkumar G, Parida L, Calafell F, Bertranpetit J, Genographic Consortium (2012) Recombination gives a new insight in the effective population size and the history of the old world human populations. Mol Biol Evol 29(1):25–30
Paigen K, Petkov P (2010) Mammalian recombination hot spots: properties, control and evolution. Nat Rev Genet 11(3):221–233
Sikora M et al (2011) A genomic analysis identifies a novel component in the genetic structure of sub-Saharan African populations. Eur J Hum Genet 19(1):84–88
Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, Hirbo JB, Awomoyi AA, Bodo JM, Doumbo O, Ibrahim M, Juma AT, Kotze MJ, Lema G, Moore JH, Mortensen H, Nyambo TB, Omar SA, Powell K, Pretorius GS, Smith MW, Thera MA, Wambebe C, Weber JL, Williams SM (2009) The genetic structure and history of Africans and African Americans. Science 324(5930):1035–1044
Underhill PA, Kivisild T (2007) Use of y chromosome and mitochondrial DNA population structure in tracing human migrations. Annu Rev Genet 41:539–536
Acknowledgments
Ongoing work on evolutionary genetics is supported by grant BFU2016-77961-P (AEI/FEDER, UE) awarded by the Ministerio de Economía y Competitividad (Spain) and with the support of the Secretaria d’Universitats i Recerca del Departament d’Economia i Coneixement de la Generalitat de Catalunya (GRC 2014 SGR 866).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Japan KK
About this chapter

Cite this chapter
Montanucci, L., Bertranpetit, J. (2017). Recombination. In: Saitou, N. (eds) Evolution of the Human Genome I. Evolutionary Studies. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56603-8_6
Download citation
DOI: https://doi.org/10.1007/978-4-431-56603-8_6
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56601-4
Online ISBN: 978-4-431-56603-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)