
Abstract
Neural stem/progenitor cells (NSCs) are defined as cells with the potential for self-renewal and differentiation into neurons, astrocytes, and oligodendrocytes. These cells can be derived from several sources, including embryonic stem cells and fetal tissue. NSCs have been found to exist not only in the developing brain but also in the mature mammalian brain. NSCs were initially cultured as floating neurospheres in the presence of epidermal growth factor from adult and embryonic murine forebrain. Cell transplantation using these cells has evolved as a promising experimental treatment approach for stroke. Additionally, the activation of endogenous neural stem/progenitor cells has recently been employed for stroke treatment. This review provides an introduction to neural stem/progenitor cells and briefly describes some advances in neural stem cell transplantation for stroke.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Cell Biology
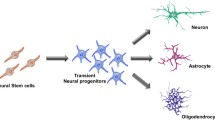
As initially observed by the pioneering neuroscientist Santiago Ramon y Cajal, the mature central nervous system (CNS) was thought to be distinguished from the developing nervous system by the lack of growth and cellular regeneration; it was believed that nerve paths were something fixed, ended, and immutable and had no regeneration potential in the adult CNS [1]. However, recent advances in neuroscience have revealed the falsehoods in this myth. In 1992, Reynolds, Weiss, and colleagues for the first time isolated neural stem cells (NSCs) and propagated them in the presence of epidermal growth factor (EGF) to give rise to large cellular spheres that they termed “neurospheres” [2, 3]. Neurons and glial cells are derived from common immature NSCs, which are defined as self-renewing and multipotential cells (Fig. 3.1). NSCs have been found to exist not only in the developing brain but also in the mature mammalian brain. Cultured NSCs derived from murine embryonic brains can be propagated by incubation in serum-free medium containing EGF and subsequently differentiated into neurons and astrocytes by incubating in low-serum medium (e.g., 1 % fetal bovine serum-containing medium without EGF) [4].

NSC and neurosphere method
NSCs exist in at least two regions of the adult brain – the subventricular zone of the lateral ventricle and the subgranular zone of the hippocampus. Newborn neurons are incorporated into existing functional networks and are thought to have important innate and adaptive roles in cognition, behavior, and tissue repair [5]. Notch signaling, which is highly active in quiescent NSCs in these areas, plays a pivotal role in maintaining the undifferentiated and quiescent state of NSCs [6–8]. Interestingly, NSCs give rise to their own niche cells through asymmetric segregation of Notch ligand Delta-like 1 during mitosis, a process that may contribute to initialization of activated NSCs to return to a basal NSC state (undifferentiated and quiescent) [9]. Conversely, transcription factors including basic helix-loop-helix (bHLH) transcription factors regulate NSC proliferation and differentiation of each cell type [10]. Proneural bHLH genes, such as Ascl 1 (as Mash 1) and Neurogenin 2, promote neuronal fate determination and suppress astrocytic gene expression [11, 12]. The bHLH gene Olig 2 regulates oligodendrocyte specification, whereas the bHLH genes Hes 1 and Hes 5 maintain NSCs by repressing proneural gene expression [13, 14]. In addition, Ascl 1 and Olig 2 regulate oligodendrocyte and motor neuron development, respectively [13, 14]. A recent report showed that oscillatory control of these factors determines NSC multipotency and fate [15].
NSCs first expand by rapid cell division to generate a large number of different types of neurons during the early stage of brain development. After this neurogenic period, NSCs mostly lose their neurogenic potential and begin to preferentially generate glial cells during postnatal stages (astrogenic phase). Early stage NSCs have a greater capacity to proliferate and self-renew than late-stage NSCs [16]. This suggests that NSCs lose their neurogenic potential during development, which might be a disadvantage for neuronal repair in adult CNS. Kishi et al. found that neocortical NSC chromatin becomes globally condensed in a stage-dependent manner and that high-mobility group A (HMGA) proteins, which are chromatin architectural proteins, are necessary for the open chromatin state in early stage NSCs [17]. They also found that reduced HMGA protein levels and resultant global chromatin condensation are involved in restriction of the NSC differentiation potential during neocortical development [17]. Thus, HMGA proteins are capable of reprogramming late-stage NSCs into cells with early stage-specific capacities.
Developmental studies and experimental data have enabled us to determine that the terminal cell differentiation state is reversible and that altering the balance of specific transcription factors could be a powerful strategy for inducing pluripotency [18]. It has recently been demonstrated that induced neural stem cells (iNSCs) can be obtained from rodent and human somatic cells, such as fibroblasts, through forced expression of defined transcription factors [Sox2, Klf4, and Myc (also known as c-Myc) and Pou3f4 (also known as Brn4)] [19]. To date, two different approaches have been successfully used to obtain iNSCs: a direct method and an indirect method that involves an intermediate destabilized state. The possibility to induce characterized iNSCs from human cells, e.g., fibroblasts, has opened new horizons for research in human disease modeling and cellular therapeutic applications in the neurological field [20].
2 Ischemia-Induced NSC Activation
In vitro studies have shown that hypoxia enhances proliferation of cultured NSCs and modifies the ability of the cells to differentiate [21–24]. Conversely, reduced glucose has been shown to suppress proliferation and increase differentiation of murine neural stem cells [25]. It is now well known that endogenous neurogenesis occurs in certain brain areas after cerebral ischemia, such as the subgranular zone of the dentate gyrus in the hippocampus [26], subventricular zone of the lateral ventricle in the striatum [27], and cortical layer [28]. Some evidence indicates that these neurons reestablish connections and contribute to functional recovery [29, 30]. These new neurons migrate into the impaired lesion, where they express markers of projection neurons. However, the majority of new neurons die during the first weeks after stroke and are only capable of replacing a small fraction of necrotic mature neurons [31]. Recently, electrical stimulation has been reported to elicit NSC activation and strengthen intrinsic neurogenesis as well as chemical stimulation, which could be suitable for the clinical application to stroke, because it is well established and its potential complications are manageable [32].
3 NSC Transplantation for Stroke
3.1 Interaction Between Transplanted NSC and Host Brain
Transplantation of NSCs has been proposed as a promising therapeutic strategy in almost all neurological disorders, including Parkinson’s disease [33], Huntington’s disease [34], Alzheimer’s disease [35], multiple sclerosis [36], amyotrophic lateral sclerosis [37], spinal cord injury [38], and ischemic stroke [39], which are characterized by the failure of CNS endogenous repair mechanisms to restore damaged tissue and rescue lost functions [40]. If the use of NSC transplantation is to be translated to clinical use, it is important to understand the mechanisms of action for improved recovery. The initial hypothesis assumed that NSCs would replace lost neurons and circuits. However, evidence for widespread afferent and efferent neuronal projections is lacking. NSCs prevent neuronal-programmed cell death and glial scar formation mainly via paracrine secretion of nerve growth factor, brain-derived neurotrophic factor, ciliary neurotrophic factor, and glial cell-derived neurotrophic factor. Recent preclinical data confirmed that transplanted NSCs may exert a “bystander” neuroprotective effect. Results also identified a series of molecules – immunomodulatory substances, neurotrophic growth factors, stem cell regulators, and guidance molecules secreted from NSCs, which are temporally and spatially orchestrated by environmental cues [41]. The bystander effect is a multistep process that depends on the timing of cell injection and route of cell transplantation [42]. Once injected, NSCs migrate and home to injured sites [43, 44], likely due to constitutively expressed chemokine receptors, such as CXCR4, cell adhesion molecules, and integrins, which allows the NSCs to follow chemoattractant gradients and reach damaged lesion sites [45]. Following migration to the injured areas, transplanted NSCs survive in close proximity to blood vessels (Fig. 3.2), where they interact with inflammatory cells, endothelial cells, astrocytes, and microglia. If the NSCs are transplanted into a non-injured brain, NSC migration does not occur [43, 44]. Conversely, NSCs have the potential to integrate into the injured brain after differentiation into appropriate cells. However, this remains undetermined and it is unclear whether this contributes to functional recovery. The major concern in utilizing these cells is the capacity of NSCs to form tumors, although tumorigenicity is less for fetal-derived NSCs than for embryonic-derived NSCs [46].

NSCs survive in close proximity to blood vessels. Human NSCs (a red, SCS212) and NSC-derived astrocytes (b red, hGFAP) attached to vessels (green)
3.2 Endogenous Brain Repair After NSC Transplantation
3.2.1 Angiogenesis/Neovascularization
Transplanted NSCs migrate toward infarct lesions along existing vessels. Chemoattractants, such as stromal cell-derived factor-1 [45] and monocyte chemoattractant protein 1 [47], are reported to be critical factors associated with cell migration and homing to lesions, although the interaction between transplanted NSCs and existing vessels has not been fully elucidated. Nevertheless, the increased vascularization in the peri-infarct area after stroke is associated with functional recovery [48, 49]. Subacute NSC transplantation enhances neovascularization, and stem cell-induced vascular endothelial growth factor (VEGF) plays a critical role, as well as an anti-inflammatory effect [42]. Moreover, these vascular events correspond with two patterns of functional recovery: an early mode of recovery independent of neovascularization and delayed recovery that is NSC secreted and VEGF dependent and coincides with increased vascularization [42].
Transplanted NSCs upregulate expression of tight junction proteins, such as occludin, claudin 5, and Zo-1, and contribute to blood-barrier integrity by reducing leakage [42]. Although the functional role for neovessels has not been fully established, in addition to tissue perfusion, neovessels express trophic factors that remodel damaged tissues in the brain after ischemia, form new synapses, and attract endogenous neuroblasts originating in the subventricular zone [50].
3.2.2 Immunomodulation
Inflammation also plays an important role in ischemic stroke. Experimentally and clinically, the brain responds to ischemic injury with an acute and prolonged inflammatory process characterized by rapid activation of resident microglia, production of proinflammatory mediators, and infiltration of various types of inflammatory cells into the ischemic brain tissue. However, these cellular events collaboratively contribute to secondary brain injury.
Interestingly, experimental stroke leads to splenic atrophy and spleen-derived, proinflammatory, monocyte, and macrophage mobilization into the circulation, as well as subsequent accumulation in the ischemic brain. The decreased splenic size inversely correlates with the extent of infarct volume [51, 52]. Therefore, removal of the spleen might be effective for reducing infarct volume after stroke.
Transplanted NSCs have an anti-inflammatory effect even after 2–3 weeks poststroke, and interestingly, this effect is associated with the development of neovessels [42]. Similarly to other stem cell types, NSCs exert immunomodulatory effects outside the brain upon systemic transplantation, occurring within secondary lymphoid organs [53]. NSC-secreted leukemia inhibitory factor inhibits differentiation of pathogenic Th17 cells through the extracellular signal-regulated MAP kinase suppression of the cytokine signaling 3 inhibitory signaling cascade that, in turn, antagonizes interleukin 6-mediated phosphorylation of signal transducer and activator of transcription 3, both of which are required for Th17 cell differentiation in peripheral lymphoid organs [54].
3.2.3 Axonal Sprouting, Dendritic Branching, and Synaptogenesis
Following ischemia, enhanced axonal sprouting takes place in the vicinity of the lesion, which extends from the intact cortex toward the deafferented cortical area [55, 56]. In rats, NSC grafts demonstrated increased corticocortical, corticostriatal, corticothalamic, and corticospinal axonal rewiring from the contralesional hemisphere, with transcallosal and corticospinal axonal sprouting correlating with functional recovery [57, 58]. Functional imaging has also shown similar remapping of the brain after stroke, indicating recruitment of both ipsi- and contralesional brain areas at least during the first few weeks following injury [59, 60].
Chronic changes in dendritic structural plasticity after stroke have also been reported with increased contralesional layer V dendritic branching peaking at 18 days poststroke, while ipsilesional layer III branching decreases at 9 weeks poststroke [61, 62]. NSCs enhance dendritic branching, length, and arborization at 3 weeks poststroke in layer V cortical neurons in both the ipsi- and contralesional cortex [57]. In vitro and in vivo studies have demonstrated that VEGF, thrombospondins 1 and 2, and slit act as mediators and are partially responsible for the NSC-induced effects on dendritic sprouting, axonal plasticity, and axonal transport [57, 63].
Some studies have shown that NSC transplantation enhances synaptophysin immunoreactivity in the ischemic boundary area after transplantation, suggesting that NSC transplantation enhances synaptogenesis [64–66]. Satisfactory functional recovery as a result of transplantation has been associated with increased expression of synaptogenesis markers [65]. Daadi et al. showed that NSCs increase expression of synaptic markers and enhance axonal reorganization in injured areas at 4 weeks after transplantation [67]. This was also confirmed with initial patch-clamp recording [67] and electron microscopy [66].
3.3 Modification of NSC Grafts for Transplantation
One of the main problems with NSC transplantation is the massive graft cell death, which is possibly due to a hostile host brain environment and reduced the effectiveness of this approach. It has been reported that only 1–3 % of grafted cells survive in the ischemic brain after grafting [68, 69], mainly due to inflammatory responses in the host brain after ischemia. To address these issues, approaches to modify NSCs for longer survival have been proposed. Minocycline-preconditioned NSCs have been reported to tolerate oxidative stress after ischemic reperfusion injury and express higher levels of paracrine factors [70]. Genetic manipulation of NSCs to overexpress copper/zinc-superoxide dismutase (SOD1) was also reported to enhance graft survival in an animal model with intracerebral hemorrhage [71]. This strategy could be a highly effective approach, although its safety should be validated.
4 Activation of Endogenous Neural Stem/Progenitor Cells
Animal studies have demonstrated that stem cell transplantation reduces ischemic brain injury by increasing endogenous neurogenesis and angiogenesis [50, 72, 73], even in the aging brain. Functional recovery has also been achieved using cell transplantation therapy, and results show that transplanted NSCs influence the host brain by increasing endogenous striatal neurogenesis [50]. It is important to note that graft-evoked neurogenesis varies depending on graft location and stroke type [74]. Nevertheless, it remains unclear how much stroke-induced or transplanted NSC-induced neurogenesis contributes to recovery or endogenous angiogenesis, axonal sprouting, dendritic branching, and synaptogenesis.
References
Ramon y Cajal S. Degeneration and regeneration of the nervous system. New York: Haffner Publishing Co; 1928. p. 2.
Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255(5052):1707–10.
Reynolds BA, Tetzlaff W, Weiss S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J Neurosci. 1992;12(11):4565–74.
Weiss S, Reynolds BA, Vescovi AL, Morshead C, Craig CG, van der Kooy D. Is there a neural stem cell in the mammalian forebrain? Trends Neurosci. 1996;19(9):387–93.
Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70(4):687–702. doi:10.1016/j.neuron.2011.05.001.
Aguirre A, Rubio ME, Gallo V. Notch and EGFR pathway interaction regulates neural stem cell number and self-renewal. Nature. 2010;467(7313):323–7. doi:10.1038/nature09347.
Andreu-Agullo C, Morante-Redolat JM, Delgado AC, Farinas I. Vascular niche factor PEDF modulates Notch-dependent stemness in the adult subependymal zone. Nat Neurosci. 2009;12(12):1514–23. doi:10.1038/nn.2437.
Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442(7104):823–6. doi:10.1038/nature04940.
Kawaguchi D, Furutachi S, Kawai H, Hozumi K, Gotoh Y. Dll1 maintains quiescence of adult neural stem cells and segregates asymmetrically during mitosis. Nat Commun. 2013;4:1880. doi:10.1038/ncomms2895.
Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci. 2002;3(7):517–30. doi:10.1038/nrn874.
Nieto M, Schuurmans C, Britz O, Guillemot F. Neural bHLH genes control the neuronal versus glial fate decision in cortical progenitors. Neuron. 2001;29(2):401–13.
Sun Y, Nadal-Vicens M, Misono S, Lin MZ, Zubiaga A, Hua X, et al. Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell. 2001;104(3):365–76.
Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109(1):61–73.
Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, et al. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109(1):75–86.
Imayoshi I, Isomura A, Harima Y, Kawaguchi K, Kori H, Miyachi H, et al. Oscillatory control of factors determining multipotency and fate in mouse neural progenitors. Science. 2013;342(6163):1203–8. doi:10.1126/science.1242366.
Nagao M, Campbell K, Burns K, Kuan CY, Trumpp A, Nakafuku M. Coordinated control of self-renewal and differentiation of neural stem cells by Myc and the p19ARF-p53 pathway. J Cell Biol. 2008;183(7):1243–57. doi:10.1083/jcb.200807130.
Kishi Y, Fujii Y, Hirabayashi Y, Gotoh Y. HMGA regulates the global chromatin state and neurogenic potential in neocortical precursor cells. Nat Neurosci. 2012;15(8):1127–33. doi:10.1038/nn.3165.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. doi:10.1016/j.cell.2006.07.024.
Kim SM, Flasskamp H, Hermann A, Arauzo-Bravo MJ, Lee SC, Lee SH, et al. Direct conversion of mouse fibroblasts into induced neural stem cells. Nat Protoc. 2014;9(4):871–81. doi:10.1038/nprot.2014.056.
Ruggieri M, Riboldi G, Brajkovic S, Bucchia M, Bresolin N, Comi GP, et al. Induced neural stem cells: methods of reprogramming and potential therapeutic applications. Prog Neurobiol. 2014;114:15–24. doi:10.1016/j.pneurobio.2013.11.001.
Horie N, So K, Moriya T, Kitagawa N, Tsutsumi K, Nagata I, et al. Effects of oxygen concentration on the proliferation and differentiation of mouse neural stem cells in vitro. Cell Mol Neurobiol. 2008;28(6):833–45. doi:10.1007/s10571-007-9237-y.
Morrison SJ, Csete M, Groves AK, Melega W, Wold B, Anderson DJ. Culture in reduced levels of oxygen promotes clonogenic sympathoadrenal differentiation by isolated neural crest stem cells. J Neurosci. 2000;20(19):7370–6.
Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B, et al. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci. 2000;20(19):7377–83.
Shingo T, Sorokan ST, Shimazaki T, Weiss S. Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J Neurosci. 2001;21(24):9733–43.
Horie N, Moriya T, Mitome M, Kitagawa N, Nagata I, Shinohara K. Lowered glucose suppressed the proliferation and increased the differentiation of murine neural stem cells in vitro. FEBS Lett. 2004;571(1–3):237–42. doi:10.1016/j.febslet.2004.06.085.
Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N, et al. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell. 2002;110(4):429–41.
Yoshikawa G, Momiyama T, Oya S, Takai K, Tanaka J, Higashiyama S, et al. Induction of striatal neurogenesis and generation of region-specific functional mature neurons after ischemia by growth factors. Laboratory investigation. J Neurosurg. 2010;113(4):835–50. doi:10.3171/2010.2.JNS09989.
Nakagomi T, Taguchi A, Fujimori Y, Saino O, Nakano-Doi A, Kubo S, et al. Isolation and characterization of neural stem/progenitor cells from post-stroke cerebral cortex in mice. Eur J Neurosci. 2009;29(9):1842–52. doi:10.1111/j.1460-9568.2009.06732.x.
Kokaia Z, Lindvall O. Neurogenesis after ischaemic brain insults. Curr Opin Neurobiol. 2003;13(1):127–32.
Jin K, Wang X, Xie L, Mao XO, Greenberg DA. Transgenic ablation of doublecortin-expressing cells suppresses adult neurogenesis and worsens stroke outcome in mice. Proc Natl Acad Sci U S A. 2010;107(17):7993–8. doi:10.1073/pnas.1000154107.
Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8(9):963–70. doi:10.1038/nm747.
Huang Y, Li Y, Chen J, Zhou H, Tan S. Electrical stimulation elicits neural stem cells activation: new perspectives in CNS repair. Front Hum Neurosci. 2015;9:586. doi:10.3389/fnhum.2015.00586.
Redmond Jr DE, Bjugstad KB, Teng YD, Ourednik V, Ourednik J, Wakeman DR, et al. Behavioral improvement in a primate Parkinson’s model is associated with multiple homeostatic effects of human neural stem cells. Proc Natl Acad Sci U S A. 2007;104(29):12175–80. doi:10.1073/pnas.0704091104.
Ryu JK, Kim J, Cho SJ, Hatori K, Nagai A, Choi HB, et al. Proactive transplantation of human neural stem cells prevents degeneration of striatal neurons in a rat model of Huntington disease. Neurobiol Dis. 2004;16(1):68–77. doi:10.1016/j.nbd.2004.01.016.
Zhang Q, Wu HH, Wang Y, Gu GJ, Zhang W, Xia R. Neural stem cell transplantation decreases neuroinflammation in a transgenic mouse model of Alzheimer’s disease. J Neurochem. 2015. doi:10.1111/jnc.13413.
Pluchino S, Quattrini A, Brambilla E, Gritti A, Salani G, Dina G, et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422(6933):688–94. doi:10.1038/nature01552.
Feldman EL, Boulis NM, Hur J, Johe K, Rutkove SB, Federici T, et al. Intraspinal neural stem cell transplantation in amyotrophic lateral sclerosis: phase 1 trial outcomes. Ann Neurol. 2014;75(3):363–73. doi:10.1002/ana.24113.
Ziv Y, Avidan H, Pluchino S, Martino G, Schwartz M. Synergy between immune cells and adult neural stem/progenitor cells promotes functional recovery from spinal cord injury. Proc Natl Acad Sci U S A. 2006;103(35):13174–9. doi:10.1073/pnas.0603747103.
Bliss T, Guzman R, Daadi M, Steinberg GK. Cell transplantation therapy for stroke. Stroke. 2007;38:817–26.
De Feo D, Merlini A, Laterza C, Martino G. Neural stem cell transplantation in central nervous system disorders: from cell replacement to neuroprotection. Curr Opin Neurol. 2012;25(3):322–33. doi:10.1097/WCO.0b013e328352ec45.
Horie N, Hiu T, Nagata I. Stem cell transplantation enhances endogenous brain repair after experimental stroke. Neurol Med Chir. 2015;55(2):107–12. doi:10.2176/nmc.ra.2014-0271.
Horie N, Pereira MP, Niizuma K, Sun G, Keren-Gill H, Encarnacion A, et al. Transplanted stem cell-secreted vascular endothelial growth factor effects poststroke recovery, inflammation, and vascular repair. Stem Cells. 2011;29(2):274–85. doi:10.1002/stem.584.
Bliss TM, Kelly S, Shah AK, Foo WC, Kohli P, Stokes C, et al. Transplantation of hNT neurons into the ischemic cortex: cell survival and effect on sensorimotor behavior. J Neurosci Res. 2006;83:1004–14.
Kelly S, Bliss TM, Shah AK, Sun GH, Ma M, Foo WC, et al. Transplanted human fetal neural stem cells survive, migrate, and differentiate in ischemic rat cerebral cortex. Proc Natl Acad Sci U S A. 2004;101:11839–44.
Imitola J, Raddassi K, Park KI, Mueller FJ, Nieto M, Teng YD, et al. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci U S A. 2004;101(52):18117–22. doi:10.1073/pnas.0408258102.
Seminatore C, Polentes J, Ellman D, Kozubenko N, Itier V, Tine S, et al. The postischemic environment differentially impacts teratoma or tumor formation after transplantation of human embryonic stem cell-derived neural progenitors. Stroke. 2010;41(1):153–9. doi:10.1161/STROKEAHA.109.563015.
Yan YP, Sailor KA, Lang BT, Park SW, Vemuganti R, Dempsey RJ. Monocyte chemoattractant protein-1 plays a critical role in neuroblast migration after focal cerebral ischemia. J Cereb Blood Flow Metab. 2007;27(6):1213–24. doi:10.1038/sj.jcbfm.9600432.
Krupinski J, Kaluza J, Kumar P, Wang M, Kumar S. Prognostic value of blood vessel density in ischaemic stroke. Lancet. 1993;342(8873):742.
Senior K. Angiogenesis and functional recovery demonstrated after minor stroke. Lancet. 2001;358(9284):817. doi:10.1016/S0140-6736(01)06014-7.
Mine Y, Tatarishvili J, Oki K, Monni E, Kokaia Z, Lindvall O. Grafted human neural stem cells enhance several steps of endogenous neurogenesis and improve behavioral recovery after middle cerebral artery occlusion in rats. Neurobiol Dis. 2013;52:191–203. doi:10.1016/j.nbd.2012.12.006.
Vendrame M, Gemma C, Pennypacker KR, Bickford PC, Davis Sanberg C, Sanberg PR, et al. Cord blood rescues stroke-induced changes in splenocyte phenotype and function. Exp Neurol. 2006;199(1):191–200. doi:10.1016/j.expneurol.2006.03.017.
Pennypacker KR, Offner H. The role of the spleen in ischemic stroke. J Cereb Blood Flow Metab. 2015;35(2):186–7. doi:10.1038/jcbfm.2014.212.
Lee ST, Chu K, Jung KH, Kim SJ, Kim DH, Kang KM, et al. Anti-inflammatory mechanism of intravascular neural stem cell transplantation in haemorrhagic stroke. Brain. 2008;131(Pt 3):616–29. doi:10.1093/brain/awm306.
Cao W, Yang Y, Wang Z, Liu A, Fang L, Wu F, et al. Leukemia inhibitory factor inhibits T helper 17 cell differentiation and confers treatment effects of neural progenitor cell therapy in autoimmune disease. Immunity. 2011;35(2):273–84. doi:10.1016/j.immuni.2011.06.011.
Carmichael ST. Themes and strategies for studying the biology of stroke recovery in the poststroke epoch. Stroke. 2008;39(4):1380–8. doi:10.1161/STROKEAHA.107.499962.
Carmichael ST, Wei L, Rovainen CM, Woolsey TA. New patterns of intracortical projections after focal cortical stroke. Neurobiol Dis. 2001;8(5):910–22. doi:10.1006/nbdi.2001.0425.
Andres RH, Horie N, Slikker W, Keren-Gill H, Zhan K, Sun G, et al. Human neural stem cells enhance structural plasticity and axonal transport in the ischaemic brain. Brain. 2011;134(Pt 6):1777–89. doi:10.1093/brain/awr094.
Li Y, Chen J, Zhang CL, Wang L, Lu D, Katakowski M, et al. Gliosis and brain remodeling after treatment of stroke in rats with marrow stromal cells. Glia. 2005;49(3):407–17. doi:10.1002/glia.20126.
Dijkhuizen RM, Singhal AB, Mandeville JB, Wu O, Halpern EF, Finklestein SP, et al. Correlation between brain reorganization, ischemic damage, and neurologic status after transient focal cerebral ischemia in rats: a functional magnetic resonance imaging study. J Neurosci. 2003;23(2):510–7.
Takatsuru Y, Fukumoto D, Yoshitomo M, Nemoto T, Tsukada H, Nabekura J. Neuronal circuit remodeling in the contralateral cortical hemisphere during functional recovery from cerebral infarction. J Neurosci. 2009;29(32):10081–6. doi:10.1523/JNEUROSCI.1638-09.2009.
Gonzalez CL, Kolb B. A comparison of different models of stroke on behaviour and brain morphology. Eur J Neurosci. 2003;18(7):1950–62.
Jones TA, Schallert T. Overgrowth and pruning of dendrites in adult rats recovering from neocortical damage. Brain Res. 1992;581(1):156–60.
Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, et al. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab. 2008;28(10):1722–32. doi:10.1038/jcbfm.2008.65.
Gutierrez-Fernandez M, Rodriguez-Frutos B, Ramos-Cejudo J, Teresa Vallejo-Cremades M, Fuentes B, Cerdan S, et al. Effects of intravenous administration of allogenic bone marrow- and adipose tissue-derived mesenchymal stem cells on functional recovery and brain repair markers in experimental ischemic stroke. Stem Cell Res Ther. 2013;4(1):11. doi:10.1186/scrt159.
Zhang L, Li Y, Zhang C, Chopp M, Gosiewska A, Hong K. Delayed administration of human umbilical tissue-derived cells improved neurological functional recovery in a rodent model of focal ischemia. Stroke. 2011;42(5):1437–44. doi:10.1161/STROKEAHA.110.593129.
Ding X, Li Y, Liu Z, Zhang J, Cui Y, Chen X, et al. The sonic hedgehog pathway mediates brain plasticity and subsequent functional recovery after bone marrow stromal cell treatment of stroke in mice. J Cereb Blood Flow Metab. 2013;33(7):1015–24. doi:10.1038/jcbfm.2013.50.
Daadi MM, Lee SH, Arac A, Grueter BA, Bhatnagar R, Maag AL, et al. Functional engraftment of the medial ganglionic eminence cells in experimental stroke model. Cell Transplant. 2009;18(7):815–26. doi:10.3727/096368909X470829.
Nakagomi N, Nakagomi T, Kubo S, Nakano-Doi A, Saino O, Takata M, et al. Endothelial cells support survival, proliferation, and neuronal differentiation of transplanted adult ischemia-induced neural stem/progenitor cells after cerebral infarction. Stem Cells. 2009;27(9):2185–95. doi:10.1002/stem.161.
Hicks AU, Lappalainen RS, Narkilahti S, Suuronen R, Corbett D, Sivenius J, et al. Transplantation of human embryonic stem cell-derived neural precursor cells and enriched environment after cortical stroke in rats: cell survival and functional recovery. Eur J Neurosci. 2009;29(3):562–74. doi:10.1111/j.1460-9568.2008.06599.x.
Sakata H, Niizuma K, Yoshioka H, Kim GS, Jung JE, Katsu M, et al. Minocycline-preconditioned neural stem cells enhance neuroprotection after ischemic stroke in rats. J Neurosci. 2012;32(10):3462–73. doi:10.1523/JNEUROSCI.5686-11.2012.
Wakai T, Sakata H, Narasimhan P, Yoshioka H, Kinouchi H, Chan PH. Transplantation of neural stem cells that overexpress SOD1 enhances amelioration of intracerebral hemorrhage in mice. J Cereb Blood Flow Metab. 2014;34(3):441–9. doi:10.1038/jcbfm.2013.215.
Tang Y, Wang J, Lin X, Wang L, Shao B, Jin K, et al. Neural stem cell protects aged rat brain from ischemia-reperfusion injury through neurogenesis and angiogenesis. J Cereb Blood Flow Metab. 2014;34(7):1138–47. doi:10.1038/jcbfm.2014.61.
Hsieh JY, Wang HW, Chang SJ, Liao KH, Lee IH, Lin WS, et al. Mesenchymal stem cells from human umbilical cord express preferentially secreted factors related to neuroprotection, neurogenesis, and angiogenesis. PLoS ONE. 2013;8(8), e72604. doi:10.1371/journal.pone.0072604.
Zhang P, Li J, Liu Y, Chen X, Lu H, Kang Q, et al. Human embryonic neural stem cell transplantation increases subventricular zone cell proliferation and promotes peri-infarct angiogenesis after focal cerebral ischemia. Neuropathology. 2011;31(4):384–91. doi:10.1111/j.1440-1789.2010.01182.x.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Japan KK
About this chapter
Cite this chapter
Horie, N. (2017). Neural Stem Cells/Neuronal Progenitor Cells. In: Houkin, K., Abe, K., Kuroda, S. (eds) Cell Therapy Against Cerebral Stroke. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56059-3_3
Download citation
DOI: https://doi.org/10.1007/978-4-431-56059-3_3
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56057-9
Online ISBN: 978-4-431-56059-3
eBook Packages: MedicineMedicine (R0)