
Abstract
Cone dysfunction syndromes (CDS) are a heterogeneous group of hereditary retinal disorders characterized by impaired visual acuity, defective color vision, nystagmus, and photophobia, which occur at birth or during early infancy. In contrast to the cone or cone-rod dystrophies, CDS are classified as stationary disorders, and which include congenital achromatopsia (ACHM, also known as rod monochromacy) with an autosomal recessive inheritance pattern. A total of six genes (CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, and ATF6) have been identified as causes of ACHM. However, only the mutations CNGA3, CNGB3, and PDE6C have been identified in the few cases of achromatopsia that have been reported in Japanese patients. Thus, this chapter describes the detailed clinical and molecular genetic findings for ACHM in Japanese patients.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
12.1 Introduction
Cone dysfunction syndromes (CDS) are a heterogeneous group of hereditary retinal disorders characterized by impaired visual acuity, defective color vision, nystagmus, and photophobia which occur at birth or early infancy [1, 2]. In contrast to cone or cone-rod dystrophies, CDS are classified as stationary disorders, which include congenital achromatopsia (ACHM, also known as rod monochromacy) and blue-cone monochromatism (BCM) [1, 2]. To date, the six genes that have been identified as causes of ACHM include cyclic nucleotide-gated channel alpha-3 (CNGA3) [3], cyclic nucleotide-gated channel beta-3 (CNGB3) [4, 5], guanine nucleotide-binding protein, alpha-transducing activity polypeptide 2 (GNAT2) [6, 7], phosphodiesterase 6C (PDE6C) [8], phosphodiesterase 6H (PDE6H) [9], and activating transcription factor 6 (ATF6) [10, 11]. This chapter describes genotype-phenotype associations found in ACHM, especially in Japanese patients.
12.3 Clinical Features
ACHM is a congenital and basically stationary disorder characterized by low visual acuity, pendular nystagmus, photophobia, and severe color vision defects. Although the fundus appearance is usually normal, macular pigmentary changes and macular atrophy have been described. Visual field testing shows a small central scotoma in most patients. Full-field electroretinography (ERG) shows normal rod responses but severely reduced cone responses. Spectral-domain optical coherence tomography (SD-OCT) can detect an abnormal structure of the outer retinal layer including disruption of the inner segment ellipsoid line.
12.4 Molecular Genetic Aspects
To date, the six genes identified as the causes of ACHM include CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, and ATF6. The proteins encoded by five of these genes (CNGA3, CNGB3, GNAT2, PDE6C, PDE6H) are exclusively expressed in the cone photoreceptors, where they are involved in the cone phototransduction cascade. A previous study found that CNGB3 mutations account for half of the studied ACHM cases (48.2%), while CNGA3 mutations account for 28.7%, GNAT2 mutations account for 2.2%, and PDE6C mutations account for only 1.4% [13]. On the other hand, other studies [8, 14, 15] have reported that mutations in CNGB3 account for 87% of the ACHM cases, with CNGA3 accounting for 5%, and PDE6C for less than 1%. However, there were no mutations found for GNAT2. Out of the few cases of achromatopsia reported in Japan, only the CNGA3, CNGB3, and PDE6C mutations were identified in these Japanese patients [16–18]. Therefore, the mutation spectrum of ACHM in the Japanese population remains unknown.
12.5 Clinical Aspects (Case Studies)
This study examined two Japanese families with ACHM in whom the pathogenic mutations were identified.
12.6 Family 1
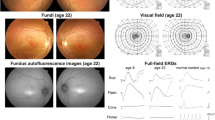
A 22-year-old female patient was referred for evaluation of photophobia and visual loss in both eyes [17]. Childhood history included nystagmus and low visual acuity. Decimal best-corrected visual acuity (BCVA) was 0.1 in both eyes, with refractions of −2.75 cylinder 10 deg. in the right eye and +1.25 sphere, −3.00 cylinder 270 deg. in the left eye. Ophthalmoscopy showed no specific findings in the optic disks or maculae (Fig. 12.1). Time-domain OCT assessment by retinal mapping disclosed a 20% thinner parafoveal thickness compared to a normal individual, even though the foveal thickness was normal (Fig. 12.1). Goldmann visual field tests showed bilateral 5-deg. central scotomas with the I-2e isopters, while the visual fields with the V-4e, I-4e, I-3e, I-2e, and I-2c targets were within normal limits in both eyes. This patient was only able to identify the first plate in the Ishihara test. The Farnsworth Panel D-15 showed there was confusion close to the scotopic axis. Although full-field ERG showed normal responses in the rod and maximal (mixed rod-plus-cone) responses, there was an extreme reduction in the cone and 30-Hz flicker responses (Fig. 12.2). Spectral sensitivity on a white background was measured in the patient and in two individuals who exhibited normal color vision using a three-channel Maxwellian view optical system as previously described [19]. The spectral sensitivity measurements for the two individuals with normal color vision showed curves with three peaks that consisted of short wavelength-sensitive (S), middle wavelength-sensitive (M), and long wavelength-sensitive (L) cones. However, the patient’s curve revealed only one peak at around 500 nm (Fig. 12.3), which was comparable to the spectral luminous efficiency function V′(λ) for scotopic vision that fits the absorption spectrum of human rhodopsin. The patient’s parents and sister reported no ocular complaints.

Ophthalmoscopy in the patient of Family 1 found no specific findings in the optic disks or the maculae (a). Time-domain OCT assessment by retinal mapping disclosed a 20% thinner parafoveal thickness compared to a normal individual, even though the foveal thickness was normal (b)

Full-field electroretinography in the patient of Family 1 demonstrated there were normal responses for the rod and maximal (mixed rod-plus-cone) responses, but extremely reduced responses for the cone and 30-Hz flicker responses

Spectral sensitivity on a white background was measured in the patient of Family 1 and in two individuals with normal color vision using a three-channel Maxwellian view optical system. The two individuals with normal color vision showed curves with three peaks that consisted of short wavelength-sensitive (S), middle wavelength-sensitive (M), and long wavelength-sensitive (L) cones. However, the patient’s curve revealed only one peak at around 500 nm, which is comparable to the spectral luminous efficiency function V′(λ) for scotopic vision that fits the absorption spectrum of human rhodopsin
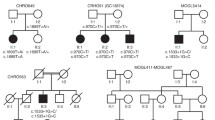
The mutation analysis using Sanger sequencing identified compound heterozygous CNGA3 mutations (c.1306C>T; p. R436W, c.1898T>C; p. L633P) in the patient (Fig. 12.4) [17]. In the familial analysis, her father and sister carried the p. R436W mutation, while her mother carried the p. L633P mutation. The L633 is not only a phylogenetically conserved amino acid residue among mammalian orthologs, but it is also one of the most important hydrophobic residues in the CLZ domain downstream of the cGMP-binding site.

Mutation analysis performed for the patient of Family 1 detected compound heterozygous CNGA3 mutations (c.1306C>T; p. R436W, c.1898T>C; p. L633P)
12.7 Family 2
A 7-year-old sister (patient 1) and her 5-year-old brother (patient 2) were referred to our department because of poor visual acuity [19]. Their parents had a consanguineous marriage. Since infancy, both patients have had hemeralopia, photophobia, and pendular nystagmus. At the time of their first visit to our hospital, the BCVA was OD, 0.1, and OS, 0.1; refractions were OD, +3.00, 0.5 X 180, and OS, +2.50, −0.75 X 180 (patient 1), and +7.00, −0.5 X 180 in both eyes (patient 2). Goldmann visual field testing that was done several years after their first visit showed there similar findings between the patients. Both patients 1 and 2 had bilateral relative central (5 deg) scotomas of the I-3c and I-2e isopters, respectively, while the peripheral visual fields with V-4e, III-4e, and I-4e isopters were normal. Both patients were only able to recognize the first plate in the Ishihara color vision test. The Farnsworth Panel D-15 indicated that both patients exhibited confusion close to a scotopic axis. The Nagel model I anomaloscope examination showed color matches along the rod line of the Rayleigh equation. Ophthalmoscopy showed that while patient 1 had atrophic-appearing macular lesions, patient 2 had no specific retinal findings. Full-field ERG showed normal rod and maximal responses, but there was an absent 30-Hz flicker response in both eyes. On the other hand, the cone ERGs differed, with the responses absent in patient 1, while the amplitude of the a- and b-waves were reduced in patient 2. Therefore, patients 1 and 2 were diagnosed with complete and incomplete ACHM, respectively. Subsequently, both patients have undergone longitudinal follow-ups.
At the age of 30, the BCVA for patient 1 was 0.2 (with moderate myopia) in her right eye and 0.1 (with moderate myopia) in her left eye. Funduscopy showed atrophic chorioretinal macular scarring in the right eye and macular atrophy in the left eye. FAI revealed complete loss of autofluorescence in the macular lesions of both eyes. SD-OCT revealed severe macular thinning of all layers in both eyes and retinal and choroidal excavation in the right eye. Visual field testing showed there were bilateral central scotomas (5–10 deg) of the I-3e or I-2e isopters, even though the peripheral visual fields of the I-5e and I-4e isopters were normal. There was a slight broadening of the central scotomas relative to that seen at 11 years of age. At the age of 26, the BCVA for patient 2 was 0.2 (with high hyperopia) in each eye. Fundus autofluorescence imaging revealed hyper-autofluorescent areas of the maculae in both eyes. SD-OCT revealed retinal thinning with loss of the outer retinal layer in each macula (Fig. 12.5).

Spectral-domain optical coherence tomography performed in patient 2 of Family 2 revealed retinal thinning with a loss of the outer retinal layer in each macula
In order to identify disease-causing variants in the mutation analysis, whole-exome sequencing was performed in each of the family members (patient 1, patient 2, and their parents). In each patient, we identified a novel homozygous mutation (c.1771G>A, p. E591K) in exon 14 of the PDE6C gene (Fig. 12.6) [18]. There were no pathogenic variants detected in the CNGA3, CNGB3, GNAT2, PDE6H, or ATF6 genes. Interestingly, there was one known disease-causing mutation (p. G79R) in OPN1SW, which is associated with congenital tritan color vision deficiencies. The father and patient 1 each had the p. G79R mutation heterozygously, while the mother and patient 2 had neither mutation [18]. Since spectral ERG indicated that the father exhibited blue-yellow color vision deficiencies and no S-cone response, he was diagnosed with congenital tritan deficiencies [18]. The different phenotypes (complete and incomplete ACHM) between the patients might be explained by a “direct effect” or “possible modifier effect” related to the OPN1SW mutation (p. G79R) that was found in patient 1, who had complete ACHM.

A novel homozygous mutation (c.1771G>A, p. E591K) was identified in exon 14 of the PDE6C gene in patients 1 and 2 of Family 2
12.8 Summary and Perspective
This chapter summarized the detailed clinical and molecular genetic findings for Japanese ACHM patients [17, 18]. However, there have been only three reports in which mutations (CNGA3, CNGB3, and PDE6C) were identified in Japanese ACHM patients [16–18]. Unlike ACHM patients of European descent, the mutation spectrum has yet to be clarified in the Japanese population. Therefore, a large cohort that investigates the genotype-phenotype correlations in ACHM in the Japanese population will need to be undertaken in the future.
References
Michaelides M, Hunt DM, Moore AT. The cone dysfunction syndromes. Br J Ophthalmol. 2004;88:291–7.
Aboshiha J, Dubis AM, Carroll J, Hardcastle AJ, Michaelides M. The cone dysfunction syndromes. Br J Ophthalmol. 2015; doi:10.1136/bjophthalmol-2014-306505.
Kohl S, Marx T, Giddings I, Jagle H, Jacobson SG, Apfelstedt-Sylla E, Zrenner E, Sharpe LT, Wissinger B. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet. 1998;19:257–9.
Sundin OH, Yang JM, Li Y, Zhu D, Hurd JN, Mitchell TN, Silva ED, Maumenee IH. Genetic basis of total colourblindness among the Pingelapese islanders. Nat Genet. 2000;25:289–93.
Kohl S, Baumann B, Broghammer M, Jagle H, Sieving P, Kellner U, Spegal R, Anastasi M, Zrenner E, Sharpe LT, Wissinger B. Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet. 2000;9:2107–16.
Kohl S, Baumann B, Rosenberg T, Kellner U, Lorenz B, Vadala M, Jacobson SG, Wissinger B. Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia. Am J Hum Genet. 2002;71:422–5.
Aligianis IA, Forshew T, Johnson S, Michaelides M, Johnson CA, Trembath RC, Hunt DM, Moore AT, Maher ER. Mapping of a novel locus for achromatopsia (ACHM4) to 1p and identification of a germline mutation in the alpha subunit of cone transducin (GNAT2). J Med Genet. 2002;39:656–60.
Thiadens AA, den Hollander AI, Roosing S, Nabuurs SB, Zekveld-Vroon RC, Collin RW, De Baere E, Koenekoop RK, van Schooneveld MJ, Strom TM, van Lith-Verhoeven JJ, Lotery AJ, van Moll-Ramirez N, Leroy BP, van den Born LI, Hoyng CB, Cremers FP, Klaver CC. Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am J Hum Genet. 2009;85:240–7.
Kohl S, Coppieters F, Meire F, Schaich S, Roosing S, Brennenstuhl C, Bolz S, van Genderen MM, Riemslag FC, Lukowski R, den Hollander AI, Cremers FP, De Baere E, Hoyng CB, Wissinger B. A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am J Hum Genet. 2012;91:527–32.
Kohl S, Zobor D, Chiang WC, Weisschuh N, Staller J, Menendez IG, Chang S, Beck SC, Garcia Garrido M, Sothilingam V, Seeliger MW, Stanzial F, Benedicenti F, Inzana F, Heon E, Vincent A, Beis J, Strom TM, Rudolph G, Roosing S, Hollander AI, Cremers FP, Lopez I, Ren H, Moore AT, Webster AR, Michaelides M, Koenekoop RK, Zrenner E, Kaufman RJ, Tsang SH, Wissinger B, Lin JH. Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat Genet. 2015;47:757–65.
Ansar M, Santos-Cortez RL, Saqib MA, Zulfiqar F, Lee K, Ashraf NM, Ullah E, Wang X, Sajid S, Khan FS, Amin-Ud-Din M, University of Washington Center for Mendelian Genomics, Smith JD, Shendure J, Bamshad MJ, Nickerson DA, Hameed A, Riazuddin S, Ahmed ZM, Ahmad W, Leal SM. Mutation of ATF6 causes autosomal recessive achromatopsia. Hum Genet. 2015;134:941–50.
Kohl S, Hamel C. Clinical utility gene card for: achromatopsia – update 2013. Eur J Hum Genet. 2013; doi:10.1038/ejhg.2013.44.
Grau T, Artemyev NO, Rosenberg T, Dollfus H, Haugen OH, Cumhur Sener E, Jurklies B, Andreasson S, Kernstock C, Larsen M, Zrenner E, Wissinger B, Kohl S. Decreased catalytic activity and altered activation properties of PDE6C mutants associated with autosomal recessive achromatopsia. Hum Mol Genet. 2011;20:719–30.
Thiadens AA, Slingerland NW, Roosing S, van Schooneveld MJ, van Lith-Verhoeven JJ, van Moll-Ramirez N, van den Born LI, Hoyng CB, Cremers FP, Klaver CC. Genetic etiology and clinical consequences of complete and incomplete achromatopsia. Ophthalmology. 2009;116(1984–1989):e1981.
Thiadens AA, Roosing S, Collin RW, van Moll-Ramirez N, van Lith-Verhoeven JJ, van Schooneveld MJ, den Hollander AI, van den Born LI, Hoyng CB, Cremers FP, Klaver CC. Comprehensive analysis of the achromatopsia genes CNGA3 and CNGB3 in progressive cone dystrophy. Ophthalmology. 2010;117(825–830):e821.
Okada A, Ueyama H, Toyoda F, Oda S, Ding WG, Tanabe S, Yamade S, Matsuura H, Ohkubo I, Kani K. Functional role of hCngb3 in regulation of human cone cng channel: effect of rod monochromacy-associated mutations in hCNGB3 on channel function. Invest Ophthalmol Vis Sci. 2004;45:2324–32.
Goto-Omoto S, Hayashi T, Gekka T, Kubo A, Takeuchi T, Kitahara K. Compound heterozygous CNGA3 mutations (R436W, L633P) in a Japanese patient with congenital achromatopsia. Vis Neurosci. 2006;23:395–402.
Katagiri S, Hayashi T, Yoshitake K, Sergeev Y, Akahori M, Furuno M, Nishino J, Ikeo K, Tsunoda K, Tsuneoka H, Iwata T. Congenital achromatopsia and macular atrophy caused by a novel recessive PDE6C mutation (p.E591K). Ophthalmic Genet. 2015;36:137–44.
Hayashi T, Kozaki K, Kitahara K, Kubo A, Nishio Y, Omoto S, Nakamura Y, Watanabe A, Toda K, Ueoka Y. Clinical heterogeneity between two Japanese siblings with congenital achromatopsia. Vis Neurosci. 2004;21:413–20.
Acknowledgments
This study was supported by grants from the Ministry of Health, Labour and Welfare of Japan and the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grant-in-Aid for Scientific Research (C) 25462738).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Japan KK
About this chapter
Cite this chapter
Hayashi, T. (2017). Cone Dysfunction Syndrome in the Japanese Population. In: Prakash, G., Iwata, T. (eds) Advances in Vision Research, Volume I. Essentials in Ophthalmology. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56511-6_12
Download citation
DOI: https://doi.org/10.1007/978-4-431-56511-6_12
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56509-3
Online ISBN: 978-4-431-56511-6
eBook Packages: MedicineMedicine (R0)