
Abstract
The hepatotropic hepatitis C virus (HCV) belongs to the Flaviviridae family and chronically infects 130–150 million people worldwide. The severe consequences the virus has for liver health, especially if left untreated, and the lack of a vaccine continue to make HCV a relevant global health problem. A considerable challenge in studying HCV is the virus’ host tropism, which is limited almost exclusively to humans and chimpanzees. The lack of suitable and ethical animal model systems has hindered our abilities to mechanistically decipher interactions of HCV with its mammalian host and to develop vaccines. However, encouraging advances, especially in the refinement of humanized mouse models, have created new opportunities for studying HCV pathogenesis and host antiviral responses in vivo. Additionally, the discovery of hepaciviruses in other organisms and advances in induced pluripotent stem cell technologies have created further avenues for exploration. The ultimate goal is to develop tractable small animal models for HCV, which optimally recapitulate all parts of the viral life cycle and present with clinically relevant manifestations of viral hepatitis. Such new models would undoubtedly shed light on both the biology and clinical consequences of chronic hepatitis C infection.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
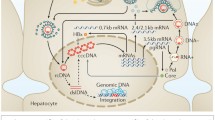
Hepatitis C virus (HCV) is an enveloped, positive-sense, single-stranded RNA virus belonging to the genus Hepacivirus in the Flaviviridae family. While approximately 150 million people are infected with HCV worldwide, this is likely an underestimate as almost twice as many individuals in the United States may carry the virus, many unknowingly (Edlin 2011). HCV causes persistent infection in 70–80 % of those who become exposed to the virus. While the acute disease is usually asymptomatic, chronic carriers left untreated frequently develop fibrosis, cirrhosis and, in some cases, hepatocellular carcinoma. Treatment for HCV has evolved rapidly in recent years and it is now possible to cure the majority of patients with largely well-tolerated therapies that include a combination of pegylated interferon (IFN)-α, ribavirin and direct acting antiviral (DAA) drugs. However, despite their potency, it remains to be seen whether even the newest DAAs will drastically reduce the global burden of disease due to the high associated costs, logistical challenges of mass deployment and risk of drug resistance. A vaccine, which would prevent infection or delay the onset of pathogenesis during a chronic infection, does not exist. Development of effective therapies has been delayed by the lack of both suitable cell culture systems and animal models. While hepaciviruses similar to HCV have been found in a variety of species, including dogs, horses and outbred mice, HCV appears to have a much more limited host range. Robust infection has only been described in humans and experimentally infected chimpanzees, but some studies have provided evidence for transient and intermittent viremia in a more exotic mammal, tree shrews. The narrow host range of HCV is not completely understood but can in part be explained by differences between species in the sequences of essential host factors at the level of entry as well as in innate antiviral responses. This growing understanding of the barriers to interspecies transmission has aided the development of inbred models with inheritable susceptibility to HCV. As will be discussed here, genetic host adaptation has been and continues to be part of a multipronged approach to develop more tractable animal models for studying HCV infection, immunity and pathogenesis (Fig. 1).

Host adaptation and viral adaptation approaches to create new animal models for the study of hepatitis C. Host adaptation through transplantation of human hepatocytes to create HEAL mice or human liver chimeric mice and/or hematopoietic stem cells (HSCs) to (co-) engraft components of a human immune system (left column). Genetic humanization can be accomplished by identification and expression of human-specific factors or by ablation of restriction factors (middle column). Cell culture passaging strategies are used to adapt HCV to murine or simian hosts (right column). iPS Induced pluripotent stem cells, ES embryonic stem cells, HSC hematopoietic stem cells, HEAL human ectopic artificial liver
2 Hepatitis C Virus Infection in Non-Human Primates
For many years, studies of hepatitis C were limited to experimentally infected chimpanzees or patient volunteers. While chimpanzees have been instrumental in analyzing HCV infection (reviewed in (Bukh 2004), Fig. 2), studies in this species are challenging due to high costs, genetic heterogeneity, small cohort sizes, limited access to relevant tissue compartments, the inability to genetically manipulate large apes and growing ethical concerns.

Phylogenetic relationship of members of the hepacivirus genus and susceptible host species. Phylogenetic tree of the hepaciviruses is adapted from (Pfaender et al. 2014a) and (Firth et al. 2014) and is based on the nucleotide sequence analysis of the NS3 protease domain and the complete NS5B gene. GHV Guereza hepacivirus, GBV-B George Barker virus B, NPHV non-primate hepacivirus, CHV canine hepacivirus, NrHV Norway rat hepacivirus, RHV rodent hepacivirus, BHV bat hepacivirus
The other non-human primate (NHP) species tested for susceptibility to HCV infection – including cynomolgus monkeys, rhesus monkeys, Japanese monkeys, Green monkeys, Doguera (Abe et al. 1993) and Chacma baboons (Sithebe et al. 2002), cottontop tamarins (Garson et al. 1997) and marmosets – do not seem to support infection. The blocks in HCV transmission in these and potentially other NHP species are not well defined and are likely due to a combination of factors. For example, simian orthologs of essential host factors required for the viral life cycle may be absent or incompatible. Similarly, dominant restriction factors, as observed with HIV, may actively antagonize uptake, replication and/or viral assembly and release. In addition, differences in the kinetics and magnitude of antiviral defenses in many cells may interfere with viral RNA replication. This may be a result of less efficient viral evasion mechanisms that usually enable HCV to establish persistent infection in human cells. For example, in various primate species, differences in the amino acid sequence of the mitochondrial antiviral signal protein (MAVS, also known as IPS1, VISA or Cardif) prevent its proteolytic cleavage by the HCV NS3/4A protease, leaving host antiviral signaling intact (Patel et al. 2012). These incompatibilities may also result in more effective cellular and humoral immune responses in NHPs, contributing to clearance of HCV.
However, these blocks do not appear absolute as stem cell-derived hepatocyte-like cells from pig-tailed macaques (Macaca nemestrina) (Sourisseau et al. 2013) and primary rhesus macaque (Macaca mulatta) hepatocytes can support the entire HCV life cycle (Scull et al. 2015). Additionally, HCV RNA replication in rhesus macaque hepatocytes is enhanced upon blunting antiviral immunity, which is consistent with the observation that the rhesus MAVS ortholog is not cleaved by HCV NS3/4A (Scull et al. 2015). Nonetheless, HCV can establish persistent replication in simianized mice, i.e. immunocompromised xenorecipients engrafted with rhesus hepatocytes (Scull et al. 2015). Yet it still remains to be shown whether rhesus or pig-tailed macaques are actually susceptible to HCV in vivo and if viral persistence can be achieved.
Tree shrews (Tupaia belangeri) – once designated as small, squirrel-like primates but now classified in the separate order Scandentia – have been shown to support intermittent, transient viremia, becoming more permissive to HCV infection when immunosuppressed (Xie et al. 1998). In follow-up studies, acute infection did progress to persistent viremia (Xu et al. 2007; Amako et al. 2010), resulting in clinically symptomatic liver disease, including steatosis, fibrosis and cirrhosis after 3 years (Amako et al. 2010). While these more recent data are promising and may enable studies of HCV immunity and pathogenesis, there are still limitations to their utility as tree shrews are an outbred, genetically diverse organism and few reagents are available for investigating their immune response to viral infection.
3 Potential Surrogate Models: Non-Primate Hepaciviruses
With such a limited host tropism, other closely related viruses have been considered as a proxy for studying HCV. The best characterized of these viruses, GB viruses, named after the surgeon George Barker (Deinhardt et al. 1967), have been used in NHP studies. GB virus B was able to cause hepatitis in marmosets (Callithrix jacchus) (Simons et al. 1995; Lanford et al. 2003) (Fig. 2) as well as other New World monkeys, including tamarins (Saguinus spp.) (Karayiannis et al. 1989; Schaluder et al. 1995) and owl monkeys (Aotus trivirgatus) (Bukh et al. 2001). GBV-B belongs to the genus Hepacivirus in the Flaviviridae family and has the same overall genome organization as HCV. However, the polyproteins of HCV and GBV-B share only 28 % amino acid identity and differ even more in their 5′ and 3′ non-coding regions. The discovery of a GBV-B-like virus, called guereza hepacivirus (GHV) after the colobus species it was identified in, is the first hepacivirus found in a wild NHP and has led to further questions concerning the evolution of hepaciviruses (Lauck et al. 2013).
Other related viruses of either the Pegivirus genus, also a part of the Flaviviridae family, or Hepacivirus genus have been identified in dogs (Kapoor et al. 2011), horses (Burbelo et al. 2012; Kapoor et al. 2013a), wild mice (Kapoor et al. 2013b), bats(Quan et al. 2013) and rats (Firth et al. 2014). Of the non-primate hepaciviruses (NPHV), those observed in horses are the most genetically similar to HCV (Pfaender et al. 2014b) (Fig. 2). However, it remains to be shown whether these viruses indeed cause hepatitis in experimentally inoculated animals before it can be determined whether they might be surrogates for modeling HCV. Importantly, there is currently no experimental evidence of NPHV transmission between horses and humans (Pfaender et al. 2015).
4 Rodent Models
Mice are widely used in biomedical research, with many existing analytical tools for dissecting their responses to infection. Furthermore, mice of genetically defined backgrounds are available which are amenable to genetic manipulations. In the following sections, we will summarize the previously established rodent models and discuss some of the recent developments that have been explored to model HCV infection and pathogenesis in rodents.
4.1 HCV Transgenic Mice
HCV does not readily infect mice, and thus early attempts to model aspects of HCV pathogenesis in mice were performed by expressing individual or multiple HCV gene products (Table 1 and reviewed in Kremsdorf and Brezillon (2007)). However, depending on the mouse background, the HCV gene product(s) expressed, and the promoter driving expression of these proteins, the histopathological features observed in these mice differed considerably. When HCV core was expressed under the control of a hepatitis B virus (HBV) promoter, animals developed severe liver disease, culminating in hepatocarcinogenesis (Moriya et al. 1997, 1998). In contrast, driving HCV core and/or E1/E2 expression with a major urinary protein (MUP) or CMV promoter produced a less pronounced and more variable disease phenotype (Pasquinelli et al. 1997; Chiyo et al. 2011; Satoh et al. 2010; Naas et al. 2005; Benali-Furet et al. 2005; Chang et al. 2008, 2009; Lerat et al. 2009; Tanaka et al. 2008; Kamegaya et al. 2005; Jeannot et al. 2012). Likewise, NS5A expression was directly cytopathic in some transgenic lines (Wang et al. 2009), but, when under the control of an apoE or MUP promoter, liver pathologies were not observed (Majumder et al. 2003). Similarly, expression of the HCV serine protease NS3/NS4A or NS4B in mouse models has not been shown to induce liver injury (Desai et al. 2011; Frelin et al. 2006; Wang et al. 2006).
In addition to modeling aspects of HCV-induced liver disease, expression of HCV proteins has been utilized for studying HCV-specific adaptive immune responses in the liver (Disson et al. 2004; Alonzi et al. 2004; Tsukiyama-Kohara et al. 2011; Wegert et al. 2009; Tumurbaatar et al. 2007; Ernst et al. 2007; Furutani et al. 2006; Takaku et al. 2003; Naas et al. 2010; Kriegs et al. 2009; Kanda et al. 2009). Pre-natal expression of HCV proteins in mice causes the murine immune system to become tolerized to viral gene products, but this tolerance can be disrupted via DNA vaccination, which primes CD8 T cells to target hepatocytes expressing HCV NS3/4A (Ahlen et al. 2009). This model lends itself to testing T cell based vaccine candidates.
Undoubtedly, HCV transgenic mice have helped analyze HCV immune responses and viral pathogenesis, but a number of factors still diminish their utility. Transgene copy numbers, and consequently levels and distribution of HCV protein expression, can vary considerably due to random integration in the mouse genome. When driven by strong viral or cellular promoters, expression of HCV gene products can surpass the levels of viral proteins that would be reached by actual infection. Furthermore, interpreting data acquired in transgenic mice is further complicated since HCV proteins are being expressed outside of the inflammatory context of acute and chronic viral infection.
4.2 Xenotransplantation Models
To overcome some of the challenges of HCV transgenic mice, xenotransplantation models have been established in which the murine host is rendered susceptible to HCV infection by xenoengraftment of permissive human cells in the mouse liver (Fig. 1). As described below, a variety of approaches have been taken to accomplish this goal.
4.2.1 Engraftment of Human Hepatoma Cells In Vivo
The simplest mouse models of xenoengraftment are made by intrahepatic injection of human hepatoma cells. To quantify RNA replication and responses to antiviral treatment such as interferon-α (IFN-α) in vivo, Huh7 cells containing an HCV replicon expressing luciferase have been injected into severe combined immunodeficient (SCID)/beige mice and analyzed by whole-body bioluminescence imaging (Zhu et al. 2006). This system is simple with minimal intra- and interexperimental variation but is not a bona fide infection model.
To actually enable the study of anti-HCV immune responses, immunocompetent fetal rats have been tolerized in utero to Huh7 cells and, after birth, transplanted with a larger number of the same cells (Wu et al. 2005). Remarkably, Huh7 cells, which are usually not readily susceptible to HCV infection in vitro, supported viremia of a patient-derived genotype 1a isolate. However, the low levels of observed viremia, complex nature of these experiments, and potential inability of rat T cells to recognize HCV antigens due to the presence of human leukocyte antigen (HLA) on the transplanted Huh7 cells make this model less than ideal.
4.2.2 Ectopic Liver Implantation Models
While it is more desirable to engraft hepatocytes instead of hepatoma cells in the human parenchyma, this is not readily accomplished with primary human hepatocytes. However, pieces of human liver and even artificial human liver organoids have been successfully implanted in ectopic sites. In the so-called “Trimera mouse” (Ilan et al. 2002), small pieces of human liver were maintained under the kidney capsule or the ear of SCID mice. When the liver tissue was taken from HCV positive donors or naïve tissue was infected with HCV prior to transplantation, viremia was maintained for several weeks. This model has been subsequently used to assess the efficacy of neutralizing antibodies (Eren et al. 2006), but the technically and logistically challenging experimental set-up, fairly rapid graft failure and low levels of HCV viremia have hampered the utility of the model.
To overcome the need for primary liver tissue, bioengineering approaches have been undertaken to reconstruct increasingly more complex tissue organoids suitable for transplantation. Human ectopic artificial livers (HEALs) have been created where cryopreserved primary human hepatocytes are supported by polymeric scaffolds, which aid maintenance of the microenvironment and thus stablize these cells. While simpler, polyethylene glycol (PEG)-based polymers were used initially (Chen et al. 2011), newer models allow for even greater control of the scaffold architecture, improving vascularization and, consequently, hepatocyte survival (Miller et al. 2012; Stevens et al. 2013). In mice engrafted intraperitoneally with HEALs (Fig. 1), humanized liver functions could be monitored for several weeks but susceptibility to hepatotropic pathogens, including HCV, has yet to be shown.
4.2.3 Human Liver Chimeric Mice
The most commonly used and best characterized humanized xenotransplantation models for HCV are human liver chimeric mice (Fig. 1). Suitable xenorecipient strains are immunodeficient to avoid graft rejection and also have endogenous liver injury to both promote hepatocyte proliferation and give the donor hepatocytes a growth advantage over the mouse hepatocytes. Donor cells, including hepatoma cell lines, primary hepatocytes and, more recently, stem-cell derived hepatocytes, are injected intrasplenically. Traveling via the portal venous system, the donor cells pass through the liver sinusoidal endothelial cells and form clusters that expand upon induction of liver injury. This can be done via partial hepatectomy or treatment with hepatotoxic chemicals, like retrorsine and carbon tetrachloride. Genetic approaches have also been utilized as they allow for more control over the severity of the liver injury and can limit hepatotoxicity to specifically mouse hepatocytes.
Robust engraftment of human hepatocytes has been shown in a number of immunodeficient liver injury models, including Alb-uPA (Meuleman et al. 2005; Mercer et al. 2001), FAH−/− (Bissig et al. 2010; de Jong et al. 2014), AFC8 (Washburn et al. 2011), MUP-uPA (Tesfaye et al. 2013) and HSV-TK (Kosaka et al. 2013) mice. The resultant human liver chimeric mice are susceptible to several human-tropic pathogens, including HBV, HCV and parasites that cause malaria in humans (reviewed in (Meuleman and Leroux-Roels 2008). Additionally, these mice can be used for monitoring human-like metabolic and toxicological responses in testing antimicrobial compounds.
With the exception of AFC8 mice, all the above-mentioned xenorecipient strains have demonstrated robust human hepatic chimerism when using adult hepatocytes. Due to genetic differences between hepatocyte donors, host responses can differ. To minimize inter-experimental variations and create a renewable resource for human donor cells, stem cell-derived hepatocytes have been explored as a possible solution. Hepatocyte-like cells (HLCs) can now be routinely generated from embryonic stem (ES) or induced pluripotent stem (iPS) cells (Touboul et al. 2010; Si-Tayeb et al. 2010). These HLCs express hepatocyte-specific markers, support hepatocyte-specific metabolic functions and can be infected with HCV (Schwartz et al. 2012; Wu et al. 2012; Roelandt et al. 2012). Recent studies suggest that HLCs can also be engrafted reasonably well in vivo and support persistent HCV infection (Carpentier et al. 2014). However, engraftment efficiency seems to depend strongly on the xenorecipient strain, as immunodeficient MUP-uPA mice, but not other liver injury models, support robust in vivo expansion.
4.2.4 HCV Immunity and Pathogenesis in Humanized Xenotransplantation Models
Human liver chimeric mice are currently the only experimental models besides chimpanzees that are readily susceptible to HCV. These mice have been used to study innate host responses to HCV and for testing the efficacy of novel therapeutic regimens. To expand the use of these mice in analyzing human immune responses to HCV, protocols are being refined so that mice are co-engrafted with human hepatocytes and components of a human immune system (Fig. 1). Initial attempts co-injected a mixture of human fetal hepatoblasts, non-parenchymal cells and hematopoietic stem cells (HSCs) into AFC8 mice, yielding reasonable immune cell engraftment but low human hepatic chimerism (Washburn et al. 2011). Nonetheless, dually engrafted mice did become chronically infected following inoculation with HCV patient isolates and exhibited an HCV-specific T cell response, which appeared to be responsible for observed signs of early liver fibrosis. While these data are encouraging, protocols need to be refined further to improve dual chimerism and minimize inter- and intravariability of experiments. More recent reports have demonstrated that extensive double humanization of both the liver and immune system can be achieved with mature hepatocytes and HSCs (Gutti et al. 2014; Wilson et al. 2014). Long-term dual reconstitution, without any evidence of hepatocyte rejection by the human immune system, was sustained even when the human cells were mismatched in their major histocompatibility complex (MHC, (Gutti et al. 2014). The latter observation is consistent with the limited HLA matching in human liver transplantations, presumably due to the tolerogenic microenvironment of the liver. However, the limited function of the transplanted human immune system at least partially contributes to the lack of allogeneic graft rejection in dually engrafted humanized mice. To improve both the cellular complexity and functionality of the engrafted human immune system, several modifications are being tested. These include, but are not limited to: the expression of human orthologs of non-redundant cytokines with limited biological cross-reactivity to foster development of human immune cell lineages which currently do not develop efficiently in conventional humanized mice; expression of human MHC in the absence of mouse MHC to ensure faithful presentation of self- and virally derived peptides to human T cells and to reduce graft-versus-host-disease; co-transplantation of HSC donor-matched human thymic cortical epithelium to facilitate proper T cell selection; the improvement of lymphoid architecture organization, especially in the spleen and lymph-nodes, to allow for adequate T and B cell priming; genetic replacement of non-compatible immune cell receptors and chemokines expressed on non-hematopoietically derived cells to improve e.g. immune cell trafficking; the introduction of a human microbiome to account for effects of species-specific commensals on the immune system (reviewed in Shultz et al. 2012).
5 Genetically Humanized Mouse Models for HCV Infection
An inbred mouse model with inheritable susceptibility to HCV would overcome the technical difficulties of the xenotransplantation model (Fig. 1). The challenge is to systematically identify and overcome any restrictions to HCV infection in murine cells. HCV’s narrow host range is not completely understood, and the viral life cycle is blocked or insufficiently supported in murine cells at multiple steps. Productive HCV uptake into human hepatocytes relies on a large number of human host molecules (reviewed in Ding et al. 2014). These include glycosaminoglycans (GAGs) present on heparan sulfate proteoglycans (HSPGs), low-density-lipoprotein receptor (LDLR) (Agnello et al. 1999), CD81 (Pileri et al. 1998), scavenger receptor class B member 1 (SCARB1) (Scarselli et al. 2002), the tight junction proteins claudin-1 (CLDN1) (Evans et al. 2007) and occludin (OCLN) (Liu et al. 2009; Ploss et al. 2009), the receptor tyrosine kinases epidermal growth factor receptor (EGFR) and ephrin receptor A2 (EphA2) (Lupberger et al. 2011), the cholesterol transporter Niemann-Pick C1-like 1 (NPC1L1) (Sainz et al. 2012), transferrin receptor 1 (TfR1) (Martin and Uprichard 2013) and the cell death-inducing DFFA-like effector b (CIDEB) (Wu et al. 2014). The block of HCV entry in rodent cells can be explained by differences in critical residues in the second extracellular loops of CD81 (Flint et al. 2006; Higginbottom et al. 2000) and OCLN (Michta et al. 2010). Consequently, expression of human CD81 and OCLN, along with human or mouse SCARB1 and CLDN1, facilitates HCV uptake in mouse cells in vitro (Ploss et al. 2009). Other human entry factors appear to contribute minimally to HCV species tropism at the level of entry, but their individual roles still need to be experimentally tested.
Establishing HCV glycoprotein-mediated uptake into mouse livers adenovirally transduced with CD81 and OCLN opened the door for genetically overcoming the barrier to HCV entry in mice. Indeed, expression of human CD81 and OCLN appears sufficient for HCV entry into hepatocytes of fully immunocompetent inbred mice. This genetically humanized mouse model allows dissection of the HCV entry process in the 3D context of the liver in vivo and has been applied to test pre-clinically the efficacy of neutralizing antibodies and vaccine candidates (Giang et al. 2012; Dorner et al. 2011; de Jong et al. 2014). Transgenic mice have also been developed in which human CD81, SCARB1, CLDN1 and OCLN expression are driven by liver-specific promoters (Hikosaka et al. 2011). However, initial reports have suggested that these lines are resistant to HCV infection in vivo (Hikosaka et al. 2011). This observation is likely due to the lower level of entry factor expression in the transgenic mice and the need for a very sensitive reporter system to quantify viral entry (Dorner et al. 2011).
As an alternative to the genetic host adaptations described above, previous studies have shown that the block of HCV at the level of entry can also be overcome through viral adaptation (Fig. 1). Using an in vitro selection approach, mutations within HCV E1 and E2 that increased the affinity of the viral envelope for mouse CD81 were identified. These mutations appeared to more broadly affect the conformation of the viral envelope, as the resulting mouse CD81-adapted strain is also less dependent on human OCLN and can enter cell lines expressing only mouse CD81, SCARB1, CLDN1 and OCLN (Bitzegeio et al. 2010). It has yet to be demonstrated if this mouse-adapted HCV strain can enter mouse primary hepatocytes in vitro or in vivo.
Establishing HCV entry in vivo has some utility, but what is ultimately needed is a model that supports all steps of the viral life cycle. More than a decade ago, it was shown that HCV RNA is translated, but not readily replicated, following entry into murine cells (Dorner et al. 2011; McCaffrey et al. 2002). Subsequent studies in cell culture demonstrated that dominant negative inhibitors are not present and that the murine orthologs of host factors critical for HCV replication cooperate sufficiently with the viral replication machinery, as HCV replicons, i.e. selectable HCV RNA genomes, can replicate in murine cell lines (Zhu et al. 2003; Uprichard et al. 2006; Frentzen et al. 2011). Nevertheless, the efficiency of post-entry steps of the viral life cycle could conceivably be improved with human host factors important for HCV replication, assembly and/or egress. However, previous gain- and loss-of-function studies converged on only a few critical host factors, namely miR-122, cyclophilin A, phosphatidylinositol 4 kinase IIIα (PI4KIIIα), and apolipoprotein E (reviewed in (Bartenschlager et al. 2010)) – all of whose sequences are largely conserved between mice and humans. Thus, additional proviral factors that enhance HCV replication and/or assembly in mouse cells have yet to be identified.
Numerous studies have shown that innate antiviral responses play a critical role in limiting HCV infection in human cells, including hepatoma cell lines and human primary hepatocytes (Andrus et al. 2011; Marukian et al. 2011). Likewise, HCV replication is drastically enhanced in cell lines with strong impairments in type I and III interferon signaling, such as mouse cells lacking MAVS (Frentzen et al. 2014), protein kinase R (PKR; (Chang et al. 2006)), interferon regulatory factor 3 (IRF3; (Lin et al. 2010)) or STAT1 (Vogt et al. 2013). Known mechanisms by which HCV evades antiviral defenses, such as the cleavage of MAVS (Meylan et al. 2005) or Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF or TICAM; (Li et al. 2005)), appear to function in mouse cells (Vogt et al. 2013). However, differences in the kinetics and/or magnitude of virally induced innate defenses may restrict HCV RNA replication more efficiently in mouse compared to human cells. Consistent with these in vitro data, mice expressing human HCV entry factors crossed to genetic backgrounds impaired in antiviral innate defenses support low level HCV RNA replication (Dorner et al. 2013). In these genetically humanized mice, infectious HCV is detectable in circulation, confirming previous studies that demonstrated late stages of the HCV life cycle are supported in mouse cells if sufficient ApoE is present (Long et al. 2011).
Recapitulating the entire HCV lifecycle in inbred immunocompetent mice is an important next step in developing a mouse model suitable for mechanistic studies of HCV immunity and pathogenesis. The previously published model required immune-suppression to establish low-level viremia. However, more recent work suggests that this may be strain-dependent. In fact, mice expressing human CD81, SCARB1, CLDN1 and OCLN on the fully immunocompetent ICR mouse background not only supported persistent infection with various HCV isolates very efficiently but also developed clinically apparent liver disease (Chen et al. 2014). While these data are somewhat difficult to reconcile with most previously published literature, it is conceivable that a fortuitous allele combination in the genetically variable outbred ICR stock favors susceptibility to HCV.
6 Summary and Outlook
The advent of highly potent DAAs holds promise to effectively treat the great majority of patients. However, current treatments are very expensive and mandate strict adherence to dosing to prevent the outgrowth of resistant viral variants. To provide simpler and more cost-effective interventions and to optimally prevent infections, a HCV vaccine may ultimately be needed. Testing and prioritization of immunotherapies and vaccines is delayed by the lack of (a) readily accessible animal model(s). More tractable in vivo platforms could also be deployed to answer questions of basic virology, HCV pathogenesis, and correlates of protective immunity (Fig. 3). A variety of partially complementary approaches are currently being pursued to develop better small animal models for HCV infection. While most other NHP species besides chimpanzees were thought to be resistant to HCV infection, recent studies show that the HCV life cycle can be established in HLCs derived from pig-tailed macaques and primary hepatocytes from rhesus macaques. This suggests that certain NHP species may indeed be permissive to HCV infection. In addition, the discovery of hepaciviruses genetically closely related to HCV in outbred mice, rats, dogs and horses may provide further avenues for studying HCV. The barriers of HCV’s narrow host tropism are now better understood and have spurred a combination of viral adaptations and/or genetic host humanizations to establish inbred rodent models with inheritable susceptibility to HCV infection. Xenotransplantation approaches are being continuously refined, and it has become possible to reproducibly generate human liver chimeric mice at fairly high throughput. These mice can then be used to analyze all aspects of the viral life cycle with genetically diverse HCV isolates. Improvements in protocols yielding HLCs from directed differentiation of ES and iPS cells hold promise to develop renewable hepatocyte sources of genetically defined backgrounds. Furthermore, these advances may enable the generation of humanized mouse avatars engrafted with patient-specific hepatocytes to model clinically relevant disease phenotypes. In proof-of-concept studies, human liver and components of a human immune system were robustly engrafted in a single xenorecipient, paving the way for modeling HCV-associated hepatitis, including relevant co-infections with HBV and/or HIV. Undoubtedly, as new techniques and protocols are perfected, it will remain important to continue evaluating the ability of any new HCV model to faithfully recapitulate aspects of HCV pathogenesis and its consequences in humans.

Comparison of different animal models for hepatitis C
Abbreviations
- AFC8:
-
Transgenic construct in which a FK506 binding protein/caspase 8 fusion protein is driven by a mouse albumin promoter
- apoE:
-
Apolipoprotein E
- Cardif:
-
Caspase activation and recruitment domain adaptor-inducing interferon- β
- CD:
-
Cluster of differentiation
- CIDEB:
-
Cell death-inducing DFFA-like effector b
- CLND1:
-
Claudin-1
- CMV:
-
Cytomegalovirus
- DAA:
-
Directly acting antiviral
- EGRF:
-
Epidermal growth factor receptor
- EphA2:
-
Ephrin receptor A2
- ER:
-
Endoplasmatic reticulum
- ES:
-
Embryonic stem cell
- FAH:
-
Fumarylacetoacetate hydrolase
- GAG:
-
Glycosaminoglycan
- GBV-B:
-
George Barker virus B
- HBV:
-
Hepatitis B virus
- HCC:
-
Hepatocellular Carcinoma
- HCV:
-
Hepatitis C virus
- HEAL:
-
Human ectopic artificial liver
- HLA:
-
Human leukocyte antigen
- HLC:
-
Hepatocyte-like cell
- HSC:
-
Hematopoietic stem cell
- HSPG:
-
Heparan sulfate proteoglycan
- Huh:
-
Human hepatoma
- IFN:
-
Interferon
- IL-1:
-
Interleukin 1
- iPS:
-
Induced pluripotent stem cell
- IPS-1:
-
IFN-β promoter stimulator-1
- IRF3:
-
Interferon regulatory factor 3
- LDLR:
-
Low-density-lipoprotein receptor
- MAVS:
-
Mitochondrial antiviral signal protein
- miR-122:
-
MicroRNA-122
- MUP:
-
Major urinary protein
- NHP:
-
Non-human primate
- NPC1L1:
-
Niemann-Pick C1-like 1
- NPHV:
-
Non-primate hepacivirus
- NS:
-
Non-structural protein
- OCLN:
-
occludin
- PI4KIIIα:
-
phosphatidylinositol 4 kinase IIIα
- PKR:
-
Protein kinase R
- SCARB1:
-
Scavenger receptor class B member 1
- SCID:
-
Severe combined immunodeficiency
- STAT1:
-
Signal transducer and activator of transcribtion 1
- TfR1:
-
Transferrin receptor 1
- TICAM:
-
Toll/IL-1 receptor domain-containing adaptor molecule
- TNFα:
-
Tumor necrosis factor α
- TRIF:
-
Toll/IL-1 receptor domain-containing adaptor inducing IFN-β
- uPA:
-
Urokinase-type plasminogen activator
- VISA:
-
Virus-induced signaling adapter
References
Abe K, Kurata T, Teramoto Y, Shiga J, Shikata T (1993) Lack of susceptibility of various primates and woodchucks to hepatitis C virus. J Med Primatol 22(7–8):433–434
Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX (1999) Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci U S A 96(22):12766–12771
Ahlen G, Derk E, Weiland M, Jiao J, Rahbin N, Aleman S, Peterson DL, Pokrovskaja K, Grander D, Frelin L, Sallberg M (2009) Cleavage of the IPS-1/Cardif/MAVS/VISA does not inhibit T cell-mediated elimination of hepatitis C virus non-structural 3/4A-expressing hepatocytes. Gut 58(4):560–569. doi:gut.2007.147264 [pii] 10.1136/gut.2007.147264
Alonzi T, Agrati C, Costabile B, Cicchini C, Amicone L, Cavallari C, Rocca CD, Folgori A, Fipaldini C, Poccia F, Monica NL, Tripodi M (2004) Steatosis and intrahepatic lymphocyte recruitment in hepatitis C virus transgenic mice. J Gen Virol 85(Pt 6):1509–1520
Amako Y, Tsukiyama-Kohara K, Katsume A, Hirata Y, Sekiguchi S, Tobita Y, Hayashi Y, Hishima T, Funata N, Yonekawa H, Kohara M (2010) Pathogenesis of hepatitis C virus infection in Tupaia belangeri. J Virol 84(1):303–311. doi:JVI.01448-09 [pii] 10.1128/JVI.01448-09
Andrus L, Marukian S, Jones CT, Catanese MT, Sheahan TP, Schoggins JW, Barry WT, Dustin LB, Trehan K, Ploss A, Bhatia SN, Rice CM (2011) Expression of paramyxovirus V proteins promotes replication and spread of hepatitis C virus in cultures of primary human fetal liver cells. Hepatology 54(6):1901–1912. doi:10.1002/hep.24557
Bartenschlager R, Cosset FL, Lohmann V (2010) Hepatitis C virus replication cycle. J Hepatol 53(3):583–585. doi:10.1016/j.jhep.2010.04.015
Benali-Furet NL, Chami M, Houel L, De Giorgi F, Vernejoul F, Lagorce D, Buscail L, Bartenschlager R, Ichas F, Rizzuto R, Paterlini-Brechot P (2005) Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 24(31):4921–4933
Bissig KD, Wieland SF, Tran P, Isogawa M, Le TT, Chisari FV, Verma IM (2010) Human liver chimeric mice provide a model for hepatitis B and C virus infection and treatment. J Clin Invest 120(3):924–930. doi:40094 [pii] 10.1172/JCI40094
Bitzegeio J, Bankwitz D, Hueging K, Haid S, Brohm C, Zeisel MB, Herrmann E, Iken M, Ott M, Baumert TF, Pietschmann T (2010) Adaptation of hepatitis C virus to mouse CD81 permits infection of mouse cells in the absence of human entry factors. PLoS Pathog 6:e1000978. doi:10.1371/journal.ppat.1000978
Bukh J (2004) A critical role for the chimpanzee model in the study of hepatitis C. Hepatology 39(6):1469–1475. doi:10.1002/hep.20268
Bukh J, Apgar CL, Govindarajan S, Purcell RH (2001) Host range studies of GB virus-B hepatitis agent, the closest relative of hepatitis C virus, in New World monkeys and chimpanzees. J Med Virol 65(4):694–697. doi:10.1002/jmv.2092 [pii]
Burbelo PD, Dubovi EJ, Simmonds P, Medina JL, Henriquez JA, Mishra N, Wagner J, Tokarz R, Cullen JM, Iadarola MJ, Rice CM, Lipkin WI, Kapoor A (2012) Serology enabled discovery of genetically diverse hepaciviruses in a new host. J Virol. doi:JVI.00250-12 [pii] 10.1128/JVI.00250-12
Carpentier A, Tesfaye A, Chu V, Nimgaonkar I, Zhang F, Lee SB, Thorgeirsson SS, Feinstone SM, Liang TJ (2014) Engrafted human stem cell-derived hepatocytes establish an infectious HCV murine model. J Clin Invest 124(11):4953–4964. doi:10.1172/JCI75456
Chang KS, Cai Z, Zhang C, Sen GC, Williams BR, Luo G (2006) Replication of hepatitis C virus (HCV) RNA in mouse embryonic fibroblasts: protein kinase R (PKR)-dependent and PKR-independent mechanisms for controlling HCV RNA replication and mediating interferon activities. J Virol 80(15):7364–7374. doi:80/15/7364 [pii] 10.1128/JVI.00586-06
Chang ML, Chen JC, Chang MY, Yeh CT, Lin WP, Liang CK, Huang SF, Dang KN, Chiu CT, Lin DY (2008) Acute expression of hepatitis C core protein in adult mouse liver: mitochondrial stress and apoptosis. Scand J Gastroenterol 43(6):747–755. doi:789872799 [pii] 10.1080/00365520701875987
Chang ML, Chen TH, Chang MY, Yeh CT (2009) Cell cycle perturbation in the hepatocytes of HCV core transgenic mice following common bile duct ligation is associated with enhanced p21 expression. J Med Virol 81(3):467–472. doi:10.1002/jmv.21403
Chen AA, Thomas DK, Ong LL, Schwartz RE, Golub TR, Bhatia SN (2011) Humanized mice with ectopic artificial liver tissues. Proc Natl Acad Sci U S A 108(29):11842–11847. doi:10.1073/pnas.1101791108
Chen J, Zhao Y, Zhang C, Chen H, Feng J, Chi X, Pan Y, Du J, Guo M, Cao H, Chen H, Wang Z, Pei R, Wang Q, Pan L, Niu J, Chen X, Tang H (2014) Persistent hepatitis C virus infections and hepatopathological manifestations in immune-competent humanized mice. Cell Res 24(9):1050–1066. doi:10.1038/cr.2014.116
Chiyo T, Sekiguchi S, Hayashi M, Tobita Y, Kanegae Y, Saito I, Kohara M (2011) Conditional gene expression in hepatitis C virus transgenic mice without induction of severe liver injury using a non-inflammatory Cre-expressing adenovirus. Virus Res 160(1–2):89–97. doi:10.1016/j.virusres.2011.05.019
de Jong YP, Dorner M, Mommersteeg MC, Xiao JW, Balazs AB, Robbins JB, Winer BY, Gerges S, Vega K, Labitt RN, Donovan BM, Giang E, Krishnan A, Chiriboga L, Charlton MR, Burton DR, Baltimore D, Law M, Rice CM, Ploss A (2014) Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci Transl Med 6(254):254ra129. doi:10.1126/scitranslmed.3009512
Deinhardt F, Holmes AW, Capps RB, Popper H (1967) Studies on the transmission of human viral hepatitis to marmoset monkeys. I. Transmission of disease, serial passages, and description of liver lesions. J Exp Med 125(4):673–688
Desai MM, Gong B, Chan T, Davey RA, Soong L, Kolokoltsov AA, Sun J (2011) Differential, type I interferon-mediated autophagic trafficking of hepatitis C virus proteins in mouse liver. Gastroenterology 141(2):674–685, 685 e671–676. doi:S0016-5085(11)00615-9 [pii] 10.1053/j.gastro.2011.04.060
Ding Q, von Schaewen M, Ploss A (2014) The impact of hepatitis C virus entry on viral tropism. Cell Host Microbe 16(5):562–568. doi:10.1016/j.chom.2014.10.009
Disson O, Haouzi D, Desagher S, Loesch K, Hahne M, Kremer EJ, Jacquet C, Lemon SM, Hibner U, Lerat H (2004) Impaired clearance of virus-infected hepatocytes in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology 126(3):859–872. doi:S0016508503020183 [pii]
Dorner M, Horwitz JA, Robbins JB, Barry WT, Feng Q, Mu K, Jones CT, Schoggins JW, Catanese MT, Burton DR, Law M, Rice CM, Ploss A (2011) A genetically humanized mouse model for hepatitis C virus infection. Nature 474(7350):208–211. doi:nature10168 [pii] 10.1038/nature10168
Dorner M, Horwitz JA, Donovan BM, Labitt RN, Budell WC, Friling T, Vogt A, Catanese MT, Satoh T, Kawai T, Akira S, Law M, Rice CM, Ploss A (2013) Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature 501(7466):237–241. doi:10.1038/nature12427
Edlin BR (2011) Perspective: test and treat this silent killer. Nature 474(7350):S18–S19. doi:474S18a [pii] 10.1038/474S18a
Eren R, Landstein D, Terkieltaub D, Nussbaum O, Zauberman A, Ben-Porath J, Gopher J, Buchnick R, Kovjazin R, Rosenthal-Galili Z, Aviel S, Ilan E, Shoshany Y, Neville L, Waisman T, Ben-Moshe O, Kischitsky A, Foung SK, Keck ZY, Pappo O, Eid A, Jurim O, Zamir G, Galun E, Dagan S (2006) Preclinical evaluation of two neutralizing human monoclonal antibodies against hepatitis C virus (HCV): a potential treatment to prevent HCV reinfection in liver transplant patients. J Virol 80(6):2654–2664. doi:80/6/2654 [pii] 10.1128/JVI.80.6.2654-2664.2006
Ernst E, Schonig K, Bugert JJ, Blaker H, Pfaff E, Stremmel W, Encke J (2007) Generation of inducible hepatitis C virus transgenic mouse lines. J Med Virol 79(8):1103–1112. doi:10.1002/jmv.20911
Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM (2007) Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446(7137):801–805. doi:nature05654 [pii] 10.1038/nature05654
Firth C, Bhat M, Firth MA, Williams SH, Frye MJ, Simmonds P, Conte JM, Ng J, Garcia J, Bhuva NP, Lee B, Che X, Quan PL, Lipkin WI (2014) Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. mBio 5(5):e01933-01914. doi:10.1128/mBio.01933-14
Flint M, von Hahn T, Zhang J, Farquhar M, Jones CT, Balfe P, Rice CM, McKeating JA (2006) Diverse CD81 proteins support hepatitis C virus infection. J Virol 80(22):11331–11342
Frelin L, Brenndorfer ED, Ahlen G, Weiland M, Hultgren C, Alheim M, Glaumann H, Rozell B, Milich DR, Bode JG, Sallberg M (2006) The hepatitis C virus and immune evasion: non-structural 3/4A transgenic mice are resistant to lethal tumour necrosis factor alpha mediated liver disease. Gut 55(10):1475–1483. doi:gut.2005.085050 [pii] 10.1136/gut.2005.085050
Frentzen A, Huging K, Bitzegeio J, Friesland M, Haid S, Gentzsch J, Hoffmann M, Lindemann D, Zimmer G, Zielecki F, Weber F, Steinmann E, Pietschmann T (2011) Completion of hepatitis C virus replication cycle in heterokaryons excludes dominant restrictions in human non-liver and mouse liver cell lines. PLoS Pathog 7(4):e1002029. doi:10.1371/journal.ppat.1002029
Frentzen A, Anggakusuma, Gurlevik E, Hueging K, Knocke S, Ginkel C, Brown RJ, Heim M, Dill MT, Kroger A, Kalinke U, Kaderali L, Kuehnel F, Pietschmann T (2014) Cell entry, efficient RNA replication, and production of infectious hepatitis C virus progeny in mouse liver-derived cells. Hepatology 59(1):78–88. doi:10.1002/hep.26626
Furutani T, Hino K, Okuda M, Gondo T, Nishina S, Kitase A, Korenaga M, Xiao SY, Weinman SA, Lemon SM, Sakaida I, Okita K (2006) Hepatic iron overload induces hepatocellular carcinoma in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology 130(7):2087–2098. doi:10.1053/j.gastro.2006.02.060
Garson JA, Whitby K, Watkins P, Morgan AJ (1997) Lack of susceptibility of the cottontop tamarin to hepatitis C infection. J Med Virol 52(3):286–288. doi:10.1002/(SICI)1096-9071(199707)52:3<286::AID-JMV9>3.0.CO;2-Z [pii]
Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, Rice CM, Ploss A, Burton DR, Law M (2012) Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc Natl Acad Sci USA 109(16):6205–6210. doi:1114927109 [pii] 10.1073/pnas.1114927109
Gutti TL, Knibbe JS, Makarov E, Zhang J, Yannam GR, Gorantla S, Sun Y, Mercer DF, Suemizu H, Wisecarver JL, Osna NA, Bronich TK, Poluektova LY (2014) Human hepatocytes and hematolymphoid dual reconstitution in treosulfan-conditioned uPA-NOG mice. Am J Pathol 184(1):101–109. doi:10.1016/j.ajpath.2013.09.008
Higginbottom A, Quinn ER, Kuo CC, Flint M, Wilson LH, Bianchi E, Nicosia A, Monk PN, McKeating JA, Levy S (2000) Identification of amino acid residues in CD81 critical for interaction with hepatitis C virus envelope glycoprotein E2. J Virol 74(8):3642–3649
Hikosaka K, Noritake H, Kimura W, Sultana N, Sharkar MT, Tagawa Y, Uezato T, Kobayashi Y, Wakita T, Miura N (2011) Expression of human factors CD81, claudin-1, scavenger receptor, and occludin in mouse hepatocytes does not confer susceptibility to HCV entry. Biomed Res 32(2):143–150. doi:JST.JSTAGE/biomedres/32.143 [pii]
Ilan E, Arazi J, Nussbaum O, Zauberman A, Eren R, Lubin I, Neville L, Ben-Moshe O, Kischitzky A, Litchi A, Margalit I, Gopher J, Mounir S, Cai W, Daudi N, Eid A, Jurim O, Czerniak A, Galun E, Dagan S (2002) The hepatitis C virus (HCV)-Trimera mouse: a model for evaluation of agents against HCV. J Infect Dis 185(2):153–161. doi:JID010683 [pii] 10.1086/338266
Jeannot E, Boorman GA, Kosyk O, Bradford BU, Shymoniak S, Tumurbaatar B, Weinman SA, Melnyk SB, Tryndyak V, Pogribny IP, Rusyn I (2012) Increased incidence of aflatoxin B1-induced liver tumors in hepatitis virus C transgenic mice. Int J Cancer J Int Cancer 130(6):1347–1356. doi:10.1002/ijc.26140
Kamegaya Y, Hiasa Y, Zukerberg L, Fowler N, Blackard JT, Lin W, Choe WH, Schmidt EV, Chung RT (2005) Hepatitis C virus acts as a tumor accelerator by blocking apoptosis in a mouse model of hepatocarcinogenesis. Hepatology 41(3):660–667. doi:10.1002/hep.20621
Kanda T, Steele R, Ray R, Ray RB (2009) Inhibition of intrahepatic gamma interferon production by hepatitis C virus nonstructural protein 5A in transgenic mice. J Virol 83(17):8463–8469. doi:JVI.00751-09 [pii] 10.1128/JVI.00751-09
Kapoor A, Simmonds P, Gerold G, Qaisar N, Jain K, Henriquez JA, Firth C, Hirschberg DL, Rice CM, Shields S, Lipkin WI (2011) Characterization of a canine homolog of hepatitis C virus. Proc Natl Acad Sci U S A 108(28):11608–11613. doi:1101794108 [pii] 10.1073/pnas.1101794108
Kapoor A, Simmonds P, Cullen JM, Scheel TK, Medina JL, Giannitti F, Nishiuchi E, Brock KV, Burbelo PD, Rice CM, Lipkin WI (2013a) Identification of a pegivirus (GB virus-like virus) that infects horses. J Virol 87(12):7185–7190. doi:10.1128/JVI.00324-13
Kapoor A, Simmonds P, Scheel TK, Hjelle B, Cullen JM, Burbelo PD, Chauhan LV, Duraisamy R, Sanchez Leon M, Jain K, Vandegrift KJ, Calisher CH, Rice CM, Lipkin WI (2013b) Identification of rodent homologs of hepatitis C virus and pegiviruses. mBio 4(2):e00216-00213. doi:10.1128/mBio.00216-13
Karayiannis P, Petrovic LM, Fry M, Moore D, Enticott M, McGarvey MJ, Scheuer PJ, Thomas HC (1989) Studies of GB hepatitis agent in tamarins. Hepatology 9(2):186–192
Kosaka K, Hiraga N, Imamura M, Yoshimi S, Murakami E, Nakahara T, Honda Y, Ono A, Kawaoka T, Tsuge M, Abe H, Hayes CN, Miki D, Aikata H, Ochi H, Ishida Y, Tateno C, Yoshizato K, Sasaki T, Chayama K (2013) A novel TK-NOG based humanized mouse model for the study of HBV and HCV infections. Biochem Biophys Res Commun 441(1):230–235. doi:10.1016/j.bbrc.2013.10.040
Kremsdorf D, Brezillon N (2007) New animal models for hepatitis C viral infection and pathogenesis studies. World J Gastroenterol: WJG 13(17):2427–2435
Kriegs M, Burckstummer T, Himmelsbach K, Bruns M, Frelin L, Ahlen G, Sallberg M, Hildt E (2009) The hepatitis C virus non-structural NS5A protein impairs both the innate and adaptive hepatic immune response in vivo. J Biol Chem 284(41):28343–28351. doi:10.1074/jbc.M109.038877
Lanford RE, Chavez D, Notvall L, Brasky KM (2003) Comparison of tamarins and marmosets as hosts for GBV-B infections and the effect of immunosuppression on duration of viremia. Virology 311(1):72–80
Lauck M, Sibley SD, Lara J, Purdy MA, Khudyakov Y, Hyeroba D, Tumukunde A, Weny G, Switzer WM, Chapman CA, Hughes AL, Friedrich TC, O’Connor DH, Goldberg TL (2013) A novel hepacivirus with an unusually long and intrinsically disordered NS5A protein in a wild Old World primate. J Virol 87(16):8971–8981. doi:10.1128/JVI.00888-13
Lerat H, Kammoun HL, Hainault I, Merour E, Higgs MR, Callens C, Lemon SM, Foufelle F, Pawlotsky JM (2009) Hepatitis C virus proteins induce lipogenesis and defective triglyceride secretion in transgenic mice. J Biol Chem 284(48):33466–33474. doi:M109.019810 [pii] 10.1074/jbc.M109.019810
Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M Jr, Lemon SM (2005) Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A 102(8):2992–2997
Lin LT, Noyce RS, Pham TN, Wilson JA, Sisson GR, Michalak TI, Mossman KL, Richardson CD (2010) Replication of subgenomic hepatitis C virus replicons in mouse fibroblasts is facilitated by deletion of interferon regulatory factor 3 and expression of liver-specific microRNA 122. J Virol 84(18):9170–9180. doi:JVI.00559-10 [pii] 10.1128/JVI.00559-10
Liu S, Yang W, Shen L, Turner JR, Coyne CB, Wang T (2009) Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J Virol 83(4):2011–2014. doi:JVI.01888-08 [pii] 10.1128/JVI.01888-08
Long G, Hiet MS, Windisch MP, Lee JY, Lohmann V, Bartenschlager R (2011) Mouse hepatic cells support assembly of infectious hepatitis C virus particles. Gastroenterology 141(3):1057–1066. doi:10.1053/j.gastro.2011.06.010
Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF (2011) EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 17(5):589–595. doi:nm.2341 [pii] 10.1038/nm.2341
Majumder M, Steele R, Ghosh AK, Zhou XY, Thornburg L, Ray R, Phillips NJ, Ray RB (2003) Expression of hepatitis C virus non-structural 5A protein in the liver of transgenic mice. FEBS Lett 555(3):528–532. doi:S0014579303013371 [pii]
Martin DN, Uprichard SL (2013) Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc Natl Acad Sci U S A 110(26):10777–10782. doi:10.1073/pnas.1301764110
Marukian S, Andrus L, Sheahan TP, Jones CT, Charles ED, Ploss A, Rice CM, Dustin LB (2011) Hepatitis C virus induces interferon-lambda and interferon-stimulated genes in primary liver cultures. Hepatology 54(6):1913–1923. doi:10.1002/hep.24580
McCaffrey AP, Ohashi K, Meuse L, Shen S, Lancaster AM, Lukavsky PJ, Sarnow P, Kay MA (2002) Determinants of hepatitis C translational initiation in vitro, in cultured cells and mice. Mol Ther 5(6):676–684
Mercer DF, Schiller DE, Elliott JF, Douglas DN, Hao C, Rinfret A, Addison WR, Fischer KP, Churchill TA, Lakey JR, Tyrrell DL, Kneteman NM (2001) Hepatitis C virus replication in mice with chimeric human livers. Nat Med 7(8):927–933
Meuleman P, Leroux-Roels G (2008) The human liver-uPA-SCID mouse: a model for the evaluation of antiviral compounds against HBV and HCV. Antivir Res 80(3):231–238. doi:10.1016/j.antiviral.2008.07.006
Meuleman P, Libbrecht L, De Vos R, de Hemptinne B, Gevaert K, Vandekerckhove J, Roskams T, Leroux-Roels G (2005) Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology 41(4):847–856. doi:10.1002/hep.20657
Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437(7062):1167–1172
Michta ML, Hopcraft SE, Narbus CM, Kratovac Z, Israelow B, Sourisseau M, Evans MJ (2010) Species-specific regions of occludin required by hepatitis C virus for cell entry. J Virol 84(22):11696–11708. doi:JVI.01555-10 [pii] 10.1128/JVI.01555-10
Miller JS, Stevens KR, Yang MT, Baker BM, Nguyen DH, Cohen DM, Toro E, Chen AA, Galie PA, Yu X, Chaturvedi R, Bhatia SN, Chen CS (2012) Rapid casting of patterned vascular networks for perfusable engineered three-dimensional tissues. Nat Mater 11(9):768–774. doi:10.1038/nmat3357
Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Ishibashi K, Matsuura Y, Miyamura T, Koike K (1997) Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J Gen Virol 78(Pt 7):1527–1531
Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, Matsuura Y, Kimura S, Miyamura T, Koike K (1998) The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med 4(9):1065–1067. doi:10.1038/2053
Naas T, Ghorbani M, Alvarez-Maya I, Lapner M, Kothary R, De Repentigny Y, Gomes S, Babiuk L, Giulivi A, Soare C, Azizi A, Diaz-Mitoma F (2005) Characterization of liver histopathology in a transgenic mouse model expressing genotype 1a hepatitis C virus core and envelope proteins 1 and 2. J Gen Virol 86(Pt 8):2185–2196. doi:86/8/2185 [pii] 10.1099/vir.0.80969-0
Naas T, Ghorbani M, Soare C, Scherling N, Muller R, Ghorbani P, Diaz-Mitoma F (2010) Adoptive transfer of splenocytes to study cell-mediated immune responses in hepatitis C infection using HCV transgenic mice. Comp Hepatol 9:7. doi:1476-5926-9-7 [pii] 10.1186/1476-5926-9-7
Pasquinelli C, Shoenberger JM, Chung J, Chang KM, Guidotti LG, Selby M, Berger K, Lesniewski R, Houghton M, Chisari FV (1997) Hepatitis C virus core and E2 protein expression in transgenic mice. Hepatology 25(3):719–727
Patel MR, Loo YM, Horner SM, Gale M, Jr., Malik HS (2012) Convergent evolution of escape from hepaciviral antagonism in primates. PLoS Biol 10(3):e1001282. doi:10.1371/journal.pbio.1001282 PBIOLOGY-D-11-03907 [pii]
Pfaender S, Brown RJP, Pietschmann T, Steinmann E (2014a) Natural reservoirs for homologs of hepatitis C virus. Emerg Microbes Infec 3 doi:Artn E21. doi:10.1038/Emi.2014.19
Pfaender S, Cavalleri JM, Walter S, Doerrbecker J, Campana B, Brown RJ, Burbelo PD, Postel A, Hahn K, Kusuma A, Riebesehl N, Baumgartner W, Becher P, Heim MH, Pietschmann T, Feige K, Steinmann E (2014b) Clinical course of infection and viral tissue tropism of hepatitis C virus-like non-primate hepaciviruses. Hepatology. doi:10.1002/hep.27440
Pfaender S, Walter S, Todt D, Behrendt P, Doerrbecker J, Wolk B, Engelmann M, Gravemann U, Seltsam A, Steinmann J, Burbelo PD, Klawonn F, Feige K, Pietschmann T, Cavalleri JM, Steinmann E (2015) Assessment of cross-species transmission of hepatitis C virus-related non-primate hepacivirus in a population of humans at high risk of exposure. J Gen Virol 96(9):2636–2642. doi:10.1099/vir.0.000208
Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S (1998) Binding of hepatitis C virus to CD81. Science 282(5390):938–941
Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM (2009) Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457(7231):882–886. doi:nature07684 [pii] 10.1038/nature07684
Quan PL, Firth C, Conte JM, Williams SH, Zambrana-Torrelio CM, Anthony SJ, Ellison JA, Gilbert AT, Kuzmin IV, Niezgoda M, Osinubi MO, Recuenco S, Markotter W, Breiman RF, Kalemba L, Malekani J, Lindblade KA, Rostal MK, Ojeda-Flores R, Suzan G, Davis LB, Blau DM, Ogunkoya AB, Alvarez Castillo DA, Moran D, Ngam S, Akaibe D, Agwanda B, Briese T, Epstein JH, Daszak P, Rupprecht CE, Holmes EC, Lipkin WI (2013) Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc Natl Acad Sci U S A 110(20):8194–8199. doi:10.1073/pnas.1303037110
Roelandt P, Obeid S, Paeshuyse J, Vanhove J, Van Lommel A, Nahmias Y, Nevens F, Neyts J, Verfaillie CM (2012) Human pluripotent stem cell-derived hepatocytes support complete replication of hepatitis C virus. J Hepatol 57(2):246–251. doi:10.1016/j.jhep.2012.03.030
Sainz B Jr, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL (2012) Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med 18(2):281–285. doi:10.1038/nm.2581
Satoh K, Takahashi H, Matsuda C, Tanaka T, Miyasaka M, Zeniya M, Kohara M (2010) Natural killer cells target HCV core proteins during the innate immune response in HCV transgenic mice. J Med Virol 82(9):1545–1553. doi:10.1002/jmv.21859
Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A (2002) The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 21(19):5017–5025
Schaluder GG, Dawson GJ, Simons JN, Pilot-Matias TJ, Gutierrez RA, Heynen CA, Knigge MF, Kurpiewski GS, Buijk SL, Leary TP et al (1995) Molecular and serologic analysis in the transmission of the GB hepatitis agents. J Med Virol 46(1):81–90
Schwartz RE, Trehan K, Andrus L, Sheahan TP, Ploss A, Duncan SA, Rice CM, Bhatia SN (2012) Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc Natl Acad Sci USA 109(7):2544–2548. doi:1121400109 [pii] 10.1073/pnas.1121400109
Scull MA, Shi C, de Jong YP, Gerold G, Ries M, von Schaewen M, Donovan BM, Labitt RN, Horwitz JA, Gaska JM, Hrebikova G, Xiao JW, Flatley B, Fung C, Chiriboga L, Walker CM, Evans DT, Rice CM, Ploss A (2015) Hepatitis C virus infects rhesus macaque hepatocytes and simianized mice. Hepatology 62(1):57–67. doi:10.1002/hep.27773
Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL (2012) Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol 12(11):786–798. doi:10.1038/nri3311
Simons JN, Pilot-Matias TJ, Leary TP, Dawson GJ, Desai SM, Schlauder GG, Muerhoff AS, Erker JC, Buijk SL, Chalmers ML et al (1995) Identification of two flavivirus-like genomes in the GB hepatitis agent. Proc Natl Acad Sci U S A 92(8):3401–3405
Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S, Duncan SA (2010) Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 51(1):297–305. doi:10.1002/hep.23354
Sithebe NP, Kew MC, Mphahlele MJ, Paterson AC, Lecatsas G, Kramvis A, de Klerk W (2002) Lack of susceptibility of Chacma baboons (Papio ursinus orientalis) to hepatitis C virus infection. J Med Virol 66(4):468–471. doi:10.1002/jmv.2167 [pii]
Sourisseau M, Goldman O, He W, Gori JL, Kiem HP, Gouon-Evans V, Evans MJ (2013) Hepatic cells derived from induced pluripotent stem cells of pigtail macaques support hepatitis C virus infection. Gastroenterology 145(5):966–969 e967. doi:10.1053/j.gastro.2013.07.026
Stevens KR, Ungrin MD, Schwartz RE, Ng S, Carvalho B, Christine KS, Chaturvedi RR, Li CY, Zandstra PW, Chen CS, Bhatia SN (2013) InVERT molding for scalable control of tissue microarchitecture. Nat Commun 4:1847. doi:10.1038/ncomms2853
Takaku S, Nakagawa Y, Shimizu M, Norose Y, Maruyama I, Wakita T, Takano T, Kohara M, Takahashi H (2003) Induction of hepatic injury by hepatitis C virus-specific CD8+ murine cytotoxic T lymphocytes in transgenic mice expressing the viral structural genes. Biochem Biophys Res Commun 301(2):330–337. doi:S0006291X02030188 [pii]
Tanaka N, Moriya K, Kiyosawa K, Koike K, Aoyama T (2008) Hepatitis C virus core protein induces spontaneous and persistent activation of peroxisome proliferator-activated receptor alpha in transgenic mice: implications for HCV-associated hepatocarcinogenesis. Int J Cancer J Int Cancer 122(1):124–131. doi:10.1002/ijc.23056
Tesfaye A, Stift J, Maric D, Cui Q, Dienes HP, Feinstone SM (2013) Chimeric mouse model for the infection of hepatitis B and C viruses. PLoS ONE 8(10):e77298. doi:10.1371/journal.pone.0077298
Touboul T, Hannan NR, Corbineau S, Martinez A, Martinet C, Branchereau S, Mainot S, Strick-Marchand H, Pedersen R, Di Santo J, Weber A, Vallier L (2010) Generation of functional hepatocytes from human embryonic stem cells under chemically defined conditions that recapitulate liver development. Hepatology 51(5):1754–1765. doi:10.1002/hep.23506
Tsukiyama-Kohara K, Sekiguchi S, Kasama Y, Salem NE, Machida K, Kohara M (2011) Hepatitis C virus-related lymphomagenesis in a mouse model. ISRN Hematol 2011:167501. doi:10.5402/2011/167501
Tumurbaatar B, Sun Y, Chan T, Sun J (2007) Cre-estrogen receptor-mediated hepatitis C virus structural protein expression in mice. J Virol Methods 146(1–2):5–13. doi:10.1016/j.jviromet.2007.05.025
Uprichard SL, Chung J, Chisari FV, Wakita T (2006) Replication of a hepatitis C virus replicon clone in mouse cells. Virol J 3:89. doi:1743-422X-3-89 [pii] 10.1186/1743-422X-3-89
Vogt A, Scull MA, Friling T, Horwitz JA, Donovan BM, Dorner M, Gerold G, Labitt RN, Rice CM, Ploss A (2013) Recapitulation of the hepatitis C virus life-cycle in engineered murine cell lines. Virology 444(1–2):1–11. doi:10.1016/j.virol.2013.05.036
Wang AG, Moon HB, Kim JM, Hwang SB, Yu DY, Lee DS (2006) Expression of hepatitis C virus nonstructural 4B in transgenic mice. Exp Mol Med 38(3):241–246. doi:200606307 [pii]
Wang AG, Lee DS, Moon HB, Kim JM, Cho KH, Choi SH, Ha HL, Han YH, Kim DG, Hwang SB, Yu DY (2009) Non-structural 5A protein of hepatitis C virus induces a range of liver pathology in transgenic mice. J Pathol 219(2):253–262. doi:10.1002/path.2592
Washburn ML, Bility MT, Zhang L, Kovalev GI, Buntzman A, Frelinger JA, Barry W, Ploss A, Rice CM, Su L (2011) A humanized mouse model to study hepatitis C virus infection, immune response, and liver disease. Gastroenterology 140(4):1334–1344. doi:10.1053/j.gastro.2011.01.001
Wegert M, La Monica N, Tripodi M, Adler G, Dikopoulos N (2009) Impaired interferon type I signalling in the liver modulates the hepatic acute phase response in hepatitis C virus transgenic mice. J Hepatol 51(2):271–278. doi:S0168-8278(09)00249-9 [pii] 10.1016/j.jhep.2009.03.014
Wilson EM, Bial J, Tarlow B, Bial G, Jensen B, Greiner DL, Brehm MA, Grompe M (2014) Extensive double humanization of both liver and hematopoiesis in FRGN mice. Stem Cell Res 13(3PA):404–412. doi:10.1016/j.scr.2014.08.006
Wu GY, Konishi M, Walton CM, Olive D, Hayashi K, Wu CH (2005) A novel immunocompetent rat model of HCV infection and hepatitis. Gastroenterology 128(5):1416–1423. doi:S0016508505003951 [pii]
Wu X, Robotham JM, Lee E, Dalton S, Kneteman NM, Gilbert DM, Tang H (2012) Productive hepatitis C virus infection of stem cell-derived hepatocytes reveals a critical transition to viral permissiveness during differentiation. PLoS Pathog 8(4):e1002617. doi:10.1371/journal.ppat.1002617
Wu X, Lee EM, Hammack C, Robotham JM, Basu M, Lang J, Brinton MA, Tang H (2014) Cell death-inducing DFFA-like effector b is required for hepatitis C virus entry into hepatocytes. J Virol 88(15):8433–8444. doi:10.1128/JVI.00081-14
Xie ZC, Riezu-Boj JI, Lasarte JJ, Guillen J, Su JH, Civeira MP, Prieto J (1998) Transmission of hepatitis C virus infection to tree shrews. Virology 244(2):513–520
Xu X, Chen H, Cao X, Ben K (2007) Efficient infection of tree shrew (Tupaia belangeri) with hepatitis C virus grown in cell culture or from patient plasma. J Gen Virol 88(Pt 9):2504–2512. doi:88/9/2504 [pii] 10.1099/vir.0.82878-0
Zhu Q, Guo JT, Seeger C (2003) Replication of hepatitis C virus subgenomes in nonhepatic epithelial and mouse hepatoma cells. J Virol 77(17):9204–9210
Zhu Q, Oei Y, Mendel DB, Garrett EN, Patawaran MB, Hollenbach PW, Aukerman SL, Weiner AJ (2006) Novel robust hepatitis C virus mouse efficacy model. Antimicrob Agents Chemother 50(10):3260–3268. doi:50/10/3260 [pii] 10.1128/AAC.00413-06
Acknowledgements
The authors thank Qiang Ding and Florian Douam for edits and critical discussion of the manuscript. Work in the laboratory is in part supported by grants from the National Institutes of Health (2 R01 AI079031-05A1, 1 R01 AI107301-01, 1 R56 AI106005-01), a Research Scholar Award from the American Cancer Society (RSG-15-048-01-MPC) and the Grand Challenge Program of Princeton University. M.v.S. is a recipient of a fellowship from the German Research Foundation (Deutsche Forschungsgemeinschaft). JMG is supported by co-funding from NIAID on iNRSA 5T32GM007388. We apologize to all colleagues whose work could not be cited due to space constraints.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
von Schaewen, M., Gaska, J.M., Ploss, A. (2016). New Animal Models for Hepatitis C. In: Miyamura, T., Lemon, S., Walker, C., Wakita, T. (eds) Hepatitis C Virus I. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56098-2_12
Download citation
DOI: https://doi.org/10.1007/978-4-431-56098-2_12
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56096-8
Online ISBN: 978-4-431-56098-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)