
Abstract
Helicobacter pylori is a highly successful human pathogen, able to establish a chronic infection in the harsh environment of the stomach. These bacteria express a variety of virulence factors that promote their survival under acidic conditions, motility and spatial orientation in gastric mucus and adherence to epithelial cells. Other pathogenicity-associated mechanisms contribute to chronic gastritis by inducing pro-inflammatory responses and by manipulating cellular responses in host cells. Although H. pylori elicits a strong inflammatory response, the immune system fails to clear the infection. The pathogen employs a range of evasion strategies to dampen or reduce host immune responses. These strategies enable H. pylori to establish an equilibrium with its host, so that the vast majority of the chronically infected individuals do not develop severe disease. However, in a subset of patients, disturbance of this equilibrium in favour of the pathogen may lead to the development of gastroduodenal ulceration, mucosa-associated lymphoid tissue (MALT) lymphoma or adenocarcinoma.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Helicobacter pylori is one of the most successful human pathogens, colonising more than 50 % of the world’s population (Suerbaum and Josenhans 2007). The infection is usually acquired in early childhood (Weyermann et al. 2009) and, in the absence of aggressive antibiotic therapy, typically persists for life (Suerbaum and Josenhans 2007). Most of the colonised individuals develop asymptomatic chronic gastritis, but in 10–20 % of cases, H. pylori infection is associated with the development of severe gastroduodenal disease, including peptic ulcers, gastric MALT lymphoma or adenocarcinoma (Kusters et al. 2006). H. pylori infection is the strongest known risk factor for gastric adenocarcinoma, and in 1994 the International Agency for Research on Cancer classified H. pylori as a class I carcinogen. The clinical outcome of H. pylori infection depends on a complex interplay of many factors, including the virulence determinants expressed by the colonising strain(s), the genetic background of the host and environmental factors.
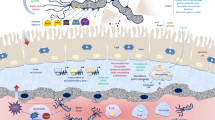
H. pylori strains show extensive genetic diversity, which is a consequence of the high mutation (Bjorkholm et al. 2001) and recombination frequencies (Suerbaum et al. 1998) in this bacterium. H. pylori has undergone a long co-evolution with its human host. Indeed, it is estimated that the association of H. pylori with modern humans predates by some 40,000 years the human migration from East Africa that occurred approximately 60,000 years ago (Moodley et al. 2012) (see Chap. 9 for more details). During this long association with humans, H. pylori has evolved sophisticated mechanisms to persistently colonise its host and avoid elimination by the immune system. These mechanisms range from colonisation factors that allow the bacterium to survive the harsh acidic environment of the stomach and establish persistent infection in the gastric mucosa, to complex strategies involving virulence factors that enable H. pylori to evade and manipulate both innate and adaptive immune responses. The lack of disease progression in the vast majority of persistently colonised individuals points to a delicate balance between the host and the pathogen. This chapter summarises the bacterial factors and biological processes that enable H. pylori to establish persistent colonisation and chronic inflammation of the human gastric mucosa. A more in-depth analysis of several of these products and processes will be provided in the chapters to follow.
2 H. pylori Colonisation and Adherence
2.1 Escape from the Stomach Lumen
In order to reach its ideal ecological niche, H. pylori must survive the extremely acidic environment of the stomach lumen and penetrate the outer mucous gel layer of the stomach. Once in the mucus, H. pylori resides in a very specific niche with an external pH of approximately 5–6. The bacterium is, however, able to increase the pH of its immediate surrounds, as well as of its cytosol and periplasm, by producing urease, which hydrolyses urea to ammonium ions and carbon dioxide (Marshall et al. 1990). Urease is composed of UreA and UreB subunits (Labigne et al. 1991) which are assembled into a catalytically active, nickel-containing dodecamer via the actions of accessory proteins UreE, UreF, UreG and UreH (Mobley et al. 1995). Urease activity is up-regulated under acidic conditions, by a proton-gated urea channel formed by the inner membrane protein, UreI, allowing rapid entry of urea into the bacteria (Skouloubris et al. 1998; Weeks et al. 2000). H. pylori can further tightly control urease activity in response to both an acidic pH and increasing concentrations of nickel ions. This occurs via up-regulation of urease gene expression by the acid-responsive signalling regulon (ArsRS) (Pflock et al. 2006) and the nickel response regulator, NikR (van Vliet et al. 2002), respectively. H. pylori mutants lacking either urease activity (through disruption of ureB) or a functional UreI were shown to be defective for colonisation in animal models of infection (Eaton et al. 2002; Skouloubris et al. 1998), thus demonstrating the essential role of urease in H. pylori pathogenesis (see also Chap. 2).
Having overcome the acidic lumen, H. pylori must next confront the viscous mucous gel covering the gastric epithelium. Gastric mucus varies in its viscoelastic properties, from a soft gel at acidic pH to a viscous solution at neutral pH. H. pylori is well adapted to this environment and is able to move rapidly, in a corkscrew like manner, through highly viscous solutions that impede the motility of rod-shaped organisms, such as Escherichia coli (Hazell et al. 1986). The spiral cell shape of H. pylori is believed to enhance its ability to penetrate the mucus, and mutants lacking a helical twist show a colonisation defect. Production and maintenance of the spiral morphology require coordinated activity of multiple enzyme networks that modify the peptidoglycan composition of the cell wall (Bonis et al. 2010; Sycuro et al. 2012).
In addition to its spiral shape, H. pylori uses flagella-mediated motility to move through both the gastric lumen and mucus to reach and maintain itself close to the epithelial surface. The importance of motility for H. pylori is illustrated by the fact that mutants lacking flagella were unable to colonise the gnotobiotic piglet model of infection (Eaton et al. 1996). H. pylori bacteria have two to five sheathed, unipolar flagella which are composed of the basal body, the hook/universal joint and the filament. The flagellar filament is composed of repeating units of two flagellins, FlaA (53 kilodalton, kDa) and FlaB (54 kDa) (Suerbaum et al. 1993). The flaA and flaB genes are not co-located on the chromosome, nor is their transcription co-regulated (Suerbaum et al. 1993). The flagellar system comprises a network of over 40 mostly unclustered and temporally regulated genes, whose transcription is hierarchical and tightly controlled by the three RNA polymerase sigma factors of H. pylori: σ28 (FliA), σ54 (RpoN) and σ80 (Douillard et al. 2009; Josenhans et al. 2002; McGowan et al. 2003; Niehus et al. 2004). H. pylori also has an anti-sigma factor, FlgM, which acts as an antagonist to FlaA (Colland et al. 2001; Niehus et al. 2004). Flagellar assembly requires interaction with the peptidoglycan layer through which the flagella has to be extruded. The peptidoglycan-degrading enzymes of the lytic transglycosylase family, Slt and MltD, are required for full motility in H. pylori (Roure et al. 2012). Inactivation of mltD, but not slt, was shown to have a significant impact on H. pylori colonisation in vivo (Roure et al. 2012).
Motile bacteria use chemotaxis for spatial orientation, coupling control of flagellar rotation with environmental sensing (Wadhams and Armitage 2004). H. pylori uses four methyl-accepting chemoreceptor proteins (TlpA, TlpB, TlpC and TlpD) to sense the external stimuli and repellent ligands. This information is then relayed via CheW, to a histidine kinase, CheA, which phosphorylates the response regulator, CheY. The phosphorylated CheY interacts with the flagellar motor to alter the rotational direction of the flagellum. H. pylori mutants lacking cheA, cheW or cheY are non-chemotactic and show colonisation defects (Terry et al. 2005). In addition, H. pylori encodes a novel chemotaxis regulator, ChePep, which preferentially localises to the flagellar pole. H. pylori chePep mutants cannot control the rotation of their flagella, but are motile. They are attenuated for colonisation of the stomach and fail to establish bacterial colonies deep in the gastric glands of mice. Interestingly, ChePep homologues are present and functionally conserved in ε-proteobacteria, but not in other bacterial classes (Howitt et al. 2011).
Microarray analyses of H. pylori showed that both exposure to low pH in vitro and infection of the gerbil stomach in vivo resulted in increased expression of many of the genes involved in motility and chemotaxis (Merrell et al. 2003; Scott et al. 2007). These findings are consistent with data showing that exposure of H. pylori to an acidic environment leads to a large increase in both the proportion and the speed of motile bacteria (Merrell et al. 2003). H. pylori exploits the pH gradient of the stomach, which ranges from pH 1.8 in the lumen to a near-neutral pH at the mucus-mucosal interface, to guide it to the epithelial surface. This pH-tactic behaviour is dependent on the chemotaxis receptor, TlpB (Croxen et al. 2006). H. pylori tlpB mutants were shown to be motile but could not colonise interleukin-12 p40 (IL-12 p40)-deficient C57BL/6 mice (Croxen et al. 2006). Expression of tlpB is regulated at the posttranscriptional level by an abundant small RNA (sRNA), regulator of polymeric G-repeats (RepG), which targets a homopolymeric G-repeat in the leader region of the tlpB mRNA. The length of this G-repeat, which varies from 6 to 16 guanine residues in different H. pylori strains, influences both the level and type (repression or activation) of regulation. There is also evidence that the length of the tlpB G-repeat can change during infection, suggesting that differential expression of tlpB may be involved in host adaptation (Pernitzsch et al. 2014).
2.2 Adhesion of H. pylori to Gastric Epithelial Cells
Although most of the H. pylori in the mucosa are free-swimming, some 20 % of the bacteria adhere to the surface of the epithelial cells (Hessey et al. 1990). Binding of H. pylori to gastric epithelial cells involves the interactions between specific bacterial adhesins and their cognate receptors on the surfaces of host cells. The known H. pylori adhesins all belong to the major outer membrane protein (OMP) family 1 (Alm et al. 2000). This family of proteins is further divided into the H elicobacter outer membrane porins (Hop) and Hop-related (Hor) subgroups (Alm et al. 2000; Odenbreit et al. 2009). All but one of the adhesins identified to date are members of the Hop family. The best characterised of these adhesins are the blood group antigen-binding ( BabA) and sialic acid-binding ( SabA) proteins, the outer inflammatory protein A (OipA) and the adherence-associated lipoproteins, AlpA and AlpB.
BabA binds to the human fucosylated Lewisb antigen (Leb) and related terminal fucose residues on blood group O (H antigen), A and B antigens present on the surface of gastric epithelial cells (Aspholm-Hurtig et al. 2004; Gerhard et al. 1999; Ilver et al. 1998). BabA binding affinity for O, A and B antigens correlates with the blood group expressed by the human host, supporting the notion of adaptation to the host during persistent infection and transmission between hosts (Aspholm-Hurtig et al. 2004). In contrast, SabA binds to sialyl-Lewis x (sLex) and Lewis a (sLea) antigens whose synthesis is up-regulated during H. pylori-induced inflammation (Mahdavi et al. 2002). SabA also binds to erythrocytes (Aspholm et al. 2006) and may play a role in inflammation, by binding and activating neutrophils (Unemo et al. 2005). More details on the effects of BabA and SabA on gastroduodenal disease can be found in Chap. 6.
OipA (formerly called HopH) is involved in the adherence of H. pylori to gastric cell lines (Dossumbekova et al. 2006; Yamaoka et al. 2004), but its host receptor has yet to be identified. Expression of functional OipA, which is regulated by slipped-strand mispairing within a CT-rich region present at the 5′ terminus of oipA, promoted IL-8 induction in vitro. Conversely, oipA inactivation in cagPAI+ clinical isolates resulted in approximately 50 % lower IL-8 responses in epithelial cells (Yamaoka et al. 2004). The notion that OipA has a role in IL-8 production in vivo is supported by the observation that OipA expression was significantly associated with high levels of IL-8 in the gastric mucosa of infected patients (Yamaoka et al. 2002). The molecular basis for the effect of OipA on IL-8 production was investigated in a study of the IL-8 promoter in gastric cell cultures (Yamaoka et al. 2004). This study showed that maximal induction of IL-8 transcription required activation of an interferon-stimulated responsive element (ISRE)-like element by the interferon regulatory factor (IRF)-1 (Yamaoka et al. 2004). Moreover, OipA was reported to selectively induce phosphorylation of signal transducer and activator of transcription 1 (STAT1), an upstream mediator of IRF-1 signalling. These results were recapitulated in vivo, where it was found that STAT1 phosphorylation in human gastric biopsy specimens correlated with the presence of functional OipA in the infecting H. pylori strain (Yamaoka et al. 2004). It has also been proposed that OipA plays a role in IL-1β, IL-17 and TNF expression and inflammation in the stomach (Sugimoto et al. 2009). Analysis of clinical isolates showed that the presence of functional OipA is associated with high H. pylori density, severe neutrophil infiltration, duodenal ulcer disease and gastric cancer (Yamaoka et al. 2002, 2006; Franco et al. 2008). The role of OipA in gastric disease is supported by data showing that inactivation of oipA reduces the incidence of cancer in Mongolian gerbils and decreases nuclear translocation of β-catenin (Franco et al. 2008), a cellular protein important for cell adhesion and regulation of genes implicated in carcinogenesis. In vitro studies with murine dendritic cells (DCs) showed that OipA suppresses DC maturation and decreases production of IL-10 (Teymournejad et al. 2014); however, the biological significance of this finding requires further investigation.
AlpA and AlpB, which are encoded by the alpA/B operon, are also required for specific adhesion of H. pylori to human gastric epithelial cells (Odenbreit et al. 1999). Furthermore, these proteins were shown to be important for colonisation in guinea pig (de Jonge et al. 2004) and murine (Lu et al. 2007) models of infection. Analysis of 200 clinical strains showed that AlpA and AlpB were expressed in all strains, suggesting that these adhesins are likely to have important functions (Odenbreit et al. 2009). The target of both AlpA and AlpB is laminin, a component of the host extracellular matrix (Senkovich et al. 2011).
3 Major H. pylori Virulence Factors Involved in Pathogenesis
3.1 cag Pathogenicity Island (cagPAI)
The cagPAI is a horizontally acquired insertion element of 40-kilobases (kb), consisting of approximately 31 genes, whose presence in a functional form is associated with an increased risk of severe gastroduodenal disease (Covacci et al. 1999; see also Chap. 4). cagPAI encodes a bacterial type IV secretion system (T4SS) and its only known effector protein, CagA, which translocates into gastric epithelial cells (Censini et al. 1996; Fischer et al. 2001; Odenbreit et al. 2000). The presence of a cagPAI appears to influence the topography of colonisation within the stomach, as cagPAI- H. pylori strains were mostly present in the mucous gel layer or near the apical surface of epithelial cells, whereas the cagPAI+ strains were found closely adjacent to gastric epithelial cells or in the intercellular epithelial spaces (Camorlinga-Ponce et al. 2004).
The H. pylori T4SS is induced by contact with the host cell and forms a large complex spanning the inner and outer membranes of the bacterium, with a pilus-like structure that protrudes from the bacterial surface (Rohde et al. 2003). It is currently unclear how the H. pylori T4SS is able to deliver not only CagA but also H. pylori cell wall peptidoglycan, into the host cell. The internalised peptidoglycan is recognised by the cytoplasmic pathogen-recognition molecule, nucleotide-binding oligomerisation domain-containing protein 1 (NOD1) (Viala et al. 2004). NOD1 sensing of H. pylori peptidoglycan triggers in epithelial cells a pro-inflammatory signalling cascade, characterised by the translocation of nuclear factor-κB (NF-κB) to the nucleus (Viala et al. 2004) and activation of the mitogen-activated protein kinases (MAPKs), p38 and extracellular signal-regulated kinase (ERK), leading to induction of CXC chemokine responses (Allison et al. 2009). H. pylori can also activate this pro-inflammatory signalling cascade via the actions of outer membrane vesicles (OMVs) which deliver peptidoglycan to cytosolic NOD1 (Hutton et al. 2010; Kaparakis et al. 2010). Both T4SS- and OMV-dependent activations of the NOD1 signalling pathway involve cholesterol-rich microdomains, or lipid rafts, in host-cell membranes (Hutton et al. 2010; Kaparakis et al. 2010). Interestingly, CagA translocation into host cells also requires the presence of functional lipid raft domains (Jimenez-Soto et al. 2009; Lai et al. 2008). H. pylori T4SS delivery of CagA (Jimenez-Soto et al. 2009; Kwok et al. 2007) and peptidoglycan (Hutton et al. 2010; Kaparakis et al. 2010) into cells was shown to be dependent upon binding of the cagPAI-encoded protein, CagL, to its cognate host-cell receptor, α5β1 integrin. CagA itself was shown to interact with the host factors, β1 integrin (Jimenez-Soto et al. 2009; Kwok et al. 2007) and phosphatidylserine (Murata-Kamiya et al. 2010).
Once CagA has translocated into epithelial cells, it localises to the plasma membrane and undergoes tyrosine phosphorylation within the EPIYA motif that is found in tandemly arranged segments located in the C-terminal half of the protein. The number and organisation of these segments differ between H. pylori strains and are thought to contribute to differences in strain pathogenicity (Argent et al. 2004; Higashi et al. 2002). There are four distinct EPIYA segments (A to D), each of which contains a single EPIYA motif, with the EPIYA A, B and C segments predominating in H. pylori isolates from Western countries and EPIYA A, B and D segments predominating in the generally more virulent East Asian isolates (Higashi et al. 2002). The cellular kinases responsible for phosphorylating the EPIYA motifs within CagA are oncoproteins belonging to the Src and Abl family kinases (Poppe et al. 2007; Selbach et al. 2002; Tammer et al. 2007).
CagA translocation and tyrosine phosphorylation lead to a perturbation of mammalian signal transduction cascades, morphological effects such as cell cytoskeletal rearrangement, elongation and scattering that has been designated the “hummingbird” phenotype, as well as modification of cellular functions (Selbach et al. 2003; Tammer et al. 2007; Tsutsumi et al. 2003). These in vitro observations are recapitulated in vivo, with the finding that CagA is actively translocated to gastric epithelial cells and tyrosine-phosphorylated and binds Src homology region 2 (SH2) domain-containing phosphatase-2 (SHP-2) in inflamed human gastric mucosa (Tsutsumi et al. 2003). The ability of CagA to perturb host-cell functions is dependent on its SHP-2 binding activity, which is determined by the number and sequences of tyrosine phosphorylation sites (Higashi et al. 2002). It should be noted that non-phosphorylated CagA also contributes to pathogenesis, through interactions that lead to induction of pro-inflammatory and mitogenic responses, suppression of apoptosis, loss of cell polarity and disruption of gastric barrier function (see Chap. 4 for a detailed discussion).
3.2 Vacuolating Cytotoxin VacA
VacA is a pore-forming toxin secreted by a classical autotransporter pathway (see Chap. 5 for more details). The protein is synthesised as a 140 kDa precursor, which is processed to produce a 33-amino acid signal peptide, the mature 88 kDa secreted toxin, an approximately 12 kDa secreted peptide and a C-terminal domain that remains associated with the bacteria (Cover and Blaser 1992; Schmitt and Haas 1994; Telford et al. 1994). The secreted VacA can undergo spontaneous proteolytic cleavage into the N-terminal p33 and C-terminal p55 fragments that are thought to represent the functional domains of VacA (Torres et al. 2005) and that remain non-covalently associated (Cover et al. 1997; Lupetti et al. 1996; Telford et al. 1994). The p33 domain is important for membrane channel formation (McClain et al. 2001; Ye et al. 1999), whereas the p55 domain is required for binding to host cells (Garner and Cover 1996). Both domains are required for toxin oligomerisation (Gangwer et al. 2007; Genisset et al. 2006). Active VacA induces structural and functional changes in epithelial cells in vitro, the most noticeable being formation of large intracellular vacuoles, the phenotype that gave the toxin its name (Leunk et al. 1988).
Most of the secreted VacA was shown to bind to cultured epithelial cells and to use lipid rafts as entry sites so as to be internalised by clathrin-independent endocytosis (Gauthier et al. 2005). A number of studies have indicated that VacA may also exert an antagonistic effect on CagA functions (see Chap. 5 for a detailed discussion). Once intracellular, VacA causes a wide range of alterations to the host cell. The large membrane-bound vacuoles induced by VacA in the cytoplasm of gastric cells originate from the late endosomal pathway and are a consequence of disruption of the late endosomal/lysosomal compartments (Papini et al. 1994). However, the role of these cytoplasmic vacuoles in H. pylori pathogenesis is unclear. As discussed in Sect. 6 below, VacA induces apoptosis of gastric epithelial cells, independently of its vacuolating activity and also promotes autophagy in these cells.
All H. pylori strains encode vacA, yet display considerable heterogeneity in their production of the vacuolating cell phenotype (Atherton et al. 1995). This diversity is largely due to polymorphisms in the vacA gene. The highest level of sequence diversity is found in the signal (s), intermediate (i) and middle (m) regions of vacA and forms the basis of a classification system. The signal sequences of the s1 and s2 alleles of VacA are processed at different sites, such that the mature s2 toxin contains a 12-amino acid hydrophilic extension at its N-terminus, which abolishes its cytotoxic activity and reduces its ability to form membrane channels, without abrogating toxin secretion (Letley et al. 2003; McClain et al. 2001). The i region, defined as either i1 or i2, is important for toxicity (Rhead et al. 2007; Winter et al. 2014); however, its role in VacA functions is not yet known. The m region of VacA contains the cell-binding site, with m1-type toxins having higher binding affinities for host cells than m2-type toxins and also showing different cell-type specificities (Pagliaccia et al. 1998; Wang et al. 2001).
H. pylori strains with the s1/m1 vacA alleles have higher levels of vacuolating activity in vitro than those carrying s1/m2 alleles (Atherton et al. 1995). Epidemiological studies are consistent with these in vitro observations, as H. pylori strains that encode s1 and m1 vacA alleles are associated with a higher risk of gastric carcinoma than strains with s2 and m2 alleles. Furthermore, s1/m1 vacA genotypes are strongly associated with peptic ulcers (Atherton et al. 1995, 1997; Strobel et al. 1998). The i1 allele of vacA shows a strong association with gastric adenocarcinoma (Rhead et al. 2007; Winter et al. 2014). Interestingly, in the murine model of infection, H. pylori bacteria producing the s2/i2 form of VacA colonised mice more efficiently than those producing the s1/i1 form of VacA or those lacking VacA, potentially suggesting a different biological role for the weakly active s2/i2 toxin. Strains producing more active VacA induced more severe and extensive metaplasia and inflammation in the mouse stomach than strains producing s2/i2 toxin (Winter et al. 2014). Thus, specific vacA alleles may contribute to the pathogenicity and clinical outcomes of H. pylori infection.
3.3 Other Putative Autotransporter Proteins of H. pylori
H. pylori genomes contain three vacA-like genes encoding proteins of 260–348 kDa (Tomb et al. 1997). The C-terminal regions of these proteins show homology to the C-terminal region of VacA, which is a β-barrel domain that is required for secretion of VacA through an autotransporter (type V) pathway. On the basis of this similarity, three proteins are predicted to be autotransporters: immunomodulating autotransporter (ImaA), flagella-associated autotransporter A (FaaA) and VacA-like protein C (VlpC) (Radin et al. 2013; Sause et al. 2012). These proteins are all present on the surface of H. pylori (Radin et al. 2013; Sause et al. 2012). However, whereas ImaA and VlpC localise to a bacterial pole, FaaA localises to a sheath overlying the flagellar filament and bulb and is important for flagellar morphology and function. The faaA mutant strain shows decreased motility, reduced flagellar stability and an increased proportion of flagella in nonpolar sites (Radin et al. 2013).
Expression levels of imaA, faaA and vlpC were up-regulated during colonisation of the mouse stomach (Radin et al. 2013; Sause et al. 2012). imaA was identified as belonging to the ArsRS regulon and thus its increased expression in vivo is likely a response to gastric acid (Sause et al. 2012). The mechanism(s) by which faaA and vlpC expression levels are regulated remain(s) unknown. Consistent with the idea that ImaA, FaaA and VlpC may have important roles in colonisation, competition experiments in mice showed that mutants for each of these autotransporters were outcompeted by wild-type bacteria in vivo (Radin et al. 2013; Sause et al. 2012). Indeed, a single challenge study confirmed that an H. pylori faaA mutant was attenuated in its ability to colonise, when compared with the wild-type strain; however, this was apparent during the early (4 days post-infection) but not late (1 month post-infection) stages of infection (Radin et al. 2013; Sause et al. 2012). It was suggested that FaaA may be important early in the infection process, when fully formed and functional flagella are required for H. pylori entry into the mucous layer (Radin et al. 2013; Sause et al. 2012). Similar to the findings above, mice challenged with H. pylori imaA mutant bacteria alone or in competition with wild-type bacteria demonstrated that this mutant also had a colonisation defect in vivo (Sause et al. 2012). In this case, however, ImaA reduced expression of inflammatory chemokines and cytokines in infected stomachs and cultured epithelial cells, suggesting that this autrotransporter may be important for dampening host immune responses (Sause et al. 2012). The immunomodulatory activity of ImaA was observed in H. pylori strains that harbour a cagPAI, suggesting that ImaA down-regulates the inflammatory responses triggered by the T4SS (Sause et al. 2012). Interestingly, ImaA exhibits some similarity to the bacterial integrin-binding protein, invasin (Sause et al. 2012). As the T4SS pilus is known to mediate its pro-inflammatory effects through binding to α5β1 integrin (Hutton et al. 2010; Kwok et al. 2007), it was suggested that ImaA and the T4SS may compete for integrin binding (Sause et al. 2012). Thus, in the absence of ImaA, the T4SS is better able to deliver the pro-inflammatory effectors, CagA and peptidoglycan (Sause et al. 2012).
3.4 γ-Glutamyl Transpeptidase
γ-Glutamyl transpeptidase (GGT) is produced by all strains of H. pylori (Chevalier et al. 1999). It plays an important role in the amino acid metabolism of H. pylori, by synthesising glutamate from both glutamine and glutathione, neither of which can be assimilated from the environment by this bacterium (Shibayama et al. 2007). H. pylori GGT hydrolyses glutamine and glutathione outside the cell, with the resulting products of this reaction being glutamate and ammonia and glutamate and cysteinylglycine, respectively. The glutamate produced is transported by a Na+-dependent reaction into H. pylori cells (Shibayama et al. 2007). The enzyme is catalytically active even at the low pH of the gastric mucosa and its expression appears to be constitutive (Chevalier et al. 1999). GGT is synthesised as a 60 kDa inactive proenzyme that undergoes autocatalytic processing to form an enzymatically active heterodimer of 40 and 20 kDa subunits (Chevalier et al. 1999; Shibayama et al. 2003).
Several lines of evidence indicate that GGT is a virulence factor of H. pylori. Although deletion of ggt did not impair the in vitro growth of H. pylori (Chevalier et al. 1999), ggt mutants were attenuated for colonisation of mice and gnotobiotic piglets (Chevalier et al. 1999; McGovern et al. 2001; Oertli et al. 2013). The degree of attenuation appears to depend on the H. pylori strain and/or the experimental animals used. It has been suggested that GGT may be associated with H. pylori-induced peptic ulcer disease (PUD) in that H. pylori isolates from patients with PUD showed significantly higher levels of GGT activity than those from patients with non-ulcer dyspepsia (Gong et al. 2010). Definitive evidence for this suggestion is, however, currently lacking.
GGT is thought to contribute to the H. pylori-induced damage to the gastric epithelial cells by promoting apoptosis and by modulating the immune response. Purified, enzymatically active GGT induced apoptosis and reduced viability in AGS gastric epithelial cells (Shibayama et al. 2003). Furthermore, GGT was shown to induce production of H2O2, leading to DNA damage, apoptosis and activation of inflammatory pathways (Gong et al. 2010). It has been suggested that GGT might contribute to the damaging effect of H. pylori on gastric cells by depleting glutamine and glutathione, which are important nutrients for maintenance of healthy gastrointestinal tissue (Shibayama et al. 2007). As discussed in Sect. 4.3 and in more detail in Chap. 5, GGT also exerts immunomodulatory effects on T-cells.
3.5 High Temperature Requirement A (HtrA) Serine Protease
H. pylori HtrA belongs to a family of serine proteases that is widely conserved in both single and multicellular organisms. This family of proteins can be distinguished from other serine proteases by their sequence homology and oligomeric structure, as well as by the presence of a protease domain and one or two carboxy terminal PDZ (post synaptic density protein, Drosophila disc large tumour suppressor, Dlg1, and zonula occludens-1 protein) domains (Clausen et al. 2011). HtrA serine proteases are involved in important cellular processes, including bacterial virulence (Clausen et al. 2011). The HtrA secreted by H. pylori (Bumann et al. 2002) cleaves the extracellular domain of the cell adhesion protein E-cadherin, present on the surface of host cells, resulting in the loss of cell-cell contact and enabling the bacterial entry into the intracellular space of epithelial cells (Hoy et al. 2010). H. pylori HtrA cleavage of E-cadherin was highly efficient at physiological and high temperatures and at pH 5–8, with highest activity observed at pH 6–7 (Hoy et al. 2013). Expression of HtrA was up-regulated by oxidative stress (Huang and Chiou 2011) and environmental acidification (Merrell et al. 2003). These characteristics of HtrA might aid H. pylori in colonising the gastric environment. Interestingly, HtrA also appears to be essential for H. pylori survival in vitro (Hoy et al. 2010; Salama et al. 2004), suggesting that the protein may have functions other than the cleavage of E-cadherin. Indeed, many HtrAs play an important role in protein quality control, with some also acting as chaperones to stabilise specific proteins (Clausen et al. 2011).
3.6 Other Pro-inflammatory Virulence Factors of H. pylori
Various new bacterial virulence factors are emerging as putative contributors to H. pylori pathogenesis in human gastric mucosa. For example, it has been suggested that the TNFα-inducing protein (Tipα, HP0596) contributes to H. pylori oncogenicity. Tipα is a homodimeric protein that has been shown to be important for colonisation of mouse gastric mucosa (Godlewska et al. 2008) and is secreted independently of the T4SS (Suganuma et al. 2005). This protein binds specifically to nucleolin, a cell surface receptor on gastric epithelial cells (Watanabe et al. 2010), whereupon it is internalised into the cytosol and then nucleus (Suganuma et al. 2008). In a mouse gastric epithelial cell line (MGT-40), Tipα was shown to induce expression of chemokine genes, such as Ccl2, Ccl7, Ccl20, Cxcl1, Cxcl2, Cxcl5 and Cxcl10 (Kuzuhara et al. 2007). Tipα was also shown to induce epithelial-mesenchymal transition in human gastric cancer cell lines (Watanabe et al. 2014).
Duodenal ulcer-promoting gene A (dupA) was originally found to be associated with an increased risk for duodenal ulcers and a reduced risk for gastric atrophy and cancer (Lu et al. 2005). Subsequent studies, however, indicated that this association held in some geographical regions, but not in others (Abadi et al. 2012; Alam et al. 2012; Arachchi et al. 2007; Argent et al. 2007; Gomes et al. 2008; Imagawa et al. 2010; Nguyen et al. 2010; Shiota et al. 2010). dupA is associated with increased IL-8 production from gastric mucosa in vivo (Hussein et al. 2010; Lu et al. 2005). This gene is located in a plasticity region and encodes a protein that is functionally homologous to the T4SS ATPase protein, VirB4. In some H. pylori genomes, dupA is located adjacent to homologues of other vir genes, and a complete dupA cluster was predicted to form a third T4SS, tfs3a (Kersulyte et al. 2009). The presence of a complete dupA cluster was found to significantly increase the risk of duodenal ulcer compared to H. pylori infection with an incomplete dupA cluster or without the dupA gene (Jung et al. 2012). These data suggest that the epidemiological studies into the role of dupA in pathogenicity should be revisited, with the focus on the presence of an intact dupA cluster. Indeed, the extensive genetic diversity of H. pylori, which contributes to its success as a pathogen, also increases the difficulty of delineating the molecular basis of the pathogenesis of H. pylori-induced diseases.
4 Avoidance and Modulation of the Host Immune Response
Although H. pylori is an extracellular pathogen, this bacterium is able to disrupt epithelial integrity (Amieva et al. 2003). Thus, H. pylori products are likely to enter the lamina propria and come into contact with immune cells (Necchi et al. 2007). Although controversial, there is also some evidence to suggest that the bacterium can invade and replicate within intracellular compartments of epithelial cells, macrophages and DCs (Ricci et al. 2011). H. pylori induces vigorous inflammatory host responses, with a large influx of neutrophils, macrophages, DCs and lymphocytes, but the immune system fails to clear the infection, attesting to the success of the various sophisticated strategies used by the pathogen to evade and subvert this system. These strategies include: evasion of the innate immune system, modulation of phagocytosis and neutrophil functions, inhibition of lymphocyte proliferation, and skewing of T-cell-mediated adaptive immune responses toward tolerogenicity (Baldari et al. 2005).
4.1 Evasion of Detection by the Innate Immune System
Host cells are able to detect conserved components of microorganisms, known as microbe-associated molecular patterns (MAMPs), via pattern recognition receptors (PRRs). The best characterised PRRs are the Toll-like receptors (TLRs), which recognise specific classes of MAMPs and respond by activating intracellular signalling pathways that lead to activation of the master transcriptional regulator, NF-κB, and pro-inflammatory gene expression. H. pylori has developed several strategies that largely allow it to avoid detection by TLRs (Takeda and Akira 2005). The best understood of these strategies involve lipopolysaccharide (LPS) and flagellin.
LPS consists of an O side chain, a core oligosaccharide and lipid A, which is anchored to the bacterial membrane. The lipid A of H. pylori LPS is predominantly tetraacylated, whereas the LPS of E. coli, which is 1,000-fold more biologically active than that of H. pylori, is hexaacylated (Moran et al. 1997). Pathogenic bacteria can evade the host innate immune system by concealing or removing the negatively charged phosphate groups present on the lipid A disaccharide backbone. The resultant net loss of negative surface charges makes the bacterial membrane more resistant to cathelicidin antimicrobial peptides (CAMPs). CAMPs, which are found in macrophages and neutrophils and at the mucosal surface, are an important component of the host innate immune response and a link between the innate and adaptive immune systems (Diamond et al. 2009). The low biological activity of H. pylori LPS has been shown to be due to the removal of phosphate groups from the 1′- and 4′-positions of lipid A by two lipid phosphatases, LpxE and LpxF, respectively (Cullen et al. 2011). Dephosphorylated LPS is attenuated for TLR4 activation and highly resistant to CAMPs. Importantly, dephosphorylation of lipid A by LpxE and LpxF is required for effective colonisation and survival of H. pylori in mice (Cullen et al. 2011). Studies indicated that H. pylori LPS initiates inflammatory signalling in human epithelial cells via TLR2, rather than the more classical sensor of Gram-negative LPS, TLR4 (Smith et al. 2011; Yokota et al. 2007). TLR2 recognises a variety of microbial components, including lipoproteins, lipoteichoic acid and atypical LPS molecules whose structures differ from those recognised by TLR4 in the number of acyl chains in the lipid A moiety (Takeda and Akira 2005).
Recognition of many but not all bacterial flagellins by TLR5, which is present on the membrane of various cell types, including epithelial cells, leads to activation of the innate immune system (Takeda and Akira 2005). However, the major flagellin of H. pylori, FlaA, is much less well recognised by TLR5 than the flagellins of other enteric mucosal pathogens, such as Salmonella typhimurium (Gewirtz et al. 2004). Moreover, unlike the flagellins of Escherichia and Salmonella, FlaA is not released from the bacteria. The evolutionarily conserved recognition sequence for TLR5 is located in the N-terminal D1 domain of bacterial flagellin, within a region that is required for flagellar filament assembly and motility. Substitution of amino acids 89–96 of the flagellin (FliC) from S. typhimurium, with the corresponding amino acids from H. pylori FlaA, abolishes its recognition by TLR5 but also renders the bacteria non-motile (Andersen-Nissen et al. 2005). H. pylori has preserved its motility by selecting for compensatory changes in other regions of FlaA, suggesting that avoidance of detection by TLR5 is important for the persistence of H. pylori at mucosal sites (Andersen-Nissen et al. 2005).
4.2 Modulation of Phagocytosis and Neutrophil Function
The engulfment and killing of microorganisms by the process known as phagocytosis are an important part of host innate defence against many pathogens. The role of macrophages in H. pylori pathogenesis, however, remains very controversial. Indeed, professional phagocytes appear to be ineffective in killing H. pylori. Reduced levels of H. pylori opsonisation by phagocytes have been attributed to its urease activity (Rokita et al. 1998) and the environmental conditions in the stomach (Berstad et al. 1997). Furthermore, cagPAI+ H. pylori strains are able to retard phagocytosis in a cagPAI-dependent but CagA-, VacA- and urease-independent manner (Allen et al. 2000; Ramarao et al. 2000). Following their engulfment, these more virulent H. pylori strains stimulate rapid and extensive homotypic phagosome fusion, leading to formation of megasomes containing large numbers of viable H. pylori. Formation of these megasomes, which were shown to be stable for 24 h, requires live, metabolically active H. pylori (Allen et al. 2000). cagPAI+ H. pylori strains induce the recruitment and retention of coronin 1 protein on phagosomes and prevent phagosome fusion with lysosomes (Zheng and Jones 2003). This inhibition of phagosome maturation is dependent on VacA and urease (Schwartz and Allen 2006; Zheng and Jones 2003). Although H. pylori strains that do not encode cagPAI, and which express a non-toxigenic form of VacA, are capable of subverting bacterial killing by macrophages for up to 24 h, their survival is inferior to that shown by cagPAI+ bacteria (Zheng and Jones 2003). The ability of toxigenic alleles of VacA to modulate autophagy may also contribute to the survival of H. pylori in macrophages, by allowing the surviving phagocytosed bacteria to escape killing (Raju et al. 2012). Despite the in vitro evidence for H. pylori survival in macrophages, further investigations are required in in vivo models to confirm the biological relevance of these observations.
One mechanism by which H. pylori has been shown to be capable of interfering with its phagocytosis by antigen-presenting cells is via the actions of a cholesterol-α-glucosyltransferase (HP0421). This enzyme, also known as type 1 capsular polysaccharide biosynthesis protein J (CapJ), catalyses the conversion of cholesterol to cholesteryl α-glucosides (Lebrun et al. 2006; Wunder et al. 2006). Although H. pylori is auxotrophic for cholesterol, its envelope contains high concentrations of cholesteryl glucosides (Tannaes and Bukholm 2005). The pathogen extracts cholesterol from the plasma membranes of epithelial cells, but excessive cholesterol promotes phagocytosis of the bacteria by antigen-presenting cells, thereby enhancing T-cell activation. Conversely, α-glucosylation of cholesterol by cholesterol-α-glucosyltransferase abrogates phagocytosis of H. pylori and T-cell activation (Wunder et al. 2006). In addition to these effects, cholesterol glucosylation by CapJ is important for tight binding of H pylori to gastric epithelial cells and for the assembly of a functional T4SS, as a capJ mutant was impaired in its ability to translocate CagA into the cytosol of host cells (Wang et al. 2012).
4.3 Inhibition of Lymphocyte Proliferation
H pylori uses the secreted proteins VacA (Gebert et al. 2003) and GGT (Shibayama et al. 2007), to inhibit lymphocyte activation and proliferation. VacA is able to inhibit proliferation of primary human B lymphocytes, as well as CD4+ and CD8+ T-cells (Torres et al. 2007). The VacA receptor on human immune cells is the β2 (CD18) integrin subunit (Sewald et al. 2008). In transformed Jurkat T-cells, VacA was shown to down-regulate transcription of IL-2, required for efficient lymphocyte activation and proliferation (Gebert et al. 2003). VacA does this by blocking nuclear translocation of the global regulator of immune response genes, nuclear factor of activated T-cells (NFAT). In activated primary human T-cells, VacA has also been shown to inhibit IL-2-driven cell-cycle progression independently of IL-2 secretion, by blocking the activation of regulatory proteins important for G1 cell-cycle transition (Oswald-Richter et al. 2006; Sundrud et al. 2004). Interestingly, murine T-cells, splenocytes and CD4+ T-cells do not significantly respond to VacA and this resistance is, at least in part, due to the impaired binding of VacA to murine cells (Algood et al. 2007; Sewald et al. 2008).
In common with H. pylori VacA, GGT inhibits proliferation of stimulated primary T-cells and peripheral blood mononuclear cells (PBMCs), but without affecting secretion of IL-2 (or IFN-γ) and without induction of apoptosis (Oertli et al. 2013; Schmees et al. 2007). The inhibition of lymphocyte proliferation involves induction of cell-cycle arrest in G1 phase through disruption of Ras-dependent signalling and requires the structural integrity of the catalytic domain of GGT and the presence of glutamine (Oertli et al. 2013; Schmees et al. 2007). It has been suggested that the inhibitory effect of GGT on T-cells is mediated indirectly by the formation of metabolites during transpeptidation (Oertli et al. 2013; Schmees et al. 2007).
4.4 Skewing of Adaptive Immune Responses Toward Tolerogenicity
H. pylori bacteria can manipulate adaptive immune responses to promote their persistence. One mechanism by which this may occur is via the preferential induction of regulatory T-cell (Treg) responses. This is evident in heavily colonised but asymptomatic carriers, who show Treg-predominant responses (Robinson et al. 2008). Similar findings were observed in a mouse model, in which depletion of Tregs led to spontaneous clearance of the infection (Arnold et al. 2011). H. pylori-induced disease in humans was associated with low Treg responses and significantly higher levels of gastric T-helper 1 (Th1) and Th2 cells, whereas in disease-free infected subjects the balance was shifted toward elevated Tregs and a reduced T-helper response (Robinson et al. 2008). Thus, it was suggested that disease is a consequence of an inadequate regulatory response to H. pylori infection (Robinson et al. 2008).
DCs play a crucial role as a link between innate and adaptive immunity, being exquisitely adept at acquiring, processing and presenting antigens to T-cells. DCs present antigens in a way that promotes tolerance, at least in part, via regulation of Treg responses (Maldonado and von Andrian 2010). Priming by tolerogenic DCs converts naive T-cells into FoxP3+ Tregs through antigen presentation in the absence of co-stimulatory signals or cytokines (Maldonado and von Andrian 2010). H. pylori is able to reprogram DCs toward a tolerogenic phenotype in vitro and in vivo. Indeed, DCs that have been exposed to H. pylori appear to preferentially prime Tregs over pro-inflammatory Th1 and Th17 responses and also fail to produce pro-inflammatory cytokines (Kao et al. 2010; Oertli et al. 2012; Wang et al. 2010). The importance of DCs in the development of H. pylori-specific immune tolerance is highlighted by the finding that systemic depletion of DCs breaks tolerance and facilitates clearance of the bacteria (Oertli et al. 2012). H. pylori VacA and GGT proteins play critical, non-redundant and non-synergistic roles in the tolerising effects of this pathogen on murine DCs in vitro and in vivo, by mechanisms that are independent of their suppressive activities on T-cells. Isogenic H. pylori mutants lacking either GGT or VacA are incapable of preventing LPS-induced DC maturation, fail to drive DC tolerisation and are attenuated for mouse colonisation (Oertli et al. 2013). Furthermore, vacA mutants induce stronger Th1 and Th17 responses and more severe gastric pathology (Oertli et al. 2013). VacA and GGT were reported to induce the expression of miR-155 and Foxp3 in human lymphocytes via a cAMP-dependent pathway (Fassi Fehri et al. 2010). Both VacA and GGT promote efficient induction of Tregs in vivo, while VacA is required to prevent allergen-induced asthma. The immunomodulatory effects of GGT are dependent on its enzymatic activity, whereas those of VacA are not linked to its vacuolating cytotoxicity, as strains expressing the toxigenic (s1/m1) or non-toxigenic (s2/m2) forms of VacA are equally tolerogenic in vitro (Oertli et al. 2013).
5 Mitigation of Inflammatory Responses
H. pylori infection leads to chronic gastritis, which generates reactive oxygen species (ROS) and nitric oxide (NO). The pathogen limits the bactericidal effects of these pro-inflammatory mediators, enabling it to chronically colonise its host. The inflammatory response induced by H. pylori generates large amounts of ROS, which encompass superoxide anions, hydroxyl radicals and hydrogen peroxide. H. pylori survive these oxidative stress conditions using a variety of stress resistance proteins. These include catalase (KatA), superoxide dismutase (SodB) and three peroxidases, an alkylhydroperoxide reductase (AhpC) and two thiolperoxidases (Tpx and bacterioferritin comigratory protein, Bcp), which catalyse the breakdown of hydrogen peroxide, superoxide and organic peroxides, respectively. H. pylori also encodes the neutrophil activating protein (NapA), which sequesters toxic levels of iron, and NADPH quinone reductase, MdaB (Stent et al. 2012). Furthermore, H. pylori bacteria respond to inactivation of important oxidative stress resistance proteins by increasing the expression of their oxidative stress resistance proteins, including KatA, SodB and NapA (Olczak et al. 2005). Disruption of katA, sodB, ahpC, tpx, bcp or mdaB in H. pylori results in an oxidative stress-sensitive phenotype that severely affects the ability of mutants to colonise the stomach (Harris et al. 2003; Olczak et al. 2002, 2003; Seyler et al. 2001; Wang and Maier 2004; Wang et al. 2005).
Infection by H. pylori leads to up-regulation of inducible nitric oxide synthase (iNOS) in the gastric mucosa, leading to mucosal damage. Data obtained using cultured macrophages indicate that this induction of iNOS is dependent on the urease released from H. pylori (Gobert et al. 2002). H. pylori bacteria mitigate the bactericidal effects of NO, which is generated during the conversion of L-arginine to L-citrulline by iNOS, via the actions of an arginase, RocF (Gobert et al. 2001). The constitutively produced RocF inhibits production of NO by competing with the host for L-arginine, which is hydrolysed to L-ornithine and urea; the latter is then used as a substrate by urease. Loss of RocF activity leads to significant NO-dependent killing of H. pylori in vitro (Gobert et al. 2001). However, rocF is not essential for H. pylori colonisation of wild-type (McGee et al. 1999) or arginase II knockout mice (Kim et al. 2011). RocF activity is stimulated by Trx1 (HP0824), one of the two thioredoxins of H. pylori. Trx1 acts as a chaperone, converting denatured or suboptimally folded RocF into its catalytically active structure and reversing the damage caused by reactive oxygen and nitrogen intermediates (McGee et al. 2006).
6 Modulation of Apoptosis and Autophagy by H. pylori
Apoptosis and autophagy are intricately connected but opposing processes that can be induced in response to cellular stress and must be finely balanced to regulate cell death and survival. Perturbation of this balance can lead to pathologies such as cancer. H. pylori is capable of inducing and inhibiting both apoptosis and autophagy. The major known H. pylori virulence factors involved in these processes are VacA, CagA and GGT.
6.1 Apoptosis
VacA induces apoptosis of gastric epithelial cells by targeting the mitochondria, where it accumulates in the inner membrane and causes depolarisation, outer membrane permeability, cytochrome C release and mitochondrial fragmentation (Cover et al. 2003; Galmiche et al. 2000; Willhite et al. 2003; Yamasaki et al. 2006). The ability of VacA to form anion-selective membrane channels is required for cytochrome C release, mitochondrial outer membrane permeabilisation (MOMP) and cell death (Willhite and Blanke 2004). VacA induces the activation of the pro-apoptotic proteins, Bax and Bcl-2 homologous antagonist/killer (Bak), thus leading to apoptosis (Yamasaki et al. 2006). VacA-mediated MOMP and activation of Bak require the mitochondrial recruitment and hyperactivation of dynamin-related protein 1 (Drp1), a critical regulator of mitochondrial fission within cells (Jain et al. 2011). GGT induces apoptosis in gastric epithelial cells via a mitochondria-mediated pathway (Kim et al. 2007) and by inducing the loss of survivin, an inhibitor of apoptosis (Valenzuela et al. 2013). Recently, another putative H. pylori virulence factor, HP0986 ( TNFR1 interacting endonuclease A, TieA), was reported to actively induce apoptosis in cultured human and murine macrophages via a Fas-mediated pathway (Alvi et al. 2011).
There is also evidence that H. pylori can prevent or block apoptosis. In a transgenic mouse model, H. pylori CagA was shown to interact with the apoptosis-stimulating protein of p53 (ASPP2) and thereby inhibit apoptosis by promoting proteasomal degradation of the p53 tumour suppressor (Buti et al. 2011). Host cells that had been co-cultured with H. pylori and then treated with the p53-activating drug doxorubicin were more resistant to apoptosis than cells not exposed to the bacterium (Buti et al. 2011). In a separate study, it was reported that CagA is also able to mediate activation of the pro-survival factor ERK and the anti-apoptotic protein, myeloid leukaemia cell differentiation protein 1 (Mcl-1) (Mimuro et al. 2007). Using the Mongolian gerbil model, the authors showed that H. pylori is able to activate the ERK-Mcl-1 pathway in vivo so as to suppress apoptosis in gastric pit cells, thereby leading to gland hyperplasia and persistent bacterial colonisation (Mimuro et al. 2007). In agreement with these findings, another group demonstrated H. pylori CagA-dependent induction of Mcl-1 expression via up-regulation of a known negative regulator of Mcl-1, the tumour suppressor microRNA (miRNA) miR-320 (Noto et al. 2013). Consistent with the Mongolian gerbil data, H. pylori was shown to induce Mcl-1 expression in a CagA-dependent manner in the murine gastric mucosa, as well as in tissues from a human population at high risk for gastric cancer (Noto et al. 2013). Moreover, Mcl-1 epithelial expression levels increased at each stage of neoplastic progression in gastric tissues from human subjects infected with cagA + strains of H. pylori (Noto et al. 2013). Thus, down-regulation of miR-320 and subsequent induction of Mcl-1 by cagA + H. pylori strains suppresses apoptosis, potentially promoting H. pylori persistence within the gastric mucosa but also possibly gastric carcinogenesis.
6.2 Autophagy
Autophagy is a tightly controlled major catabolic pathway in eukaryotes, which is required for the lysosomal/vacuolar degradation of cytoplasmic proteins and organelles. H. pylori induces autophagy in gastric epithelial cells (Tang et al. 2012; Terebiznik et al. 2009), as well as in professional phagocytes (Wang et al. 2010). VacA is necessary and sufficient to induce autophagy in gastric epithelial cells, with the induction of autophagy responsible for a decrease in the levels of intracellular VacA and vacuole biogenesis within intoxicated cells. Thus, autophagy may represent a mechanism by which host cells limit VacA-mediated damage (Terebiznik et al. 2009). In the AZ-521 human gastric epithelial cell line, VacA-induced autophagy was shown to be mediated by VacA binding to and internalisation via the low-density-lipoprotein receptor-related protein 1 (LRP1) (Yahiro et al. 2012). Knockdown of LRP1 abrogated VacA internalisation and significantly down-regulated autophagy in vitro (Yahiro et al. 2012). LRP1 is also required for the induction of autophagy-mediated degradation of CagA in response to m1 forms of VacA in AGS gastric epithelial cells. Signalling through p53 degradation is involved in m1VacA-induced autophagy in these cells (Tsugawa et al. 2012).
In addition to the effects of VacA in promoting autophagy, this cytotoxin can also block autophagy. Indeed, prolonged exposure of human gastric epithelial cells to VacA was found to disrupt toxin-induced autophagy, by blocking the maturation of autolysosomes (Raju et al. 2012). VacA alters the degradative properties of the endocytic pathway by subverting the sorting and activation of cathepsin enzymes (Satin et al. 1997). VacA-induced autolysosomes lack the key lysosomal hydrolase, cathepsin D, and so cannot complete the process of degrading their cargo, leading to accumulation of ROS and the signalling adaptor, p62 (Raju et al. 2012). Interestingly, impairment of autophagy and the accumulation of p62 lead to enhanced tumourigenicity (Moscat and Diaz-Meco 2009). Further work is required to determine whether VacA subversion of autophagy may promote gastric cancer development.
Finally, H. pylori can modulate autophagy through a mechanism involving the microRNA, MIR30B (Tang et al. 2012). Expression of MIR30B was elevated in gastric mucosal tissues from infected patients, as well as during infection of gastric epithelial cell lines, and this effect was a specific response to H. pylori. Moreover, elevated MIR30B expression in human gastric tissues was inversely correlated with the mRNA levels of the genes encoding two of the proteins that are important in regulating autophagy, autophagy-related protein 12 (ATG12) and BCL2-interacting coiled-coil protein (BECN1). Inhibition of autophagy by MIR30B was demonstrated to increase the number of VacA-dependent large vacuoles and enhanced the intracellular survival of the pathogen, demonstrating that autophagy is involved in regulating the levels of intracellular VacA (Tang et al. 2012). Taken together, these data show that H. pylori VacA is able to use different strategies to interfere with autophagy in gastric epithelial cells.
7 Conclusions and Outlook
H. pylori is arguably one of the most successful human pathogens, persistently colonising the gastric mucosa of more than 50 % of the world’s population. This pathogen induces vigorous inflammatory host responses. However, due to the many effective strategies employed by the bacterium to subvert host immune responses, the infection cannot be easily cleared. H. pylori uses its numerous virulence factors to establish chronic infection, alter cellular signalling cascades, cause damage to the mucosa and modulate host immune responses. The multitude of virulence factors, together with their complex interactions and allelic variations, render it difficult to dissect the individual contributions of these factors to the chronicity of H. pylori infection and its long-term consequences. Moreover H. pylori is not only highly heterogeneous but also genetically unstable, adding to the difficulty in studying the virulence mechanisms of this human pathogen. As discussed here, H. pylori can alter cell proliferation, apoptosis and autophagy processes, as well as down-regulate cellular tumour suppressor genes. All together, these changes contribute to oncogenesis and the development of more severe gastric disease. Although the last decade has seen great advances in our understanding of the virulence mechanisms of H. pylori, much of this knowledge has been gained from experiments conducted in vitro or in animal models and awaits confirmation from clinical and epidemiological studies. Such studies must encompass populations from diverse geographic locations, as both bacterial and host polymorphisms are likely to contribute to the pathogenesis of H. pylori infection in humans.
References
Abadi AT, Taghvaei T, Wolfram L, Kusters JG (2012) Infection with Helicobacter pylori strains lacking dupA is associated with an increased risk of gastric ulcer and gastric cancer development. J Med Microbiol 61:23–30
Alam J, Maiti S, Ghosh P, De R, Chowdhury A, Das S, Macaden R, Devarbhavi H, Ramamurthy T, Mukhopadhyay AK (2012) Significant association of the dupA gene of Helicobacter pylori with duodenal ulcer development in a South-east Indian population. J Med Microbiol 61:1295–1302
Algood HM, Torres VJ, Unutmaz D, Cover TL (2007) Resistance of primary murine CD4+ T cells to Helicobacter pylori vacuolating cytotoxin. Infect Immun 75:334–341
Allen LA, Schlesinger LS, Kang B (2000) Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J Exp Med 191:115–128
Allison CC, Kufer TA, Kremmer E, Kaparakis M, Ferrero RL (2009) Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J Immunol 183:8099–8109
Alm RA, Bina J, Andrews BM, Doig P, Hancock RE, Trust TJ (2000) Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect Immun 68:4155–4168
Alvi A, Ansari SA, Ehtesham NZ, Rizwan M, Devi S, Sechi LA, Qureshi IA, Hasnain SE, Ahmed N (2011) Concurrent proinflammatory and apoptotic activity of a Helicobacter pylori protein (HP986) points to its role in chronic persistence. PLoS ONE 6:e22530
Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S (2003) Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300:1430–1434
Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, Logan SM, Aderem A (2005) Evasion of Toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci U S A 102:9247–9252
Arachchi HS, Kalra V, Lal B, Bhatia V, Baba CS, Chakravarthy S, Rohatgi S, Sarma PM, Mishra V, Das B, Ahuja V (2007) Prevalence of duodenal ulcer-promoting gene (dupA) of Helicobacter pylori in patients with duodenal ulcer in North Indian population. Helicobacter 12:591–597
Argent RH, Kidd M, Owen RJ, Thomas RJ, Limb MC, Atherton JC (2004) Determinants and consequences of different levels of CagA phosphorylation for clinical isolates of Helicobacter pylori. Gastroenterology 127:514–523
Argent RH, Burette A, Miendje Deyi VY, Atherton JC (2007) The presence of dupA in Helicobacter pylori is not significantly associated with duodenal ulceration in Belgium, South Africa, China, or North America. Clin Infect Dis 45:1204–1206
Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, Sparwasser T, Muller A (2011) Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 140:199–209
Aspholm M, Olfat FO, Norden J, Sonden B, Lundberg C, Sjostrom R, Altraja S, Odenbreit S, Haas R, Wadstrom T, Engstrand L, Semino-Mora C, Liu H, Dubois A, Teneberg S, Arnqvist A, Boren T (2006) SabA is the H. pylori hemagglutinin and is polymorphic in binding to sialylated glycans. PLoS Pathog 2:e110
Aspholm-Hurtig M, Dailide G, Lahmann M, Kalia A, Ilver D, Roche N, Vikstrom S, Sjostrom R, Linden S, Backstrom A, Lundberg C, Arnqvist A, Mahdavi J, Nilsson UJ, Velapatino B, Gilman RH, Gerhard M, Alarcon T, Lopez-Brea M, Nakazawa T, Fox JG, Correa P, Dominguez-Bello MG, Perez-Perez GI, Blaser MJ, Normark S, Carlstedt I, Oscarson S, Teneberg S, Berg DE, Boren T (2004) Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science 305:519–522
Atherton JC, Cao P, Peek RM Jr, Tummuru MK, Blaser MJ, Cover TL (1995) Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem 270:17771–17777
Atherton JC, Peek RM Jr, Tham KT, Cover TL, Blaser MJ (1997) Clinical and pathological importance of heterogeneity in vacA, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology 112:92–99
Baldari CT, Lanzavecchia A, Telford JL (2005) Immune subversion by Helicobacter pylori. Trends Immunol 26:199–207
Berstad AE, Brandtzaeg P, Stave R, Halstensen TS (1997) Epithelium related deposition of activated complement in Helicobacter pylori associated gastritis. Gut 40:196–203
Bjorkholm B, Sjolund M, Falk PG, Berg OG, Engstrand L, Andersson DI (2001) Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc Natl Acad Sci U S A 98:14607–14612
Bonis M, Ecobichon C, Guadagnini S, Prevost MC, Boneca IG (2010) A M23B family metallopeptidase of Helicobacter pylori required for cell shape, pole formation and virulence. Mol Microbiol 78:809–819
Bumann D, Aksu S, Wendland M, Janek K, Zimny-Arndt U, Sabarth N, Meyer TF, Jungblut PR (2002) Proteome analysis of secreted proteins of the gastric pathogen Helicobacter pylori. Infect Immun 70:3396–3403
Buti L, Spooner E, Van der Veen AG, Rappuoli R, Covacci A, Ploegh HL (2011) Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc Natl Acad Sci U S A 108:9238–9243
Camorlinga-Ponce M, Romo C, Gonzalez-Valencia G, Munoz O, Torres J (2004) Topographical localisation of cagA positive and cagA negative Helicobacter pylori strains in the gastric mucosa; an in situ hybridisation study. J Clin Pathol 57:822–828
Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A (1996) cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U S A 93:14648–14653
Chevalier C, Thiberge JM, Ferrero RL, Labigne A (1999) Essential role of Helicobacter pylori γ-glutamyltranspeptidase for the colonization of the gastric mucosa of mice. Mol Microbiol 31:1359–1372
Clausen T, Kaiser M, Huber R, Ehrmann M (2011) HTRA proteases: regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol 12:152–162
Colland F, Rain JC, Gounon P, Labigne A, Legrain P, De Reuse H (2001) Identification of the Helicobacter pylori anti-sigma 28 factor. Mol Microbiol 41:477–487
Covacci A, Telford JL, Del Giudice G, Parsonnet J, Rappuoli R (1999) Helicobacter pylori virulence and genetic geography. Science 284:1328–1333
Cover TL, Blaser MJ (1992) Purification and characterization of the vacuolating toxin from Helicobacter pylori. J Biol Chem 267:10570–10575
Cover TL, Hanson PI, Heuser JE (1997) Acid-induced dissociation of VacA, the Helicobacter pylori vacuolating cytotoxin, reveals its pattern of assembly. J Cell Biol 138:759–769
Cover TL, Krishna US, Israel DA, Peek RM Jr (2003) Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res 63:951–957
Croxen MA, Sisson G, Melano R, Hoffman PS (2006) The Helicobacter pylori chemotaxis receptor TlpB (HP0103) is required for pH taxis and for colonization of the gastric mucosa. J Bacteriol 188:2656–2665
Cullen TW, Giles DK, Wolf LN, Ecobichon C, Boneca IG, Trent MS (2011) Helicobacter pylori versus the host: remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PLoS Pathog 7:e1002454
de Jonge R, Durrani Z, Rijpkema SG, Kuipers EJ, van Vliet AH, Kusters JG (2004) Role of the Helicobacter pylori outer-membrane proteins AlpA and AlpB in colonization of the guinea pig stomach. J Med Microbiol 53:375–379
Diamond G, Beckloff N, Weinberg A, Kisich KO (2009) The roles of antimicrobial peptides in innate host defense. Curr Pharm Des 15:2377–2392
Dossumbekova A, Prinz C, Mages J, Lang R, Kusters JG, Van Vliet AH, Reindl W, Backert S, Saur D, Schmid RM, Rad R (2006) Helicobacter pylori HopH (OipA) and bacterial pathogenicity: genetic and functional genomic analysis of hopH gene polymorphisms. J Infect Dis 194:1346–1355
Douillard FP, Ryan KA, Hinds J, O’Toole PW (2009) Effect of FliK mutation on the transcriptional activity of the {sigma}54 sigma factor RpoN in Helicobacter pylori. Microbiology 155:1901–1911
Eaton KA, Suerbaum S, Josenhans C, Krakowka S (1996) Colonization of gnotobiotic piglets by Helicobacter pylori deficient in two flagellin genes. Infect Immun 64:2445–2448
Eaton KA, Gilbert JV, Joyce EA, Wanken AE, Thevenot T, Baker P, Plaut A, Wright A (2002) In vivo complementation of ureB restores the ability of Helicobacter pylori to colonize. Infect Immun 70:771–778
Fassi Fehri L, Koch M, Belogolova E, Khalil H, Bolz C, Kalali B, Mollenkopf HJ, Beigier-Bompadre M, Karlas A, Schneider T, Churin Y, Gerhard M, Meyer TF (2010) Helicobacter pylori induces miR-155 in T cells in a cAMP-Foxp3-dependent manner. PLoS One 5:e9500
Fischer W, Puls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R (2001) Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol 42:1337–1348
Franco AT, Johnston E, Krishna U, Yamaoka Y, Israel DA, Nagy TA, Wroblewski LE, Piazuelo MB, Correa P, Peek RM Jr (2008) Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res 68:379–387
Galmiche A, Rassow J, Doye A, Cagnol S, Chambard JC, Contamin S, de Thillot V, Just I, Ricci V, Solcia E, Van Obberghen E, Boquet P (2000) The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J 19:6361–6370
Gangwer KA, Mushrush DJ, Stauff DL, Spiller B, McClain MS, Cover TL, Lacy DB (2007) Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc Natl Acad Sci U S A 104:16293–16298
Garner JA, Cover TL (1996) Binding and internalization of the Helicobacter pylori vacuolating cytotoxin by epithelial cells. Infect Immun 64:4197–4203
Gauthier NC, Monzo P, Kaddai V, Doye A, Ricci V, Boquet P (2005) Helicobacter pylori VacA cytotoxin: a probe for a clathrin-independent and Cdc42-dependent pinocytic pathway routed to late endosomes. Mol Biol Cell 16:4852–4866
Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R (2003) Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301:1099–1102
Genisset C, Galeotti CL, Lupetti P, Mercati D, Skibinski DA, Barone S, Battistutta R, de Bernard M, Telford JL (2006) A Helicobacter pylori vacuolating toxin mutant that fails to oligomerize has a dominant negative phenotype. Infect Immun 74:1786–1794
Gerhard M, Lehn N, Neumayer N, Boren T, Rad R, Schepp W, Miehlke S, Classen M, Prinz C (1999) Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci U S A 96:12778–12783
Gewirtz AT, Yu Y, Krishna US, Israel DA, Lyons SL, Peek RM Jr (2004) Helicobacter pylori flagellin evades toll-like receptor 5-mediated innate immunity. J Infect Dis 189:1914–1920
Gobert AP, McGee DJ, Akhtar M, Mendz GL, Newton JC, Cheng Y, Mobley HL, Wilson KT (2001) Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proc Natl Acad Sci U S A 98:13844–13849
Gobert AP, Mersey BD, Cheng Y, Blumberg DR, Newton JC, Wilson KT (2002) Cutting edge: urease release by Helicobacter pylori stimulates macrophage inducible nitric oxide synthase. J Immunol 168:6002–6006
Godlewska R, Pawlowski M, Dzwonek A, Mikula M, Ostrowski J, Drela N, Jagusztyn-Krynicka EK (2008) Tip-α (hp0596 gene product) is a highly immunogenic Helicobacter pylori protein involved in colonization of mouse gastric mucosa. Curr Microbiol 56:279–286
Gomes LI, Rocha GA, Rocha AM, Soares TF, Oliveira CA, Bittencourt PF, Queiroz DM (2008) Lack of association between Helicobacter pylori infection with dupA-positive strains and gastroduodenal diseases in Brazilian patients. Int J Med Microbiol 298:223–230
Gong M, Ling SS, Lui SY, Yeoh KG, Ho B (2010) Helicobacter pylori γ-glutamyl transpeptidase is a pathogenic factor in the development of peptic ulcer disease. Gastroenterology 139:564–573
Harris AG, Wilson JE, Danon SJ, Dixon MF, Donegan K, Hazell SL (2003) Catalase (KatA) and KatA-associated protein (KapA) are essential to persistent colonization in the Helicobacter pylori SS1 mouse model. Microbiology 149:665–672
Hazell SL, Lee A, Brady L, Hennessy W (1986) Campylobacter pyloridis and gastritis: association with intercellular spaces and adaptation to an environment of mucus as important factors in colonization of the gastric epithelium. J Infect Dis 153:658–663
Hessey SJ, Spencer J, Wyatt JI, Sobala G, Rathbone BJ, Axon AT, Dixon MF (1990) Bacterial adhesion and disease activity in Helicobacter associated chronic gastritis. Gut 31:134–138
Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, Hatakeyama M (2002) Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci U S A 99:14428–14433
Howitt MR, Lee JY, Lertsethtakarn P, Vogelmann R, Joubert LM, Ottemann KM, Amieva MR (2011) ChePep controls Helicobacter pylori Infection of the gastric glands and chemotaxis in the Epsilonproteobacteria. MBio 2:e00098-11
Hoy B, Lower M, Weydig C, Carra G, Tegtmeyer N, Geppert T, Schroder P, Sewald N, Backert S, Schneider G, Wessler S (2010) Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep 11:798–804
Hoy B, Brandstetter H, Wessler S (2013) The stability and activity of recombinant Helicobacter pylori HtrA under stress conditions. J Basic Microbiol 53:402–409
Huang CH, Chiou SH (2011) Proteomic analysis of upregulated proteins in Helicobacter pylori under oxidative stress induced by hydrogen peroxide. Kaohsiung J Med Sci 27:544–553
Hussein NR, Argent RH, Marx CK, Patel SR, Robinson K, Atherton JC (2010) Helicobacter pylori dupA is polymorphic, and its active form induces proinflammatory cytokine secretion by mononuclear cells. J Infect Dis 202:261–269
Hutton ML, Kaparakis-Liaskos M, Turner L, Cardona A, Kwok T, Ferrero RL (2010) Helicobacter pylori exploits cholesterol-rich microdomains for induction of NF-κB-dependent responses and peptidoglycan delivery in epithelial cells. Infect Immun 78:4523–4531
Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Boren T (1998) Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 279:373–377
Imagawa S, Ito M, Yoshihara M, Eguchi H, Tanaka S, Chayama K (2010) Helicobacter pylori dupA and gastric acid secretion are negatively associated with gastric cancer development. J Med Microbiol 59:1484–1489
Jain P, Luo ZQ, Blanke SR (2011) Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc Natl Acad Sci U S A 108:16032–16037
Jimenez-Soto LF, Kutter S, Sewald X, Ertl C, Weiss E, Kapp U, Rohde M, Pirch T, Jung K, Retta SF, Terradot L, Fischer W, Haas R (2009) Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD-independent manner. PLoS Pathog 5:e1000684
Josenhans C, Niehus E, Amersbach S, Horster A, Betz C, Drescher B, Hughes KT, Suerbaum S (2002) Functional characterization of the antagonistic flagellar late regulators FliA and FlgM of Helicobacter pylori and their effects on the H. pylori transcriptome. Mol Microbiol 43:307–322
Jung SW, Sugimoto M, Shiota S, Graham DY, Yamaoka Y (2012) The intact dupA cluster is a more reliable Helicobacter pylori virulence marker than dupA alone. Infect Immun 80:381–387
Kao JY, Zhang M, Miller MJ, Mills JC, Wang B, Liu M, Eaton KA, Zou W, Berndt BE, Cole TS, Takeuchi T, Owyang SY, Luther J (2010) Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology 138:1046–1054
Kaparakis M, Turnbull L, Carneiro L, Firth S, Coleman HA, Parkington HC, Le Bourhis L, Karrar A, Viala J, Mak J, Hutton ML, Davies JK, Crack PJ, Hertzog PJ, Philpott DJ, Girardin SE, Whitchurch CB, Ferrero RL (2010) Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol 12:372–385
Kersulyte D, Lee W, Subramaniam D, Anant S, Herrera P, Cabrera L, Balqui J, Barabas O, Kalia A, Gilman RH, Berg DE (2009) Helicobacter pylori’s plasticity zones are novel transposable elements. PLoS One 4:e6859
Kim KM, Lee SG, Park MG, Song JY, Kang HL, Lee WK, Cho MJ, Rhee KH, Youn HS, Baik SC (2007) γ-glutamyltranspeptidase of Helicobacter pylori induces mitochondria-mediated apoptosis in AGS cells. Biochem Biophys Res Commun 355:562–567
Kim SH, Langford ML, Boucher JL, Testerman TL, McGee DJ (2011) Helicobacter pylori arginase mutant colonizes arginase II knockout mice. World J Gastroenterol 17:3300–3309
Kusters JG, van Vliet AH, Kuipers EJ (2006) Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev 19:449–490
Kuzuhara T, Suganuma M, Kurusu M, Fujiki H (2007) Helicobacter pylori-secreting protein Tipα is a potent inducer of chemokine gene expressions in stomach cancer cells. J Cancer Res Clin Oncol 133:287–296
Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, Misselwitz R, Berger J, Sewald N, Konig W, Backert S (2007) Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 449:862–866
Labigne A, Cussac V, Courcoux P (1991) Shuttle cloning and nucleotide sequences of Helicobacter pylori genes responsible for urease activity. J Bacteriol 173:1920–1931
Lai CH, Chang YC, Du SY, Wang HJ, Kuo CH, Fang SH, Fu HW, Lin HH, Chiang AS, Wang WC (2008) Cholesterol depletion reduces Helicobacter pylori CagA translocation and CagA-induced responses in AGS cells. Infect Immun 76:3293–3303
Lebrun AH, Wunder C, Hildebrand J, Churin Y, Zahringer U, Lindner B, Meyer TF, Heinz E, Warnecke D (2006) Cloning of a cholesterol-α-glucosyltransferase from Helicobacter pylori. J Biol Chem 281:27765–27772
Letley DP, Rhead JL, Twells RJ, Dove B, Atherton JC (2003) Determinants of non-toxicity in the gastric pathogen Helicobacter pylori. J Biol Chem 278:26734–26741
Leunk RD, Johnson PT, David BC, Kraft WG, Morgan DR (1988) Cytotoxic activity in broth-culture filtrates of Campylobacter pylori. J Med Microbiol 26:93–99
Lu H, Hsu PI, Graham DY, Yamaoka Y (2005) Duodenal ulcer promoting gene of Helicobacter pylori. Gastroenterology 128:833–848
Lu H, Wu JY, Beswick EJ, Ohno T, Odenbreit S, Haas R, Reyes VE, Kita M, Graham DY, Yamaoka Y (2007) Functional and intracellular signaling differences associated with the Helicobacter pylori AlpAB adhesin from Western and East Asian strains. J Biol Chem 282:6242–6254
Lupetti P, Heuser JE, Manetti R, Massari P, Lanzavecchia S, Bellon PL, Dallai R, Rappuoli R, Telford JL (1996) Oligomeric and subunit structure of the Helicobacter pylori vacuolating cytotoxin. J Cell Biol 133:801–807
Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, Altraja S, Wadstrom T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB, Arnqvist A, Hammarstrom L, Boren T (2002) Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 297:573–578
Maldonado RA, von Andrian UH (2010) How tolerogenic dendritic cells induce regulatory T cells. Adv Immunol 108:111–165
Marshall BJ, Barrett LJ, Prakash C, McCallum RW, Guerrant RL (1990) Urea protects Helicobacter (Campylobacter) pylori from the bactericidal effect of acid. Gastroenterology 99:697–702
McClain MS, Cao P, Iwamoto H, Vinion-Dubiel AD, Szabo G, Shao Z, Cover TL (2001) A 12-amino-acid segment, present in type s2 but not type s1 Helicobacter pylori VacA proteins, abolishes cytotoxin activity and alters membrane channel formation. J Bacteriol 183:6499–6508
McGee DJ, Radcliff FJ, Mendz GL, Ferrero RL, Mobley HL (1999) Helicobacter pylori rocF is required for arginase activity and acid protection in vitro but is not essential for colonization of mice or for urease activity. J Bacteriol 181:7314–7322
McGee DJ, Kumar S, Viator RJ, Bolland JR, Ruiz J, Spadafora D, Testerman TL, Kelly DJ, Pannell LK, Windle HJ (2006) Helicobacter pylori thioredoxin is an arginase chaperone and guardian against oxidative and nitrosative stresses. J Biol Chem 281:3290–3296
McGovern KJ, Blanchard TG, Gutierrez JA, Czinn SJ, Krakowka S, Youngman P (2001) γ-Glutamyltransferase is a Helicobacter pylori virulence factor but is not essential for colonization. Infect Immun 69:4168–4173
McGowan CC, Necheva AS, Forsyth MH, Cover TL, Blaser MJ (2003) Promoter analysis of Helicobacter pylori genes with enhanced expression at low pH. Mol Microbiol 48:1225–1239
Merrell DS, Goodrich ML, Otto G, Tompkins LS, Falkow S (2003) pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect Immun 71:3529–3539
Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M, Nagai T, Fujita Y, Nagamatsu K, Ishijima N, Koyasu S, Haas R, Sasakawa C (2007) Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe 2:250–263
Mobley HL, Garner RM, Bauerfeind P (1995) Helicobacter pylori nickel-transport gene nixA: synthesis of catalytically active urease in Escherichia coli independent of growth conditions. Mol Microbiol 16:97–109
Moodley Y, Linz B, Bond RP, Nieuwoudt M, Soodyall H, Schlebusch CM, Bernhoft S, Hale J, Suerbaum S, Mugisha L, van der Merwe SW, Achtman M (2012) Age of the association between Helicobacter pylori and man. PLoS Pathog 8:e1002693
Moran AP, Lindner B, Walsh EJ (1997) Structural characterization of the lipid A component of Helicobacter pylori rough- and smooth-form lipopolysaccharides. J Bacteriol 179:6453–6463
Moscat J, Diaz-Meco MT (2009) p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 137:1001–1004
Murata-Kamiya N, Kikuchi K, Hayashi T, Higashi H, Hatakeyama M (2010) Helicobacter pylori exploits host membrane phosphatidylserine for delivery, localization, and pathophysiological action of the CagA oncoprotein. Cell Host Microbe 7:399–411
Necchi V, Candusso ME, Tava F, Luinetti O, Ventura U, Fiocca R, Ricci V, Solcia E (2007) Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology 132:1009–1023
Nguyen LT, Uchida T, Tsukamoto Y, Kuroda A, Okimoto T, Kodama M, Murakami K, Fujioka T, Moriyama M (2010) Helicobacter pylori dupA gene is not associated with clinical outcomes in the Japanese population. Clin Microbiol Infect 16:1264–1269
Niehus E, Gressmann H, Ye F, Schlapbach R, Dehio M, Dehio C, Stack A, Meyer TF, Suerbaum S, Josenhans C (2004) Genome-wide analysis of transcriptional hierarchy and feedback regulation in the flagellar system of Helicobacter pylori. Mol Microbiol 52:947–961
Noto JM, Piazuelo MB, Chaturvedi R, Bartel CA, Thatcher EJ, Delgado A, Romero-Gallo J, Wilson KT, Correa P, Patton JG, Peek RM Jr (2013) Strain-specific suppression of microRNA-320 by carcinogenic Helicobacter pylori promotes expression of the antiapoptotic protein Mcl-1. Am J Physiol Gastrointest Liver Physiol 305:G786–G796
Odenbreit S, Till M, Hofreuter D, Faller G, Haas R (1999) Genetic and functional characterization of the alpAB gene locus essential for the adhesion of Helicobacter pylori to human gastric tissue. Mol Microbiol 31:1537–1548
Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R (2000) Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287:1497–1500
Odenbreit S, Swoboda K, Barwig I, Ruhl S, Boren T, Koletzko S, Haas R (2009) Outer membrane protein expression profile in Helicobacter pylori clinical isolates. Infect Immun 77:3782–3790
Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, Maxeiner J, Hansson M, Taube C, Quiding-Jarbrink M, Muller A (2012) DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest 122:1082–1096
Oertli M, Noben M, Engler DB, Semper RP, Reuter S, Maxeiner J, Gerhard M, Taube C, Muller A (2013) Helicobacter pylori γ-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc Natl Acad Sci U S A 110:3047–3052
Olczak AA, Olson JW, Maier RJ (2002) Oxidative-stress resistance mutants of Helicobacter pylori. J Bacteriol 184:3186–3193
Olczak AA, Seyler RW Jr, Olson JW, Maier RJ (2003) Association of Helicobacter pylori antioxidant activities with host colonization proficiency. Infect Immun 71:580–583
Olczak AA, Wang G, Maier RJ (2005) Up-expression of NapA and other oxidative stress proteins is a compensatory response to loss of major Helicobacter pylori stress resistance factors. Free Radic Res 39:1173–1182
Oswald-Richter K, Torres VJ, Sundrud MS, VanCompernolle SE, Cover TL, Unutmaz D (2006) Helicobacter pylori VacA toxin inhibits human immunodeficiency virus infection of primary human T cells. J Virol 80:11767–11775
Pagliaccia C, de Bernard M, Lupetti P, Ji X, Burroni D, Cover TL, Papini E, Rappuoli R, Telford JL, Reyrat JM (1998) The m2 form of the Helicobacter pylori cytotoxin has cell type-specific vacuolating activity. Proc Natl Acad Sci U S A 95:10212–10217
Papini E, de Bernard M, Milia E, Bugnoli M, Zerial M, Rappuoli R, Montecucco C (1994) Cellular vacuoles induced by Helicobacter pylori originate from late endosomal compartments. Proc Natl Acad Sci U S A 91:9720–9724
Pernitzsch SR, Tirier SM, Beier D, Sharma CM (2014) A variable homopolymeric G-repeat defines small RNA-mediated posttranscriptional regulation of a chemotaxis receptor in Helicobacter pylori. Proc Natl Acad Sci U S A 111:E501–E510
Pflock M, Finsterer N, Joseph B, Mollenkopf H, Meyer TF, Beier D (2006) Characterization of the ArsRS regulon of Helicobacter pylori, involved in acid adaptation. J Bacteriol 188:3449–3462
Poppe M, Feller SM, Romer G, Wessler S (2007) Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene 26:3462–3472
Radin JN, Gaddy JA, Gonzalez-Rivera C, Loh JT, Algood HM, Cover TL (2013) Flagellar localization of a Helicobacter pylori autotransporter protein. MBio 4:e00613–00612
Raju D, Hussey S, Ang M, Terebiznik MR, Sibony M, Galindo-Mata E, Gupta V, Blanke SR, Delgado A, Romero-Gallo J, Ramjeet MS, Mascarenhas H, Peek RM, Correa P, Streutker C, Hold G, Kunstmann E, Yoshimori T, Silverberg MS, Girardin SE, Philpott DJ, El Omar E, Jones NL (2012) Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology 142:1160–1171
Ramarao N, Gray-Owen SD, Backert S, Meyer TF (2000) Helicobacter pylori inhibits phagocytosis by professional phagocytes involving type IV secretion components. Mol Microbiol 37:1389–1404
Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA, Eshagh Hosseini M, Atherton JC (2007) A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology 133:926–936
Ricci V, Romano M, Boquet P (2011) Molecular cross-talk between Helicobacter pylori and human gastric mucosa. World J Gastroenterol 17:1383–1399
Robinson K, Kenefeck R, Pidgeon EL, Shakib S, Patel S, Polson RJ, Zaitoun AM, Atherton JC (2008) Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut 57:1375–1385
Rohde M, Puls J, Buhrdorf R, Fischer W, Haas R (2003) A novel sheathed surface organelle of the Helicobacter pylori cag type IV secretion system. Mol Microbiol 49:219–234
Rokita E, Makristathis A, Presterl E, Rotter ML, Hirschl AM (1998) Helicobacter pylori urease significantly reduces opsonization by human complement. J Infect Dis 178:1521–1525
Roure S, Bonis M, Chaput C, Ecobichon C, Mattox A, Barriere C, Geldmacher N, Guadagnini S, Schmitt C, Prevost MC, Labigne A, Backert S, Ferrero RL, Boneca IG (2012) Peptidoglycan maturation enzymes affect flagellar functionality in bacteria. Mol Microbiol 86:845–856
Salama NR, Shepherd B, Falkow S (2004) Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J Bacteriol 186:7926–7935
Satin B, Norais N, Telford J, Rappuoli R, Murgia M, Montecucco C, Papini E (1997) Effect of Helicobacter pylori vacuolating toxin on maturation and extracellular release of procathepsin D and on epidermal growth factor degradation. J Biol Chem 272:25022–25028
Sause WE, Castillo AR, Ottemann KM (2012) The Helicobacter pylori autotransporter ImaA (HP0289) modulates the immune response and contributes to host colonization. Infect Immun 80:2286–2296
Schmees C, Prinz C, Treptau T, Rad R, Hengst L, Voland P, Bauer S, Brenner L, Schmid RM, Gerhard M (2007) Inhibition of T-cell proliferation by Helicobacter pylori γ-glutamyl transpeptidase. Gastroenterology 132:1820–1833
Schmitt W, Haas R (1994) Genetic analysis of the Helicobacter pylori vacuolating cytotoxin: structural similarities with the IgA protease type of exported protein. Mol Microbiol 12:307–319
Schwartz JT, Allen LA (2006) Role of urease in megasome formation and Helicobacter pylori survival in macrophages. J Leukoc Biol 79:1214–1225
Scott DR, Marcus EA, Wen Y, Oh J, Sachs G (2007) Gene expression in vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface. Proc Natl Acad Sci U S A 104:7235–7240
Selbach M, Moese S, Hauck CR, Meyer TF, Backert S (2002) Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J Biol Chem 277:6775–6778
Selbach M, Moese S, Hurwitz R, Hauck CR, Meyer TF, Backert S (2003) The Helicobacter pylori CagA protein induces cortactin dephosphorylation and actin rearrangement by c-Src inactivation. EMBO J 22:515–528
Senkovich OA, Yin J, Ekshyyan V, Conant C, Traylor J, Adegboyega P, McGee DJ, Rhoads RE, Slepenkov S, Testerman TL (2011) Helicobacter pylori AlpA and AlpB bind host laminin and influence gastric inflammation in gerbils. Infect Immun 79:3106–3116
Sewald X, Gebert-Vogl B, Prassl S, Barwig I, Weiss E, Fabbri M, Osicka R, Schiemann M, Busch DH, Semmrich M, Holzmann B, Sebo P, Haas R (2008) Integrin subunit CD18 Is the T-lymphocyte receptor for the Helicobacter pylori vacuolating cytotoxin. Cell Host Microbe 3:20–29
Seyler RW Jr, Olson JW, Maier RJ (2001) Superoxide dismutase-deficient mutants of Helicobacter pylori are hypersensitive to oxidative stress and defective in host colonization. Infect Immun 69:4034–4040
Shibayama K, Kamachi K, Nagata N, Yagi T, Nada T, Doi Y, Shibata N, Yokoyama K, Yamane K, Kato H, Iinuma Y, Arakawa Y (2003) A novel apoptosis-inducing protein from Helicobacter pylori. Mol Microbiol 47:443–451
Shibayama K, Wachino J, Arakawa Y, Saidijam M, Rutherford NG, Henderson PJ (2007) Metabolism of glutamine and glutathione via γ-glutamyltranspeptidase and glutamate transport in Helicobacter pylori: possible significance in the pathophysiology of the organism. Mol Microbiol 64:396–406
Shiota S, Matsunari O, Watada M, Hanada K, Yamaoka Y (2010) Systematic review and meta-analysis: the relationship between the Helicobacter pylori dupA gene and clinical outcomes. Gut Pathog 2:13
Skouloubris S, Thiberge JM, Labigne A, De Reuse H (1998) The Helicobacter pylori UreI protein is not involved in urease activity but is essential for bacterial survival in vivo. Infect Immun 66:4517–4521
Smith SM, Moran AP, Duggan SP, Ahmed SE, Mohamed AS, Windle HJ, O’Neill LA, Kelleher DP (2011) Tribbles 3: a novel regulator of TLR2-mediated signaling in response to Helicobacter pylori lipopolysaccharide. J Immunol 186:2462–2471
Stent A, Every AL, Sutton P (2012) Helicobacter pylori defense against oxidative attack. Am J Physiol Gastrointest Liver Physiol 302:G579–G587
Strobel S, Bereswill S, Balig P, Allgaier P, Sonntag HG, Kist M (1998) Identification and analysis of a new vacA genotype variant of Helicobacter pylori in different patient groups in Germany. J Clin Microbiol 36:1285–1289
Suerbaum S, Josenhans C (2007) Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat Rev Microbiol 5:441–452
Suerbaum S, Josenhans C, Labigne A (1993) Cloning and genetic characterization of the Helicobacter pylori and Helicobacter mustelae flaB flagellin genes and construction of H. pylori flaA- and flaB-negative mutants by electroporation-mediated allelic exchange. J Bacteriol 175:3278–3288
Suerbaum S, Smith JM, Bapumia K, Morelli G, Smith NH, Kunstmann E, Dyrek I, Achtman M (1998) Free recombination within Helicobacter pylori. Proc Natl Acad Sci U S A 95:12619–12624
Suganuma M, Kurusu M, Suzuki K, Nishizono A, Murakami K, Fujioka T, Fujiki H (2005) New tumor necrosis factor-α-inducing protein released from Helicobacter pylori for gastric cancer progression. J Cancer Res Clin Oncol 131:305–313
Suganuma M, Yamaguchi K, Ono Y, Matsumoto H, Hayashi T, Ogawa T, Imai K, Kuzuhara T, Nishizono A, Fujiki H (2008) TNF-α-inducing protein, a carcinogenic factor secreted from H. pylori, enters gastric cancer cells. Int J Cancer 123:117–122
Sugimoto M, Ohno T, Graham DY, Yamaoka Y (2009) Gastric mucosal interleukin-17 and -18 mRNA expression in Helicobacter pylori-induced Mongolian gerbils. Cancer Sci 100:2152–2159
Sundrud MS, Torres VJ, Unutmaz D, Cover TL (2004) Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc Natl Acad Sci U S A 101:7727–7732
Sycuro LK, Wyckoff TJ, Biboy J, Born P, Pincus Z, Vollmer W, Salama NR (2012) Multiple peptidoglycan modification networks modulate Helicobacter pylori’s cell shape, motility, and colonization potential. PLoS Pathog 8:e1002603
Takeda K, Akira S (2005) Toll-like receptors in innate immunity. Int Immunol 17:1–14
Tammer I, Brandt S, Hartig R, Konig W, Backert S (2007) Activation of Abl by Helicobacter pylori: a novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology 132:1309–1319
Tang B, Li N, Gu J, Zhuang Y, Li Q, Wang HG, Fang Y, Yu B, Zhang JY, Xie QH, Chen L, Jiang XJ, Xiao B, Zou QM, Mao XH (2012) Compromised autophagy by MIR30B benefits the intracellular survival of Helicobacter pylori. Autophagy 8:1045–1057
Tannaes T, Bukholm G (2005) Cholesteryl-6-O-acyl-α-D-glucopyranoside of Helicobacter pylori relate to relative lysophospholipid content. FEMS Microbiol Lett 244:117–120
Telford JL, Ghiara P, Dell’Orco M, Comanducci M, Burroni D, Bugnoli M, Tecce MF, Censini S, Covacci A, Xiang Z et al (1994) Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J Exp Med 179:1653–1658
Terebiznik MR, Raju D, Vazquez CL, Torbricki K, Kulkarni R, Blanke SR, Yoshimori T, Colombo MI, Jones NL (2009) Effect of Helicobacter pylori’s vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy 5:370–379
Terry K, Williams SM, Connolly L, Ottemann KM (2005) Chemotaxis plays multiple roles during Helicobacter pylori animal infection. Infect Immun 73:803–811
Teymournejad O, Mobarez AM, Hassan ZM, Moazzeni SM, Ahmadabad HN (2014) In vitro suppression of dendritic cells by Helicobacter pylori OipA. Helicobacter 19:136–143
Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC (1997) The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547
Torres VJ, Ivie SE, McClain MS, Cover TL (2005) Functional properties of the p33 and p55 domains of the Helicobacter pylori vacuolating cytotoxin. J Biol Chem 280:21107–21114
Torres VJ, VanCompernolle SE, Sundrud MS, Unutmaz D, Cover TL (2007) Helicobacter pylori vacuolating cytotoxin inhibits activation-induced proliferation of human T and B lymphocyte subsets. J Immunol 179:5433–5440
Tsugawa H, Suzuki H, Saya H, Hatakeyama M, Hirayama T, Hirata K, Nagano O, Matsuzaki J, Hibi T (2012) Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells. Cell Host Microbe 12:764–777
Tsutsumi R, Higashi H, Higuchi M, Okada M, Hatakeyama M (2003) Attenuation of Helicobacter pylori CagA x SHP-2 signaling by interaction between CagA and C-terminal Src kinase. J Biol Chem 278:3664–3670
Unemo M, Aspholm-Hurtig M, Ilver D, Bergstrom J, Boren T, Danielsson D, Teneberg S (2005) The sialic acid binding SabA adhesin of Helicobacter pylori is essential for nonopsonic activation of human neutrophils. J Biol Chem 280:15390–15397
Valenzuela M, Bravo D, Canales J, Sanhueza C, Diaz N, Almarza O, Toledo H, Quest AF (2013) Helicobacter pylori-induced loss of survivin and gastric cell viability is attributable to secreted bacterial γ-glutamyl transpeptidase activity. J Infect Dis 208:1131–1141
van Vliet AH, Poppelaars SW, Davies BJ, Stoof J, Bereswill S, Kist M, Penn CW, Kuipers EJ, Kusters JG (2002) NikR mediates nickel-responsive transcriptional induction of urease expression in Helicobacter pylori. Infect Immun 70:2846–2852
Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Memet S, Huerre MR, Coyle AJ, DiStefano PS, Sansonetti PJ, Labigne A, Bertin J, Philpott DJ, Ferrero RL (2004) Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 5:1166–1174
Wadhams GH, Armitage JP (2004) Making sense of it all: bacterial chemotaxis. Nat Rev Mol Cell Biol 5:1024–1037
Wang G, Maier RJ (2004) An NADPH quinone reductase of Helicobacter pylori plays an important role in oxidative stress resistance and host colonization. Infect Immun 72:1391–1396
Wang WC, Wang HJ, Kuo CH (2001) Two distinctive cell binding patterns by vacuolating toxin fused with glutathione S-transferase: one high-affinity m1-specific binding and the other lower-affinity binding for variant m forms. Biochemistry 40:11887–11896
Wang G, Olczak AA, Walton JP, Maier RJ (2005) Contribution of the Helicobacter pylori thiol peroxidase bacterioferritin comigratory protein to oxidative stress resistance and host colonization. Infect Immun 73:378–384
Wang YH, Gorvel JP, Chu YT, Wu JJ, Lei HY (2010) Helicobacter pylori impairs murine dendritic cell responses to infection. PLoS One 5:e10844
Wang HJ, Cheng WC, Cheng HH, Lai CH, Wang WC (2012) Helicobacter pylori cholesteryl glucosides interfere with host membrane phase and affect type IV secretion system function during infection in AGS cells. Mol Microbiol 83:67–84
Watanabe T, Tsuge H, Imagawa T, Kise D, Hirano K, Beppu M, Takahashi A, Yamaguchi K, Fujiki H, Suganuma M (2010) Nucleolin as cell surface receptor for tumor necrosis factor-α inducing protein: a carcinogenic factor of Helicobacter pylori. J Cancer Res Clin Oncol 136:911–921
Watanabe T, Takahashi A, Suzuki K, Kurusu-Kanno M, Yamaguchi K, Fujiki H, Suganuma M (2014) Epithelial-mesenchymal transition in human gastric cancer cell lines induced by TNF-α-inducing protein of Helicobacter pylori. Int J Cancer 134:2373–2382
Weeks DL, Eskandari S, Scott DR, Sachs G (2000) A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science 287:482–485
Weyermann M, Rothenbacher D, Brenner H (2009) Acquisition of Helicobacter pylori infection in early childhood: independent contributions of infected mothers, fathers, and siblings. Am J Gastroenterol 104:182–189
Willhite DC, Blanke SR (2004) Helicobacter pylori vacuolating cytotoxin enters cells, localizes to the mitochondria, and induces mitochondrial membrane permeability changes correlated to toxin channel activity. Cell Microbiol 6:143–154
Willhite DC, Cover TL, Blanke SR (2003) Cellular vacuolation and mitochondrial cytochrome c release are independent outcomes of Helicobacter pylori vacuolating cytotoxin activity that are each dependent on membrane channel formation. J Biol Chem 278:48204–48209
Winter JA, Letley DP, Cook KW, Rhead JL, Zaitoun AA, Ingram RJ, Amilon KR, Croxall NJ, Kaye PV, Robinson K, Atherton JC (2014) A role for the vacuolating cytotoxin, VacA, in colonization and Helicobacter pylori-induced metaplasia in the stomach. J Infect Dis 210:954–963
Wunder C, Churin Y, Winau F, Warnecke D, Vieth M, Lindner B, Zahringer U, Mollenkopf HJ, Heinz E, Meyer TF (2006) Cholesterol glucosylation promotes immune evasion by Helicobacter pylori. Nat Med 12:1030–1038
Yahiro K, Satoh M, Nakano M, Hisatsune J, Isomoto H, Sap J, Suzuki H, Nomura F, Noda M, Moss J, Hirayama T (2012) Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by Helicobacter pylori VacA. J Biol Chem 287:31104–31115
Yamaoka Y, Kikuchi S, el-Zimaity HM, Gutierrez O, Osato MS, Graham DY (2002) Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology 123:414–424
Yamaoka Y, Kudo T, Lu H, Casola A, Brasier AR, Graham DY (2004) Role of interferon-stimulated responsive element-like element in interleukin-8 promoter in Helicobacter pylori infection. Gastroenterology 126:1030–1043
Yamaoka Y, Ojo O, Fujimoto S, Odenbreit S, Haas R, Gutierrez O, El-Zimaity HM, Reddy R, Arnqvist A, Graham DY (2006) Helicobacter pylori outer membrane proteins and gastroduodenal disease. Gut 55:775–781
Yamasaki E, Wada A, Kumatori A, Nakagawa I, Funao J, Nakayama M, Hisatsune J, Kimura M, Moss J, Hirayama T (2006) Helicobacter pylori vacuolating cytotoxin induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and cell death, independent of vacuolation. J Biol Chem 281:11250–11259
Ye D, Willhite DC, Blanke SR (1999) Identification of the minimal intracellular vacuolating domain of the Helicobacter pylori vacuolating toxin. J Biol Chem 274:9277–9282
Yokota S, Ohnishi T, Muroi M, Tanamoto K, Fujii N, Amano K (2007) Highly-purified Helicobacter pylori LPS preparations induce weak inflammatory reactions and utilize Toll-like receptor 2 complex but not Toll-like receptor 4 complex. FEMS Immunol Med Microbiol 51:140–148
Zheng PY, Jones NL (2003) Helicobacter pylori strains expressing the vacuolating cytotoxin interrupt phagosome maturation in macrophages by recruiting and retaining TACO (coronin 1) protein. Cell Microbiol 5:25–40
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Praszkier, J., Sutton, P., Ferrero, R.L. (2016). Virulence Mechanisms of Helicobacter pylori: An Overview. In: Backert, S., Yamaoka, Y. (eds) Helicobacter pylori Research. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55936-8_3
Download citation
DOI: https://doi.org/10.1007/978-4-431-55936-8_3
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55934-4
Online ISBN: 978-4-431-55936-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)