
Abstract
The roundworm Caenorhabditis elegans is one of the most popular model organisms for research on aging because of its short lifespan and genetic tractability. Studies using C. elegans have identified many genes and pathways that regulate aging, several of which are conserved in other species, including mammals. In this chapter, we describe longevity-regulatory pathways including insulin/IGF-1 (insulin-like growth factor 1) signaling, TOR (target of rapamycin) signaling, autophagy, mitochondrial respiration, and HIF-1 (hypoxia-inducible factor 1) pathways. We also review the effects of dietary restriction, a key environmental factor that influences aging, on longevity-regulatory genetic factors. In addition, we illustrate the roles of two important C. elegans tissues, those of the sensory neural and reproductive systems, in regulating longevity at the molecular level. For each of the subtopics, we explain how changes in the expression of genes involved in each pathway and system alter longevity. We also speculate on the evolutionary significance of the genes and pathways that affect longevity. Given the conserved nature of longevity regulation, the dissection of the roles of these genetic factors in determining the C. elegans lifespan will provide important clues for understanding the secrets of human aging.
Y.L., S.W.A.A., M.A., M.S., A.B.H., D.-E.J., H.G.S., W.H., D.L., and K.S. contributed equally.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- C. elegans
- Aging
- Insulin/IGF-1
- Target of rapamycin
- Dietary restriction
- Autophagy
- Hypoxia-inducible factor
- Mitochondria
- Sensory neurons
- Reproductive system
1 Introduction
For a long time, the lifespans of living organisms were believed to be limited by the passive, age-dependent degeneration of tissues, which eventually leads to death. However, the findings of studies conducted in the past two decades tell us otherwise. Scientists have shown that organismal lifespans are subjected to active regulation by many genes and pathways. Although the exact mechanisms remain unclear, we now know that genetic factors influence the rate of organismal aging, in response to changes in environmental signals as well as physiologic inputs.
The small roundworm Caenorhabditis elegans has been exploited as a fundamental tool for research on aging, revealing crucial lifespan-regulatory pathways. One of the best advantages of C. elegans as a model for research on aging is that the C. elegans lifespan is only a few weeks. In addition, C. elegans undergoes clear age-dependent physiologic and behavioral changes and possesses many lifespan-regulatory pathways that are conserved across phyla. In fact, many evolutionarily conserved genes and pathways that affect organismal longevity were first identified in C. elegans.
The first lifespan-regulatory pathway identified in C. elegans was the insulin/ insulin-like growth factor 1 (IGF-1) signaling (IIS) pathway. Subsequent research led to the identification of target of rapamycin (TOR) signaling, dietary restriction (DR), steroid signaling, autophagy, reduced mitochondrial respiration, the hypoxia inducible factor 1 (HIF-1) pathway, and the sensory and reproductive systems as major lifespan-regulatory pathways in C. elegans. The identification of genes acting in those pathways and systems shed light on ensuing research in more complex organisms by revealing that many of the pathways are indeed evolutionarily conserved. In this chapter, we will review the roles and mechanisms by which key genes in lifespan-regulatory pathways modulate C. elegans lifespan. We include a table with an extensive list of C. elegans longevity-influencing genes, many of which are not described in the text due to space limits (Table 8.1). Furthermore, we speculate regarding the physiologic natures of the lifespan-regulatory pathways and possible reasons why these pathways modulate aging in C. elegans.
2 Longevity-Regulatory Pathways in C. elegans
2.1 The Insulin/Insulin-Like Growth Factor 1 Signaling Pathway
The insulin/IGF-1 signaling (IIS) pathway is one of the most highly characterized and evolutionarily conserved pathways that regulate aging (Fig. 8.1). In C. elegans, IIS is presumably initiated by the modulation of the activity of DAF-2, an insulin/IGF-1 receptor homolog, upon the binding of insulin-like peptides (ILPs). C. elegans expresses 40 ILPs, some of which are predicted to be DAF-2 agonists (e.g., ins-7) or antagonists (e.g., ins-18) (Kawano et al. 2000; Murphy et al. 2003, 2007; Fernandes de Abreu et al. 2014). The up-regulation of DAF-2 leads to the activation of a phosphoinositide 3-kinase (PI3K) cascade, which in turn regulates several transcription factors that affect lifespan (reviewed in Kaletsky and Murphy 2010; Murphy and Hu 2013).

Lifespan regulation by the insulin/IGF-1 signaling pathway in C. elegans . Insulin-like peptides (ILPs) bind to insulin/IGF-1 receptor DAF-2 to regulate its phosphorylation. The reduction of DAF-2 activity leads to decreased binding of IST-1, the insulin receptor substrate, resulting in the inactivation of phosphoinositide-3 kinase (AGE-1), which is responsible for the conversion of PI(4,5)P2 to PI(3,4,5)P3. This event decreases the activities of phosphoinositide-dependent kinase 1 (PDK-1) and the AKT-1 and AKT-2 (AKT-1/2) kinases. This reduces the phosphorylation of the DAF-16/FOXO and SKN-1/NRF2 transcription factors, increasing their activities. The inhibition of DAF-2 also increases the activity of heat shock transcription factor 1 (HSF-1). The activation of DAF-16, SKN-1, and HSF-1 leads to longevity by transcriptionally regulating the expression of downstream longevity genes
Many components of IIS regulate lifespan. For example, mutations in daf-2 can double the lifespan (Kenyon et al. 1993). The importance of DAF-2 as a longevity-regulatory factor is highlighted by the findings that mouse and human DAF-2 homologs are associated with longevity (reviewed in Kenyon 2010). Mutations in age-1, which encodes the catalytic subunit of PI3K (Morris et al. 1996), greatly extend lifespan as well (Friedman and Johnson 1988). Other genes in the IIS pathway whose genetic perturbation extends lifespan include ins-7 and daf-28, two ILP genes; ist-1 (insulin receptor substrate homolog); pdk-1, which encodes a phosphoinositide-dependent kinase (PDK); and akt-1 and akt-2, two C. elegans Akt/PKB homologs (Murphy et al. 2003; Paradis and Ruvkun 1998; Malone et al. 1996; Paradis et al. 1999; Wolkow et al. 2002). The transcription factors that act downstream of IIS, including DAF-16/FOXO, heat shock factor 1 (HSF-1) and SKN-1/NRF2, are required for longevity in animals with reduced IIS (Kenyon et al. 1993; Lin et al. 1997; Ogg et al. 1997; Hsu et al. 2003; Morley and Morimoto 2004; Tullet et al. 2008). Among those, DAF-16 is the best characterized longevity-promoting transcription factor, and its human homolog FOXO3A is also linked to longevity (reviewed in Kenyon 2010). DAF-16 is inactivated by phosphorylation via AKT-1 and AKT-2, and the dephosphorylation of DAF-16 leads to DAF-16 activation via nuclear localization (Lin et al. 2001; Henderson and Johnson 2001; Lee et al. 2001). The activation of DAF-16 leads to the induction of various genes that are crucial for longevity, including molecular chaperones, antioxidants, antimicrobials, and stress resistance genes (reviewed in Kaletsky and Murphy 2010; Murphy and Hu 2013). Many other longevity-regulatory genes involved in IIS have been identified using genetic, genomic, and proteomic approaches (Table 8.1). It will be important to characterize the relationships among those many factors to ascertain their impact on lifespan regulation.
What is the physiologic interpretation of the effects of IIS on longevity? IIS influences not only longevity but also other physiologic processes including larval development, fat metabolism, immunity, and stress resistance (reviewed in Kaletsky and Murphy 2010; Murphy and Hu 2013). Increased resistance to various stresses, including heat (Lithgow et al. 1995), osmotic stress (Lamitina and Strange 2005), reactive oxygen species (ROS) (Honda and Honda 1999), hypoxia (Scott et al. 2002) and endoplasmic reticulum (ER) stress (Henis-Korenblit et al. 2010), and proteotoxicity (Morley et al. 2002; Hsu et al. 2003) will confer survival benefits to C. elegans in general. At the cellular level, the stress resistance phenotypes might contribute to longevity by promoting cellular maintenance capabilities. In addition, animals with reduced IIS have higher chances to survive infection by various pathogens because of enhanced innate immunity (Garsin et al. 2003). This appears to lengthen lifespan in C. elegans, because the main cause of death in aged C. elegans in laboratory is infection by E. coli, the worm’s bacterial food (Garigan et al. 2002). Moreover, somatic cells in C. elegans with reduced IIS have the characteristics of germline stem cells, which are robustly protected from various stresses (Curran et al. 2009). Overall, C. elegans appears to employ endocrine IIS to enhance cellular protection and maintenance in the face of harsh environmental conditions, which may lead to longevity.
2.2 Lifespan-Regulating Genes in the Target of Rapamycin (TOR) Signaling Pathway
The TOR pathway is another evolutionarily well-conserved signaling pathway that influences aging (Fig. 8.2). TOR is a serine/threonine kinase that plays various physiologic roles, including involvement in cellular growth, metabolism, protein synthesis and autophagy, and aging, in response to changes in nutrient status (Stanfel et al. 2009; Kapahi et al. 2010; Evans et al. 2011; Laplante and Sabatini 2012; Johnson et al. 2013). TOR interacts with other proteins such as the regulatory-associated protein raptor and the rapamycin-insensitive companion rictor. Those interactions determine the formation of TOR complex 1 (TORC1) and TOR complex 2 (TORC2), respectively. Although both complexes are linked to longevity, the signaling pathway of TORC1 is characterized in more detail than that of TORC2. TORC1 exerts its effects by regulating downstream targets, including ribosomal protein S6 kinase, which promotes protein synthesis via the phosphorylation of ribosomal protein subunit 6.

Dietary restriction increases lifespan through AMPK, TOR signaling, and autophagy. Dietary restriction extends lifespan through energy-sensing pathways in general. Dietary restriction increases the activity of AMP-activated protein kinase (AMPK) while down-regulating target of rapamycin (TOR) kinase. Activated AMPK extends lifespan partly by increasing autophagy. Reduced TOR signaling leads to the activation of autophagy via PHA-4/FOXA and the reduction of translation via the down-regulation of ribosomal S6 kinase 1 (RSKS-1)
The inhibition of various TOR pathway genes extends the lifespan of C. elegans. The RNAi knockdown or mutation of let-363/TOR, daf-15/RAPTOR, rsks-1 (ribosomal protein S6 kinase), ribosomal subunits, or translational initiation factors increases the lifespan of C. elegans (Vellai et al. 2003; Jia et al. 2004; Ching et al. 2010; Chen et al. 2013b; Seo et al. 2013; Hansen et al. 2007; Pan et al. 2007; Syntichaki et al. 2007; Curran and Ruvkun 2007). In addition, treatment with rapamycin, the inhibitor of TOR, extends lifespan in C. elegans (Robida-Stubbs et al. 2012). Reduced translation underlies the longevity caused by the inhibition of TOR signaling (Hansen et al. 2007; Pan et al. 2007). The inhibition of the translation initiation factors ife-2/eIF4E and ifg-1/eIF4G, which are predicted to be regulated by TOR, extends lifespan (Pan et al. 2007; Wang et al. 2010; Rogers et al. 2011; Hansen et al. 2007; Syntichaki et al. 2007; Curran and Ruvkun 2007). Several longevity-promoting transcription factors including pha-4/FoxA (Sheaffer et al. 2008), HSF-1 (Seo et al. 2013), SKN-1 (Robida-Stubbs et al. 2012), and DAF-16 (Seo et al. 2013; Robida-Stubbs et al. 2012; Hansen et al. 2007) mediate the longevity caused by reduced TOR signaling. In addition, AMP-activated protein kinase (AMPK), a nutrient-sensing and longevity-promoting kinase, is required for the extended lifespan of rsks-1 mutants (Selman et al. 2009). Thus, reduced TORC1 signaling appears to lead to decreased translation, which in turn up-regulates various downstream longevity factors.
How does TOR signaling modulate longevity by influencing mRNA translation rates? Because protein synthesis requires large amounts of energy and metabolic resources such as ATP and amino acids, thrifty usage of proteins may be a cost-effective way for organisms to use resources for maintenance. Additionally, a slowed rate of protein synthesis may give organisms a chance to increase overall protein quality, because protein repair and degradation systems can be efficiently act on a relatively small amount of proteins. In addition to reducing translation, the inhibition of TOR signaling enhances autophagy-related processes (reviewed in Green et al. 2014). This in turn removes and/or recycles damaged proteins and organelles by selectively transporting them to lysosomes, which can promote healthy cellular environments. Thus, the inhibition of TOR signaling may benefit longevity by enhancing protein quality and reducing proteotoxicity during aging.
2.3 Genes That Mediate Dietary Restriction-Induced Longevity
Dietary restriction, which is defined as the restriction of food intake without malnutrition, extends lifespan in various species (reviewed in Piper et al. 2011). In C. elegans, diverse DR regimens have been used, such as genetic mutations that decrease feeding rates, the dilution or deprivation of food (bacteria) concentrations, intermittent fasting, and culture in axenic media that contain sparse nutrients (summarized in Greer and Brunet 2009). Interestingly, different genes and pathways mediate the effects of diverse DR regimens on longevity in C. elegans (Fig. 8.2, Table 8.1). The most notable genetic factors that mediate the longevity conferred by DR are cellular energy sensors. Dietary restriction decreases ATP levels and subsequently increases the AMP/ATP ratio, and this in turn activates the energy sensor AMPK (Hardie 2014). The catalytic α subunit of AMPK in C. elegans, AAK-2, is necessary and sufficient for DR-induced longevity (Greer and Brunet 2009; Greer et al. 2007). Target of rapamycin (TOR), another major energy sensor, is also implicated as a mediator of DR-induced longevity (Hansen et al. 2007). The inhibition of let-363, C. elegans TOR, extends lifespan, perhaps by mimicking DR. The lifespan extension conferred by intermittent fasting is mediated by RHEB-1 (GTPase), an activator of TOR (Honjoh et al. 2009). One possible mechanism by which reduced TOR levels mediate DR-induced longevity is the up-regulation of autophagy (reviewed in Green et al. 2014), which helps cellular maintenance during DR. Thus, DR appears to increase lifespan by regulating energy sensors to maximize cellular maintenance under conditions where energy is scarce.
DR alters the activities of longevity-promoting transcription factors. The AMPK activated upon DR up-regulates DAF-16 to mediate longevity (Greer et al. 2007). Dietary restriction reduces TOR signaling, which leads to changes in the activity of transcription factors including PHA-4 (Panowski et al. 2007; Sheaffer et al. 2008), hypoxia-inducible factor 1 (HIF-1) (Chen et al. 2009), HSF-1 (Seo et al. 2013; Steinkraus et al. 2008), DAF-16 (Robida-Stubbs et al. 2012; Seo et al. 2013), and SKN-1 (Bishop and Guarente 2007; Robida-Stubbs et al. 2012). SIR-2.1 (sirtuin: NAD-dependent protein deacetylase) (Wang and Tissenbaum 2006) and PNC-1 (a key component of the NAD+ salvage pathway) (Moroz et al. 2014) mediate DR-induced longevity. NHR-62 (nuclear receptor) mediates the lifespan-increasing effects of DR by controlling fat metabolism and autophagy (Heestand et al. 2013). WWP-1 (E3 ligase) and UBC-18 (E2 conjugating enzyme), components of the ubiquitin system, extend lifespan upon DR by degrading kruppel-like factor 1 (KLF-1) (Carrano et al. 2014; Carrano et al. 2009).
Under DR, organisms decrease their rates of growth and reproduction to preserve resources for survival until food conditions become more favorable. These maintenance responses appear to promote stress resistances and eventually lead to longevity. In C. elegans, various factors, including cellular energy sensors, in diverse longevity pathways mediate DR-induced longevity in a diet regimen-dependent manner. Thus, these factors may sense various DR cues and transmit longevity signals to different pathways. It will be important to dissect the mechanisms by which these diverse factors or pathways interact with each other for lifespan extension in response to DR.
2.4 Autophagy-Related Genes Required for Longevity
Autophagy is a process that promotes the degradation of cellular components to recycle macromolecules and organelles (reviewed in Levine and Klionsky 2004). The appropriate clearance of damaged cellular components mediated by autophagy is one of the crucial requirements for lifespan extension in C. elegans. Autophagy was first identified in mammals, and subsequent genetic studies using yeast identified many autophagy-related genes (ATGs). Autophagy begins with the induction of membrane changes (regulated by unc-51/ATG1), vesicle nucleation (regulated by bec-1/ATG6, vps-34/VPS34), vesicle expansion (regulated by atg-7/ATG7, lgg-1/ATG8, lgg-3/ATG12), and eventually retrieval (regulated by atg-18/ATG18). Those ATGs are well-conserved across species including C. elegans.
Many lifespan-regulating factors such as IIS, TOR, DR, and reproductive pathways have been shown to modulate autophagy in C. elegans. The role of autophagy in promoting longevity was first shown for reduced IIS (Melendez et al. 2003). daf-2 mutants display increased levels of autophagy and require autophagy-related genes, including bec-1/ATG6 and lgg-3/APG12, for longevity (Melendez et al. 2003; Hars et al. 2007). Acting downstream of IIS (Apfeld et al. 2004), AMPK contributes to the up-regulation of autophagy in daf-2 mutants (Egan et al. 2011). Although DAF-16 is not required for increased autophagy in daf-2 mutants (Hansen et al. 2008), overexpression of the DAF-16 is sufficient to induce autophagy (Jia et al. 2009).
Dietary restriction induces autophagy, and essential autophagy genes are required for the longevity caused by genetic mimesis of DR (Jia and Levine 2007; Hansen et al. 2008). Dietary restriction appears to reduce TOR signaling (reviewed in Johnson et al. 2013) and up-regulate the transcription factors PHA-4 and TFEB/hlh-30 to mediate autophagy-induced longevity (Hansen et al. 2008; Lapierre et al. 2013a). Overall, increased autophagy is required for the longevity caused by multiple signaling pathways, most if not all of which are sensitive to nutrient conditions. Thus, autophagy may provide the nutrients and energy required for longevity pathways. Because evidence supporting the hypothesis that enhanced autophagy per se is sufficient for longevity is scarce, it seems likely that autophagy is a limiting factor for longevity.
2.5 Longevity Caused by Reduced Mitochondrial Function
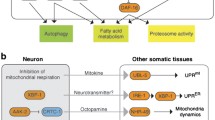
Mitochondria are crucial for many physiologic processes including energy production. Interestingly, a mild inhibition of the mitochondrial electron transport chain (ETC) generally promotes longevity in C. elegans (Fig. 8.3) (reviewed in Hwang et al. 2012). One of the first long-lived mitochondrial ETC mutants that were identified was the clk-1 (demethoxyubiquinone hydroxylase) mutant (Wong et al. 1995; Braeckman et al. 1999; Ewbank et al. 1997). Other long-lived ETC mutants include the isp-1 (iron-sulfur protein) mutants, which are defective in mitochondrial complex III (Feng 2001), and the nuo-6 (NADH ubiquinone oxidoreductase) mutants, which are defective in mitochondrial complex I (Yang and Hekimi 2010b). In addition, RNAi knockdown of many genes that encode mitochondrial ETC components leads to longevity (Dillin et al. 2002b; Lee et al. 2003; Hansen et al. 2005).

Lifespan-regulatory pathways of reduced mitochondrial respiration and HIF-1. Impaired mitochondrial respiration extends lifespan by causing a global change in gene expression via non-mitochondrial mediators. These mediators include nuclear transcription factors, AMPK (AMP-activated protein kinase), the apoptotic signaling pathway, and the mitochondrial unfolded protein response (UPRmito). In addition, increased reactive oxygen species (ROS) levels mediate longevity in respiratory mutants through AMPK and HIF-1. HIF-1 is also stabilized upon hypoxia via EGL-9 (proline hydroxylase) and VHL-1 (ubiquitin E3 ligase)
Several key downstream factors and signaling pathways are known to mediate the longevity of mitochondrial ETC mutants. These include FSTR-1/-2, AMPK, HIF-1, CEH-23 (homeobox domain transcription factor), the mitochondrial unfolded protein response (UPRmito) genes, CEP-1 (p53 homolog), components of the apoptotic signaling pathway, and the TAF-4/TFIID complex (Baruah et al. 2014; Yee et al. 2014; Khan et al. 2013; Walter et al. 2011; Durieux et al. 2011; Lee et al. 2010; Ventura et al. 2009; Cristina et al. 2009; Curtis et al. 2006; Hwang et al. 2014). The inhibition of the ETC appears to modulate signaling from the mitochondria to other cellular organelles including the nucleus. For example, reduced mitochondrial respiration leads to changes in global gene expression, which contribute to a long lifespan (Yee et al. 2014; Cristina et al. 2009). The global changes in gene expression appear to be mediated by transcription factors such as HIF-1 and CEP-1 (Hwang et al. 2014; Baruah et al. 2014).
Many long-lived ETC mutants display increased ROS levels, which actually contribute to longevity (Hwang et al. 2014; Lee et al. 2010; Yang and Hekimi 2010a; Van Raamsdonk and Hekimi 2012). Long-lived mitochondrial mutants have increased mitochondrial ROS levels (Yang and Hekimi 2010a; Hwang et al. 2014; Lee et al. 2010), and antioxidant treatment suppresses this longevity (Yang and Hekimi 2010a; Van Raamsdonk and Hekimi 2012). Thus, increased ROS levels seem to contribute to the long lifespan of ETC mutants. Furthermore, increased HIF-1 and AMPK activities in response to elevated ROS levels mediate this ROS-induced longevity (Hwang et al. 2014; Lee et al. 2010). These findings invite a revision of the free radical theory of aging (Harman 1956, 1972), which proposes that ROS cause aging and therefore shorten lifespan.
Another key parallel signaling pathway required for the longevity of ETC mutants is the UPRmito. The UPRmito is a stress response that relays signals from the mitochondria to the nucleus to induce mitochondrial chaperon proteins (reviewed in Haynes et al. 2013). Impaired ETC function in one tissue (e.g., neurons) activates the UPRmito and relays yet unidentified longevity signals to other tissues (e.g., intestinal cells) to extend lifespan (Durieux et al. 2011). However, the activation of the UPRmito is not sufficient to promote longevity (Bennett et al. 2014).
Since the first long-lived mitochondrial respiratory clk-1 mutants were identified, numerous studies have been conducted to reveal the molecular mechanisms underlying this lifespan regulation. Only recently, scientists started to understand the paradox of how reduced ETC delays aging and increases lifespan. Interestingly, simple and small animal species that have high respiration rates tend to live shorter lives, whereas complex and large species with low respiration rates tend to live longer lives (reviewed in Kenyon 2010). Perhaps the longevity displayed by C. elegans ETC mutants mimics the evolution of longevity among species.
2.6 The Regulation of Lifespan by the Hypoxia-Inducible Factor 1-Regulatory Pathway
Hypoxia-inducible factor 1 (HIF-1) is a key transcription factor that regulates responses to conditions of low oxygen (Fig. 8.3) (reviewed in Powell-Coffman 2010; Semenza 2012). Under normal oxygen conditions, HIF-1 is hydroxylated by the proline hydroxylase EGL-9 and ubiquitinated by von Hippel-Lindau-1 (VHL-1), an E3 ligase component. Under conditions of low oxygen, EGL-9 cannot hydroxylate HIF-1, leading to the stabilization of HIF-1 and the induction of HIF-1 target genes. HIF-1 modulates various biological processes, including lifespan- and aging-related processes in C. elegans.
The up-regulation of HIF-1, by the genetic inhibition of VHL-1 or EGL-9 (Lee et al. 2010; Mehta et al. 2009; Muller et al. 2009), or by the overexpression of hif-1, increases lifespan (Zhang et al. 2009). The activation of HIF-1 also contributes to the longevity conferred by mitochondrial ROS in a positive-feedback fashion and through the modulation of iron-metabolism genes (Lee et al. 2010; Hwang et al. 2014). Interestingly, HIF-1 also regulates lifespan in a temperature-dependent manner, possibly through IIS (Lee et al. 2010; Leiser et al. 2011; Chen et al. 2009; Zhang et al. 2009), and mediates DR-induced longevity (Chen et al. 2009). Overall, HIF-1 appears to act as a sensor and mediator for various lifespan-regulatory signals such as oxygen concentration, mitochondrial ROS, temperature changes, and nutrient levels.
Because HIF-1 is one of the recently identified factors that regulate aging in C. elegans, the mechanisms by which HIF-1 increases lifespan remain elusive. Different from vertebrate models, the availability of viable hif-1, vhl-1, and egl-9 mutants has made C. elegans a unique and important model organism to study the role of HIF-1 in aging. Future studies regarding HIF-1, including tissue-specific roles and the functional characterization of upstream and downstream factors, will provide mechanistic insights into how this evolutionarily conserved transcription factor exerts its effects on longevity.
2.7 Sensory Neuronal Regulation of Longevity
C. elegans is equipped with a sensory nervous system that perceives environmental changes. Intriguingly, sensory neurons modulate lifespan in C. elegans (Fig. 8.4), and this phenomenon is also observed in Drosophila and mice (Linford et al. 2011; Jeong et al. 2012; Riera et al. 2014). Structural perturbations of a subset of ciliated sensory neurons, including the genetic disruption of che-2/IFT80, daf-10/IFT122, daf-19/RFX2, or osm-5/IFT88, increase lifespan in C. elegans (Apfeld and Kenyon 1999; Alcedo and Kenyon 2004). Many C. elegans mutants that have defects in sensory signal transduction also live long. The genetic inhibition of str-2, a putative sensory G protein-coupled receptor, or of kin-29 (SIK3 kinase), which regulates the expression of subsets of neuronal sensory receptors, lengthens lifespan (Lanjuin and Sengupta 2002; Alcedo and Kenyon 2004). The inhibition of G proteins that act downstream of sensory receptors such as gpa-1, gpa-5, gpa-9, and odr-3 extends lifespan as well (Lans and Jansen 2007; Alcedo and Kenyon 2004). The genetic modulation of downstream cation channels, including tax-4 (cyclic nucleotide-gated channel subunit), ocr-2 and osm-9 (neuronal transient receptor potential vanilloid (TRPV) channels), and cold-sensitive trpa-1 (TRPA channel), can increase lifespan (Apfeld and Kenyon 1999; Lee and Ashrafi 2008; Lee and Kenyon 2009; Xiao et al. 2013; Riera et al. 2014). Thus, the inhibition of the sensory neural structure or function generally increases lifespan in C. elegans.

Pathways that act downstream of sensory neurons to modulate lifespan. The inhibition of thermosensory neurons decreases C. elegans lifespan at high temperature (25 °C) by decreasing the expression of DAF-9 (cytochrome P450) in distal tissues such as hypodermis and XXX cells, resulting in the activation of DAF-12 (nuclear receptor). The perturbation of chemosensory neurons presumably decreases insulin/IGF-1 signaling, which promotes the nuclear localization and activation of DAF-16/FOXO to enhance longevity
The longevity caused by sensory impairment appears to be mediated at least partly by the IIS pathway. Defects in sensory neurons promote the nuclear localization and transcriptional activation of DAF-16 (Lin et al. 2001; Xiao et al. 2013; Gaglia et al. 2012). In addition, the long lifespans caused by sensory mutations are largely suppressed by daf-16 mutations (Apfeld and Kenyon 1999; Hahm et al. 2009; Lanjuin and Sengupta 2002; Lee and Ashrafi 2008; Xiao et al. 2013; Alcedo and Kenyon 2004; Lans and Jansen 2007). Thus, the disruption of sensory neurons increases lifespan in C. elegans through the activation of DAF-16. In addition, the long lifespan of sensory daf-10/IFT122 mutants requires the induction of mct-1, a putative monocarboxylate transporter (Gaglia et al. 2012). This suggests that the transportation of hormones or small molecules modulates lifespan by acting downstream of the sensory perturbation. In contrast to these long-lived sensory mutants, which mostly have chemosensory defects, mutants that have defects in thermosensory AFD neurons are short lived at high temperatures (25 °C) (Lee and Kenyon 2009). This lifespan regulation is mediated by steroid signaling, composed of DAF-9 (cytochrome P450) and DAF-12 (nuclear receptor) in multiple tissues, including hypodermis and endocrine XXX cells (Lee and Kenyon 2009). How these various sensory modalities affect lifespan by employing different downstream factors is currently unclear.
It is intriguing that the inhibition of a small number of sensory neurons can have a large effect on the organismal lifespan. Several signaling pathways that regulate aging, including those involved in IIS, TOR, DR, and autophagy, are concerned with nutrient and food availability. Because foods have smells and tastes as well as nutrients, the sensory neurons may be an intrinsic factor that acts at the upstream end of longevity signaling pathways that are linked with food availability. In fact, sensory cues can directly influence lifespan via sensory neurons in C. elegans and Drosophila (Libert et al. 2007; Maier et al. 2010). Hence, one can speculate that sensory neurons monitor environmental changes such as food availability and temperature fluctuations and modulate physiologic processes that eventually affect lifespan.
2.8 Lifespan Regulation by the Reproductive System
Organismal longevity is frequently associated with reduced reproduction. In C. elegans, the removal of the germline promotes longevity (Fig. 8.5) (Hsin and Kenyon 1999). This phenomenon does not result from a simple trade-off between longevity and reproduction, because the removal of the somatic gonad together with germline does not result in longevity (Hsin and Kenyon 1999). Instead, when the germline is removed, the somatic gonad actively promotes longevity by sending signals that modulate steroid signaling, DAF-16 activities, and fat metabolism.

The components of the reproductive system, which regulates longevity. The removal of germline cells increases lifespan by transmitting longevity signals from the somatic gonad to intestinal cells in C. elegans. This gonadal signaling increases the synthesis of dafachronic acid (DA), which results in the activation of DAF-12 (nuclear receptor). In addition, signals from the gonad enhance the nuclear localization and transcriptional activity of DAF-16/FOXO. Moreover, the gonadal signaling modulates fat metabolism by up-regulating the NHR-49, 80 (nuclear receptors: NHRs) and LIPL-4 (lipase) to promote longevity
The DAF-12 is one of the key components in steroid signaling (Antebi et al. 2000) and is required for longevity in germline-ablated worms (Hsin and Kenyon 1999). The activity of DAF-12 is regulated by dafachronic acids (DAs), which are bile acid-like steroid ligands (Motola et al. 2006). DAs are synthesized from cholesterol by multiple enzymatic components such as DAF-36 (Rieske-like oxygenase), DHS-16 (3-hydroxysteroid dehydrogenase), and DAF-9 (Rottiers et al. 2006; Wollam et al. 2012; Jia et al. 2002; Gerisch et al. 2001). Those components, as well as DAs, contribute to the longevity induced by the lack of the germline resulting from laser ablation or glp-1 (germ line proliferation 1) mutations. For example, mutations in daf-36, dhs-16, or daf-9 suppress the long lifespan induced by the lack of germ cells (Rottiers et al. 2006; Wollam et al. 2012; Gerisch et al. 2001). Dafachronic acids are ligands of DAF-12 that promote lifespan extension in animals lacking germline cells (Gerisch et al. 2007; Yamawaki et al. 2010; Mahanti et al. 2014), although treatment with DAs is not sufficient to increase lifespan in wild-type worms (Gerisch et al. 2007; Yamawaki et al. 2010). Thus, the loss of the germline leads to the production of high levels of DAs in the somatic gonad, which activate DAF-12 and promote longevity.
Another component that mediates the longevity conferred by germline loss is DAF-16, which is activated by germline loss and is required for the lifespan extension associated with germline loss (Hsin and Kenyon 1999; Lin et al. 2001). Upon germline removal, intestinal DAF-16 translocates from the cytosol to the nucleus (Lin et al. 2001). This process is mediated by DAF-9, DAF-12, and KRI-1/KRIT1/CCM1, independently of IIS (Berman and Kenyon 2006). Moreover, the transcriptional activity of nuclear DAF-16 is regulated by several factors such as TCER-1/TCERG-1, PHI-62 (a predicted RNA-binding protein), and FTT-2/14-3-3 (Ghazi et al. 2009; McCormick et al. 2012). Thus, the loss of the germline enhances the transcriptonal activity of DAF-16 to induce longevity genes and increases lifespan.
Fat metabolism also plays key roles in the regulation of lifespan by the reproductive system. Oil red O fat staining and Coherent Anti-Stokes Raman Scattering (CARS) microscopy indicate that germline loss increases fat storage (O’Rourke et al. 2009; Lapierre et al. 2013b). Moreover, several factors that regulate fat metabolism are required for the longevity conferred by germline loss, including NHR-49 and NHR-80, nuclear receptors that regulate fat metabolism (Goudeau et al. 2011; Ratnappan et al. 2014). The gonadal signaling is also mediated by the induction of lipl-4 (a triglyceride lipase) that functions to increase lifespan (Wang et al. 2008; Lapierre et al. 2011). Thus, changes in fat metabolism contribute to the extension of lifespan by germline loss.
In C. elegans, the somatic gonad seems to relay longevity signals to other body parts upon sensing the loss of the germline. When the germ cells are compromised, the somatic gonad sends signals that may help the survival of the soma, and the animals may resume reproduction under conditions that favor reproduction. This may help the animals balance the whole system between the maintenance of somatic health and the promotion of reproduction. Interestingly, the regulation of lifespan by the reproductive system is also observed in other species, including Drosophila (Flatt et al. 2008) and mice (Cargill et al. 2003). Thus, the elucidation of the mechanisms by which the reproductive system regulates longevity will provide useful information regarding how reproduction and aging have evolved in an interlocked manner in complex organisms such as humans.
3 Conclusions
In this chapter, we reviewed representative pathways and genes that influence longevity in C. elegans. More than 20 years of research using C. elegans has provided invaluable information about the genetics of aging. Importantly, many of the genes that regulate aging in C. elegans are implicated in the longevity of mammals, including humans. For example, the identification of IIS as a longevity pathway in C. elegans has led the way for the discovery of FOXO3A variants in long-lived humans. In addition, the dissection of TOR signaling as a target of anti-aging medicine has helped the emergence of rapamycin as a lifespan-extending drug in mice (Harrison et al. 2009). Overall, it is indisputable that the aging research using C. elegans has provided pivotal clues to the basis for slowing human aging and delaying the onset of age-related diseases.
Organismal lifespan is highly plastic and subject to changes in environmental and internal conditions. Under normal conditions, lifespan-regulatory pathways support growth, reproduction, and other essential cellular functions such as translation and energy production. However, under harsh conditions, including low food availability and the presence of various stressors, those pathways appear to shift from growth and reproduction to protective states, which eventually lead to longevity. For example, a reduction in food availability alters the function of multiple lifespan-regulatory pathways, including TOR, IIS, and autophagy, to promote longevity. The loss of the germline extends lifespan, likely by sending longevity signals from the reproductive organs to other body parts to support the health of somatic tissues. Decreases in sensory perception can extend lifespan, probably by transmitting cues for environmental stresses, including low food availability, into internal longevity signals. These examples can help us to interpret how organisms switch their physiologic status between growth/reproduction and maintenance/protection at the molecular level upon changes in extrinsic and intrinsic conditions.
Many of the genes and pathways described in this chapter have emerged as promising targets for anti-aging medicine. However, most of these pathways are tightly linked to one another. Hence, it will be difficult to predict the exact physiologic outcome of intervention in one pathway, which may lead to alterations in the function of other vital pathways. In this regard, a major challenge in aging research may be to unravel the complex network among the lifespan-regulatory pathways. In addition, some factors that increase longevity come with expenses including reduced fitness. Therefore, it will be important to uncouple the longevity from adverse side effects by elucidating precise mechanisms. Because many of the pathways that regulate aging are evolutionarily conserved, findings regarding C. elegans longevity genes will likely impact on aging research in mammals as well. Thus, deciphering the whole network among these pathways and genes in C. elegans will eventually help us to achieve longer and healthier lifespans in humans.
References
Ailion M, Inoue T, Weaver CI, Holdcraft RW, Thomas JH (1999) Neurosecretory control of aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A 96(13):7394–7397
Alam H, Williams TW, Dumas KJ, Guo C, Yoshina S, Mitani S, Hu PJ (2010) EAK-7 controls development and life span by regulating nuclear DAF-16/FoxO activity. Cell Metab 12(1):30–41. doi:10.1016/j.cmet.2010.05.004
Alcedo J, Kenyon C (2004) Regulation of C. elegans longevity by specific gustatory and olfactory neurons. Neuron 41(1):45–55
Antebi A, Yeh WH, Tait D, Hedgecock EM, Riddle DL (2000) daf-12 encodes a nuclear receptor that regulates the Dauer diapause and developmental age in C. elegans. Genes Dev 14(12):1512–1527
Apfeld J, Kenyon C (1999) Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature 402(6763):804–809. doi:10.1038/45544
Apfeld J, O'Connor G, McDonagh T, DiStefano PS, Curtis R (2004) The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev 18(24):3004–3009
Araiz C, Château MT, Galas S (2008) 14-3-3 regulates life span by both DAF-16-dependent and-independent mechanisms in Caenorhabditis elegans. Exp Gerontol 43(6):505–519. doi:10.1016/j.exger.2008.03.001
Arantes-Oliveira N, Apfeld J, Dillin A, Kenyon C (2002) Regulation of life-span by germ-line stem cells in Caenorhabditis elegans. Science 295(5554):502–505. doi:10.1126/science.1065768
Baker BM, Nargund AM, Sun T, Haynes CM (2012) Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet 8(6), e1002760. doi:10.1371/journal.pgen.1002760
Baruah A, Chang H, Hall M, Yuan J, Gordon S, Johnson E, Shtessel LL, Yee C, Hekimi S, Derry WB, Lee SS (2014) CEP-1, the Caenorhabditis elegans p53 homolog, mediates opposing longevity outcomes in mitochondrial electron transport chain mutants. PLoS Genet 10(2), e1004097. doi:10.1371/journal.pgen.1004097
Bennett CF, Vander Wende H, Simko M, Klum S, Barfield S, Choi H, Pineda VV, Kaeberlein M (2014) Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat Commun 5:3483. doi:10.1038/ncomms4483
Berdichevsky A, Viswanathan M, Horvitz HR, Guarente L (2006) C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell 125(6):1165–1177. doi:10.1016/j.cell.2006.04.036
Berman JR, Kenyon C (2006) Germ-cell loss extends C. elegans life span through regulation of DAF-16 by kri-1 and lipophilic-hormone signaling. Cell 124(5):1055–1068. doi:10.1016/j.cell.2006.01.039
Bishop NA, Guarente L (2007) Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature 447(7144):545–549. doi:10.1038/nature05904
Boehm M, Slack F (2005) A developmental timing microRNA and its target regulate life span in C. elegans. Science 310(5756):1954–1957. doi:10.1126/science.1115596
Boulias K, Horvitz HR (2012) The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO. Cell Metab 15(4):439–450. doi:10.1016/j.cmet.2012.02.014
Braeckman BP, Houthoofd K, De Vreese A, Vanfleteren JR (1999) Apparent uncoupling of energy production and consumption in long-lived Clk mutants of Caenorhabditis elegans. Curr Biol 9(9):493–496
Brisbin S, Liu J, Boudreau J, Peng J, Evangelista M, Chin-Sang I (2009) A role for C. elegans Eph RTK signaling in PTEN regulation. Dev Cell 17(4):459–469. doi:10.1016/j.devcel.2009.08.009
Brock TJ, Browse J, Watts JL (2006) Genetic regulation of unsaturated fatty acid composition in C. elegans. PLoS Genet 2(7), e108. doi:10.1371/journal.pgen.0020108
Budovskaya YV, Wu K, Southworth LK, Jiang M, Tedesco P, Johnson TE, Kim SK (2008) An elt-3/elt-5/elt-6 GATA transcription circuit guides aging in C. elegans. Cell 134(2):291–303. doi:10.1016/j.cell.2008.05.044
Burnett C, Valentini S, Cabreiro F, Goss M, Somogyvari M, Piper MD, Hoddinott M, Sutphin GL, Leko V, McElwee JJ, Vazquez-Manrique RP, Orfila AM, Ackerman D, Au C, Vinti G, Riesen M, Howard K, Neri C, Bedalov A, Kaeberlein M, Soti C, Partridge L, Gems D (2011) Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 477(7365):482–485. doi:10.1038/nature10296
Cabreiro F, Ackerman D, Doonan R, Araiz C, Back P, Papp D, Braeckman BP, Gems D (2011) Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radic Biol Med 51(8):1575–1582. doi:10.1016/j.freeradbiomed.2011.07.020
Cargill SL, Carey JR, Muller HG, Anderson G (2003) Age of ovary determines remaining life expectancy in old ovariectomized mice. Aging Cell 2(3):185–190
Carrano AC, Liu Z, Dillin A, Hunter T (2009) A conserved ubiquitination pathway determines longevity in response to diet restriction. Nature 460(7253):396–399. doi:10.1038/nature08130
Carrano AC, Dillin A, Hunter T (2014) A Kruppel-like factor downstream of the E3 ligase WWP-1 mediates dietary-restriction-induced longevity in Caenorhabditis elegans. Nat Commun 5:3772. doi:10.1038/ncomms4772
Chamoli M, Singh A, Malik Y, Mukhopadhyay A (2014) A novel kinase regulates dietary restriction-mediated longevity in Caenorhabditis elegans. Aging Cell 13(4):641–655. doi:10.1111/acel.12218
Chen D, Thomas EL, Kapahi P (2009) HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans. PLoS Genet 5(5), e1000486. doi:10.1371/journal.pgen.1000486
Chen AT, Guo C, Dumas KJ, Ashrafi K, Hu PJ (2013a) Effects of Caenorhabditis elegans sgk-1 mutations on lifespan, stress resistance, and DAF-16/FoxO regulation. Aging Cell 12(5):932–940. doi:10.1111/acel.12120
Chen D, Li PW, Goldstein BA, Cai W, Thomas EL, Chen F, Hubbard AE, Melov S, Kapahi P (2013b) Germline signaling mediates the synergistically prolonged longevity produced by double mutations in daf-2 and rsks-1 in C. elegans. Cell Rep 5(6):1600–1610. doi:10.1016/j.celrep.2013.11.018
Chiang WC, Ching TT, Lee HC, Mousigian C, Hsu AL (2012) HSF-1 regulators DDL-1/2 link insulin-like signaling to heat-shock responses and modulation of longevity. Cell 148(1–2):322–334. doi:10.1016/j.cell.2011.12.019
Ching TT, Paal AB, Mehta A, Zhong L, Hsu AL (2010) drr-2 encodes an eIF4H that acts downstream of TOR in diet-restriction-induced longevity of C. elegans. Aging Cell 9(4):545–557. doi:10.1111/j.1474-9726.2010.00580.x
Cristina D, Cary M, Lunceford A, Clarke C, Kenyon C (2009) A regulated response to impaired respiration slows behavioral rates and increases lifespan in Caenorhabditis elegans. PLoS Genet 5(4), e1000450. doi:10.1371/journal.pgen.1000450
Curran SP, Ruvkun G (2007) Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet 3(4), e56. doi:10.1371/journal.pgen.0030056
Curran SP, Wu X, Riedel CG, Ruvkun G (2009) A soma-to-germline transformation in long-lived Caenorhabditis elegans mutants. Nature 459(7250):1079–1084. doi:10.1038/nature08106
Curtis R, O'Connor G, DiStefano PS (2006) Aging networks in Caenorhabditis elegans: AMP-activated protein kinase (aak-2) links multiple aging and metabolism pathways. Aging Cell 5(2):119–126. doi:10.1111/j.1474-9726.2006.00205.x
de Jong L, Meng Y, Dent J, Hekimi S (2004) Thiamine pyrophosphate biosynthesis and transport in the nematode Caenorhabditis elegans. Genetics 168(2):845–854. doi:10.1534/genetics.104.028605
de Lencastre A, Pincus Z, Zhou K, Kato M, Lee SS, Slack FJ (2010) MicroRNAs both promote and antagonize longevity in C. elegans. Curr Biol 20(24):2159–2168. doi:10.1016/j.cub.2010.11.015
Dillin A, Crawford DK, Kenyon C (2002a) Timing requirements for insulin/IGF-1 signaling in C. elegans. Science 298(5594):830–834. doi:10.1126/science.1074240
Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C (2002b) Rates of behavior and aging specified by mitochondrial function during development. Science 298(5602):2398–2401. doi:10.1126/science.1077780
Dorman JB, Albinder B, Shroyer T, Kenyon C (1995) The age-1 and daf-2 genes function in a common pathway to control the lifespan of Caenorhabditis elegans. Genetics 141(4):1399–1406
Durieux J, Wolff S, Dillin A (2011) The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144(1):79–91. doi:10.1016/j.cell.2010.12.016
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331(6016):456–461. doi:10.1126/science.1196371
Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC (2005) Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 308(5725):1181–1184. doi:10.1126/science.1109083
Essers Paul B, Nonnekens J, Goos Yvonne J, Betist Marco C, Viester Marjon D, Mossink B, Lansu N, Korswagen Hendrik C, Jelier R, Brenkman Arjan B, MacInnes Alyson W (2015) A long noncoding RNA on the ribosome is required for lifespan extension. Cell Rep 10(3):339–345. doi:10.1016/j.celrep.2014.12.029
Evans DS, Kapahi P, Hsueh WC, Kockel L (2011) TOR signaling never gets old: aging, longevity and TORC1 activity. Ageing Res Rev 10(2):225–237. doi:10.1016/j.arr.2010.04.001
Ewald CY, Landis JN, Abate JP, Murphy CT, Blackwell TK (2014) Dauer-independent insulin/IGF-1-signalling implicates collagen remodelling in longevity. Nature. doi:10.1038/nature14021
Ewbank JJ, Barnes TM, Lakowski B, Lussier M, Bussey H, Hekimi S (1997) Structural and functional conservation of the Caenorhabditis elegans timing gene clk-1. Science 275(5302):980–983
Feng J, Bussiere F, Hekimi S (2001) Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell 1(5):633–644
Fernandes de Abreu DA, Caballero A, Fardel P, Stroustrup N, Chen Z, Lee K, Keyes WD, Nash ZM, Lopez-Moyado IF, Vaggi F, Cornils A, Regenass M, Neagu A, Ostojic I, Liu C, Cho Y, Sifoglu D, Shen Y, Fontana W, Lu H, Csikasz-Nagy A, Murphy CT, Antebi A, Blanc E, Apfeld J, Zhang Y, Alcedo J, Ch'ng Q (2014) An insulin-to-insulin regulatory network orchestrates phenotypic specificity in development and physiology. PLoS Genet 10(3), e1004225. doi:10.1371/journal.pgen.1004225
Fisher AL, Lithgow GJ (2006) The nuclear hormone receptor DAF-12 has opposing effects on Caenorhabditis elegans lifespan and regulates genes repressed in multiple long-lived worms. Aging Cell 5(2):127–138. doi:10.1111/j.1474-9726.2006.00203.x
Flatt T, Min KJ, D'Alterio C, Villa-Cuesta E, Cumbers J, Lehmann R, Jones DL, Tatar M (2008) Drosophila germ-line modulation of insulin signaling and lifespan. Proc Natl Acad Sci U S A 105(17):6368–6373. doi:10.1073/pnas.0709128105
Friedman DB, Johnson TE (1988) Three mutants that extend both mean and maximum life span of the nematode, Caenorhabditis elegans, define the age-1 gene. J Gerontol 43(4):B102–B109
Gaglia MM, Jeong DE, Ryu EA, Lee D, Kenyon C, Lee SJ (2012) Genes that act downstream of sensory neurons to influence longevity, Dauer formation, and pathogen responses in Caenorhabditis elegans. PLoS Genet 8(12), e1003133. doi:10.1371/journal.pgen.1003133
Garigan D, Hsu AL, Fraser AG, Kamath RS, Ahringer J, Kenyon C (2002) Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics 161(3):1101–1112
Garsin DA, Villanueva JM, Begun J, Kim DH, Sifri CD, Calderwood SB, Ruvkun G, Ausubel FM (2003) Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science 300 (5627):1921. doi:10.1126/science.1080147
Gems D, Sutton AJ, Sundermeyer ML, Albert PS, King KV, Edgley ML, Larsen PL, Riddle DL (1998) Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics 150(1):129–155
Gerisch B, Weitzel C, Kober-Eisermann C, Rottiers V, Antebi A (2001) A hormonal signaling pathway influencing C. elegans metabolism, reproductive development, and life span. Dev Cell 1(6):841–851
Gerisch B, Rottiers V, Li D, Motola DL, Cummins CL, Lehrach H, Mangelsdorf DJ, Antebi A (2007) A bile acid-like steroid modulates Caenorhabditis elegans lifespan through nuclear receptor signaling. Proc Natl Acad Sci U S A 104(12):5014–5019. doi:10.1073/pnas.0700847104
Gharbi H, Fabretti F, Bharill P, Rinschen MM, Brinkkotter S, Frommolt P, Burst V, Schermer B, Benzing T, Muller RU (2013) Loss of the Birt-Hogg-Dube gene product folliculin induces longevity in a hypoxia-inducible factor-dependent manner. Aging Cell 12(4):593–603. doi:10.1111/acel.12081
Ghazi A, Henis-Korenblit S, Kenyon C (2007) Regulation of Caenorhabditis elegans lifespan by a proteasomal E3 ligase complex. Proc Natl Acad Sci U S A 104(14):5947–5952. doi:10.1073/pnas.0700638104
Ghazi A, Henis-Korenblit S, Kenyon C (2009) A transcription elongation factor that links signals from the reproductive system to lifespan extension in Caenorhabditis elegans. PLoS Genet 5(9), e1000639. doi:10.1371/journal.pgen.1000639
Goudeau J, Bellemin S, Toselli-Mollereau E, Shamalnasab M, Chen Y, Aguilaniu H (2011) Fatty acid desaturation links germ cell loss to longevity through NHR-80/HNF4 in C. elegans. PLoS Biol 9(3), e1000599. doi:10.1371/journal.pbio.1000599
Green DR, Galluzzi L, Kroemer G (2014) Metabolic control of cell death. Science 345(6203):1250256
Greer EL, Brunet A (2009) Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell 8(2):113–127. doi:10.1111/j.1474-9726.2009.00459.x
Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A (2007) An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol 17(19):1646–1656. doi:10.1016/j.cub.2007.08.047
Hahm JH, Kim S, Paik YK (2009) Endogenous cGMP regulates adult longevity via the insulin signaling pathway in Caenorhabditis elegans. Aging Cell 8(4):473–483. doi:10.1111/j.1474-9726.2009.00495.x
Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, Lee SS (2005) A systematic RNAi screen for longevity genes in C. elegans. Genes Dev 19(13):1544–1555. doi:10.1101/gad.1308205
Hansen M, Hsu AL, Dillin A, Kenyon C (2005) New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet 1(1):119–128. doi:10.1371/journal.pgen.0010017
Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C (2007) Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 6(1):95–110. doi:10.1111/j.1474-9726.2006.00267.x
Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C (2008) A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet 4(2), e24. doi:10.1371/journal.pgen.0040024
Hardie DG (2014) AMPK-sensing energy while talking to other signaling pathways. Cell Metab 20(6):939–952. doi:10.1016/j.cmet.2014.09.013
Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J Gerontol 11(3):298–300
Harman D (1972) The biologic clock: the mitochondria? J Am Geriatr Soc 20(4):145–147
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460(7253):392–395. doi:10.1038/nature08221
Hars ES, Qi H, Ryazanov AG, Jin S, Cai L, Hu C, Liu LF (2007) Autophagy regulates ageing in C. elegans. Autophagy 3(2):93–95
Hashimoto Y, Ookuma S, Nishida E (2009) Lifespan extension by suppression of autophagy genes in Caenorhabditis elegans. Genes Cells 14(6):717–726. doi:10.1111/j.1365-2443.2009.01306.x
Haynes CM, Fiorese CJ, Lin YF (2013) Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends Cell Biol 23(7):311–318. doi:10.1016/j.tcb.2013.02.002
Heestand BN, Shen Y, Liu W, Magner DB, Storm N, Meharg C, Habermann B, Antebi A (2013) Dietary restriction induced longevity is mediated by nuclear receptor NHR-62 in Caenorhabditis elegans. PLoS Genet 9(7), e1003651. doi:10.1371/journal.pgen.1003651
Henderson ST, Johnson TE (2001) daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr Biol 11(24):1975–1980
Henis-Korenblit S, Zhang P, Hansen M, McCormick M, Lee SJ, Cary M, Kenyon C (2010) Insulin/IGF-1 signaling mutants reprogram ER stress response regulators to promote longevity. Proc Natl Acad Sci U S A 107(21):9730–9735. doi:10.1073/pnas.1002575107
Hertweck M, Gobel C, Baumeister R (2004) C. elegans SGK-1 is the critical component in the Akt/PKB kinase complex to control stress response and life span. Dev Cell 6(4):577–588
Honda Y, Honda S (1999) The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J 13(11):1385–1393
Honjoh S, Yamamoto T, Uno M, Nishida E (2009) Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature 457(7230):726–730. doi:10.1038/nature07583
Honnen SJ, Buchter C, Schroder V, Hoffmann M, Kohara Y, Kampkotter A, Bossinger O (2012) C. elegans VANG-1 modulates life span via insulin/IGF-1-like signaling. PLoS One 7(2), e32183. doi:10.1371/journal.pone.0032183
Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J (2013) Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497(7450):451–457. doi:10.1038/nature12188
Hsin H, Kenyon C (1999) Signals from the reproductive system regulate the lifespan of C. elegans. Nature 399(6734):362–366. doi:10.1038/20694
Hsu AL, Murphy CT, Kenyon C (2003) Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300(5622):1142–1145. doi:10.1126/science.1083701
Huang X, Zhang H, Zhang H (2011) The zinc-finger protein SEA-2 regulates larval developmental timing and adult lifespan in C. elegans. Development 138(10):2059–2068. doi:10.1242/dev.057109
Hwang AB, Jeong DE, Lee SJ (2012) Mitochondria and organismal longevity. Curr Genomics 13(7):519–532. doi:10.2174/138920212803251427
Hwang AB, Ryu EA, Artan M, Chang HW, Kabir MH, Nam HJ, Lee D, Yang JS, Kim S, Mair WB, Lee C, Lee SS, Lee SJ (2014) Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc Natl Acad Sci U S A 111(42):E4458–E4467. doi:10.1073/pnas.1411199111
Jeong DE, Artan M, Seo K, Lee SJ (2012) Regulation of lifespan by chemosensory and thermosensory systems: findings in invertebrates and their implications in mammalian aging. Front Genet 3:218. doi:10.3389/fgene.2012.00218
Jia K, Albert PS, Riddle DL (2002) DAF-9, a cytochrome P450 regulating C. elegans larval development and adult longevity. Development 129(1):221–231
Jia K, Chen D, Riddle DL (2004) The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 131(16):3897–3906. doi:10.1242/dev.01255
Jia K, Levine B (2007) Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy 3(6):597–599
Jia K, Thomas C, Akbar M, Sun Q, Adams-Huet B, Gilpin C, Levine B (2009) Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc Natl Acad Sci U S A 106(34):14564–14569. doi:10.1073/pnas.0813319106
Jin C, Li J, Green CD, Yu X, Tang X, Han D, Xian B, Wang D, Huang X, Cao X, Yan Z, Hou L, Liu J, Shukeir N, Khaitovich P, Chen CD, Zhang H, Jenuwein T, Han JD (2011) Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab 14(2):161–172. doi:10.1016/j.cmet.2011.07.001
Johnson SC, Rabinovitch PS, Kaeberlein M (2013) mTOR is a key modulator of ageing and age-related disease. Nature 493(7432):338–345. doi:10.1038/nature11861
Johnson DW, Llop JR, Farrell SF, Yuan J, Stolzenburg LR, Samuelson AV (2014) The Caenorhabditis elegans Myc-Mondo/Mad complexes integrate diverse longevity signals. PLoS Genet 10(4), e1004278. doi:10.1371/journal.pgen.1004278
Kaletsky R, Murphy CT (2010) The role of insulin/IGF-like signaling in C. elegans longevity and aging. Dis Model Mech 3(7–8):415–419. doi:10.1242/dmm.001040
Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, Kockel L (2010) With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab 11(6):453–465. doi:10.1016/j.cmet.2010.05.001
Kawano T, Ito Y, Ishiguro M, Takuwa K, Nakajima T, Kimura Y (2000) Molecular cloning and characterization of a new insulin/IGF-like peptide of the nematode Caenorhabditis elegans. Biochem Biophys Res Commun 273(2):431–436. doi:10.1006/bbrc.2000.2971
Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R (1993) A C. elegans mutant that lives twice as long as wild type. Nature 366(6454):461–464. doi:10.1038/366461a0
Kenyon CJ (2010) The genetics of ageing. Nature 464(7288):504–512. doi:10.1038/nature08980
Khan MH, Ligon M, Hussey LR, Hufnal B, Farber R 2nd, Munkacsy E, Rodriguez A, Dillow A, Kahlig E, Rea SL (2013) TAF-4 is required for the life extension of isp-1, clk-1 and tpk-1 Mit mutants. Aging 5(10):741–758
Kim SK, Budovskaya YV, Johnson TE (2013) Reconciliation of daf-2 suppression by elt-3 in Caenorhabditis elegans from Tonsaker et al. (2012) and Kim et al. (2012). Mech Ageing Dev 134(1–2):64–65. doi:10.1016/j.mad.2012.12.004
Kim Y, Sun H (2007) Functional genomic approach to identify novel genes involved in the regulation of oxidative stress resistance and animal lifespan. Aging Cell 6(4):489–503. doi:10.1111/j.1474-9726.2007.00302.x
Lamitina ST, Strange K (2005) Transcriptional targets of DAF-16 insulin signaling pathway protect C. elegans from extreme hypertonic stress. Am J Physiol Cell Physiol 288(2):C467–C474. doi:10.1152/ajpcell.00451.2004
Lanjuin A, Sengupta P (2002) Regulation of chemosensory receptor expression and sensory signaling by the KIN-29 Ser/Thr kinase. Neuron 33(3):369–381
Lans H, Jansen G (2007) Multiple sensory G proteins in the olfactory, gustatory and nociceptive neurons modulate longevity in Caenorhabditis elegans. Dev Biol 303(2):474–482. doi:10.1016/j.ydbio.2006.11.028
Lapierre LR, Gelino S, Melendez A, Hansen M (2011) Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr Biol 21(18):1507–1514. doi:10.1016/j.cub.2011.07.042
Lapierre LR, De Magalhaes Filho CD, McQuary PR, Chu CC, Visvikis O, Chang JT, Gelino S, Ong B, Davis AE, Irazoqui JE, Dillin A, Hansen M (2013a) The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun 4:2267. doi:10.1038/ncomms3267
Lapierre LR, Silvestrini MJ, Nunez L, Ames K, Wong S, Le TT, Hansen M, Melendez A (2013b) Autophagy genes are required for normal lipid levels in C. elegans. Autophagy 9(3):278–286. doi:10.4161/auto.22930
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149(2):274–293. doi:10.1016/j.cell.2012.03.017
Larsen PL, Albert PS, Riddle DL (1995) Genes that regulate both development and longevity in Caenorhabditis elegans. Genetics 139(4):1567–1583
Lakowski B, Hekimi S (1998) The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci 95(22):13091–13096
Lee BH, Ashrafi K (2008) A TRPV channel modulates C. elegans neurosecretion, larval starvation survival, and adult lifespan. PLoS Genet 4(10), e1000213. doi:10.1371/journal.pgen.1000213
Lee SJ, Kenyon C (2009) Regulation of the longevity response to temperature by thermosensory neurons in Caenorhabditis elegans. Curr Biol 19(9):715–722. doi:10.1016/j.cub.2009.03.041
Lee RY, Hench J, Ruvkun G (2001) Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr Biol 11(24):1950–1957
Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G (2003) A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet 33(1):40–48. doi:10.1038/ng1056
Lee SJ, Murphy CT, Kenyon C (2009) Glucose shortens the life span of C. elegans by downregulating DAF-16/FOXO activity and aquaporin gene expression. Cell Metab 10(5):379–391. doi:10.1016/j.cmet.2009.10.003
Lee SJ, Hwang AB, Kenyon C (2010) Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol 20(23):2131–2136. doi:10.1016/j.cub.2010.10.057
Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, Bonni A (2006) A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125(5):987–1001. doi:10.1016/j.cell.2006.03.046
Leiser SF, Begun A, Kaeberlein M (2011) HIF-1 modulates longevity and healthspan in a temperature-dependent manner. Aging Cell 10(2):318–326. doi:10.1111/j.1474-9726.2011.00672.x
Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6(4):463–477
Li J, Tewari M, Vidal M, Lee SS (2007a) The 14-3-3 protein FTT-2 regulates DAF-16 in Caenorhabditis elegans. Dev Biol 301(1):82–91
Li W, Gao B, Lee SM, Bennett K, Fang D (2007b) RLE-1, an E3 ubiquitin ligase, regulates C. elegans aging by catalyzing DAF-16 polyubiquitination. Dev Cell 12(2):235–246. doi:10.1016/j.devcel.2006.12.002
Li J, Ebata A, Dong Y, Rizki G, Iwata T, Lee SS (2008) Caenorhabditis elegans HCF-1 functions in longevity maintenance as a DAF-16 regulator. PLoS Biol 6(9), e233. doi:10.1371/journal.pbio.0060233
Libert S, Zwiener J, Chu X, Vanvoorhies W, Roman G, Pletcher SD (2007) Regulation of Drosophila life span by olfaction and food-derived odors. Science 315(5815):1133–1137. doi:10.1126/science.1136610
Lin K, Dorman JB, Rodan A, Kenyon C (1997) daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278(5341):1319–1322
Lin K, Hsin H, Libina N, Kenyon C (2001) Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet 28(2):139–145. doi:10.1038/88850
Linford NJ, Kuo TH, Chan TP, Pletcher SD (2011) Sensory perception and aging in model systems: from the outside in. Annu Rev Cell Dev Biol 27:759–785. doi:10.1146/annurev-cellbio-092910-154240
Lithgow GJ, White TM, Melov S, Johnson TE (1995) Thermotolerance and extended life-span conferred by single-gene mutations and induced by thermal stress. Proc Natl Acad Sci U S A 92(16):7540–7544
Lithgow GJ, Kapahi P (2011) Life span extension via eIF4G inhibition is mediated by posttranscriptional remodeling of stress response gene expression in C. elegans. Cell Metab 14(1):55–66. doi:10.1016/j.cmet.2011.05.010
Lucanic M, Held JM, Vantipalli MC, Klang IM, Graham JB, Gibson BW, Lithgow GJ, Gill MS (2011) N-acylethanolamine signalling mediates the effect of diet on lifespan in Caenorhabditis elegans. Nature 473(7346):226–229. doi:10.1038/nature10007
Magner DB, Wollam J, Shen Y, Hoppe C, Li D, Latza C, Rottiers V, Hutter H, Antebi A (2013) The NHR-8 nuclear receptor regulates cholesterol and bile acid homeostasis in C. elegans. Cell Metab 18(2):212–224. doi:10.1016/j.cmet.2013.07.007
Mahanti P, Bose N, Bethke A, Judkins JC, Wollam J, Dumas KJ, Zimmerman AM, Campbell SL, Hu PJ, Antebi A, Schroeder FC (2014) Comparative metabolomics reveals endogenous ligands of DAF-12, a nuclear hormone receptor, regulating C. elegans development and lifespan. Cell Metab 19(1):73–83. doi:10.1016/j.cmet.2013.11.024
Maier W, Adilov B, Regenass M, Alcedo J (2010) A neuromedin U receptor acts with the sensory system to modulate food type-dependent effects on C. elegans lifespan. PLoS Biol 8(5), e1000376. doi:10.1371/journal.pbio.1000376
Malone EA, Inoue T, Thomas JH (1996) Genetic analysis of the roles of daf-28 and age-1 in regulating Caenorhabditis elegans Dauer formation. Genetics 143(3):1193–1205
Masse I, Molin L, Billaud M, Solari F (2005) Lifespan and Dauer regulation by tissue-specific activities of Caenorhabditis elegans DAF-18. Dev Biol 286(1):91–101. doi:10.1016/j.ydbio.2005.07.010
Matsunaga Y, Gengyo-Ando K, Mitani S, Iwasaki T, Kawano T (2012) Physiological function, expression pattern, and transcriptional regulation of a Caenorhabditis elegans insulin-like peptide, INS-18. Biochem Biophys Res Commun 423(3):478–483. doi:10.1016/j.bbrc.2012.05.145
Maures TJ, Greer EL, Hauswirth AG, Brunet A (2011) The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell 10(6):980–990. doi:10.1111/j.1474-9726.2011.00738.x
McCormick M, Chen K, Ramaswamy P, Kenyon C (2012) New genes that extend Caenorhabditis elegans' lifespan in response to reproductive signals. Aging Cell 11(2):192–202. doi:10.1111/j.1474-9726.2011.00768.x
Mehta R, Steinkraus KA, Sutphin GL, Ramos FJ, Shamieh LS, Huh A, Davis C, Chandler-Brown D, Kaeberlein M (2009) Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science 324(5931):1196–1198. doi:10.1126/science.1173507
Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B (2003) Autophagy genes are essential for Dauer development and life-span extension in C. elegans. Science 301(5638):1387–1391. doi:10.1126/science.1087782
Mihaylova VT, Borland CZ, Manjarrez L, Stern MJ, Sun H (1999) The PTEN tumor suppressor homolog in Caenorhabditis elegans regulates longevity and Dauer formation in an insulin receptor-like signaling pathway. Proc Natl Acad Sci U S A 96(13):7427–7432
Morley JF, Morimoto RI (2004) Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell 15(2):657–664. doi:10.1091/mbc.E03-07-0532
Morley JF, Brignull HR, Weyers JJ, Morimoto RI (2002) The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A 99(16):10417–10422. doi:10.1073/pnas.152161099
Moroz N, Carmona JJ, Anderson E, Hart AC, Sinclair DA, Blackwell TK (2014) Dietary restriction involves NAD(+) -dependent mechanisms and a shift toward oxidative metabolism. Aging Cell 13(6):1075–1085. doi:10.1111/acel.12273
Morris JZ, Tissenbaum HA, Ruvkun G (1996) A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 382(6591):536–539. doi:10.1038/382536a0
Motola DL, Cummins CL, Rottiers V, Sharma KK, Li T, Li Y, Suino-Powell K, Xu HE, Auchus RJ, Antebi A, Mangelsdorf DJ (2006) Identification of ligands for DAF-12 that govern Dauer formation and reproduction in C. elegans. Cell 124(6):1209–1223. doi:http://dx.doi.org/10.1016/j.cell.2006.01.037
Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, Guarente L, Auwerx J (2013) The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154(2):430–441. doi:10.1016/j.cell.2013.06.016
Mukhopadhyay A, Deplancke B, Walhout AJ, Tissenbaum HA (2005) C. elegans tubby regulates life span and fat storage by two independent mechanisms. Cell Metab 2(1):35–42. doi:10.1016/j.cmet.2005.06.004
Muller RU, Fabretti F, Zank S, Burst V, Benzing T, Schermer B (2009) The von Hippel Lindau tumor suppressor limits longevity. J Am Soc Nephrol 20(12):2513–2517. doi:10.1681/asn.2009050497
Murphy CT, Hu PJ (2013) Insulin/insulin-like growth factor signaling in C. elegans. WormBook:1–43. doi:10.1895/wormbook.1.164.1
Murphy CT, Lee SJ, Kenyon C (2007) Tissue entrainment by feedback regulation of insulin gene expression in the endoderm of Caenorhabditis elegans. Proc Natl Acad Sci U S A 104(48):19046–19050. doi:10.1073/pnas.0709613104
Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424(6946):277–283. doi:10.1038/nature01789
Nanji M, Hopper NA, Gems D (2005) LET-60 RAS modulates effects of insulin/IGF-1 signaling on development and aging in Caenorhabditis elegans. Aging Cell 4(5):235–245. doi:10.1111/j.1474-9726.2005.00166.x
Neumann-Haefelin E, Qi W, Finkbeiner E, Walz G, Baumeister R, Hertweck M (2008) SHC-1/p52Shc targets the insulin/IGF-1 and JNK signaling pathways to modulate life span and stress response in C. elegans. Genes Dev 22(19):2721–2735. doi:10.1101/gad.478408
Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G (1997) The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389(6654):994–999. doi:10.1038/40194
Oh SW, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis RJ, Tissenbaum HA (2005) JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc Natl Acad Sci U S A 102(12):4494–4499. doi:10.1073/pnas.0500749102
Okuyama T, Inoue H, Ookuma S, Satoh T, Kano K, Honjoh S, Hisamoto N, Matsumoto K, Nishida E (2010) The ERK-MAPK pathway regulates longevity through SKN-1 and insulin-like signaling in Caenorhabditis elegans. J Biol Chem 285(39):30274–30281. doi:10.1074/jbc.M110.146274
O'Rourke EJ, Ruvkun G (2013) MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat Cell Biol 15(6):668–676. doi:10.1038/ncb2741
O'Rourke EJ, Soukas AA, Carr CE, Ruvkun G (2009) C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab 10(5):430–435. doi:10.1016/j.cmet.2009.10.002
Padmanabhan S, Mukhopadhyay A, Narasimhan SD, Tesz G, Czech MP, Tissenbaum HA (2009) A PP2A regulatory subunit regulates C. elegans insulin/IGF-1 signaling by modulating AKT-1 phosphorylation. Cell 136(5):939–951. doi:10.1016/j.cell.2009.01.025
Paek J, Lo JY, Narasimhan SD, Nguyen TN, Glover-Cutter K, Robida-Stubbs S, Suzuki T, Yamamoto M, Blackwell TK, Curran SP (2012) Mitochondrial SKN-1/Nrf mediates a conserved starvation response. Cell Metab 16(4):526–537. doi:10.1016/j.cmet.2012.09.007
Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P (2007) Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell 6(1):111–119. doi:10.1111/j.1474-9726.2006.00266.x
Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A (2007) PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature 447(7144):550–555. doi:10.1038/nature05837
Paradis S, Ailion M, Toker A, Thomas JH, Ruvkun G (1999) A PDK1 homolog is necessary and sufficient to transduce AGE-1 PI3 kinase signals that regulate diapause in Caenorhabditis elegans. Genes Dev 13(11):1438–1452
Paradis S, Ruvkun G (1998) Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev 12(16):2488–2498
Park SK, Link CD, Johnson TE (2010) Life-span extension by dietary restriction is mediated by NLP-7 signaling and coelomocyte endocytosis in C. elegans. FASEB J 24(2):383–392. doi:10.1096/fj.09-142984, fj.09-142984 [pii]
Petrascheck M, Ye X, Buck LB (2007) An antidepressant that extends lifespan in adult Caenorhabditis elegans. Nature 450(7169):553–556
Pierce SB, Costa M, Wisotzkey R, Devadhar S, Homburger SA, Buchman AR, Ferguson KC, Heller J, Platt DM, Pasquinelli AA, Liu LX, Doberstein SK, Ruvkun G (2001) Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes Dev 15(6):672–686. doi:10.1101/gad.867301
Piper MD, Partridge L, Raubenheimer D, Simpson SJ (2011) Dietary restriction and aging: a unifying perspective. Cell Metab 14(2):154–160. doi:10.1016/j.cmet.2011.06.013
Possik E, Jalali Z, Nouet Y, Yan M, Gingras MC, Schmeisser K, Panaite L, Dupuy F, Kharitidi D, Chotard L, Jones RG, Hall DH, Pause A (2014) Folliculin regulates ampk-dependent autophagy and metabolic stress survival. PLoS Genet 10(4), e1004273. doi:10.1371/journal.pgen.1004273
Powell-Coffman JA (2010) Hypoxia signaling and resistance in C. elegans. Trends Endocrinol Metab 21(7):435–440. doi:10.1016/j.tem.2010.02.006
Rahman MM, Stuchlick O, El-Karim EG, Stuart R, Kipreos ET, Wells L (2010) Intracellular protein glycosylation modulates insulin mediated lifespan in C.elegans. Aging 2(10):678–690
Ratnappan R, Amrit FR, Chen SW, Gill H, Holden K, Ward J, Yamamoto KR, Olsen CP, Ghazi A (2014) Germline signals deploy NHR-49 to modulate fatty-acid beta-oxidation and desaturation in somatic tissues of C. elegans. PLoS Genet 10(12), e1004829. doi:10.1371/journal.pgen.1004829
Rea SL, Ventura N, Johnson TE (2007) Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol 5(10), e259. doi:10.1371/journal.pbio.0050259
Riera CE, Huising MO, Follett P, Leblanc M, Halloran J, Van Andel R, de Magalhaes Filho CD, Merkwirth C, Dillin A (2014) TRPV1 pain receptors regulate longevity and metabolism by neuropeptide signaling. Cell 157(5):1023–1036. doi:10.1016/j.cell.2014.03.051
Rizki G, Iwata TN, Li J, Riedel CG, Picard CL, Jan M, Murphy CT, Lee SS (2011) The evolutionarily conserved longevity determinants HCF-1 and SIR-2.1/SIRT1 collaborate to regulate DAF-16/FOXO. PLoS Genet 7(9), e1002235. doi:10.1371/journal.pgen.1002235
Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK (2012) TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab 15(5):713–724. doi:10.1016/j.cmet.2012.04.007
Rogers AN, Chen D, McColl G, Czerwieniec G, Felkey K, Gibson BW, Hubbard A, Melov S, Lithgow GJ, Kapahi P (2011) Life span extension via eIF4G inhibition is mediated by posttranscriptional remodeling of stress response gene expression in C. elegans. Cell Metab 14(1):55–66. doi:10.1016/j.cmet.2011.05.010
Rottiers V, Motola DL, Gerisch B, Cummins CL, Nishiwaki K, Mangelsdorf DJ, Antebi A (2006) Hormonal control of C. elegans dauer formation and life span by a Rieske-like oxygenase. Dev Cell 10(4):473–482. doi:10.1016/j.devcel.2006.02.008
Samuelson AV, Klimczak RR, Thompson DB, Carr CE, Ruvkun G (2007) Identification of Caenorhabditis elegans genes regulating longevity using enhanced RNAi-sensitive strains. Cold Spring Harb Symp Quant Biol 72:489–497. doi:10.1101/sqb.2007.72.068
Schiavi A, Torgovnick A, Kell A, Megalou E, Castelein N, Guccini I, Marzocchella L, Gelino S, Hansen M, Malisan F, Condo I, Bei R, Rea SL, Braeckman BP, Tavernarakis N, Testi R, Ventura N (2013) Autophagy induction extends lifespan and reduces lipid content in response to frataxin silencing in C. elegans. Exp Gerontol 48(2):191–201. doi:10.1016/j.exger.2012.12.002
Schmeisser K, Mansfeld J, Kuhlow D, Weimer S, Priebe S, Heiland I, Birringer M, Groth M, Segref A, Kanfi Y, Price NL, Schmeisser S, Schuster S, Pfeiffer AF, Guthke R, Platzer M, Hoppe T, Cohen HY, Zarse K, Sinclair DA, Ristow M (2013) Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat Chem Biol 9(11):693–700. doi:10.1038/nchembio.1352
Schreiber MA, Pierce-Shimomura JT, Chan S, Parry D, McIntire SL (2010) Manipulation of behavioral decline in Caenorhabditis elegans with the Rag GTPase raga-1. PLoS Genet 6(5), e1000972. doi:10.1371/journal.pgen.1000972
Scott BA, Avidan MS, Crowder CM (2002) Regulation of hypoxic death in C. elegans by the insulin/IGF receptor homolog DAF-2. Science 296(5577):2388–2391. doi:10.1126/science.1072302
Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, Withers DJ (2009) Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326(5949):140–144. doi:10.1126/science.1177221
Semenza GL (2012) Hypoxia-inducible factors in physiology and medicine. Cell 148(3):399–408. doi:10.1016/j.cell.2012.01.021
Seo K, Choi E, Lee D, Jeong DE, Jang SK, Lee SJ (2013) Heat shock factor 1 mediates the longevity conferred by inhibition of TOR and insulin/IGF-1 signaling pathways in C. elegans. Aging Cell 12(6):1073–1081. doi:10.1111/acel.12140
Shaw WM, Luo S, Landis J, Ashraf J, Murphy CT (2007) The C. elegans TGF-beta dauer pathway regulates longevity via insulin signaling. Curr Biol 17(19):1635–1645. doi:10.1016/j.cub.2007.08.058
Sheaffer KL, Updike DL, Mango SE (2008) The target of rapamycin pathway antagonizes pha-4/FoxA to control development and aging. Curr Biol 18(18):1355–1364. doi:10.1016/j.cub.2008.07.097
Shen Y, Wollam J, Magner D, Karalay O, Antebi A (2012) A steroid receptor-microRNA switch regulates life span in response to signals from the gonad. Science 338(6113):1472–1476. doi:10.1126/science.1228967
Smith-Vikos T, de Lencastre A, Inukai S, Shlomchik M, Holtrup B, Slack FJ (2014) MicroRNAs mediate dietary-restriction-induced longevity through PHA-4/FOXA and SKN-1/Nrf transcription factors. Curr Biol 24(19):2238–2246. doi:10.1016/j.cub.2014.08.013
Soukas AA, Kane EA, Carr CE, Melo JA, Ruvkun G (2009) Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev 23(4):496–511. doi:10.1101/gad.1775409
Stanfel MN, Shamieh LS, Kaeberlein M, Kennedy BK (2009) The TOR pathway comes of age. Biochim Biophys Acta 1790(10):1067–1074. doi:10.1016/j.bbagen.2009.06.007
Steinkraus KA, Smith ED, Davis C, Carr D, Pendergrass WR, Sutphin GL, Kennedy BK, Kaeberlein M (2008) Dietary restriction suppresses proteotoxicity and enhances longevity by an hsf-1-dependent mechanism in Caenorhabditis elegans. Aging Cell 7(3):394–404. doi:10.1111/j.1474-9726.2008.00385.x
Syntichaki P, Troulinaki K, Tavernarakis N (2007) eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature 445(7130):922–926. doi:10.1038/nature05603
Takahashi Y, Daitoku H, Hirota K, Tamiya H, Yokoyama A, Kako K, Nagashima Y, Nakamura A, Shimada T, Watanabe S, Yamagata K, Yasuda K, Ishii N, Fukamizu A (2011) Asymmetric arginine dimethylation determines life span in C. elegans by regulating forkhead transcription factor DAF-16. Cell Metab 13(5):505–516. doi:10.1016/j.cmet.2011.03.017
Taylor RC, Dillin A (2013) XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153(7):1435–1447. doi:10.1016/j.cell.2013.05.042
Tepper RG, Ashraf J, Kaletsky R, Kleemann G, Murphy CT, Bussemaker HJ (2013) PQM-1 complements DAF-16 as a key transcriptional regulator of DAF-2-mediated development and longevity. Cell 154(3):676–690. doi:10.1016/j.cell.2013.07.006
Thondamal M, Witting M, Schmitt-Kopplin P, Aguilaniu H (2014) Steroid hormone signalling links reproduction to lifespan in dietary-restricted Caenorhabditis elegans. Nat Commun 5:4879. doi:10.1038/ncomms5879
Thyagarajan B, Blaszczak AG, Chandler KJ, Watts JL, Johnson WE, Graves BJ (2010) ETS-4 is a transcriptional regulator of life span in Caenorhabditis elegans. PLoS Genet 6(9), e1001125. doi:10.1371/journal.pgen.1001125
Tissenbaum HA, Guarente L (2001) Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410(6825):227–230. doi:10.1038/35065638
Tonsaker T, Pratt RM, McGhee JD (2012) Re-evaluating the role of ELT-3 in a GATA transcription factor circuit proposed to guide aging in C. elegans. Mech Ageing Dev 133(1):50–53. doi:10.1016/j.mad.2011.09.006
Torgovnick A, Schiavi A, Testi R, Ventura N (2010) A role for p53 in mitochondrial stress response control of longevity in C. elegans. Exp Gerontol 45(7–8):550–557. doi:10.1016/j.exger.2010.02.007
Toth ML, Sigmond T, Borsos E, Barna J, Erdelyi P, Takacs-Vellai K, Orosz L, Kovacs AL, Csikos G, Sass M, Vellai T (2008) Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 4(3):330–338
Tsang WY, Sayles LC, Grad LI, Pilgrim DB, Lemire BD (2001) Mitochondrial respiratory chain deficiency in Caenorhabditis elegans results in developmental arrest and increased life span. J Biol Chem 276(34):32240–32246. doi:10.1074/jbc.M103999200
Tullet JM, Araiz C, Sanders MJ, Au C, Benedetto A, Papatheodorou I, Clark E, Schmeisser K, Jones D, Schuster EF, Thornton JM, Gems D (2014) DAF-16/FoxO directly regulates an atypical AMP-activated protein kinase gamma isoform to mediate the effects of insulin/IGF-1 signaling on aging in Caenorhabditis elegans. PLoS Genet 10(2), e1004109. doi:10.1371/journal.pgen.1004109
Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, Oliveira RP, Baumeister R, Blackwell TK (2008) Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 132(6):1025–1038. doi:10.1016/j.cell.2008.01.030
van der Horst A, Schavemaker JM, Pellis-van Berkel W, Burgering BM (2007) The Caenorhabditis elegans nicotinamidase PNC-1 enhances survival. Mech Ageing Dev 128(4):346–349. doi:10.1016/j.mad.2007.01.004
Van Gilst MR, Hadjivassiliou H, Jolly A, Yamamoto KR (2005) Nuclear hormone receptor NHR-49 controls fat consumption and fatty acid composition in C. elegans. PLoS Biol 3(2), e53. doi:10.1371/journal.pbio.0030053
Van Raamsdonk JM, Hekimi S (2009) Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet 5(2), e1000361. doi:10.1371/journal.pgen.1000361
Van Raamsdonk JM, Hekimi S (2012) Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci U S A 109(15):5785–5790. doi:10.1073/pnas.1116158109
Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F (2003) Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 426(6967):620. doi:10.1038/426620a
Ventura N, Rea S, Henderson ST, Condo I, Johnson TE, Testi R (2005) Reduced expression of frataxin extends the lifespan of Caenorhabditis elegans. Aging Cell 4(2):109–112
Ventura N, Rea SL, Schiavi A, Torgovnick A, Testi R, Johnson TE (2009) p53/CEP-1 increases or decreases lifespan, depending on level of mitochondrial bioenergetic stress. Aging Cell 8(4):380–393. doi:10.1111/j.1474-9726.2009.00482.x
Viswanathan M, Guarente L (2011) Regulation of Caenorhabditis elegans lifespan by sir-2.1 transgenes. Nature 477(7365):E1–E2. doi:10.1038/nature10440
Vora M, Shah M, Ostafi S, Onken B, Xue J, Ni JZ, Gu S, Driscoll M (2013) Deletion of microRNA-80 activates dietary restriction to extend C. elegans healthspan and lifespan. PLoS Genet 9(8), e1003737. doi:10.1371/journal.pgen.1003737
Walter L, Baruah A, Chang HW, Pace HM, Lee SS (2011) The homeobox protein CEH-23 mediates prolonged longevity in response to impaired mitochondrial electron transport chain in C. elegans. PLoS Biol 9(6), e1001084. doi:10.1371/journal.pbio.1001084
Wang J, Robida-Stubbs S, Tullet JM, Rual JF, Vidal M, Blackwell TK (2010) RNAi screening implicates a SKN-1-dependent transcriptional response in stress resistance and longevity deriving from translation inhibition. PLoS Genet 6(8). doi:10.1371/journal.pgen.1001048
Wang MC, O'Rourke EJ, Ruvkun G (2008) Fat metabolism links germline stem cells and longevity in C. elegans. Science 322(5903):957–960. doi:10.1126/science.1162011
Wang Y, Tissenbaum HA (2006) Overlapping and distinct functions for a Caenorhabditis elegans SIR2 and DAF-16/FOXO. Mech Ageing Dev 127(1):48–56. doi:10.1016/j.mad.2005.09.005
Wang Y, Oh SW, Deplancke B, Luo J, Walhout AJ, Tissenbaum HA (2006) C. elegans 14-3-3 proteins regulate life span and interact with SIR-2.1 and DAF-16/FOXO. Mech Ageing Dev 127(9):741–747. doi:10.1016/j.mad.2006.05.005
Wolff S, Ma H, Burch D, Maciel GA, Hunter T, Dillin A (2006) SMK-1, an essential regulator of DAF-16-mediated longevity. Cell 124(5):1039–1053. doi:10.1016/j.cell.2005.12.042
Wolkow CA, Munoz MJ, Riddle DL, Ruvkun G (2002) Insulin receptor substrate and p55 orthologous adaptor proteins function in the Caenorhabditis elegans daf-2/insulin-like signaling pathway. J Biol Chem 277(51):49591–49597. doi:10.1074/jbc.M207866200
Wollam J, Magner DB, Magomedova L, Rass E, Shen Y, Rottiers V, Habermann B, Cummins CL, Antebi A (2012) A novel 3-hydroxysteroid dehydrogenase that regulates reproductive development and longevity. PLoS Biol 10(4), e1001305. doi:10.1371/journal.pbio.1001305
Wong A, Boutis P, Hekimi S (1995) Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 139(3):1247–1259
Xiao R, Zhang B, Dong Y, Gong J, Xu T, Liu J, Xu XZ (2013) A genetic program promotes C. elegans longevity at cold temperatures via a thermosensitive TRP channel. Cell 152(4):806–817. doi:10.1016/j.cell.2013.01.020
Xu X, Kim SK (2012) The GATA transcription factor egl-27 delays aging by promoting stress resistance in Caenorhabditis elegans. PLoS Genet 8(12), e1003108. doi:10.1371/journal.pgen.1003108
Yamawaki TM, Berman JR, Suchanek-Kavipurapu M, McCormick M, Gaglia MM, Lee SJ, Kenyon C (2010) The somatic reproductive tissues of C. elegans promote longevity through steroid hormone signaling. PLoS Biol 8(8). doi:10.1371/journal.pbio.1000468
Yang CC, Chen D, Lee SS, Walter L (2011) The dynamin-related protein DRP-1 and the insulin signaling pathway cooperate to modulate Caenorhabditis elegans longevity. Aging Cell 10(4):724–728. doi:10.1111/j.1474-9726.2011.00711.x
Yang J, Chen D, He Y, Melendez A, Feng Z, Hong Q, Bai X, Li Q, Cai G, Wang J, Chen X (2013) MiR-34 modulates Caenorhabditis elegans lifespan via repressing the autophagy gene atg9. Age (Dordr) 35(1):11–22. doi:10.1007/s11357-011-9324-3
Yang W, Hekimi S (2010a) A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol 8(12), e1000556. doi:10.1371/journal.pbio.1000556
Yang W, Hekimi S (2010b) Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 9(3):433–447, doi:ACE571 [pii]
Yee C, Yang W, Hekimi S (2014) The Intrinsic Apoptosis Pathway Mediates the Pro-Longevity Response to Mitochondrial ROS in C. elegans. Cell 157(4):897–909. doi:10.1016/j.cell.2014.02.055
Zarse K, Schulz TJ, Birringer M, Ristow M (2007) Impaired respiration is positively correlated with decreased life span in Caenorhabditis elegans models of Friedreich Ataxia. FASEB J 21(4):1271–1275. doi:10.1096/fj.06-6994com
Zhang Y, Xu J, Puscau C, Kim Y, Wang X, Alam H, Hu PJ (2008) Caenorhabditis elegans EAK-3 inhibits dauer arrest via nonautonomous regulation of nuclear DAF-16/FoxO activity. Dev Biol 315(2):290–302. doi:10.1016/j.ydbio.2007.12.032
Zhang M, Poplawski M, Yen K, Cheng H, Bloss E, Zhu X, Patel H, Mobbs CV (2009a) Role of CBP and SATB-1 in aging, dietary restriction, and insulin-like signaling. PLoS Biol 7(11), e1000245. doi:10.1371/journal.pbio.1000245
Zhang Y, Shao Z, Zhai Z, Shen C, Powell-Coffman JA (2009b) The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS One 4(7), e6348. doi:10.1371/journal.pone.0006348
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Japan
About this chapter
Cite this chapter
Lee, Y. et al. (2015). Genes and Pathways That Influence Longevity in Caenorhabditis elegans . In: Mori, N., Mook-Jung, I. (eds) Aging Mechanisms. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55763-0_8
Download citation
DOI: https://doi.org/10.1007/978-4-431-55763-0_8
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55762-3
Online ISBN: 978-4-431-55763-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)