
Abstract
Sarcopenia is an age-related loss of skeletal muscle mass and strength. In this chapter, advances in the association of sarcopenia and diabetes mellitus are discussed. Falls in diabetic patients associate with decline of muscle mass or strength in the elderly. Insulin resistance impairs the protein regeneration in skeletal muscle and also induces the protein breakdown and muscle wasting, leading to development of sarcopenia. This insulin resistance suggests the most important linkage between sarcopenia and diabetes. Sarcopenia and obesity appear to have additive effects on insulin resistance and age-related changes in body composition. Loss of skeletal muscle mass affecting glucose disposal and impaired energy homeostasis affecting muscle protein content, together, might lead to a vicious cycle. Insulin resistance and inflammation leads to muscle wasting through the pathways involved in Akt/PKB, FoxOs, PGC-1α, and AMPK. The accumulation of AGEs through glucose intolerance enhanced by mitochondrial ROS with promotion of apoptosis leads to the development of muscle wasting. Exercise is known as the most efficient treatment of sarcopenia with diabetes but less information is known for nutritional replenishment or medications. Sarcopenia in diabetes mellitus would have higher physical dysfunction and mortality risks than those in nondiabetic older adults.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The aging population is increasing worldwide. The older population of those aged 65 years is expected to reach 16.2 % of the total world’s population and the “oldest-old” aged 80 years or older will increase from 1.6 to 4.4 % by 2050 [1]. In the elderly, functional decline is typically associated with the increase of body fat and the reduction of lean muscle quality; this contributes to physical disability [2] known as sarcopenia. Sarcopenia is related to frailty, glucose homeostasis impairment [3], and geriatric syndromes [4] which affect 20–50 % of adults aged over 60 [5]. Obesity and diabetes appeared to be at higher risks of processing metabolic dysfunction, contributing to the progression of sarcopenia [6] related to alterations of decreased insulin sensitivity [7], unbalanced fuel oxidation [8], and reduced muscle protein content [9].
Diabetes is currently viewed as the most common metabolic dysfunction of this century and referred to high levels of circulating glucose and overall insulin resistance. Epidemiological studies reported that the number of new diabetic patients has increased by 50 % over the last 10 years [10]. The incidence of type 2 diabetes (T2D) in people older than 60 years old was more than 20 % higher than in younger people [11], and around 80 % of people with T2D were obese [12]. Various clinical studies also indicated that obesity and insulin resistance were associated with T2D, which might play important roles in the pathogenesis of physical function decline and sarcopenia [13]. The interplay between obesity and diabetes in an age-related sarcopenia population is now considered as an important concern in geriatrics. Here, we discussed the interactions and mechanisms of sarcopenia in diabetes mellitus and considered the future perspectives of this area.
2 Evidence in Clinical Studies for Sarcopenia and Diabetes
2.1 Fall in Skeletal Muscle Function Decline with T2D
The primary function of the skeletal muscle is to generate force and to provide locomotion. Fall is one of the geriatric syndromes, which has approximately 30 % of people aged 65 and older fall each year [14] and frequently leads to the risks of injury and disability. It has been reported that increased falls in diabetic patients with hypoglycemia [15] are associated with 1 % muscle size decline per year after age 50 [16]. Further, older adults with T2D have shown an accelerated decline in leg lean mass, muscle strength, and functional capacity in comparison with normoglycemic controls [17]. In addition, in T2D patients in the elderly, leg muscle strength has been presented to be 30 % lower compared with nondiabetic controls in a 3-year-period study [18]. Therefore, T2D may be associated with increasing fall risks through loss of skeletal muscle mass and strength, especially in older adults.
2.2 Insulin Resistance and Skeletal Muscle
Insulin, which maintains blood-glucose homeostasis, induces glucose uptake into muscle tissues and mediates lipolysis in adipose tissues. Skeletal muscle plays an important role in glucose metabolism and mediates whole-body insulin-stimulated glucose uptake [19]. In T2D, insulin resistance impairs the balance between protein synthesis and degradation in skeletal muscle cells, which induces muscle wasting [20] and leads to the development of sarcopenia. This insulin resistance suggests the most important linkage between sarcopenia and T2D. T2D and muscle weakness may provide a vicious cycle: loss of skeletal muscle mass affects glucose disposal and energy homeostasis impairment affects muscle protein content.
2.3 Sarcopenic Obesity
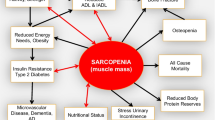
Recently, obesity incidence has also increased in the older population. Sarcopenia and obesity in elderly subjects appeared to have additive effects on insulin resistance and age-related changing in body composition compared with the obesity condition [21]. Interestingly, insulin resistance is associated with sarcopenia in both non-obese and obese individuals, but only sarcopenic obese individuals have established the associations with insulin resistance and dysglycemia, with or without diabetes [22]. This suggested that sarcopenia could be developed independently from obesity and sarcopenic obese individuals with more unfavorable physical dysfunction than individuals with neither sarcopenia nor obesity. Altogether, sarcopenic obesity could be described as having a larger amount of fat but lower lean muscle mass, excess of energy intake, physical dysfunction, impairment of insulin sensitivity, and glucose homeostasis [23]. The diagnostic criteria for sarcopenic obesity in the early stages could be helpful in the clinical observation for the reduction of health risks in the aged society. The interactions of sarcopenia, diabetes, and obesity have been summarized in Fig. 16.1.

Interaction between sarcopenia and diabetes. Insulin resistance in skeletal muscle is the most important link between sarcopenia and diabetes. Sarcopenia and obesity appear to have additive effects on insulin resistance and age-related changing in body composition. Diabetes, sarcopenia, and obesity may provide a vicious cycle: loss of skeletal muscle mass affects glucose disposal and energy homeostasis impairment affects muscle protein content. Coexistence of sarcopenia with diabetes or/and obesity increases the risk for fall or disability and cardio- and cerebrovascular diseases
3 Evidences in Basic Studies for Sarcopenia and Diabetes
3.1 Muscle Fiber-Type Switching
Evaluation of changes in skeletal muscle fiber composition during the early stages of the metabolic syndrome and diabetes is required to provide essential insights as to fiber-type distribution. Age-related sarcopenia is associated with muscle constitution in differing amounts, in individual skeletal muscle [24], and a decrease of size and number of fast type II muscle fiber has been observed [25, 26]. The EDL, or the extensor digitorum longus glycolytic muscle, is predominantly composed of type IIb fibers which has mainly reduced by 25–30 % in the cross-sectional area of the skeletal muscle of humans aged 70 [27]. In addition, insulin resistance is associated with higher proportions of glycolytic fast-twitch type IIb fibers [28]. The muscle transformation studies of the extended periods of bed rest have observed the slow-to-fast muscle transitions (myosin heavy chain, MyHCI to MyHCII) [29]. This shift in fiber-type composition may lead to the increase of the total EDL content in the aging process of muscle atrophy; however, this increase failed to prevent natural muscle aging and resulted in total skeletal muscle mass reduction in sarcopenia. The decrease in total type II fiber may have negative effects on the production of essentially muscular power in daily life, which is a pathognomonic change in sarcopenia or sarcopenia in diabetes.
3.2 Inflammation in Skeletal Muscle
Inflammation, an important mediator in the pathogenesis of insulin resistance, has been observed in both diabetes mellitus and sarcopenia. Since the muscle is the primary tissue which produces and responds to a variety of hormones and cytokines, it has been involved in modulating muscle protein degradation and myogenesis through the prevention of inflammation; further, pro-inflammation is counteracted by insulin resistance [30]. TNF-α is highly expressed in adipose tissues from obese subjects, which develops insulin resistance, induces IL-6 [31], and blocks muscle tissues differentiation leading to sarcopenia [32]. In addition, IL-6 and TNF-α activate TNF-α-related apoptosis-inducing ligand receptors (TNFR1) which stimulate inhibitor of κB kinase [33, 34], causing NF-κB activation resulting in processing protein degradation in the muscle. The effects of insulin antagonizing mediated by IL-6 on skeletal muscle have been described and chronic exposure to IL-6 caused inflammation which impaired insulin-stimulated GLUT4 translocation in skeletal muscle [35]. STAT3, signal transducer and activator of transcription 3, has been characterized in myokines signaling [36] which can link the activation of signal transducers of the Janus kinase (JAK) protein [37] and increased activity of the transcription factor C-EBPß and C-EBPδ involved in muscle wasting [38] through ActII receptor and myostatin [39] (Fig. 16.2).

Molecular mechanism of muscle wasting in diabetes. Decline of skeletal muscle mass or strength occurs under a variety of conditions and involves the regulation of muscle protein catabolism and mitochondrial dysfunction: FoxO1 and FoxO3 induce a decrease in muscle mass associated with an upregulation of ubiquitin E3 ligases MAFbx/atrogin-1 and MuRF1 expression. In cancer cachexia and sepsis, FoxOs inhibit the MAFbx/atrogin-1, MuRF1, and Bnip3 mRNA expression, which has been associated with inhibition of muscle fiber atrophy. NF-kB induces muscle breakdown by promoting protein degradations in skeletal muscle by regulation of ubiquitin-proteasome pathway (UPS). AGEs and angiotensin II via the angiotensin type 1 (AT1) receptor activate NADPH oxidase and lead to ROS production, facilitating activation of caspases, which contributes to muscle mass loss. IL-6 and TNF-α activate TNF-α-related apoptosis-inducing ligand receptors (TNFR1) which stimulate inhibitor of κB kinase (IKK) and cause NF-κB activation, thus processing the protein degradations in muscle. Signal transducer and activator of transcription 3 (STAT3) links the activation of signal transducers of the Janus kinase (JAK) protein and increases activity of the transcription factor C-EBPß and C-EBPδ involved in muscle wasting through ActII receptor and myostatin. Suppressor of cytokine signaling (SOCS3) targets IRS-1 in inflammation-induced insulin resistance. IRS-1 is rapidly degraded after IGF-1 stimulation and blocks FoxO1, leading to inhibition of atrophy. IRS-1 phosphorylation links to dephosphorylation of Akt, which mediates insulin resistance. Inactivation of tuberous sclerosis complex 1/2 (TSC1/TSC2) stimulates Akt–mTOR signaling and leads to “incomplete” autophagy in muscle wasting. AMPK inactivates mTOR and decreases the activation of ribosomal protein S6 kinase 1 (S6K1), an activator of protein synthesis, thereby increasing the rates of muscle mass loss. The autophagic regulator proteins, Atg12, LC3-II, and apoptotic genes Bnip3, promote mitochondrial disruption by activation of FoxOs during muscle atrophy
On the other hand, IL-6 or IL-15 [40, 41], which is secreted from skeletal muscle, suppresses TNF-α effects on the exercise conditions [42]. These cytokines produced from skeletal muscle are called “myokines” and exert autocrine, paracrine, or endocrine effects. The interaction between myokines production and diabetes is still unclear, but myokines can be the candidate biomarkers for metabolic disorders, including diabetes.
3.3 Molecule-Related Signaling Pathways in Muscle Wasting
Many reports have shown the molecular mechanisms for muscle wasting using animal models which fed on high-fat or sucrose diet and of diabetes. The molecules signaling pathways related to muscle wasting has been shown in this section.
3.3.1 Akt/PKB Signaling
In skeletal muscle, Akt/PKB plays a key role in insulin and PI3K/Akt/mTOR signaling pathway, which is regulating energy metabolism and protein synthesis. Previous studies have shown that SOCS3, suppressor of cytokine signaling, targeted to IRS-1 in inflammation-induced insulin resistance [43]. IRS-1 is rapidly degraded after IGF-1 stimulation and blocking of FoxO1 leads to inhibition of atrophy. The activation of Akt/PKB results in the glucose uptake through the stimulation and translocation of GLUT4, leading to the increased uptake of glucose, thus decreasing the amount of circulating glucose and regulating glucose metabolism upon insulin exposure [44, 45]. However, IRS-1 phosphorylation is linked to dephosphorylation of Akt/PKB [46], which mediates insulin resistance and further inactivates tuberous sclerosis complex 1/2 (TSC1/TSC2) [47] which stimulated Akt–mTOR signaling and autophagy in muscular dystrophy (Fig. 16.2), since signaling through IGF-1/PI3K/Akt activated the mechanistic target of rapamycin (mTOR) pathway [48] and critically mediated the fork head boxO (FoxO) transcription factors [49]. Protein synthesis in skeletal muscle is strongly related to Akt/PKB signaling, through the activation of mTOR [48], to regulate GSK3 (glycogen synthase kinase 3) activity which results in enhanced glycogen synthesis [50]. Furthermore, the defected of insulin actions may produce the increase of protein degradation [51]; this is supposed the Akt/FoxO pathway may contribute to sarcopenia-related muscle protein degradation.
3.3.2 FoxOs Transcription Factors
Decline of skeletal muscle mass or strength involves regulation of muscle protein catabolism and mitochondrial dysfunction. FoxOs are important regulators of muscle energy homeostasis and carbohydrate catabolism in the fast state [52]. FoxO1 and FoxO3 induced a decreasing of muscle mass which associated with an upregulation of ubiquitin E3 ligases MAFbx/atrogin-1 and the muscle ring-finger protein 1 (MuRF1) expression [53]. Recently, defect in autophagy-dependent signaling, an important mechanism for maintaining cell self-renew and protein turnover, is also observed in various muscular dystrophies [54]. The autophagic regulator proteins, LC3-II, Atg12, and Bnip3, promote mitochondrial disruption by activation of FoxOs during muscle atrophy [55]. Furthermore, mitochondrial dysfunction has been proposed through FoxOs, switching of muscle type from slow-twitch oxidative type I fiber to fast-twitch glycolytic fiber (MyHCI to MyHCII).
3.3.3 PGC-1α and AMPK
In sarcopenia and sarcopenic obese individuals, insulin resistance is involved in developing muscle atrophy in aging and diabetes mellitus [56]. Importantly, high-fat diet-induced insulin resistance observed the reduction of skeletal muscle mitochondrial function and decreased expression of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) [57]; thus, the hypothesis of mitochondrial dysfunction leading to sarcopenia has been in attention. The downregulation of PGC-1α by high-fat diet implicated the development of skeletal muscle insulin resistance of T2D individuals compared to nondiabetic individuals [58–61]. It was also suggested that PGC-1α plays an important role in fiber-type switching from fast-twitch glycolytic fiber into slow-twitch oxidative type I fiber [62]. These fiber-type switching effects through PGC-1α might be induced by PGC-1α1. However, muscle-specific induction of PGC-1α4, but not PGC-1α1, has shown the increase of muscle mass and strength, resistance to cancer cachexia through activating IGF-1, and suppressing myostatin gene expression [63]. Altogether, PGC-1α and its isoforms may play a crucial role in regulating mitochondrial biogenesis and muscle mass and strength.
AMP-activated protein kinase (AMPK) is a primary regulator of skeletal muscle metabolic homeostasis [64]. AMPK impacts the insulin-mediated effects on muscle protein synthesis through the interfering by the Akt/PKB signaling pathway [65] and promotes glucose uptake and oxidation through migration of GLUT4 to the cellular membrane [66]. Importantly, AMPK activation by AICAR in skeletal muscle [67, 68] improved muscle function and mitochondrial activity in muscle atrophy. In addition, AMPK inactivated mTOR and decreased the activation of ribosomal protein S6 kinase 1, an activator of protein synthesis, thereby increasing the rates of sarcopenic muscle mass loss. On the other hand, AMPK-mediated activation of FoxO3 contributed to the proteolysis with the expression of muscle atrophy F-box (MAFbx/atrogin-1) and MuRF1 [69]. AMPK also was found to promote directly the phosphorylation of PGC-1α and to induce mitochondrial biogenesis [70, 71]. Further investigations will be needed to clarify these conflicting findings on the role of AMPK in the development of sarcopenia.
3.4 ROS and Mitochondrial Dysfunction
ROS and oxidative stress have been considered as important pathogenic components of metabolic diseases [72]. Mitochondrial dysfunction produces increased amounts of ROS, resulting in oxidative damage, which includes decreased mitochondrial content and oxidative capacity and increased mitochondrial DNA mutations [73]. Activation of the renin-angiotensin system (RAS) is commonly observed in patients either with diabetes, obesity, or both. ROS production could be enhanced by angiotensin II (Ang II), which stimulated the angiotensin type 1 (AT1, G-protein-coupled) receptor and activated NADPH oxidase. In rodent model, RAS blockade increases survival rate and prevents age-related defects [74]. Thus, it is suggested that Ang II contributes to mitochondrial dysfunction in the aging process. This indicated that diabetes mellitus and age-related sarcopenia may have additive effects for ROS production. Furthermore, NF-kB induces muscle breakdown by promoting protein degradations in skeletal muscle through regulation of ubiquitin-proteasome pathway (UPS) [75]. The maintenance of mitochondrial morphology and regeneration of self-renewal could be due to the mitochondrial fusion and fission interactions. This mitochondrial fission contributes to the quality control of creating new mitochondria and removing of damaged mitochondria during high cellular stress [76]. Disruptions of either event may induce metabolic disorders and leads to developmental defects and diseases [77], suggesting that the maintenance in aging muscle cell mitochondrial morphology may prevent cell dysfunction.
3.5 AGE Accumulation and Diabetic Neuropathy
The formation of advanced glycation end products (AGEs) is generated through nonenzymatic glycation of many heterogenous compounds and the complex and diverse possibilities of reaction of glucose with proteins, lipids, and nucleic acids [78]. AGEs are regarded as key molecules to be one source of oxidative stress in aging [79]. It is well known that long-term high-fat diet and endogenously formed AGEs contribute to the progression of diabetic complications [80]. AGEs are produced endogenously via food, and the concentration of circulating AGEs increases in high-fat diet [81]. Interestingly, it has been reported that AGE accumulation was decreased in association with administration of insulin and greater in fast-twitch fibers in non-insulin diabetic animal model [82]. AGE/RAGE in aging stimulates the activation of ERK1/2 and P38 MAPK pathways and increased apoptosis transcription factor such as NF-kB [83]. In recent years, the modified protein methylglyoxal (MG) was observed to involve AGE formation in aged individuals [84, 85]. MG is believed to induce protein glycation leading to the formation of AGEs; some reports indicated that MG or AGEs administration to Sprague-Dawley rats resulted in increased glucose levels and insulin resistance [86, 87]. Moreover, the direct effects of AGEs from Tanaka’s group have shown that AGE2 or AGE3 markedly suppressed the expressions of MyoD and myogenin protein on myoblastic differentiation in C2C12 cells and significantly inhibited mRNA expression. This suggested that AGEs have direct negative effects on myogenesis [88].
T2D leads to chronic hyperglycemia and is related to the major age-related microvascular complications such as microangiopathic and macroangiopathic damage and motor neuropathies (diabetic neuropathy) [89]. Enhanced mitochondrial apoptosis has also been observed in muscle denervation and implicated in diabetes and neuropathy in human neuromuscular disorders, which exhibited significant muscle weakness and reduced functional capacity in the ankle and knee [90]. In addition, the AGE-RAGE axis and accumulation of AGEs in the peripheral nerve also play important roles in the pathogenesis of diabetic neuropathy [91], thus impairing the quality of life of diabetic patients. It is not yet established, but increasing studies suggested that accumulation of AGEs through glucose intolerance enhanced by mitochondrial ROS together with promotion of apoptosis leads to an elevated risk of developing sarcopenia.
4 Potential Treatments for Sarcopenia with Diabetes
Epidemiological and intervention studies for exercise training have strongly supported its efficacy for prevention, leading to the management of diabetes and sarcopenia. In exercising and amino acid supplementation (AAS) study [92], not only enhanced muscle mass or walking speed but also enhanced muscle strength was observed in sarcopenic women. In addition, high-intensity progressive resistance training was effective in improving glycemic control and physical activity, increasing lean mass among high-risk older adults with T2D [93]. Thus, resistance training and a combination of training and nutritional replenishment, like amino acids, might also be a beneficial intervention in sarcopenia with diabetes.
As for antidiabetic drugs, a class of thiazolidinediones (TZDs) can activate AMPK in insulin-resistant animals [94] and mediate mitochondrial effects on neurodegeneration [95]. In T2D patients, rosiglitazone [96] and pioglitazone [97] both enhanced insulin reactions and reduced plasma nonesterified fatty acids [97], which improved insulin-stimulated muscle glucose disposal. These TZDs may involve a potential role of age-related mitochondrial dysfunction, neurodegeneration diseases [98], and diabetes with sarcopenia. Indeed, a combination of pioglitazone and resistance training leads to a potentiated effect on muscle power compared with resistance training alone in older obese women [99]. Metformin also activates AMPK and enhances insulin sensitivity in skeletal muscle, thereby stimulating glucose uptake [100]; however, there has been no reliable clinical evidence so far. At any rate, insulin resistance and glucose intolerance can contribute to muscle wasting; therefore, the therapies for improving insulin resistance and glucose intolerance are supposed to have a potential to prevent sarcopenia.
5 Closing Remarks and Perspectives
We conclude that subjects with sarcopenia in diabetes mellitus would have higher physical dysfunction and mortality risks than those nondiabetic older adults. Sarcopenic obesity could be described as independent of sarcopenia and obesity, both related to insulin resistance and inflammation in diabetes mellitus. We introduce the molecular pathways underlying the pathogenesis of sarcopenia in diabetes in this chapter and, in particular, mitochondrial dysfunction and AGE accumulation might be significant targets common to sarcopenia and diabetes. Further studies will be needed to improve our knowledge on the interaction between diabetes and sarcopenia and to establish beneficial therapeutic interventions combined with exercise for slowing down and reversing the loss of muscle mass and strength in older adults with T2D.
References
United Nations (2007) World population prospects, the 2006 revision: highlights. United Nations, New York
Smalley KJ, Knerr AN, Kendrick ZV, Colliver JA, Owen OE (1990) Reassessment of body-mass indexes. Am J Clin Nutr 52(3):405–408
Dutta C, Hadley EC, Lexell J (1997) Sarcopenia and physical performance in old age: overview. Muscle Nerve 5:S5–S9
Kinney JM (2004) Nutritional frailty, sarcopenia and falls in the elderly. Curr Opin Clin Nutr Metab Care 7(1):15–20. doi:10.1097/01.mco.0000109601.04238.46
Berger MJ, Doherty TJ (2010) Sarcopenia: prevalence, mechanisms, and functional consequences. Interdiscip Top Gerontol 37:94–114. doi:10.1159/000319997
Villareal DT, Banks M, Siener C, Sinacore DR, Klein S (2004) Physical frailty and body composition in obese elderly men and women. Obes Res 12(6):913–920. doi:10.1038/oby.2004.111
Goodman MN, Ruderman NB (1979) Insulin sensitivity of rat skeletal muscle: effects of starvation and aging. Am J Physiol 236(5):E519–E523
Sial S, Coggan AR, Carroll R, Goodwin J, Klein S (1996) Fat and carbohydrate metabolism during exercise in elderly and young subjects. Am J Physiol-Endocrinol Metab 271(6):E983–E989
Dardevet D, Sornet C, Balage M, Grizard J (2000) Stimulation of in vitro rat muscle protein synthesis by leucine decreases with age. J Nutr 130(11):2630–2635
Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ et al (2011) National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet 378(9785):31–40. doi:10.1016/s0140-6736(11)60679-x
Styskal J, Van Remmen H, Richardson A, Salmon AB (2012) Oxidative stress and diabetes: what can we learn about insulin resistance from antioxidant mutant mouse models? Free Radic Biol Med 52(1):46–58. doi:10.1016/j.freeradbiomed.2011.10.441
Laaksonen DE, Niskanen L, Lakka HM, Lakka TA, Uusitupa M (2004) Epidemiology and treatment of the metabolic syndrome. Ann Med 36(5):332–346. doi:10.1080/07853890410031849
Stenholm S, Harris TB, Rantanen T, Visser M, Kritchevsky SB, Ferrucci L (2008) Sarcopenic obesity: definition, cause and consequences. Curr Opin Clin Nutr Metab Care 11(6):693–700. doi:10.1097/MCO.0b013e328312c37d
Gillespie LD, Gillespie WJ, Robertson MC, Lamb SE, Cumming RG, Rowe BH (2003) Interventions for preventing falls in elderly people. Cochrane Database Syst Rev (4):Cd000340. doi: 10.1002/14651858.cd000340
Marzetti E, Leeuwenburgh C (2006) Skeletal muscle apoptosis, sarcopenia and frailty at old age. Exp Gerontol 41(12):1234–1238. doi:10.1016/j.exger.2006.08.011
Goodpaster BH, Park SW, Harris TB, Kritchevsky SB, Nevitt M, Schwartz AV et al (2006) The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J Gerontol Ser A-Biol Sci Med Sci 61(10):1059–1064
Leenders M, Verdijk LB, van der Hoeven L, Adam JJ, van Kranenburg J, Nilwik R et al (2013) Patients with type 2 diabetes show a greater decline in muscle mass, muscle strength, and functional capacity with aging. J Am Med Dir Assoc 14(8):585–592. doi:10.1016/j.jamda.2013.02.006
Park SW, Goodpaster BH, Strotmeyer ES, Kuller LH, Broudeau R, Kammerer C et al (2007) Accelerated loss of skeletal muscle strength in older adults with type 2 diabetes: the health, aging, and body composition study. Diabetes Care 30(6):1507–1512. doi:10.2337/dc06-2537
Corcoran MP, Lamon-Fava S, Fielding RA (2007) Skeletal muscle lipid deposition and insulin resistance: effect of dietary fatty acids and exercise. Am J Clin Nutr 85(3):662–677
Russell ST, Rajani S, Dhadda RS, Tisdale MJ (2009) Mechanism of induction of muscle protein loss by hyperglycaemia. Exp Cell Res 315(1):16–25. doi:10.1016/j.yexcr.2008.10.002
Lim S, Kim JH, Yoon JW, Kang SM, Choi SH, Park YJ et al (2010) Sarcopenic obesity: prevalence and association with metabolic syndrome in the Korean Longitudinal Study on Health and Aging (KLoSHA). Diabetes Care 33(7):1652–1654. doi:10.2337/dc10-0107
Srikanthan P, Hevener AL, Karlamangla AS (2010) Sarcopenia exacerbates obesity-associated insulin resistance and dysglycemia: findings from the National Health and Nutrition Examination Survey III. PLoS One 5(5):e10805. doi:10.1371/journal.pone.0010805
de Simone G, Pasanisi F, Ferrara AL, Roman MJ, Lee ET, Contaldo F et al (2013) Relative fat-free mass deficiency and left ventricular adaptation to obesity: the strong heart study. Int J Cardiol 168(2):729–733. doi:10.1016/j.ijcard.2012.09.055
Grimby G (1995) Muscle performance and structure in the elderly as studied cross-sectionally and longitudinally. J Gerontol A Biol Sci Med Sci 50(Spec No):17–22
Blough ER, Linderman JK (2000) Lack of skeletal muscle hypertrophy in very aged male Fischer 344 x Brown Norway rats. J Appl Physiol (Bethesda, Md: 1985) 88(4):1265–1270
Holloszy JO, Chen M, Cartee GD, Young JC (1991) Skeletal muscle atrophy in old rats: differential changes in the three fiber types. Mech Ageing Dev 60(2):199–213
Porter MM, Vandervoort AA, Lexell J (1995) Aging of human muscle: structure, function and adaptability. Scand J Med Sci Sports 5(3):129–142
Lillioja S, Young AA, Culter CL, Ivy JL, Abbott WG, Zawadzki JK et al (1987) Skeletal muscle capillary density and fiber type are possible determinants of in vivo insulin resistance in man. J Clin Invest 80(2):415–424. doi:10.1172/jci113088
Ohlendieck K (2011) Skeletal muscle proteomics: current approaches, technical challenges and emerging techniques. Skelet Muscle 1(1):6. doi:10.1186/2044-5040-1-6
Pedersen BK (2010) Muscle-to-fat interaction: a two-way street? J Physiol 588(Pt 1):21. doi:10.1113/jphysiol.2009.184747
Briaud I, Lingohr MK, Dickson LM, Wrede CE, Rhodes CJ (2003) Differential activation mechanisms of Erk-1/2 and p70(S6K) by glucose in pancreatic beta-cells. Diabetes 52(4):974–983
Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, Pedersen BK (2005) Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 54(10):2939–2945
Mikkelsen HB (2010) Interstitial cells of Cajal, macrophages and mast cells in the gut musculature: morphology, distribution, spatial and possible functional interactions. J Cell Mol Med 14(4):818–832. doi:10.1111/j.1582-4934.2010.01025.x
Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A et al (1997) Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity 7(5):715–725
Benito M (2011) Tissue specificity on insulin action and resistance: past to recent mechanisms. Acta Physiol 201(3):297–312. doi:10.1111/j.1748-1716.2010.02201.x
Yu H, Jove R (2004) The STATs of cancer – new molecular targets come of age. Nat Rev Cancer 4(2):97–105. doi:10.1038/nrc1275
Boulton TG, Zhong Z, Wen Z, Darnell JE Jr, Stahl N, Yancopoulos GD (1995) STAT3 activation by cytokines utilizing gp130 and related transducers involves a secondary modification requiring an H7-sensitive kinase. Proc Natl Acad Sci U S A 92(15):6915–6919
Penner G, Gang G, Sun X, Wray C, Hasselgren PO (2002) C/EBP DNA-binding activity is upregulated by a glucocorticoid-dependent mechanism in septic muscle. Am J Physiol Regul Integr Comp Physiol 282(2):R439–R444. doi:10.1152/ajpregu.00512.2001
Toledo M, Busquets S, Ametller E, Lopez-Soriano FJ, Argiles JM (2011) Sirtuin 1 in skeletal muscle of cachectic tumour-bearing rats: a role in impaired regeneration? J Cachex Sarcopenia Muscle 2(1):57–62. doi:10.1007/s13539-011-0018-6
Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G (2001) Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab 280(5):E745–E751
Marzetti E, Carter CS, Wohlgemuth SE, Lees HA, Giovannini S, Anderson B et al (2009) Changes in IL-15 expression and death-receptor apoptotic signaling in rat gastrocnemius muscle with aging and life-long calorie restriction. Mech Ageing Dev 130(4):272–280
Pedersen BK, Steensberg A, Keller P, Keller C, Fischer C, Hiscock N et al (2003) Muscle-derived interleukin-6: lipolytic, anti-inflammatory and immune regulatory effects. Pflugers Arch: Eur J Physiol 446(1):9–16. doi:10.1007/s00424-002-0981-z
Rui L, Yuan M, Frantz D, Shoelson S, White MF (2002) SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem 277(44):42394–42398. doi:10.1074/jbc.C200444200
Huang L, Gong L, Jiang X, Xing D (2014) Photoactivation of GLUT4 translocation promotes glucose uptake via PI3-K/Akt2 signaling in 3T3-L1 adipocytes. J Innov Opt Health Sci 7(3):1–10. doi: 10.1142/s1793545813500673
Hajduch E, Alessi DR, Hemmings BA, Hundal HS (1998) Constitutive activation of protein kinase B alpha by membrane targeting promotes glucose and system A amino acid transport, protein synthesis, and inactivation of glycogen synthase kinase 3 in L6 muscle cells. Diabetes 47(7):1006–1013
Andreozzi F, Procopio C, Greco A, Mannino GC, Miele C, Raciti GA et al (2011) Increased levels of the Akt-specific phosphatase PH domain leucine-rich repeat protein phosphatase (PHLPP)-1 in obese participants are associated with insulin resistance. Diabetologia 54(7):1879–1887. doi:10.1007/s00125-011-2116-6
Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4(9):648–657. doi:10.1038/ncb839
Nader GA (2007) Muscle growth learns new tricks from an old dog. Nat Med 13(9):1016–1018. doi:10.1038/nm0907-1016
Sandri M (2008) Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 23:160–170. doi:10.1152/physiol.00041.2007
van der Velden JL, Langen RC, Kelders MC, Willems J, Wouters EF, Janssen-Heininger YM et al (2007) Myogenic differentiation during regrowth of atrophied skeletal muscle is associated with inactivation of GSK-3beta. Am J Physiol Cell Physiol 292(5):C1636–C1644. doi:10.1152/ajpcell.00504.2006
Welch AA (2014) The 5th international symposium of the nutrition society nutritional influences on age-related skeletal muscle loss. Proc Nutr Soc 73(1):16–33. doi:10.1017/s0029665113003698
Furuyama T, Kitayama K, Yamashita H, Mori N (2003) Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J 375(Pt 2):365–371. doi:10.1042/bj20030022
Kamei Y, Miura S, Suzuki M, Kai Y, Mizukami J, Taniguchi T et al (2004) Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J Biol Chem 279(39):41114–41123. doi:10.1074/jbc.M400674200
Irwin ML, Yasui Y, Ulrich CM, Bowen D, Rudolph RE, Schwartz RS et al (2003) Effect of exercise on total and intra-abdominal body fat in postmenopausal women: a randomized controlled trial. JAMA 289(3):323–330
McLoughlin TJ, Smith SM, DeLong AD, Wang H, Unterman TG, Esser KA (2009) FoxO1 induces apoptosis in skeletal myotubes in a DNA-binding-dependent manner. Am J Physiol Cell Physiol 297(3):C548–C555. doi:10.1152/ajpcell.00502.2008
Cleasby ME, Jarmin S, Eilers W, Elashry M, Andersen DK, Dickson G et al (2014) Local overexpression of the myostatin propeptide increases glucose transporter expression and enhances skeletal muscle glucose disposal. Am J Physiol-Endocrinol Metab 306(7):E814–E823. doi:10.1152/ajpendo.00586.2013
Henagan TM, Lenard NR, Gettys TW, Stewart LK (2014) Dietary quercetin supplementation in mice increases skeletal muscle PGC1 alpha expression, improves mitochondrial function and attenuates insulin resistance in a time-specific manner. Plos One 9(2):1–11. doi: 10.1371/journal.pone.0089365
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J et al (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34(3):267–273. doi:10.1038/ng1180
Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S et al (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A 100(14):8466–8471. doi:10.1073/pnas.1032913100
Crunkhorn S, Dearie F, Mantzoros C, Gami H, da Silva WS, Espinoza D et al (2007) Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J Biol Chem 282(21):15439–15450. doi:10.1074/jbc.M611214200
Sparks LM, Xie H, Koza RA, Mynatt R, Hulver MW, Bray GA et al (2005) A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes 54(7):1926–1933
Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O et al (2002) Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418(6899):797–801. doi:10.1038/nature00904
Ruas JL, White JP, Rao RR, Kleiner S, Brannan KT, Harrison BC et al (2012) A PGC-1alpha isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151(6):1319–1331. doi:10.1016/j.cell.2012.10.050
Steinberg GR, Kemp BE (2009) AMPK in health and disease. Physiol Rev 89(3):1025–1078. doi:10.1152/physrev.00011.2008
Ohanna M, Sobering AK, Lapointe T, Lorenzo L, Praud C, Petroulakis E et al (2005) Atrophy of S6K1(-/-) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat Cell Biol 7(3):286–294. doi:10.1038/ncb1231
Pehmoller C, Treebak JT, Birk JB, Chen S, Mackintosh C, Hardie DG et al (2009) Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am J Physiol Endocrinol Metab 297(3):E665–E675. doi:10.1152/ajpendo.00115.2009
Sanchez AM, Csibi A, Raibon A, Cornille K, Gay S, Bernardi H et al (2012) AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J Cell Biochem 113(2):695–710. doi:10.1002/jcb.23399
Tong JF, Yan X, Zhu MJ, Du M (2009) AMP-activated protein kinase enhances the expression of muscle-specific ubiquitin ligases despite its activation of IGF-1/Akt signaling in C2C12 myotubes. J Cell Biochem 108(2):458–468. doi:10.1002/jcb.22272
Nakashima K, Yakabe Y (2007) AMPK activation stimulates myofibrillar protein degradation and expression of atrophy-related ubiquitin ligases by increasing FOXO transcription factors in C2C12 myotubes. Biosci Biotechnol Biochem 71(7):1650–1656
Birkenfeld AL, Lee HY, Guebre-Egziabher F, Alves TC, Jurczak MJ, Jornayvaz FR et al (2011) Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab 14(2):184–195. doi:10.1016/j.cmet.2011.06.009
Jager S, Handschin C, St-Pierre J, Spiegelman BM (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A 104(29):12017–12022. doi:10.1073/pnas.0705070104
Whaley-Connell A, McCullough PA, Sowers JR (2011) The role of oxidative stress in the metabolic syndrome. Rev Cardiovasc Med 12(1):21–29. doi:10.3909/ricm0555
Rooyackers OE, Adey DB, Ades PA, Nair KS (1996) Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci U S A 93(26):15364–15369
de Cavanagh EMV, Inserra F, Ferder L (2011) Angiotensin II blockade: a strategy to slow ageing by protecting mitochondria? Cardiovasc Res 89(1):31–40. doi:10.1093/cvr/cvq285
Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG et al (2004) IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119(2):285–298. doi:10.1016/j.cell.2004.09.027
Nicolson GL, Ash ME (2014) Lipid replacement therapy: a natural medicine approach to replacing damaged lipids in cellular membranes and organelles and restoring function. Biochim Biophys Acta Biomemb 1838(6):1657–1679. doi:10.1016/j.bbamem.2013.11.010
Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL et al (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause charcot-marie-tooth neuropathy type 2A. Nat Genet 36(5):449–451. doi:10.1038/ng1341
Thorpe SR, Baynes JW (2003) Maillard reaction products in tissue proteins: new products and new perspectives. Amino Acids 25(3–4):275–281. doi:10.1007/s00726-003-0017-9
Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS et al (1994) Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem 269(13):9889–9897
Teshima Y, Takahashi N, Nishio S, Saito S, Kondo H, Fukui A et al (2014) Production of reactive oxygen species in the diabetic heart – roles of mitochondria and NADPH oxidase. Circ J 78(2):300–306. doi:10.1253/circj.CJ-13-1187
Van Puyvelde K, Mets T, Njemini R, Beyer I, Bautmans I (2014) Effect of advanced glycation end product intake on inflammation and aging: a systematic review. Nutr Rev 72(10):638–650. doi:10.1111/nure.12141
Snow LM, Thompson LV (2009) Influence of insulin and muscle fiber type in nepsilon-(carboxymethyl)-lysine accumulation in soleus muscle of rats with streptozotocin-induced diabetes mellitus. Pathobiology J Immunopathol Mol Cell Biol 76(5):227–234. doi:10.1159/000228898
Alves M, Calegari VC, Cunha DA, Saad MJ, Velloso LA, Rocha EM (2005) Increased expression of advanced glycation end-products and their receptor, and activation of nuclear factor kappa-B in lacrimal glands of diabetic rats. Diabetologia 48(12):2675–2681. doi:10.1007/s00125-005-0010-9
Thornalley PJ (2005) Dicarbonyl intermediates in the maillard reaction. Ann N Y Acad Sci 1043:111–117. doi:10.1196/annals.1333.014
Stadtman ER (2006) Protein oxidation and aging. Free Radic Res 40(12):1250–1258. doi:10.1080/10715760600918142
Dhar A, Desai KM, Wu L (2010) Alagebrium attenuates acute methylglyoxal-induced glucose intolerance in Sprague-Dawley rats. Br J Pharmacol 159(1):166–175. doi:10.1111/j.1476-5381.2009.00469.x
Falone S, D’Alessandro A, Mirabilio A, Petruccelli G, Cacchio M, Di Ilio C et al (2012) Long term running biphasically improves methylglyoxal-related metabolism, redox homeostasis and neurotrophic support within adult mouse brain cortex. PLoS One 7(2):e31401. doi:10.1371/journal.pone.0031401
Tanaka K, Kanazawa I, Yamaguchi T, Yano S, Kaji H, Sugimoto T (2014) Active vitamin D possesses beneficial effects on the interaction between muscle and bone. Biochem Biophys Res Commun 450(1):482–487. doi:10.1016/j.bbrc.2014.05.145
Brownlee M (1995) The pathological implications of protein glycation. Clin Invest Med Clin Exp 18(4):275–281
Andersen H (2012) Motor dysfunction in diabetes. Diabetes Metab Res Rev 28:89–92. doi:10.1002/dmrr.2257
El-Mesallamy HO, Hamdy NM, Ezzat OA, Reda AM (2011) Levels of soluble advanced glycation end product-receptors and other soluble serum markers as indicators of diabetic neuropathy in the foot. J Invest Med 59(8):1233–1238. doi:10.231/JIM.0b013e318231db64
Kim HK, Suzuki T, Saito K, Yoshida H, Kobayashi H, Kato H et al (2012) Effects of exercise and amino acid supplementation on body composition and physical function in community-dwelling elderly Japanese sarcopenic women: a randomized controlled trial. J Am Geriatr Soc 60(1):16–23. doi:10.1111/j.1532-5415.2011.03776.x
Castaneda C, Layne JE, Munoz-Orians L, Gordon PL, Walsmith J, Foldvari M et al (2002) A randomized controlled trial of resistance exercise training to improve glycemic control in older adults with type 2 diabetes. Diabetes Care 25(12):2335–2341
Lessard SJ, Chen ZP, Watt MJ, Hashem M, Reid JJ, Febbraio MA et al (2006) Chronic rosiglitazone treatment restores AMPKalpha2 activity in insulin-resistant rat skeletal muscle. Am J Physiol Endocrinol Metab 290(2):E251–E257. doi:10.1152/ajpendo.00096.2005
Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V et al (2005) Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol 70(2):177–188. doi:10.1016/j.bcp.2005.03.033
Miyazaki Y, He H, Mandarino LJ, DeFronzo RA (2003) Rosiglitazone improves downstream insulin receptor signaling in type 2 diabetic patients. Diabetes 52(8):1943–1950
Bajaj M, Baig R, Suraamornkul S, Hardies LJ, Coletta DK, Cline GW et al (2010) Effects of pioglitazone on intramyocellular fat metabolism in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab 95(4):1916–1923. doi:10.1210/jc.2009-0911
Colca JR, Feinstein DL (2012) Altering mitochondrial dysfunction as an approach to treating Alzheimer’s disease. Adv Pharmacol 64:155–176. doi:10.1016/b978-0-12-394816-8.00005-2, San Diego, Calif
Marsh AP, Shea MK, Vance Locke RM, Miller ME, Isom S, Miller GD et al (2013) Resistance training and pioglitazone lead to improvements in muscle power during voluntary weight loss in older adults. J Gerontol A Biol Sci Med Sci 68(7):828–836. doi:10.1093/gerona/gls258
Owen MR, Doran E, Halestrap AP (2000) Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348(Pt 3):607–614
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Sugimoto, K., Wang, CC., Rakugi, H. (2016). Sarcopenia in Diabetes Mellitus. In: Inaba, M. (eds) Musculoskeletal Disease Associated with Diabetes Mellitus. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55720-3_16
Download citation
DOI: https://doi.org/10.1007/978-4-431-55720-3_16
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55719-7
Online ISBN: 978-4-431-55720-3
eBook Packages: MedicineMedicine (R0)