
Abstract
Helicobacter pylori is an ancient companion of humans and has coevolved with the human race at least since its migration out of Africa. Consequently, it is well adapted to its exclusive niche, the mucosal surface of the human stomach, and has developed elaborate strategies to evade or suppress immunity and establish persistent infection. This chapter will discuss the most recent findings regarding innate immune recognition of H. pylori and the mechanisms that allow the bacteria to avoid detection and subsequent killing by antimicrobial peptides and other first-line defense mechanisms. Additional topics focus on the differences in adaptive immune responses that may explain the broad spectrum of disease outcomes in H. pylori-infected individuals, which range from a completely asymptomatic carrier state to often fatal gastric cancer development. The immune cell compartments driving H. pylori infection-associated immunopathology are discussed along with their main effector mechanisms. Additionally, several strategies employed by H. pylori to block the clonal expansion of specific effector T cells, and to preferentially induce the differentiation of regulatory T cells, are introduced in light of their putative role in establishing and maintaining persistent infection. A final topic deals with the consequences of H. pylori-specific immunomodulation for the risk of the carrier to develop immune-related disorders such as chronic inflammatory diseases of the lower bowel and certain allergic disease manifestations. A potential protective effect of H. pylori on such immune disorders is discussed with regard to the latest epidemiological findings in humans as well as experimental studies in animal models.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Helicobacter pylori
- Type IV secretion
- Immunomodulation
- Adaptive immunity
- Innate immune recognition
- Virulence determinants
- Persistence
1 Introduction
Helicobacter pylori exclusively infects the human gastric mucosa , where it colonizes the mucus and binds to gastric epithelial pit cells (Salama et al. 2013). The mucus layer and gastric epithelial cell monolayer of the stomach mucosa thus form the first host defense barrier against H. pylori . The host/H. pylori interaction at the mucosal surface of the stomach is characterized by a fine balance of both pro- and anti-inflammatory responses, which, in ~80 % of carriers, permits persistent infection while at the same time preventing clinically overt disease. H. pylori expresses pathogen-associated molecular patterns (PAMPs) that evade detection by the host innate immune system yet retain their biological function as structural components of the bacterial cell wall and motility apparatus. Other evolutionary adaptations allow the bacteria to suppress anti-Helicobacter immunity by directly interfering with effector T cell activation, proliferation, and function and by indirectly blocking T cells via the induction of highly suppressive regulatory T cells. The development of peptic ulcer disease and gastric premalignant and malignant lesions is now widely viewed to be the consequence of a misbalance in effector and regulatory T cell responses to the infection that arises due to a specific genetic or lifestyle predisposition of the host or a mismatch between the genetic makeup of host and pathogen. The preferential induction of regulatory over effector T cells, which is a hallmark of persistent (asymptomatic) H. pylori infection, exerts not only local but also systemic effects that remain poorly understood. One of the consequences of the systemic immunomodulation by H. pylori is the relative protection of the infected fraction of the population from allergic and chronic inflammatory diseases such as allergen-induced asthma, hay fever, ectopic dermatitis, inflammatory bowel diseases, and celiac disease, all of which are immune-related disorders. The purpose of the following chapters is the detailed discussion of the elaborate molecular adaptations of H. pylori that allow it to evade and manipulate host immune response s and to thereby establish persistent infection. The local and systemic consequences of this host/pathogen interaction are also discussed.
2 Innate Immune Responses to H. pylori
As mentioned above, the gastric epithelial cell monolayer of the stomach mucosa forms the first innate immune defense barrier against H. pylori and therefore has been studied in great detail. The direct interaction of H. pylori with epithelial cells results in the assembly of the type IV secretion system (T4SS) pilus (for more details, see Chap. 4) and the subsequent exposure to virulence factors encoded on the Cag pathogenicity island (cagPAI); among these, the CagL /integrin α5β1 interaction has proven to be directly responsible for the activation of NF-κB and production of the neutrophil chemoattractant interleukin- 8 (IL -8) (Gorrell et al. 2012; Jimenez-Soto et al. 2009; Kwok et al. 2007; Shaffer et al. 2011). In line with the well-known pro-inflammatory/immunostimulatory activity of the H. pylori T4SS, mutants lacking this activity colonize mice and gerbils at higher levels than the corresponding wild-type strains because they are controlled less efficiently; cagPAI mutants further cause significantly less preneoplastic immunopathology in rodent models (Arnold et al. 2011b; Rieder et al. 2005). In human carriers, cagPAI-positive strains (often identified serologically by their expression of the cagPAI-encoded CagA protein) are clearly associated with more severe disease and higher gastric cancer risk than cagPAI/CagA-negative strains (Blaser et al. 1995; Huang et al. 2003). Several studies suggest that in humans, the expression of cagPAI proteins appears to confer a colonization advantage through the downregulation of antimicrobial peptides: for example, it has been demonstrated that H. pylori reduces the expression of human β-defensin 1 in a T4SS- and NF-κB-dependent manner, with colonization and gastric β-defensin 1 levels being inversely correlated in H. pylori carriers (Patel et al. 2013). Another human antimicrobial peptide, β-defensin 3, which is known to be highly active against H. pylori, is initially induced by the infection in vitro in an epidermal growth factor receptor (EGFR )- and mitogen-activated protein (MAP) kinase-dependent manner but is then stably shut down through CagA-mediated activation of the Src homology domain containing protein tyrosine phosphatase 2 (SHP2) and down-modulation of the EGFR signaling pathway (Bauer et al. 2012a). The downregulation of β-defensin 3 protein could indeed be confirmed in the gastric mucosa of H. pylori-infected subjects (Bauer et al. 2012b). In line with the critical role of the cagPAI-encoded T4SS in pro-inflammatory and immune escape mechanisms, its expression and function have been shown to be fine-tuned by DNA rearrangements at direct repeats in the coding sequence of the CagY protein, an essential component of the T4SS, which is subject to immune-driven selective pressure in both mice and nonhuman primates (Barrozo et al. 2013).
Whereas cagPAI-induced responses are best characterized and understood in gastric epithelial cells, most non-P AI-mediated interactions of H. pylori with the host have been studied in cells of the innate immune system. Innate immune recognition of H. pylori is unique in that it predominantly results in anti- rather than pro-inflammatory responses; several pathogen-associated molecular patterns (PAMPs) of H. pylori have further evolved to specifically avoid excessive inflammation (Fig. 12.1). The lipopolysaccharide (LPS ) constituent of the bacterial outer membrane, which in enteropathogenic bacteria has strong immunostimulatory properties and readily activates inflammatory signaling via toll-like receptor 4 (TLR4), has evolved in H. pylori to avoid TLR4 recognition (Cullen et al. 2012; Moran et al. 1997). The relative bio-inactivity of H. pylori LPS has been attributed to its tetra-acylation (whereas E. coli LPS is hexa-acylated) (Moran et al. 1997) and to the removal of phosphate groups from the 1′ and 4′ positions of the lipid A backbone, which generates LPS that has less negative charge, escapes detection by TLR4, and resists binding by antimicrobial peptides (Cullen et al. 2012). Mutants lacking the phosphatases required for lipid A modification consequently fail to colonize mice (Cullen et al. 2012). A similarly elaborate mechanism of immune evasion has been reported for H. pylori flagellin, which is mutated in exactly those N-terminal positions that mediate binding to TLR5 in Salmonella enterica flagellin (Andersen-Nissen et al. 2005; Gewirtz et al. 2004). Additional interactions of H. pylori PAMPs with host innate immune receptors include the detection of H. pylori DNA by the endosomally localized TLR9 (Otani et al. 2012) and the binding of an as yet unidentified ligand to TLR2 (Fig. 12.1). The interactions via TLR9 and TLR2 result in predominantly anti-inflammatory responses. Lipofection of dendritic cells (DCs ) with H. pylori DNA activates TLR9 (Rad et al. 2009), with documented anti-inflammatory consequences in the first months of infection in a mouse model (Otani et al. 2012). The putative immunoregulatory properties of H. pylori DNA have in fact even been exploited successfully for therapeutic purposes: oral administration of purified DNA alleviates experimentally induced inflammatory bowel disease in mice (Luther et al. 2011), a finding that has been attributed to the unique immunoregulatory sequence 5′TTTAGGG that is overrepresented in the H. pylori genome (Owyang et al. 2012). The predominant TLR activated by H. pylori is TLR2, which drives an anti-inflammatory signature characterized by high expression levels of the regulatory cytokine interleukin- 10 (IL -10) in DCs (Rad et al. 2009) (Fig. 12.1). Whereas the H. pylori ligand for TLR2 remains unknown, it is clear from various studies conducted in vitro and in vivo that TLR2 signaling counteracts H. pylori clearance and promotes immune tolerance (Sayi et al. 2011; Sun et al. 2013). TLR2-deficient mice control H. pylori and related Helicobacter species efficiently develop more severe and accelerated immunopathology (Sayi et al. 2011; Sun et al. 2013). TLR2 proficiency of B cells and DCs is required for the differentiation of regulatory T cells, the H. pylori-induced production of IL-10 and other tolerogenic responses in vitro and in vivo, and likely accounts for the phenotype of TLR2−/− mice (Rad et al. 2009; Sayi et al. 2011; Sun et al. 2013). In addition to the mentioned PAMPs and their receptors, H. pylori RNA has also been shown to mediate innate immune recognition. 5′-triphosphorylated RNA of H. pylori is sensed by the endosomal TLR8, as well as the cytoplasmic retinoic acid-inducible gene (RIG)-like helicase receptor RIG-I, the latter leading to the production of type I interferons (IFNs) (Rad et al. 2009). Whether this pathway of innate immune detection contributes to H. pylori control or pathogenesis is currently not known.

H. pylori subverts and evades innate immune recognition. H. pylori produces several PAMPs that have evolved to evade detection by pro-inflammatory TLRs . Examples include H. pylori’s tetra-acylated LPS , which is bioinactive due to specific lipid A modifications that prevent detection by TLR4. H. pylori flagella cannot be detected by TLR5 due to mutations in the TLR5 binding site of flagellin. H. pylori DNA, enriched for the immunoregulatory sequence TTTAGGG, as well as an as yet uncharacterized PAMP (and possibly H. pylori LPS) are detected by TLRs 9 and 2, respectively; these TLRs preferentially activate anti-inflammatory signaling pathways and IL -10 expression. 5′ triphosphorylated RNA is detected by the cytosolic receptor RIG-I, which activates the transcription factors IRF3 and IRF7 to induce type I IFN expression; H. pylori RNA is likely also detected by TLR8 in endosomes. H. pylori’s fucosylated DC-SIGN ligands suppress activation of the signaling pathways downstream of DC-SIGN and activate anti-inflammatory genes. Note that not all depicted pattern recognition receptors are necessarily expressed by the same cell type; a generic cell is shown here for simplicity. DD, death domain; TIR, toll/interleukin-1 receptor domain; CARD, caspase activation and recruitment domain; MyD88, myeloid differentiation primary response gene 88; DC-SIGN, DC-specific intercellular adhesion molecule-3-grabbing non-integrin; SRC, steroid receptor coactivator
In addition to the described immune escape and immunomodulatory mechanisms involving TLRs , H. pylori has further evolved to bind the C-type lectin receptor DC-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN); however, in contrast to other pathogens expressing (mannosylated) DC-SIGN ligands, the fucosylated DC-SIGN ligands of H. pylori fail to activate the signaling cascade downstream of this receptor and instead dissociate the respective signaling complexes to suppress pro-inflammatory signaling (Gringhuis et al. 2009). Anti-inflammatory consequences have also been reported of the H. pylori-induced activation of the inflammasome . It is now clear that H. pylori activates caspase- 1 and induces the proteolytic processing of caspase-1-dependent cytokines in an NLRP3- and ASC-dependent manner (Hitzler et al. 2012b; Kim et al. 2013). Mature IL -1β and IL-18 are produced and secreted by DCs that have been exposed to H. pylori (Hitzler et al. 2012b; Kim et al. 2013); transcriptional activation of pro-IL-1β appears to depend on the cagPAI (Kim et al. 2013), whereas pro-IL-18 is known to be constitutively expressed. Interestingly, the two caspase-1-dependent cytokines fulfill opposing roles in the context of H. pylori control and pathogenesis. Whereas IL-1β is absolutely required for the differentiation and function of Th1 and Th17 cells and thus for the control of experimental infection as well as for the development of gastritis and preneoplastic immunopathology (Hitzler et al. 2012b; Kim et al. 2013), IL-18 is dispensible for immunity to H. pylori (Hitzler et al. 2012b). The phenotypes of IL-1R and IL-1β-deficient mouse strains are nicely in line with observations in humans, where promoter polymorphisms in the IL-1β gene leading to increased steady-state production of the cytokine predispose to an increased gastric cancer risk (El-Omar et al. 2000). Quite to the contrary, mice lacking IL-18 or its receptor control Helicobacter infections more efficiently because they fail to generate FoxP3-positive regulatory T cells (Tregs) and to develop immune tolerance to the infection and, as a consequence, exhibit overly active Th17 cells (Hitzler et al. 2012b; Oertli et al. 2012). The defect in Treg differentiation of IL-18/IL-18R-deficient mice has been attributed to the failure of IL-18−/− DCs to induce Treg differentiation (Oertli et al. 2012). The differential properties of caspase-1-dependent cytokines in H. pylori control and immunopathology are reminiscent of the functions of both cytokines in the lower GI tract, especially in models of experimentally induced colitis, which are driven in large part by IL-1β (Coccia et al. 2012) and restricted by IL-18 (Zaki et al. 2010).
Collectively, the data generated in recent years on the various innate immune response s to (and their manipulation by) H. pylori suggest that the bacterium effectively prevents its clearance and promotes its persistence by both evading innate immune detection and subsequent killing and by skewing innate immune responses toward anti-inflammatory and regulatory signals. The postulated >60,000 years of coexistence of H. pylori with its human host have provided the selective pressure necessary to drive these evolutionary processes; the remarkable genetic variability, high mutation rate, and natural competence of H. pylori provide the genetic setting facilitating its host adaptation.
3 Anti-Helicobacter Adaptive Immune Responses Differ in Symptomatic and Asymptomatic Carriers
Whereas approximately one-half of humanity is infected with H. pylori , only a fraction of human carriers will develop H. pylori-related gastric or duodenal disease. The remaining ~80 % of the infected population remain asymptomatic for life and in fact may never know that they are colonized. Host genetic traits (see Chap. 14 on host gene polymorphisms and their effects on disease outcome) and the specific virulence factors expressed by the infecting H. pylori strain (see Chaps. 3, 4, 5, 6, and 7) have been discussed as critical modulators influencing disease outcome. One important predictor and driver of disease that has emerged in recent years from studies conducted in human carriers as well as in mouse models is the severity and polarization of gastric H. pylori-specific T helper cell responses (Arnold et al. 2011b; Harris et al. 2008; Robinson et al. 2008). Individuals with peptic ulcer disease (PUD) have a threefold higher anti-H. pylori Th1 response and sixfold higher Th2 response than asymptomatic carrier s (Robinson et al. 2008). The PUD patients at the same time exhibited a twofold lower Treg response than asymptomatic carriers and significantly reduced IL -10 and transforming growth factor (TGF)-β levels in the gastric mucosa ; the authors concluded from this imbalance that an inadequate Treg response was associated with and possibly responsible for the development of H. pylori-associated disease (Robinson et al. 2008). Another study with a similar objective and approach compared the anti-H. pylori responses of children and adults and also found an inverse correlation between the degree of gastritis and the numbers of gastric Tregs and production of Treg-derived cytokines (Harris et al. 2008). These two seminal studies have confirmed and extended previous work showing that Tregs home to and accumulate in the gastric mucosa of infected but not uninfected individuals, where they suppress H. pylori-specific memory T cell responses (Lundgren et al. 2003, 2005a, b). Taken together, the observational studies in humans imply that asymptomatic (healthy) carriers predominantly launch Treg responses to H. pylori infection, which effectively control immunopathology and promote persistent infection , whereas symptomatic carriers presenting with disease exhibit T-effector-dominated responses (Fig. 12.2). The studies in humans are corroborated by experimental studies in mice, where Treg depletion improves infection control by deregulating Th1 and Th17 responses and at the same time promotes the development of chronic infection-associated gastritis, atrophy, and intestinal metaplasia (Arnold et al. 2011b; Hitzler et al. 2011). Treg-derived IL-10 and TGF-β are each critical for preventing T-effector cell-driven immunopathology (Arnold et al. 2011b), suggesting that the expression levels of these cytokines in the gastric mucosa are excellent indicators of disease outcome. Interestingly, the lack or neutralization of IL-10 signaling is sufficient to clear H. pylori and to induce strong T cell-dependent immunopathology (Ismail et al. 2003; Sayi et al. 2011).

Symptomatic and asymptomatic carrier s differ in terms of their anti- H. pylori T cell responses. Observational studies comparing the T cell responses of peptic ulcer disease patients and asymptomatic carriers (Robinson et al. 2008) and children vs. adults with relatively mild and severe gastritis, respectively (Harris et al. 2008), have revealed that Treg /T-effector cell ratios correlate with disease outcome. Asymptomatic carriers predominantly generate Treg responses to the infection, which suppress Th1 and Th17 cells through the production of soluble cytokines IL -10 and TGF-β and other immunosuppressive mechanisms. This scenario is modeled in C57BL/6 mice neonatally infected with H. pylori (Arnold et al. 2011b) In contrast, the gastric mucosa of symptomatic carriers is infiltrated by Th17 and Th1 cells and exposed to high levels of the signature cytokines IFN-γ, TNF-α , and IL-17, which drive chronic gastritis and promote disease progression toward chronic atrophic gastritis and hyperplasia, intestinal metaplasia , ulcers, and gastric cancer . Infection of adult C57BL/6 mice mirrors the scenario found in symptomatic carriers. H. pylori colonization levels are generally higher in asymptomatic vs. symptomatic carriers and directly proportional to gastric mucosal Treg numbers
H. pylori is typically acquired during early childhood, with the mother serving as the main source of infection in populations with low prevalence (Weyermann et al. 2009). The infection is generally contracted within the first 2 years of life (Rothenbacher et al. 2000), i.e., at a time when the immune system is immature and predisposed to develop immune tolerance rather than immunity to foreign dietary and environmental antigens and the newly acquired microbiota (Arnold et al. 2005). Animal models that take into account and aim to reflect the age at the time of H. pylori acquisition have revealed that exposure to H. pylori during the neonatal period leads to the development of Treg -mediated immune tolerance to H. pylori (Arnold et al. 2011b). Although anti-H. pylori effector T cell responses are generated normally by the neonatally infected murine host, these are under the tight control of Tregs (Fig. 12.2). Neonatally infected animals are protected against infection-induced gastric immunopathology , not just in the weeks and months following experimental infection but apparently for life (Arnold et al. 2011b). The hallmarks of the neonatal infection model are reminiscent of the Treg-predominant responses of infected children (Harris et al. 2008); it is interesting to note in this context that children not only suffer less frequently from the consequences of gastric colonization with H. pylori but also appear to benefit more in terms of their reduced allergy risk (see below). Understanding the relative role of T-effector vs. Treg responses in the balance of H. pylori clearance and immunopathology is of immediate practical relevance in H. pylori vaccinology, as protective immunity can only be achieved by vaccination strategies aimed at overcoming immune counter-regulation (Becher et al. 2010; Hitzler et al. 2011) and should ideally be sterilizing (see Chap. 24 on vaccine development against H. pylori).
4 Gastric P reneoplastic Immunopathology Is Driven by T Helper Cell Responses and T Cell-Derived Cytokines
It is now well accepted due to work conducted in experimental infection and vaccine -induced protection models that the host control of H. pylori colonization depends on CD4+ effector T cells, but not on B cells or antibodies (Akhiani et al. 2002, 2004a, b; Ermak et al. 1998; Hitzler et al. 2011; Sayi et al. 2009; Velin et al. 2005, 2009; Velin and Michetti 2010). Human volunteer infections confirm that strong T cell responses and H. pylori clearance are tightly correlated and possibly causally associated (Aebischer et al. 2008). Out of a total of 58 volunteers who were challenged with live H. pylori in a seminal study published in 2008, 13 managed to clear the infection as judged by urea breath test; eight of these had been vaccinated against H. pylori using the Salmonella typhimurium vaccine strain Ty21a expressing H. pylori urease and five had cleared the challenge infection spontaneously (Aebischer et al. 2008). H. pylori-specific T helper cells were detected in 9 of the 13 volunteers who cleared the infection successfully, but only in 6 of 45 who did not. While the study disappointingly provided little evidence for vaccination-induced protective immunity , it did show convincingly that anti-H. pylori T cell responses correlate well with clearance (Aebischer et al. 2008).
It is evident from studies using T cell-deficient mouse strains that CD4+ TCRα/β+ T cells are required for the control of H. pylori as well as for the induction of immunopathology : mice specifically lacking CD4+ TCRα/β+ T cells either due to a deletion of the major histocompatibility complex (MHC) II or to lack of the TCR β−chain fail to control experimental infections (in vaccinated as well as naive mice) and are at the same time protected against infection-associated immunopathology (Hitzler et al. 2011, 2012a; Akhiani et al. 2002; Ermak et al. 1998). It has been postulated that the CD4+ T helper cell subsets and signature cytokines that contribute to infection control in experimental models at the same time promote the immunopathology that is a hallmark of immunocompetent hosts and that manifests histologically as chronic (atrophic) gastritis (Hitzler et al. 2012a; Horvath et al. 2012; Sayi et al. 2009; Shi et al. 2010; Stoicov et al. 2009). Considerable early evidence suggests that Th1-polarized T cells are critical mediators of H. pylori control and immunopathology. Th1 cells and their signature cytokine IFN-γ have been implicated in vaccine -induced protective immunity and in infection-associated gastritis: mice lacking the p40 subunit of the Th1-polarizing cytokine IL -12 or the receptor for the Th1 signature cytokine IFN-γ fail to control H. pylori upon challenge infection and IFN-γR−/− mice further fail to develop gastritis (Akhiani et al. 2002). Similarly, mice lacking Th1 cells due to the genetic ablation of the lineage-defining transcription factor T-bet are protected from gastric atrophy and the development of gastric cancer later in life (Stoicov et al. 2009). The adoptive transfer of wild-type but not IFN-γ-deficient CD4+ T cells controls the infection and induces preneoplastic pathology in H. pylori-infected T cell-deficient recipients (Sayi et al. 2009). In hindsight, some of the early work must be interpreted with caution: for example, it is now clear that the p40 subunit of IL-12 is shared by the related cytokine IL-23, which is generally accepted to drive Th17 but not Th1 responses (Cua et al. 2003). Consequently, p40−/− mice lack Th17 as well as Th1 cells (Cua et al. 2003). More recent studies have addressed the relative contribution of Th1 and Th17 cells to infection control; neither p19−/− nor p35−/− mice – lacking the specific subunits of IL-23 and IL-12, i.e., deficient for either Th17 or Th1 cells – were protected upon vaccination with the gold standard cholera toxin adjuvant whole cell extract vaccine (Hitzler et al. 2011); the contribution of Th17 cells to H. pylori control was also confirmed by other investigators (Horvath et al. 2012). In addition to their role in H. pylori control, Th17 cells also contribute to infection-induced immunopathology, as assessed in mice lacking the IL-23-specific p19 subunit (Hitzler et al. 2012a; Horvath et al. 2012) Whether the best-studied Th17 signature cytokine IL-17 mediates the effects of Th17 cells remains controversial; neutralizing antibodies to IL-17 prevents H. pylori control and immunopathology in vaccination models (Velin et al. 2009) but have been reported to promote colonization in non-vaccinated, experimentally infected mice (Shi et al. 2010). In line with the known reciprocal negative regulation of Th1 and Th17 subsets, this effect has been attributed by some investigators to higher Th1 cytokine expression in the absence of IL-17 signaling (Otani et al. 2009). In summary, the work of many groups has documented beyond doubt that the control of H. pylori and the resulting infection-induced gastric immunopathology preceding the development of gastric cancer are inseparably linked, at least in mouse models, and mediated by both Th1 and Th17 subsets of T cells. Public health strategies aimed at reducing gastric cancer risk by eradicating H. pylori in high-risk individuals or populations thus might benefit from combining antibiotic treatment with T cell-targeting immunomodulatory therapies to accelerate mucosal healing and improve treatment outcome also in individuals with preexisting atrophy and metaplasia that do not currently benefit sufficiently from eradication therapy alone (Rokkas et al. 2007; Wong et al. 2004).
5 H. pylori Suppresses Effector T Cell Responses to Achieve P ersistent Infection
Given the critical role of effector T cells (Th1 and Th17 subsets) in controlling H. pylori , it is not surprising that the bacteria have evolved elaborate mechanisms of suppressing human T cell activity, proliferation, and clonal expansion. One key virulence factor/persistence determinant with T cell inhibitory properties is the vacuolating cytotoxin VacA . VacA was initially identified due to its ability to induce massive vacuolation in primary gastric epithelial cells and certain gastric epithelial cell lines (Smoot et al. 1996; Harris et al. 1996) and to its association with peptic ulcer disease (Atherton et al. 1995) (see Chap. 5 on VacA). It is also known to induce apoptosis in gastric epithelial cells (Cover et al. 2003), presumably via insertion into mitochondrial membranes followed by cytochrome C release (Domanska et al. 2010). Its vacuolating and pro-apoptotic activity requires a stretch of N-terminally encoded hydrophobic amino acids, which allow VacA to form hexameric pores in artificial lipid bilayers as well as in endosomal, lysosomal, and mitochondrial membranes of epithelial cells and phagocytes (McClain et al. 2001; Czajkowsky et al. 1999). VacA is expressed by all H. pylori isolates in the form of either the m1 or m2 allele, which differ in expression levels, vacuolating activity, and association with disease (Atherton et al. 1999). Mouse studies have demonstrated that VacA is not only a virulence but also a colonization factor, as mutants lacking VacA are rapidly outcompeted by the wild type in mixed infections (Salama et al. 2001) and colonized at significantly lower levels in single infections (Oertli et al. 2013). In addition to its vacuolating and pro-apoptotic effects on epithelial cells and in line with its critical role in colonization, VacA has been shown to inhibit the activation and proliferation of T and of B cells (Fig. 12.3) (Torres et al. 2007; Boncristiano et al. 2003; Gebert et al. 2003; Sundrud et al. 2004). In human primary T cells, activation upon TCR engagement is blocked at the level of the Ca2+/calmodulin-dependent phosphatase calcineurin and the nuclear translocation of the transcription factor nuclear factor of activated T cells (NFAT) (Boncristiano et al. 2003; Gebert et al. 2003). VacA activity on T cells requires the same N-terminal hydrophobic region that also mediates vacuolization (Sundrud et al. 2004) and binding to a surface receptor, the β2 integrin (CD18) (Sewald et al. 2008), which associates with CD11a/αL to form the heterodimeric lymphocyte function-associated antigen-1 (LFA-1) receptor (Fig. 12.3). VacA is taken up as LFA1 is recycled in a PKC-mediated, phosphorylation-dependent manner (Sewald et al. 2010). It seems that CD18 must be directly phosphorylated by either PKCη or PKCζ in its cytoplasmic tail to initiate VacA endocytosis and inhibition of NFAT target gene transactivation (Fig. 12.3) (Sewald et al. 2010). Murine T cells are resistant to VacA because they do not express the receptor (Sewald et al. 2008; Algood et al. 2007); rather, it appears that VacA promotes persistence in mice through a mechanism involving its interaction with DCs (see below) (Oertli et al. 2013). In humans, VacA’s inhibitory activity on T cells likely prevents the clonal expansion of H. pylori-specific, antigen-activated T cells, thereby interfering effectively with a critical branch of adaptive immune defense against this infection.

H. pylori impairs T cell-mediated immunity through the production and secretion of VacA and GGT . All strains of H. pylori express the secreted virulence factors VacA and GGT to directly inhibit T cell activation, proliferation, and effector functions. Hexameric VacA binds to the β2 integrin subunit of the heterodimeric transmembrane receptor LFA-1; the receptor complex is internalized upon protein kinase C-mediated serine/threonine phosphorylation of the β2 integrin cytoplasmic tail. Cytoplasmic VacA prevents nuclear translocation of NFAT by inhibiting its dephosphorylation by the Ca2+/calmodulin-dependent phosphatase calcineurin and thereby blocks IL -2 production and subsequent T cell activation and proliferation. GGT arrests T cells in the G1 phase of the cell cycle and thus prevents their proliferation. Note that the direct effects of VacA on T cells appear to be human specific. LFA-1, lymphocyte function-associated antigen-1; NFAT, nuclear factor of activated T cells; GGT, γ-glutamyl transpeptidase; CnA, B, calcineurin A and B subunits; CaM, calmodulin
In addition to VacA , all strains of H. pylori produce and secrete a second immunomodulatory molecule known to interfere with T cell proliferation, the gamma-glutamyl transpeptidase (GGT ). GGT has enzymatic activity and catalyzes the transfer of the γ-glutamyl moiety of glutamine or glutathione to amino acids, allowing H. pylori to convert glutamine and glutathione into glutamate, which can be taken up and incorporated into the TCA cycle. H. pylori mutants lacking GGT fail to colonize mice (Chevalier et al. 1999; Oertli et al. 2013), and this phenotype has been linked to the ability of GGT to prevent T cell proliferation (Gerhard et al. 2005; Schmees et al. 2007) (Fig. 12.3). Other parameters of T cell activation (NFAT translocation, cytokine production) are not affected by GGT; its inhibitory effect on proliferation could be linked to cell cycle arrest in the G1 phase and to the enzymatic activity of GGT (Gerhard et al. 2005; Schmees et al. 2007). Similar to VacA, GGT also exerts strong immunomodulatory effects on DCs , which acquire tolerogenic activity upon exposure to GGT-proficient H. pylori or the recombinant protein (discussed in more detail below) (Oertli et al. 2013). The inclusion of both VacA and GGT in experimental H. pylori vaccines (Malfertheiner et al. 2008) indicates that the neutralization of H. pylori’s immunomodulatory properties is widely seen as a promising intervention strategy. Further details on GGT function can be obtained in Chaps. 6 and 7.
6 H. pylori P romotes Tolerogenic DC Functions and Induces Regulatory T Cells
DCs were long known mostly for their essential role in immunity to intracellular and extracellular pathogens, to which they contribute through their unique ability to prime naive T cells to differentiate, to proliferate, and to acquire effector functions such as cytotoxicity and cytokine production. It has become clear in recent years, however, that DCs are also critically involved in the development of immune tolerance to autoantigens, allergens, and harmless antigens of the commensal human microflora (Maldonado and von Andrian 2010; Yogev et al. 2012). A major pathway driving DC-mediated immune tolerance to self-, dietary, or environmental antigens involves the thymus-independent, “peripheral” induction of highly suppressive, “inducible” Tregs, which, like their thymus-derived “natural” counterparts, typically express the lineage-defining transcription factor FoxP3, the surface marker CD25, and an array of secreted and surface-exposed regulatory molecules that may include IL -10, TGF-β, CTLA-4, PD1, GITR, and others. Tregs efficiently block effector T cell responses by direct and indirect mechanisms, and their prolonged dysregulation results in severe and often fatal autoimmunity (Bollrath and Powrie 2013; Harrison and Powrie 2013). Persistent pathogens, such as Mycobacterium tuberculosis and certain helminths, have evolved to selectively recruit or activate Tregs to or in their preferred niche to promote chronicity (McBride et al. 2013; Ottenhoff 2012; Maizels and Smith 2011). The same is true for H. pylori , which preferentially induces Tregs (Robinson et al. 2008) and relies on Treg -mediated immunosuppression to promote persistent infection (Arnold et al. 2011b) (also see above). Although difficult to study meaningfully in humans, the process of Treg induction and DC/Treg-mediated immune tolerance and suppression has received substantial attention lately and several key mechanisms have been elucidated. Multiple studies have shown in vitro and in vivo that H. pylori specifically targets DCs to promote their tolerogenic (i.e., Treg-inducing) properties and to at the same time subvert their immunogenic functions. Cultured murine and human DCs that have been exposed to and have phagocytosed live H. pylori fail to prime Th17 and Th1 responses and instead preferentially induce FoxP3 expression and suppressive activity in cocultured naive T cells (Kao et al. 2010; Kim et al. 2011; Oertli et al. 2012) (Fig. 12.4). This tolerogenic activity has been attributed to the failure of H. pylori-exposed DCs to mature, i.e., to express high levels of MHCII and co-stimulatory markers such as CD80, CD86, and CD40, as well as T helper cell-differentiating and T helper cell-activating cytokines such as IL-12 or IL-23 (Kaebisch et al. 2013; Oertli et al. 2012) (Fig. 12.4). This is true even in the presence of strong maturation signals delivered via TLR -4 or TLR-9 engagement, e.g., by E. coli LPS or CpG oligonucleotides (Oertli et al. 2012). The semi-mature DCs that result from H. pylori exposure are characterized by high expression of MHCII yet virtually no or low expression of co-stimulatory markers; similarly, these DCs fail to produce IL-12, IL-6, and TNF-α and instead secrete large amounts of the anti-inflammatory cytokine IL-10 (Oertli et al. 2012) (Fig. 12.4). Semi-mature and immature DCs have been implicated in Treg induction and tolerogenic immune response s (Maldonado and von Andrian 2010); indeed, H. pylori-exposed, semi-mature DCs are excellent inducers of Treg differentiation in vitro and in vivo (Kao et al. 2010; Oertli et al. 2012), and their forced maturation by LPS treatment is sufficient to break tolerance in vivo (Oertli et al. 2012). Consistent with the experimental results, the gastric mucosa of H. pylori-infected human carriers is populated by semi-mature DCs lacking co-stimulatory markers (Oertli et al. 2012). The H. pylori virulence and persistence factors required for the bacteria’s tolerogenic activity on DCs are under intense investigation, whereas two secreted factors, the vacuolating cytotoxin VacA and the γ-glutamyl transpeptidase GGT , have been implicated in the inhibition of murine DC maturation and tolerogenic reprogramming (Kim et al. 2011; Oertli et al. 2012); the T4SS of H. pylori seems to be equally or more important in human DCs (Kaebisch et al. 2013).

The maturation status of dendritic cells (DCs ) directs T helper cell differentiation. Immature DCs with high phagocytic activity follow chemokine gradients to sites of microbial colonization, where they actively sample antigen, which is then processed and presented in the context of MHC class II molecules on the cell surface. (a) Simultaneous stimulation of phagocytic DCs by recognition of PAMPs via cytosolic or membrane-bound pattern recognition receptors promotes DC maturation , which results in upregulation of maturation markers and co-stimulatory molecules (CD40, CD80, CD86) on the cell surface, as well as the production of T cell-differentiating and T cell-activating, as well as other pro-inflammatory cytokines (IL -12, IL-23, TNF-α , IL-6). Three signals (antigen recognition via the T cell receptor, co-stimulatory signals, and soluble cytokine signals) are required for the differentiation of naive T cells into Th1 or Th17 cells, with high levels of IL-12 driving Th1 and high levels of IL-23 driving Th17 differentiation. Both subsets are required for H. pylori control. (b) H. pylori exposure generates semi-mature DCs with high MHC class II expression but no or little co-stimulation and low levels of Th1/Th17 cell-differentiating cytokines, resulting in Treg differentiation and immunosuppression. An additional marker of semi-mature, tolerogenic DCs is their expression and secretion of IL-10
Several studies have attempted to functionally address the contribution of (tolerogenic) DCs to immune tolerance and to H. pylori -specific immunity in experimental models (Kao et al. 2010). Interestingly, the depletion of CD11c+ DCs in a genetic model taking advantage of the CD11c-driven expression of the diphtheria toxin receptor improves clearance of H. pylori upon challenge infection of vaccinated mice (Hitzler et al. 2011). Whereas prior vaccination with H. pylori extract in conjunction with either the gold standard cholera toxin adjuvant or a novel mycobacteria-derived adjuvant (CAF01) slashed H. pylori burdens by two orders of magnitude, this reduction could be further improved by another one to two orders of magnitude by the depletion of DCs (Hitzler et al. 2011). Similar effects were obtained in experimental H. pylori infection models not involving prior immunization (Hitzler et al. 2011; Oertli et al. 2012). In both settings, the depletion of DCs not only reduced bacterial burdens but also boosted all relevant correlates of (vaccine -induced) protective immunity , such as the recruitment of memory T cells, mast cells , and neutrophils to the gastric mucosa , the priming of H. pylori-specific Th1 and Th17 cells in the draining mesenteric lymph nodes, and the local production of protective cytokines including IFN-γ and IL -17 (Hitzler et al. 2011; Oertli et al. 2012). The results suggest that DCs are dispensible for immunity to H. pylori and instead are an essential element of immunoregulation, preventing control of the infection. Similar observations have been made in models of autoimmunity, in which the depletion of DCs invariably aggravates rather than improves disease severity (Yogev et al. 2012). A model of adoptive bone marrow-derived DC transfer into H. pylori-infected mice also showed that the transfer of DCs that had been exposed to H. pylori ex vivo, but not of naive DCs, efficiently induced Treg differentiation in vivo (Kao et al. 2010). Tregs induced in vivo upon Hp -DC transfer in turn suppressed anti-H. pylori-specific Th17 responses and their depletion or the depletion of IL-10 and/or TGF-β, promoted H. pylori clearance (Kao et al. 2010). All available in vivo data thus suggest that H. pylori effectively directs DCs to acquire tolerogenic properties, which drive Treg differentiation and anti-inflammatory cytokine production, suppress T-effector cell functions, and promote persistent infection (Fig. 12.4).
7 H. pylori P rotects Against Allergic and Chronic Inflammatory Diseases Through the Induction of Treg -Mediated Immune Tolerance
Public health in developed countries has been dominated by two major trends since the second half of the twentieth century. The incidence of infectious diseases has declined sharply in that time frame, whereas immunological disorders such as multiple sclerosis (MS), inflammatory bowel disease (IBD), allergic asthma and other allergic diseases, and type I diabetes have dramatically increased in incidence over the same time period (Bach 2002). The incidence of infections with H. pylori has paralleled those of other infectious agents, with childhood acquisition rates dropping in the USA from >50 to 10 % between the beginning and the end of the twentieth century (Blaser and Falkow 2009). In line with the carcinogenic properties of H. pylori infection (Parsonnet et al. 1991), a beneficial effect of this trend has been the steady decline in gastric cancer rates and associated mortality in countries from which H. pylori has disappeared (Forman 2005). The downsides (some confirmed, some debated) of the loss of H. pylori in developed countries are (a) the increase in esophageal diseases such as esophagitis , Barrett’s esophagus, and esophageal cancer with the latter having increased in incidence by over sixfold in the years from 1975 to 2000 alone (Pohl and Welch 2005) and (b) the increase in asthma and allergies (Eder et al. 2006). Numerous epidemiological studies have shown an inverse association of H. pylori infection with asthma and other allergies with respiratory tract manifestations, which was particularly strong in children and adolescents and in individuals with early onset allergies and asthma (Amberbir et al. 2011; Blaser et al. 2008; Chen and Blaser 2007, 2008; Reibman et al. 2008). Meta-analyses examining 14 and 19 published studies on the topic have confirmed the general trend (Wang et al. 2013; Zhou et al. 2013). The chronic inflammatory skin disease atopic dermatitis /eczema has also been inversely linked to H. pylori infection in studies including over 3000 German schoolchildren and almost 2000 Japanese university students (Herbarth et al. 2007; Shiotani et al. 2008). This trend was recently confirmed in a longitudinal study conducted on over 1000 Ethiopian children (Amberbir et al. 2014). Similarly, H. pylori infection seems to confer protection against IBD, as suggested by a recent meta-analysis of 23 articles examining such a possible link (Luther et al. 2011). Only 27 % of IBD patients had evidence of infection with H. pylori compared to 41 % of patients in the control group (Luther et al. 2011). A large epidemiological survey conducted on 136,000 patients in the USA found a decreased risk of celiac disease in individuals infected with H. pylori (Lebwohl et al. 2013).
Following up on the various observational studies in human populations, possible protective effects of experimental H. pylori infection have been examined in animal models of allergic asthma and IBD (Fig. 12.5). In a murine model of allergic asthma induced by ovalbumin or house dust mite antigen sensitization and challenge, H. pylori infection conferred protection against the airway hyperresponsiveness, bronchoalveolar eosinophilia , lung inflammation, and goblet cell metaplasia that are hallmarks of asthma in humans and mice (Arnold et al. 2011a). The protective effects were evident in animals that had been experimentally infected during the neonatal period (Arnold et al. 2011a), i.e., at an age when humans typically contract the infection from their mothers (Weyermann et al. 2009). Asthma protection conferred by H. pylori was abolished by antibiotic eradication therapy prior to allergen challenge and depended critically on Tregs (Fig. 12.5); the systemic depletion of Tregs abrogated asthma protection, and pure populations of Tregs were sufficient to transfer protection from neonatally infected donors to naive recipients (Arnold et al. 2011a). These results are in line with the finding that neonatal infection with H. pylori induces Treg -mediated immune tolerance to the bacteria (Arnold et al. 2011b) and that children predominantly launch Treg responses to H. pylori infection (Harris et al. 2008). Experimental models of colitis in mice also confirm the trends seen in human IBD patients (Fig. 12.5); for example, H. pylori infection effectively reduced the Th17-driven colitis induced by Salmonella typhimurium infection (Higgins et al. 2010). The protective agent of H. pylori in colitis appears to be its DNA, which exhibits a strongly biased ratio of immunoregulatory-to-immunostimulatory sequences and – when administered orally – protects against the development of acute or chronic DSS-induced colitis (Luther et al. 2011). The protective effects of H. pylori DNA were attributed to its activity on DCs , which prevents IL -12 and type I IFN production and generally favors anti- over pro-inflammatory responses (Luther et al. 2011; Owyang et al. 2012). Overall, the combined results from observational studies in humans and interventional studies in mice suggest strongly that H. pylori infection can protect against experimentally induced allergic asthma and IBD; the protective mechanisms appear to involve Tregs and DCs, which are actively induced and “tolerized” by H. pylori, respectively. It is likely that the protection against allergic and chronic inflammatory diseases that is conferred by H. pylori is a by-product of its immunomodulatory activity, which allows the bacteria to suppress Th1 and Th17 responses and to establish and maintain persistent infection . Similar effects have been reported for other persistent pathogens including helminths and Mycobacterium tuberculosis, which have been proposed to suppress allergen-specific immune response s in asthma through the induction of Tregs or through immune deviation (Obihara et al. 2007; Barlan et al. 2006). The potential use of microbial products and live attenuated bacteria or parasites in the treatment of allergies and IBD has received increasing attention lately, and some (especially helminth-derived) compounds are in clinical development (Torres et al. 2013). Whether H. pylori may be exploited in a similar fashion remains to be seen in the future.

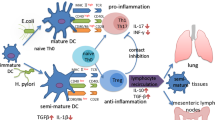
H. pylori exerts systemic immunomodulatory effects. H. pylori exclusively inhabits the gastric mucosa , but has systemic immunomodulatory effects that manifest in the airways and lower gastrointestinal tract. Tissue-resident DCs can sample H. pylori antigens in the gastric mucosa and subsequently migrate to the stomach-draining and mesenteric lymph nodes (MLNs), where they prime T cell (in particular Treg ) responses. Alternatively, soluble antigens can be transported via the lymph to the MLNs for presentation by resident DC populations. MLN-derived, H. pylori-specific Tregs enter the circulation and accumulate not only in the gastric mucosa but also at other mucosal surfaces of the body, such as those of the airways and lower bowel. According to current models, pathogenic effector T cell populations (allergen-specific Th17 and Th2 cells and colitogenic Th1 and Th17 cells) are suppressed by H. pylori-induced Tregs via soluble mediators (such as IL -10) and contact-dependent mechanisms. Abbreviations used: Th1/2/17, T helper cell subsets; Treg, regulatory T cell
8 Conclusions and Outlook
Despite having received substantial attention for three decades, several aspects of the H. pylori /host interaction remain poorly understood. Among them is the manipulation of innate and adaptive immunity by H. pylori, a feature that is central to the persistence of the bacteria and to the chronicity of H. pylori-associated diseases. Much attention has focused on gaining a better understanding of the prerequisites of successful vaccination aiming to prevent the primary infection (see Chap. 24), whereas less is known about the steady-state interaction of an established H. pylori infection with the host immune system. The asymptomatic carrier state in particular is vastly understudied, and the genetic and lifestyle parameters affecting the risk of developing H. pylori-associated gastric disease remain largely enigmatic. Although it is now clear that gastric infection-induced lesions are immunopathological in nature and at least in animal models can be attributed to the detrimental effects of T helper cells and their signature cytokines on the gastric mucosa , it appears likely that H. pylori toxins contribute to the mucosal damage. Experimental research into the pathomechanisms active during H. pylori infections is limited by the failure of most strains to colonize small rodents persistently and to the differences in gastric physiology and immunology that exist between humans and all currently used animal models (maybe with the exception of rhesus macaques). Future avenues of research in the field of H. pylori immunobiology will likely explore the relative contribution of direct (bacterially mediated) and indirect (immunopathological) effects to gastric carcinogenesis and will shed more light on the differential disease risk within and across human populations. Exciting new insights are expected as more elaborate model systems (such as gastric organoids, humanized mice, etc.) become available to the broader research community. Finally, future research directions will certainly take into account that H. pylori – beside its role as a gastric pathogen – is also an ancient member of our gastric microbiota that, together with other constituents of the microbiota of the gastrointestinal tract, has shaped the evolution of the human mucosal immune system .
References
Aebischer T, Bumann D, Epple HJ, Metzger W, Schneider T, Cherepnev G, Walduck AK, Kunkel D, Moos V, Loddenkemper C, Jiadze I, Panasyuk M, Stolte M, Graham DY, Zeitz M, Meyer TF (2008) Correlation of T cell response and bacterial clearance in human volunteers challenged with Helicobacter pylori revealed by randomised controlled vaccination with Ty21a-based Salmonella vaccines. Gut 57:1065–1072
Akhiani AA, Pappo J, Kabok Z, Schon K, Gao W, Franzen LE, Lycke N (2002) Protection against Helicobacter pylori infection following immunization is IL-12-dependent and mediated by Th1 cells. J Immunol 169:6977–6984
Akhiani AA, Schon K, Franzen LE, Pappo J, Lycke N (2004a) Helicobacter pylori-specific antibodies impair the development of gastritis, facilitate bacterial colonization, and counteract resistance against infection. J Immunol 172:5024–5033
Akhiani AA, Schon K, Lycke N (2004b) Vaccine-induced immunity against Helicobacter pylori infection is impaired in IL-18-deficient mice. J Immunol 173:3348–3356
Algood HM, Torres VJ, Unutmaz D, Cover TL (2007) Resistance of primary murine CD4+ T cells to Helicobacter pylori vacuolating cytotoxin. Infect Immun 75:334–341
Amberbir A, Medhin G, Erku W, Alem A, Simms R, Robinson K, Fogarty A, Britton J, Venn A, Davey G (2011) Effects of Helicobacter pylori, geohelminth infection and selected commensal bacteria on the risk of allergic disease and sensitization in 3-year-old Ethiopian children. Clin Exp Allergy 41:1422–1430
Amberbir A, Medhin G, Abegaz WE, Hanlon C, Robinson K, Fogarty A, Britton J, Venn A, Davey G (2014) Exposure to Helicobacter pylori infection in early childhood and the risk of allergic disease and atopic sensitization: a longitudinal birth cohort study. Clin Exp Allergy 44:563–571
Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, Logan SM, Aderem A (2005) Evasion of toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci U S A 102:9247–9252
Arnold B, Schuler T, Hammerling GJ (2005) Control of peripheral T-lymphocyte tolerance in neonates and adults. Trends Immunol 26:406–411
Arnold IC, Dehzad N, Reuter S, Martin H, Becher B, Taube C, Müller A (2011a) Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J Clin Invest 121:3088–3093
Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, Sparwasser T, Müller A (2011b) Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 140:199–209
Atherton JC, Cao P, Peek RM Jr, Tummuru MK, Blaser MJ, Cover TL (1995) Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem 270:17771–17777
Atherton JC, Sharp PM, Cover TL, Gonzalez-Valencia G, Peek RM Jr, Thompson SA, Hawkey CJ, Blaser MJ (1999) Vacuolating cytotoxin (vacA) alleles of Helicobacter pylori comprise two geographically widespread types, m1 and m2, and have evolved through limited recombination. Curr Microbiol 39:211–218
Bach JF (2002) The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med 347:911–920
Barlan I, Bahceciler NN, Akdis M, Akdis CA (2006) Bacillus Calmette-Guerin, mycobacterium bovis, as an immunomodulator in atopic diseases. Immunol Allergy Clin North Am 26:365–377
Barrozo RM, Cooke CL, Hansen LM, Lam AM, Gaddy JA, Johnson EM, Cariaga TA, Suarez G, Peek RM Jr, Cover TL, Solnick JV (2013) Functional plasticity in the type IV secretion system of Helicobacter pylori. PLoS Pathog 9:e1003189
Bauer B, Pang E, Holland C, Kessler M, Bartfeld S, Meyer TF (2012a) The Helicobacter pylori virulence effector CagA abrogates human beta-defensin 3 expression via inactivation of EGFR signaling. Cell Host Microbe 11:576–586
Bauer B, Wex T, Kuester D, Meyer T, Malfertheiner P (2012b) Differential expression of human beta defensin 2 and 3 in gastric mucosa of Helicobacter pylori-infected individuals. Helicobacter 18:6–12
Becher D, Deutscher ME, Simpfendorfer KR, Wijburg OL, Pederson JS, Lew AM, Strugnell RA, Walduck AK (2010) Local recall responses in the stomach involving reduced regulation and expanded help mediate vaccine-induced protection against Helicobacter pylori in mice. Eur J Immunol 40:2778–2790
Blaser MJ, Falkow S (2009) What are the consequences of the disappearing human microbiota? Nat Rev Microbiol 7:887–894
Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, Stemmermann GN, Nomura A (1995) Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 55:2111–2115
Blaser MJ, Chen Y, Reibman J (2008) Does Helicobacter pylori protect against asthma and allergy? Gut 57:561–567
Bollrath J, Powrie FM (2013) Controlling the frontier: regulatory T-cells and intestinal homeostasis. Semin Immunol 25:352–357
Boncristiano M, Paccani SR, Barone S, Ulivieri C, Patrussi L, Ilver D, Amedei A, D’Elios MM, Telford JL, Baldari CT (2003) The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J Exp Med 198:1887–1897
Chen Y, Blaser MJ (2007) Inverse associations of Helicobacter pylori with asthma and allergy. Arch Intern Med 167:821–827
Chen Y, Blaser MJ (2008) Helicobacter pylori colonization is inversely associated with childhood asthma. J Infect Dis 198:553–560
Chevalier C, Thiberge JM, Ferrero RL, Labigne A (1999) Essential role of Helicobacter pylori gamma-glutamyltranspeptidase for the colonization of the gastric mucosa of mice. Mol Microbiol 31:1359–1372
Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, Maloy KJ (2012) IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med 209:1595–1609
Cover TL, Krishna US, Israel DA, Peek RM Jr (2003) Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res 63:951–957
Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD (2003) Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421:744–748
Cullen TW, Giles DK, Wolf LN, Ecobichon C, Boneca IG, Trent MS (2012) Helicobacter pylori versus the host: remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PLoS Pathog 7:e1002454
Czajkowsky DM, Iwamoto H, Cover TL, Shao Z (1999) The vacuolating toxin from Helicobacter pylori forms hexameric pores in lipid bilayers at low pH. Proc Natl Acad Sci U S A 96:2001–2006
Domanska G, Motz C, Meinecke M, Harsman A, Papatheodorou P, Reljic B, Dian-Lothrop EA, Galmiche A, Kepp O, Becker L, Gunnewig K, Wagner R, Rassow J (2010) Helicobacter pylori VacA toxin/subunit p34: targeting of an anion channel to the inner mitochondrial membrane. PLoS Pathog 6:e1000878
Eder W, Ege MJ, von Mutius E (2006) The asthma epidemic. N Engl J Med 355:2226–2235
El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni JF Jr, Rabkin CS (2000) Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 404:398–402
Ermak TH, Giannasca PJ, Nichols R, Myers GA, Nedrud J, Weltzin R, Lee CK, Kleanthous H, Monath TP (1998) Immunization of mice with urease vaccine affords protection against Helicobacter pylori infection in the absence of antibodies and is mediated by MHC class II-restricted responses. J Exp Med 188:2277–2288
Forman D (2005) Re: the role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst 97:1013–1014
Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R (2003) Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301:1099–1102
Gerhard M, Schmees C, Voland P, Endres N, Sander M, Reindl W, Rad R, Oelsner M, Decker T, Mempel M, Hengst L, Prinz C (2005) A secreted low-molecular-weight protein from Helicobacter pylori induces cell-cycle arrest of T cells. Gastroenterology 128:1327–1339
Gewirtz AT, Yu Y, Krishna US, Israel DA, Lyons SL, Peek RM Jr (2004) Helicobacter pylori flagellin evades toll-like receptor 5-mediated innate immunity. J Infect Dis 189:1914–1920
Gorrell RJ, Guan J, Xin Y, Tafreshi MA, Hutton ML, McGuckin MA, Ferrero RL, Kwok T (2012) A novel NOD1- and CagA-independent pathway of interleukin-8 induction mediated by the Helicobacter pylori type IV secretion system. Cell Microbiol 15:554–570
Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Geijtenbeek TB (2009) Carbohydrate-specific signaling through the DC-SIGN signalosome tailors immunity to mycobacterium tuberculosis, HIV-1 and Helicobacter pylori. Nat Immunol 10:1081–1088
Harris PR, Cover TL, Crowe DR, Orenstein JM, Graham MF, Blaser MJ, Smith PD (1996) Helicobacter pylori cytotoxin induces vacuolation of primary human mucosal epithelial cells. Infect Immun 64:4867–4871
Harris PR, Wright SW, Serrano C, Riera F, Duarte I, Torres J, Pena A, Rollan A, Viviani P, Guiraldes E, Schmitz JM, Lorenz RG, Novak L, Smythies LE, Smith PD (2008) Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology 134:491–499
Harrison OJ, Powrie FM (2013) Regulatory T cells and immune tolerance in the intestine. Cold Spring Harb Perspect Biol 5:a018341
Herbarth O, Bauer M, Fritz GJ, Herbarth P, Rolle-Kampczyk U, Krumbiegel P, Richter M, Richter T (2007) Helicobacter pylori colonisation and eczema. J Epidemiol Community Health 61:638–640
Higgins PD, Johnson LA, Luther J, Zhang M, Sauder KL, Blanco LP, Kao JY (2010) Prior Helicobacter pylori infection ameliorates Salmonella typhimurium-induced colitis: mucosal crosstalk between stomach and distal intestine. Inflamm Bowel Dis 17:1398–1408
Hitzler I, Oertli M, Becher B, Agger EM, Müller A (2011) Dendritic cells prevent rather than promote immunity conferred by a helicobacter vaccine using a mycobacterial adjuvant. Gastroenterology 141:186–196
Hitzler I, Kohler E, Engler DB, Yazgan AS, Muller A (2012a) The role of Th cell subsets in the control of Helicobacter infections and in T cell-driven gastric immunopathology. Front Immunol 3:142
Hitzler I, Sayi A, Kohler E, Engler DB, Koch KN, Hardt WD, Muller A (2012b) Caspase-1 has both proinflammatory and regulatory properties in helicobacter infections, which are differentially mediated by its substrates IL-1beta and IL-18. J Immunol 188:3594–3602
Horvath DJ Jr, Washington MK, Cope VA, Algood HMS (2012) IL-23 contributes to control of chronic Helicobacter pylori infection and the development of T helper responses in a mouse model. Front Immun 3:56
Huang JQ, Zheng GF, Sumanac K, Irvine EJ, Hunt RH (2003) Meta-analysis of the relationship between cagA seropositivity and gastric cancer. Gastroenterology 125:1636–1644
Ismail HF, Fick P, Zhang J, Lynch RG, Berg DJ (2003) Depletion of neutrophils in IL-10(−/−) mice delays clearance of gastric Helicobacter infection and decreases the Th1 immune response to Helicobacter. J Immunol 170:3782–3789
Jimenez-Soto LF, Kutter S, Sewald X, Ertl C, Weiss E, Kapp U, Rohde M, Pirch T, Jung K, Retta SF, Terradot L, Fischer W, Haas R (2009) Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD-independent manner. PLoS Pathog 5:e1000684
Kaebisch R, Mejias-Luque R, Prinz C, Gerhard M (2013) Helicobacter pylori cytotoxin-associated gene A impairs human dendritic cell maturation and function through IL-10-mediated activation of STAT3. J Immunol 192:316–323
Kao JY, Zhang M, Miller MJ, Mills JC, Wang B, Liu M, Eaton KA, Zou W, Berndt BE, Cole TS, Takeuchi T, Owyang SY, Luther J (2010) Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology 138:1046–1054
Kim JM, Kim JS, Yoo DY, Ko SH, Kim N, Kim H, Kim YJ (2011) Stimulation of dendritic cells with Helicobacter pylori vacuolating cytotoxin negatively regulates their maturation via the restoration of E2F1. Clin Exp Immunol 166:34–45
Kim DJ, Park JH, Franchi L, Backert S, Nunez G (2013) The Cag pathogenicity island and interaction between TLR2/NOD2 and NLRP3 regulate IL-1beta production in Helicobacter pylori-infected dendritic cells. Eur J Immunol 43:2650–2658
Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, Misselwitz R, Berger J, Sewald N, Konig W, Backert S (2007) Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 449:862–866
Lebwohl B, Blaser MJ, Ludvigsson JF, Green PH, Rundle A, Sonnenberg A, Genta RM (2013) Decreased risk of celiac disease in patients with Helicobacter pylori colonization. Am J Epidemiol 178:1721–1730
Lundgren A, Suri-Payer E, Enarsson K, Svennerholm AM, Lundin BS (2003) Helicobacter pylori-specific CD4+ CD25 high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect Immun 71:1755–1762
Lundgren A, Stromberg E, Sjoling A, Lindholm C, Enarsson K, Edebo A, Johnsson E, Suri-Payer E, Larsson P, Rudin A, Svennerholm AM, Lundin BS (2005a) Mucosal FOXP3-expressing CD4+ CD25 high regulatory T cells in Helicobacter pylori-infected patients. Infect Immun 73:523–531
Lundgren A, Trollmo C, Edebo A, Svennerholm AM, Lundin BS (2005b) Helicobacter pylori-specific CD4+ T cells home to and accumulate in the human Helicobacter pylori-infected gastric mucosa. Infect Immun 73:5612–5619
Luther J, Owyang SY, Takeuchi T, Cole TS, Zhang M, Liu M, Erb-Downward J, Rubenstein JH, Chen CC, Pierzchala AV, Paul JA, Kao JY (2011) Helicobacter pylori DNA decreases pro-inflammatory cytokine production by dendritic cells and attenuates dextran sodium sulphate-induced colitis. Gut 60:1479–1486
Maizels RM, Smith KA (2011) Regulatory T cells in infection. Adv Immunol 112:73–136
Maldonado RA, von Andrian UH (2010) How tolerogenic dendritic cells induce regulatory T cells. Adv Immunol 108:111–165
Malfertheiner P, Schultze V, Rosenkranz B, Kaufmann SH, Ulrichs T, Novicki D, Norelli F, Contorni M, Peppoloni S, Berti D, Tornese D, Ganju J, Palla E, Rappuoli R, Scharschmidt BF, Del Giudice G (2008) Safety and immunogenicity of an intramuscular Helicobacter pylori vaccine in noninfected volunteers: a phase I study. Gastroenterology 135:787–795
McBride A, Konowich J, Salgame P (2013) Host defense and recruitment of Foxp3(+) T regulatory cells to the lungs in chronic Mycobacterium tuberculosis infection requires toll-like receptor 2. PLoS Pathog 9:e1003397
McClain MS, Cao P, Cover TL (2001) Amino-terminal hydrophobic region of Helicobacter pylori vacuolating cytotoxin (VacA) mediates transmembrane protein dimerization. Infect Immun 69:1181–1184
Moran AP, Lindner B, Walsh EJ (1997) Structural characterization of the lipid A component of Helicobacter pylori rough- and smooth-form lipopolysaccharides. J Bacteriol 179:6453–6463
Obihara CC, Kimpen JL, Beyers N (2007) The potential of Mycobacterium to protect against allergy and asthma. Curr Allergy Asthma Rep 7:223–230
Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, Maxeiner J, Hansson M, Taube C, Quiding-Jarbrink M, Muller A (2012) DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest 122:1082–1096
Oertli M, Noben M, Engler DB, Semper RP, Reuter S, Maxeiner J, Gerhard M, Taube C, Muller A (2013) Helicobacter pylori gamma-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc Natl Acad Sci U S A 110:3047–3052
Otani K, Watanabe T, Tanigawa T, Okazaki H, Yamagami H, Watanabe K, Tominaga K, Fujiwara Y, Oshitani N, Arakawa T (2009) Anti-inflammatory effects of IL-17A on Helicobacter pylori-induced gastritis. Biochem Biophys Res Commun 382:252–258
Otani K, Tanigawa T, Watanabe T, Nadatani Y, Sogawa M, Yamagami H, Shiba M, Watanabe K, Tominaga K, Fujiwara Y, Arakawa T (2012) Toll-like receptor 9 signaling has anti-inflammatory effects on the early phase of Helicobacter pylori-induced gastritis. Biochem Biophys Res Commun 426:342–349
Ottenhoff TH (2012) New pathways of protective and pathological host defense to mycobacteria. Trends Microbiol 20:419–428
Owyang SY, Luther J, Owyang CC, Zhang M, Kao JY (2012) Helicobacter pylori DNA’s anti-inflammatory effect on experimental colitis. Gut Microbes 3:168–171
Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK (1991) Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 325:1127–1131
Patel SR, Smith K, Letley DP, Cook KW, Memon AA, Ingram RJ, Staples E, Backert S, Zaitoun AM, Atherton JC, Robinson K (2013) Helicobacter pylori down regulates expression of human beta-defensin 1 in the gastric mucosa in a type IV secretion-dependent fashion. Cell Microbiol 15:2080–2092
Pohl H, Welch HG (2005) The role of over diagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst 97:142–146
Rad R, Ballhorn W, Voland P, Eisenacher K, Mages J, Rad L, Ferstl R, Lang R, Wagner H, Schmid RM, Bauer S, Prinz C, Kirschning CJ, Krug A (2009) Extracellular and intracellular pattern recognition receptors cooperate in the recognition of Helicobacter pylori. Gastroenterology 136:2247–2257
Reibman J, Marmor M, Filner J, Fernandez-Beros ME, Rogers L, Perez-Perez GI, Blaser MJ (2008) Asthma is inversely associated with Helicobacter pylori status in an urban population. PLoS One 3:e4060
Rieder G, Merchant JL, Haas R (2005) Helicobacter pylori cag-type IV secretion system facilitates corpus colonization to induce precancerous conditions in Mongolian gerbils. Gastroenterology 128:1229–1242
Robinson K, Kenefeck R, Pidgeon EL, Shakib S, Patel S, Polson RJ, Zaitoun AM, Atherton JC (2008) Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut 57:1375–1385
Rokkas T, Pistiolas D, Sechopoulos P, Robotis I, Margantinis G (2007) The long-term impact of Helicobacter pylori eradication on gastric histology: a systematic review and meta-analysis. Helicobacter 12(Suppl 2):32–38
Rothenbacher D, Inceoglu J, Bode G, Brenner H (2000) Acquisition of Helicobacter pylori infection in a high-risk population occurs within the first 2 years of life. J Pediatr 136:744–748
Salama NR, Otto G, Tompkins L, Falkow S (2001) Vacuolating cytotoxin of Helicobacter pylori plays a role during colonization in a mouse model of infection. Infect Immun 69:730–736
Salama NR, Hartung ML, Muller A (2013) Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat Rev Microbiol 11:385–399
Sayi A, Kohler E, Hitzler I, Arnold I, Schwendener R, Rehrauer H, Muller A (2009) The CD4+ T cell-mediated IFN-gamma response to Helicobacter infection is essential for clearance and determines gastric cancer risk. J Immunol 182:7085–7101
Sayi A, Kohler E, Toller IM, Flavell RA, Muller W, Roers A, Müller A (2011) TLR-2-activated B cells suppress Helicobacter-induced preneoplastic gastric immunopathology by inducing T regulatory-1 cells. J Immunol 186:878–890
Schmees C, Prinz C, Treptau T, Rad R, Hengst L, Voland P, Bauer S, Brenner L, Schmid RM, Gerhard M (2007) Inhibition of T-cell proliferation by Helicobacter pylori gamma-glutamyl transpeptidase. Gastroenterology 132:1820–1833
Sewald X, Gebert-Vogl B, Prassl S, Barwig I, Weiss E, Fabbri M, Osicka R, Schiemann M, Busch DH, Semmrich M, Holzmann B, Sebo P, Haas R (2008) Integrin subunit CD18 Is the T-lymphocyte receptor for the Helicobacter pylori vacuolating cytotoxin. Cell Host Microbe 3:20–29
Sewald X, Jimenez-Soto L, Haas R (2010) PKC-dependent endocytosis of the Helicobacter pylori vacuolating cytotoxin in primary T lymphocytes. Cell Microbiol 13:482–496
Shaffer CL, Gaddy JA, Loh JT, Johnson EM, Hill S, Hennig EE, McClain MS, McDonald WH, Cover TL (2011) Helicobacter pylori exploits a unique repertoire of type IV secretion system components for pilus assembly at the bacteria-host cell interface. PLoS Pathog 7:e1002237
Shi Y, Liu XF, Zhuang Y, Zhang JY, Liu T, Yin Z, Wu C, Mao XH, Jia KR, Wang FJ, Guo H, Flavell RA, Zhao Z, Liu KY, Xiao B, Guo Y, Zhang WJ, Zhou WY, Guo G, Zou QM (2010) Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J Immunol 184:5121–5129
Shiotani A, Miyanishi T, Kamada T, Haruma K (2008) Helicobacter pylori infection and allergic diseases: epidemiological study in Japanese university students. J Gastroenterol Hepatol 23:e29–e33
Smoot DT, Resau JH, Earlington MH, Simpson M, Cover TL (1996) Effects of Helicobacter pylori vacuolating cytotoxin on primary cultures of human gastric epithelial cells. Gut 39:795–799
Stoicov C, Fan X, Liu JH, Bowen G, Whary M, Kurt-Jones E, Houghton J (2009) T-bet knockout prevents Helicobacter felis-induced gastric cancer. J Immunol 183:642–649
Sun X, Zhang M, El-Zataari M, Owyang SY, Eaton KA, Liu M, Chang YM, Zou W, Kao JY (2013) TLR2 mediates Helicobacter pylori-induced tolerogenic immune response in mice. PLoS One 8:e74595
Sundrud MS, Torres VJ, Unutmaz D, Cover TL (2004) Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc Natl Acad Sci U S A 101:7727–7732
Torres VJ, VanCompernolle SE, Sundrud MS, Unutmaz D, Cover TL (2007) Helicobacter pylori vacuolating cytotoxin inhibits activation-induced proliferation of human T and B lymphocyte subsets. J Immunol 179:5433–5440
Torres J, Danese S, Colombel JF (2013) New therapeutic avenues in ulcerative colitis: thinking out of the box. Gut 62:1642–1652
Velin D, Michetti P (2010) Advances in vaccination against Helicobacter pylori. Expert Rev Gastroenterol Hepatol 4:157–166
Velin D, Bachmann D, Bouzourene H, Michetti P (2005) Mast cells are critical mediators of vaccine-induced Helicobacter clearance in the mouse model. Gastroenterology 129:142–155
Velin D, Favre L, Bernasconi E, Bachmann D, Pythoud C, Saiji E, Bouzourene H, Michetti P (2009) Interleukin-17 is a critical mediator of vaccine-induced reduction of Helicobacter infection in the mouse model. Gastroenterology 136:2237–2246 e2231
Wang Q, Yu C, Sun Y (2013) The association between asthma and Helicobacter pylori: a meta-analysis. Helicobacter 18:41–53
Weyermann M, Rothenbacher D, Brenner H (2009) Acquisition of Helicobacter pylori infection in early childhood: independent contributions of infected mothers, fathers, and siblings. Am J Gastroenterol 104:182–189
Wong BC, Lam SK, Wong WM, Chen JS, Zheng TT, Feng RE, Lai KC, Hu WH, Yuen ST, Leung SY, Fong DY, Ho J, Ching CK (2004) Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA 291:187–194
Yogev N, Frommer F, Lukas D, Kautz-Neu K, Karram K, Ielo D, von Stebut E, Probst HC, van den Broek M, Riethmacher D, Birnberg T, Blank T, Reizis B, Korn T, Wiendl H, Jung S, Prinz M, Kurschus FC, Waisman A (2012) Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD-1 receptor(+) regulatory T cells. Immunity 37:264–275
Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD (2010) The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32:379–391
Zhou X, Wu J, Zhang G (2013) Association between Helicobacter pylori and asthma: a meta-analysis. Eur J Gastroenterol Hepatol 25:460–468
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Müller, A., Hartung, M.L. (2016). Helicobacter pylori and the Host Immune Response. In: Backert, S., Yamaoka, Y. (eds) Helicobacter pylori Research. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55936-8_12
Download citation
DOI: https://doi.org/10.1007/978-4-431-55936-8_12
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55934-4
Online ISBN: 978-4-431-55936-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)