
Abstract
Inwardly rectifying potassium (Kir) channels were first identified based on their unique functional property of preferential conductance of potassium ions into the cell. The property of inward rectification differs significantly from their more widely studied Kv (voltage-gated potassium) channel relatives, which exhibit outward rectification resulting from voltage-dependent opening and closing of the channel pore. Rather, inward rectification arises from asymmetrical voltage-dependent blockade of these channels by endogenous intracellular polyamines. This distinct role of polyamines enables the physiological function of Kir channels to maintain a substantial potassium conductance when cells are at rest, but to shut down their conductance when faced with depolarizing stimuli to allow excitation events (e.g., action potentials) to take place. Functional studies of cloned Kir channels, and recent crystallographic insights, have revealed the importance of numerous side chains that line the channel and interact with polyamines as they move toward a stable binding site. The displacement of permeating K+ ions in the channel pore, coupled to polyamine migration through the pore, underlies the very steeply voltage-dependent blockade.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Andersen’s syndrome
- Electrophysiology
- Inward rectifier
- Ion channel
- Long QT
- Polyamine
- Potassium channel
- Short QT
- Spermine
1 Introduction
Inwardly rectifying potassium (Kir) currents were first characterized decades ago in skeletal and cardiac muscle fibers. Also initially referred to as ‘anomalous’ rectifiers, these currents were named for their unique characteristic of preferentially conducting inward currents, while outward currents were inhibited (Hutter and Noble 1960; Noble 1962). This trait contrasted with the outward rectification and intrinsic voltage dependence of the classic voltage-dependent K+ (Kv) currents in excitable cells first described by Hodgkin and Huxley (Hodgkin and Huxley 1952). The first inwardly rectifying potassium (Kir) channel genes were cloned in the early 1990s, with reports of cloning IRK1 (now referred to as Kir2.1 or KCNJ2) (Kubo et al. 1993a), GIRK1 (now Kir3.1 or KCNJ3) (Kubo et al. 1993b), and ROMK1 (now Kir1.1, or KCNJ1) (Ho et al. 1993). These major steps forward allowed for heterologous expression of these ion channel genes, comparison of their functional properties, and identification of the major sequence determinants of Kir channel function.
Although many chapters in this textbook are concerned with important roles of polyamines in cell growth, metabolism, and proliferation, it turns out that polyamines also play a major role in cellular electrical behavior by controlling inward rectification of Kir channels. The identification of polyamine block as the underlying physiological mechanism of inward rectification was hinted at by a few early publications describing that rectification was not an ‘intrinsic’ property of the inward rectifying conductance (Vandenberg 1987; Matsuda et al. 1987). That is, if channels were removed from the cellular environment (by excised patch-clamp recordings, or highly effective dialysis of intracellular contents), properties of inward rectification could be weakened or abolished. This was a particularly unique mechanism of ion channel regulation, because most voltage-dependent regulatory mechanisms were presumed to be caused by conformational changes of ion channel proteins themselves, rather than an extrinsic factor. Subsequent screening of active components in specific cell lysate fractions led to the identification of polyamines as the major determinant of steeply voltage-dependent inward rectification of cloned IRK1 (Kir2.1) channels (Lopatin et al. 1994). All polyamines are able to block Kir channels, although spermine and spermidine are typically the most potent, whereas shorter endogenous polyamines such as putrescine are weaker blockers. The properties of polyamine block match several important features of currents described in primary cell culture systems such as cardiomyocytes, notably the steep effective valence of blockade and the strong coupling of the voltage dependence of rectification with the reversal potential of K+ ions (Lopatin and Nichols 1996). For the remainder of this chapter, inward rectification is primarily discussed in the context of Kir2.1 channels, which are the most widely studied model of polyamine block, and also exhibit the most potent and steeply voltage-dependent polyamine block (Nichols and Lopatin 1997).
2 Characteristics of Polyamine Block
2.1 Steepness of Voltage-Dependent Block
To an electrophysiologist, one of the most striking traits of polyamine block is the very high ‘effective valence’ of the blocking process. Voltage-dependent blockers such as polyamines are unique because blocker affinity for the ion channel target is not fixed, but rather affinity varies with changes in transmembrane voltage. A common way to assess this feature experimentally is to record ionic currents in the absence and presence of a known blocker concentration, at different membrane voltages (see Fig. 18.1a). In the absence of polyamines or divalent ions such as Mg2+, Kir channels behave as ohmic resistors over a physiological range of membrane voltage (Guo and Lu 2002). However, in the presence of polyamines, current inhibition becomes very significant at depolarized voltages. The percentage of current inhibition can be calculated at each voltage, and plotted graphically to illustrate the voltage dependence of the blockade (Fig. 18.1b). In the simplest scenario of a two-state equilibrium (open ↔ blocked), voltage-dependent blockade (i.e., the fraction of control current that remains in the presence of the blocker) is sigmoidal and can be described by a Boltzmann function such as

Voltage-dependent block of Kir2.1 channels by spermine. a Currents recorded using the inside-out patch-clamp method, in symmetrical 150 mM K+ solutions. Membrane patches containing Kir2.1 channels were stepped between −100 and +30 mV in 10-mV steps, in the presence and absence of 100 μM spermine. Note the large outward currents (above the dashed ‘zero current’ line) in control conditions, but strong inhibition of outward currents by spermine. b Relative conductance is calculated by dividing the current magnitude in spermine vs control at each voltage. At negative voltages, spermine effects are very weak, whereas at positive voltages currents are strongly inhibited
In this equation, Kd (0 mV) reflects blocker affinity for the channel in the absence of any membrane potential, and zδ describes how quickly blocker affinity changes with membrane voltage: larger zδ values reflect a steeper change of blocker affinity as membrane voltage is changed. Typically, zδ is interpreted as being the product of the valence of the blocker (i.e., +4 for spermine), and the fraction of the transmembrane field traversed by the blocker en route to its final binding site (i.e., 0 if the blocker does not enter the transmembrane field, or 1 if the blocker completely traverses the transmembrane field, so 0 < δ < 1). In reality, the voltage dependence of polyamine block of Kir2.1 channels is not quite this simple, and generally exhibits multiple components (a shallow and a steep component; Fig. 18.1b), corresponding to distinct low- and high-affinity binding sites (Xie et al. 2002).
Most notably, the voltage dependence of polyamine blockade is very steep relative to other commonly used ion channel blockers, meaning that in a physiological setting where polyamines are present, strongly rectifying Kir channels switch from being conductive to strongly inhibited over a very narrow voltage range. Moreover, a notable feature of polyamine block of Kir channels is that the effective valence of spermine block is higher than the charge of the blocker. Specifically, the effective valence of spermine and spermidine block of Kir2.1 channels is typically reported to be nearly 5 (Guo and Lu 2003). Because the charge of spermine can reach a maximum of +4, this property cannot be rationalized solely on the basis of migration of the charged blocker across the transmembrane voltage difference (Woodhull 1973).
2.2 Coupling of Polyamine Blockers and Permeating Ions
The second classical feature of inwardly rectifying potassium channels is that the degree of rectification depends significantly on the reversal potential of K+, and particularly the extracellular K+ concentration (Hagiwara et al. 1976). Specifically, as the extracellular K+ concentration is increased with a constant intracellular K+ concentration (thereby shifting the K+ reversal potential to more positive voltages), the degree of rectification at a given voltage becomes less pronounced. In the simple descriptive equation for voltage-dependent block shown above, this phenomenon would be described as a K+-dependent change in Kd (0 mV). This permeant ion dependence of affinity is thought to reflect interactions between ions and blockers in the pore. An important but not always appreciated aspect of this process is the strong asymmetrical dependence on K+ concentration. Extracellular K+ has a far more dramatic impact on polyamine block than intracellular K+, although there is not a well-accepted mechanistic explanation for this asymmetry (Lopatin and Nichols 1996; Kurata et al. 2010).
3 Inwardly Rectifying Potassium Channel Ontogeny and Structures
3.1 Kir Channel Genes
Kir channels are categorized into seven families (Kir1–7) with a total of at least 15 different genes/subunits (in humans) (Kubo et al. 2005; Hibino et al. 2010). A functional Kir channel comprises four pore-forming subunits and can be homo- or heterotetrameric, with heteromerization permitted among members of the same family (e.g., Kir2.1 with Kir2.2, Kir3.1 with Kir3.4) (Hibino et al. 2010), and in some cases with obligatory auxiliary subunits as with Kir6 channel assembly with sulfonylurea receptors (Inagaki et al. 1995). A peculiarity of the Kir channel family is that although they are collectively referred to as inward rectifiers, inward rectification is not especially prominent in many of these channel types. Kir2 family channels exhibit strong rectification and are the prototypical ion channels for the study of polyamine block. Kir3 and Kir4 family channels exhibit intermediate rectification properties (i.e., weaker affinity and shallower voltage dependence relate to Kir2 channels), whereas Kir1 and Kir6 family channels are extremely insensitive to polyamine block. As is discussed later, these differences in polyamine sensitivity can be attributed to a small subset of amino acids that differ between these Kir subfamilies (Hibino et al. 2010).
3.2 Structures and Domain Architecture of Kir Channels
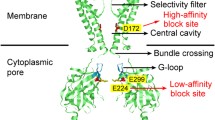
Recent atomic resolution structures of Kir channels have significantly enhanced our understanding of Kir channel function and provide an important framework to understanding their interactions with polyamines (Whorton and MacKinnon 2011; Hansen et al. 2011) (Fig. 18.2). The Kir channel transmembrane pore consists of two transmembrane α-helices from each subunit, linked via a re-entrant segment that forms a selectivity filter similar to virtually all K+-selective channels. Importantly, these channels are not intrinsically sensitive to voltage because they lack the canonical voltage-sensing domain that endows other channel types with voltage sensitivity (Yellen 2002). The transmembrane helices of each subunit comprise the outer helix (TM1) and the inner helix (TM2) that lines the pore. Most crystal structures of Kir channels have been captured in the closed state, and no fully open conformation of a Kir channel has yet been reported, although some ‘partially open’ conformations of Kir2.2 and Kir3.2 have been described (Hansen et al. 2011; Whorton and MacKinnon 2011), along with a mutant KirBac3.1 channel designed to prop open the channel pore (Bavro et al. 2012). In all cases, crystallographic evidence indicates that Kir channel opening involves widening of an aperture in the bundle-crossing region of the channel, and this is consistent with several functional studies using accessibility of MTS reagents or polyamine blockers to study gating conformations of these channels (Phillips and Nichols 2003; Phillips et al. 2003). In addition to the canonical K+ channel pore-forming domain, Kir channels comprise a large cytoplasmic domain. The transmembrane and cytoplasmic domains form a stable interface generating a single long ion-conducting pore. The cytoplasmic domain is composed of both the N- and C-termini of the protein and forms binding sites for numerous permeating ions, intracellular ligands, and polyamine blockers.

Atomic resolution structure of Kir2.2. Coordinates used were reported by Hansen et al., (2011). Two subunits have been removed to more clearly depict the central pore axis that forms the permeation pathway. The rectification controller residue, and cytoplasmic residues important for polyamine block, are highlighted as spheres
The additional K+ ions housed in the extended Kir channel pore likely play a role in generating the steep voltage dependence of the block already discussed (Xu et al. 2009), because polyamine movement through the pore requires the displacement of ions that occupy binding sites ahead of the blocker (Fig. 18.3). That is, as polyamines migrate toward their stable binding site, ions lying ahead of the blocker in the pore are forced to move through the transmembrane field, and thereby affect the voltage dependence of the process. An understanding of the details of ion-binding sites and the mechanisms that couple blocker and ion movement is important to describe the steep voltage dependence of these channel types and the observed dependence on K+ concentration gradients. Although a firm description of interactions between blockers and permeant ions remains elusive, crystal structures have also provided valuable insights into mapping specific ion-binding sites along the Kir channel pore. The selectivity filter of Kir channels closely resembles other K+-selective channels and comprises four binding sites for K+ ions formed by backbone carbonyl atoms. In addition, a commonly observed K+ ion-binding site (the ‘cavity site’) is apparent in the inner cavity region of Kir channels. Additional binding sites that are unique to Kir channels are formed by the intracellular cytoplasmic domain, with up to five ions visible in some crystal structures (Pegan et al. 2005; Nishida et al. 2007; Xu et al. 2009). Overall, the large number of ion-binding sites that have been identified in Kir channel structures is consistent with the notion that a significant component of the voltage dependence of polyamine block arises from the displacement of ions ahead of the blocker, rather than movement of the charged blocker itself through the transmembrane field (Pearson and Nichols 1998; Guo and Lu 2003).

Coupled movement of blockers and ions in Kir channel pores. Each image is a schematic of a Kir channel, depicting the progressive movement of spermine as it interacts with the cytoplasmic domain of the channel and migrates toward its stable binding site. As the blocker moves toward the deep binding site, multiple ions are displaced through the transmembrane field, generating the steep voltage dependence associated with polyamine block
3.3 Important Residues for Polyamine Binding
Kir channel proteins exhibit strong sequence conservation across all subfamilies but markedly different rectification properties and sensitivities to polyamine block (Hibino et al. 2010). For example, channels of the Kir2 subfamily exhibit a potent spermine block that is steeply voltage dependent, whereas Kir1 or Kir6 subfamily channels exhibit very weak inhibition by spermine or other polyamines. The sequence similarity between channels with significant functional differences was highly amenable to chimeric approaches, and rational site-directed mutagenesis, to identify key residues that control polyamine binding (Taglialatela et al. 1994; Wible et al. 1994; Kubo and Murata 2001; Lu 2004). These experiments have demonstrated that strong rectification in Kir2 channels can be attributed to rings of pore-lining acidic residues (generally considered to carry negative charges) in both the cytoplasmic and transmembrane domains of the channel.
3.3.1 Inner Cavity Residues That Control Polyamine Block
The most important determinant of polyamine sensitivity of Kir channels lies within the pore-lining TM2 helix, at Kir2.1 residue D172 (Wible et al. 1994; Lopatin et al. 1994) (Fig. 18.2). This residue is frequently referred to as the ‘rectification controller.’ Neutralization of this residue in polyamine-sensitive channels such as Kir2.1 significantly weakens polyamine block. Consistent with this finding, introduction of acidic residues into polyamine-insensitive channels such as Kir1.1 or Kir6.2 endows strong sensitivity to polyamine block, with steep voltage dependence (Lu and MacKinnon 1994; Shyng et al. 1997). It is noteworthy that there is not a strict positional requirement for a TM2 negative charge to introduce polyamine sensitivity: substitution of a negative charge at any pore-lining position in the transmembrane domain of Kir6.2 has been shown to introduce strong polyamine sensitivity (Kurata et al. 2004). However, among naturally occurring Kir channels, negatively charged residues are virtually never present at any position except the rectification controller D172 equivalent.
3.3.2 Cytoplasmic Domain Residues That Control Polyamine Block
There are also numerous pore-lining acidic residues in the cytoplasmic domain (Kir2.1 residues E224, D255, D259, E299), along with a pore-lining aromatic residue (Kir2.1 residue F254) that primarily control low-affinity interactions with polyamines and also alter inward rectification properties when mutated (Fig. 18.2). Broadly speaking, mutations at these positions tend to impact the kinetics of polyamine block/unblock, and also the presence of the shallow voltage-dependent component of polyamine block (Shin et al. 2005; Kurata et al. 2007) (see also Fig. 18.1). However, mutations at E224 or E299 also diminish the overall voltage dependence and affinity of polyamine block (Kubo and Murata 2001; Guo and Lu 2003). More specifically, polyamine interactions with residues in the cytoplasmic domain are thought to underlie the early rate-limiting step of polyamine block, which is followed sequentially by polyamine migration into the inner cavity. This second step (entry of polyamines into the inner cavity) is associated with displacement of significantly more charge, and is thus far more steeply voltage dependent than the weak association of polyamines with the cytoplasmic domain (Fig. 18.3) (Shin and Lu 2005; Kurata et al. 2007). However, binding of polyamines in the cytoplasmic domain has been suggested to facilitate blocker entry into the inner cavity by increasing the local polyamine concentration (Lopatin et al. 1995; Kubo and Murata 2001; Xie et al. 2002).
3.4 The Nature of the Terminal Polyamine-Binding Site
Among the most debated aspects of polyamine block of Kir channels has been the details of the ‘stable’ polyamine-binding site in the Kir channel inner cavity. Although the role of the rectification controller residue is not in question, some studies have argued for a polyamine orientation with the leading end of the blocker located in the vicinity of the rectification controller (Guo and Lu 2003; Lu 2004), whereas others have argued for a deeper binding site between the rectification controller and the selectivity filter (Chang et al. 2003; John et al. 2004; Kurata et al. 2006, 2010). Overall, the first hypothesis is motivated primarily by data demonstrating that residues within the cytoplasmic domain may have significant effects on the stability of the ‘deep’ binding equilibrium for polyamines, suggesting that they contribute to the polyamine-binding site in some way. However, a deeper binding site is indicated by data demonstrating that polyamines can be trapped in the inner cavity after the introduction of positively charged adducts (such as MTSEA or MTSET) near the cytoplasmic entrance of the inner cavity. These data suggest that spermine likely binds above the introduced adducts just one α-helical turn away from the rectification controller (toward the cytoplasmic side, at Kir2.1 position I176C, or Kir6.2 position L164C) (Kurata et al. 2010, 2013). Missing from the dialogue on polyamine binding is a crystal structure convincingly demonstrating the orientation of polyamines in the inner cavity deep binding site. Future experiments will surely continue to refine descriptions of polyamine binding.
4 Diseases Related to Polyamine-Sensitive Kir Channels
The Kir2 family are the most sensitive Kir channels to voltage-dependent polyamine block. Their physiological roles are best understood in excitable tissues such as skeletal and cardiac muscle where they maintain resting membrane potential and shut off upon depolarization to allow an action potential. Not surprisingly, loss- and gain-of-function mutations of Kir2 family channels have interesting consequences on cardiac function, and other consequences related to bone and muscle development also emerge as a result of these channel defects. Loss of function mutations in Kir2.1 result in Andersen’s syndrome, an autosomal dominant genetic disorder characterized by cardiac arrhythmias caused by prolongation of the action potential (long QT syndrome, LQT7), periodic muscle paralysis, and abnormal facial and digital bone structures (Plaster et al. 2001; Tristani-Firouzi and Etheridge 2010). Many associated mutations either disrupt channel interactions with PIP2 or impair channel trafficking (Tristani-Firouzi et al. 2002; Lopes et al. 2002).
The functional counterpoint to Kir2.1 loss-of-function is mutations that cause gain-of-function. Remarkably, mutations of Kir2.1 residue D172 that disrupt polyamine binding have been reported to cause a form of short QT syndrome (SQT3) (Priori et al. 2005). This defect results in shortening of the cardiac action potential and is also associated with arrhythmias and cardiac death. The first SQT3 mutation reported is in fact the D172N mutation that has been commonly used to study polyamine block of Kir2.1 channels, corresponding to neutralization of the rectification controller. Given the large number of residues identified as important for polyamine block (see Sect. 18.3.3), it seems likely that other Kir2 channel mutations will emerge in studies of SQT3; however, at present no additional mutations have been reported.
5 Summary
Polyamine block of Kir channels is an essential physiological mechanism of ion channel regulation that is distinct from the more commonly studied roles of polyamines in cell growth and proliferation. Intracellular polyamines enter the Kir channel pore at depolarized voltages, causing preferential blockade of outward K+ currents and allowing cellular electrical excitation to proceed. Recently identified mutations of the Kir2.1 channel cause disruption of polyamine block and are linked to cardiac arrhythmias.
References
Bavro VN, De ZR, Schmidt MR, Muniz JR, Zubcevic L, Sansom MS, Venien-Bryan C, Tucker SJ (2012) Structure of a KirBac potassium channel with an open bundle crossing indicates a mechanism of channel gating. Nat Struct Mol Biol 19:158–163
Chang HK, Yeh SH, Shieh RC (2003) The effects of spermine on the accessibility of residues in the M2 segment of Kir2.1 channels expressed in Xenopus oocytes. J Physiol 553:101–112
Guo D, Lu Z (2002) IRK1 inward rectifier K(+) channels exhibit no intrinsic rectification. J Gen Physiol 120:539–551
Guo D, Lu Z (2003) Interaction mechanisms between polyamines and IRK1 inward rectifier K+ channels. J Gen Physiol 122:485–500
Hagiwara S, Miyazaki S, Rosenthal NP (1976) Potassium current and the effect of cesium on this current during anomalous rectification of the egg cell membrane of a starfish. J Gen Physiol 67:621–638
Hansen SB, Tao X, MacKinnon R (2011) Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature (Lond) 477:495–498
Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90:291–366
Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC (1993) Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature (Lond) 362:31–38
Hodgkin AL, Huxley AF (1952) The components of membrane conductance in the giant axon of Loligo. J Physiol 116:473–496
Hutter OF, Noble D (1960) Rectifying properties of heart muscle. Nature (Lond) 188:495
Inagaki N, Gonoi T, Clement JP, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J (1995) Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270:1166–1170
John SA, Xie LH, Weiss JN (2004) Mechanism of inward rectification in Kir channels. J Gen Physiol 123:623–625
Kubo Y, Murata Y (2001) Control of rectification and permeation by two distinct sites after the second transmembrane region in Kir2.1 K+ channel. J Physiol 531:645–660
Kubo Y, Baldwin TJ, Jan YN, Jan LY (1993a) Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature (Lond) 362:127–133
Kubo Y, Reuveny E, Slesinger PA, Jan YN, Jan LY (1993b) Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel. Nature (Lond) 364:802–806
Kubo Y, Adelman JP, Clapham DE, Jan LY, Karschin A, Kurachi Y, Lazdunski M, Nichols CG, Seino S, Vandenberg CA (2005) International Union of Pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol Rev 57:509–526
Kurata HT, Phillips LR, Rose T, Loussouarn G, Herlitze S, Fritzenschaft H, Enkvetchakul D, Nichols CG, Baukrowitz T (2004) Molecular basis of inward rectification: polyamine interaction sites located by combined channel and ligand mutagenesis. J Gen Physiol 124:541–554
Kurata HT, Marton LJ, Nichols CG (2006) The polyamine binding site in inward rectifier K+ channels. J Gen Physiol 127:467–480
Kurata HT, Cheng WW, Arrabit C, Slesinger PA, Nichols CG (2007) The role of the cytoplasmic pore in inward rectification of Kir2.1 channels. J Gen Physiol 130:145–155
Kurata HT, Zhu EA, Nichols CG (2010) Locale and chemistry of spermine binding in the archetypal inward rectifier Kir2.1. J Gen Physiol 135:495–508
Kurata HT, Akrouh A, Li JB, Marton LJ, Nichols CG (2013) Scanning the topography of polyamine blocker binding in an inwardly rectifying potassium channel. J Biol Chem 288:6591–6601
Lopatin AN, Nichols CG (1996) [K+] dependence of polyamine-induced rectification in inward rectifier potassium channels (IRK1, Kir2.1). J Gen Physiol 108:105–113
Lopatin AN, Makhina EN, Nichols CG (1994) Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature (Lond) 372:366–369
Lopatin AN, Makhina EN, Nichols CG (1995) The mechanism of inward rectification of potassium channels: “long-pore plugging” by cytoplasmic polyamines. J Gen Physiol 106:923–955
Lopes CM, Zhang H, Rohacs T, Jin T, Yang J, Logothetis DE (2002) Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 34:933–944
Lu Z (2004) Mechanism of rectification in inward-rectifier K+ channels. Annu Rev Physiol 66:103–129
Lu Z, MacKinnon R (1994) Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature (Lond) 371:243–246
Matsuda H, Saigusa A, Irisawa H (1987) Ohmic conductance through the inwardly rectifying K channel and blocking by internal Mg2+. Nature (Lond) 325:156–159
Nichols CG, Lopatin AN (1997) Inward rectifier potassium channels. Annu Rev Physiol 59:171–191
Nishida M, Cadene M, Chait BT, MacKinnon R (2007) Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J 26:4005–4015
Noble D (1962) A modification of the Hodgkin–Huxley equations applicable to Purkinje fibre action and pace-maker potentials. J Physiol 160:317–352
Pearson WL, Nichols CG (1998) Block of the Kir2.1 channel pore by alkylamine analogues of endogenous polyamines. J Gen Physiol 112:351–363
Pegan S, Arrabit C, Zhou W, Kwiatkowski W, Collins A, Slesinger PA, Choe S (2005) Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci 8:279–287
Phillips LR, Nichols CG (2003) Ligand-induced closure of inward rectifier Kir6.2 channels traps spermine in the pore. J Gen Physiol 122:795–804
Phillips LR, Enkvetchakul D, Nichols CG (2003) Gating dependence of inner pore access in inward rectifier K(+) channels. Neuron 37:953–962
Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL Jr, Fish FA, Hahn A, Nitu A, Ozdemir C, Serdaroglu P, Subramony SH, Wolfe G, Fu YH, Ptacek LJ (2001) Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell 105:511–519
Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, Napolitano C, Anumonwo J, di Barletta MR, Gudapakkam S, Bosi G, Stramba-Badiale M, Jalife J (2005) A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 96:800–807
Shin HG, Lu Z (2005) Mechanism of the voltage sensitivity of IRK1 inward-rectifier K+ channel block by the polyamine spermine. J Gen Physiol 125:413–426
Shin HG, Xu Y, Lu Z (2005) Evidence for sequential ion-binding loci along the inner pore of the IRK1 inward-rectifier K+ channel. J Gen Physiol 126:123–135
Shyng S, Ferrigni T, Nichols CG (1997) Control of rectification and gating of cloned KATP channels by the Kir6.2 subunit. J Gen Physiol 110:141–153
Taglialatela M, Wible BA, Caporaso R, Brown AM (1994) Specification of pore properties by the carboxyl terminus of inwardly rectifying K+ channels. Science 264:844–847
Tristani-Firouzi M, Etheridge SP (2010) Kir 2.1 channelopathies: the Andersen–Tawil syndrome. Pflugers Arch 460:289–294
Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, Bendahhou S, Kwiecinski H, Fidzianska A, Plaster N, Fu YH, Ptacek LJ, Tawil R (2002) Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest 110:381–388
Vandenberg CA (1987) Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc Natl Acad Sci USA 84:2560–2564
Whorton MR, MacKinnon R (2011) Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147:199–208
Wible BA, Taglialatela M, Ficker E, Brown AM (1994) Gating of inwardly rectifying K+ channels localized to a single negatively charged residue. Nature (Lond) 371:246–249
Woodhull AM (1973) Ionic blockage of sodium channels in nerve. J Gen Physiol 61:687–708
Xie LH, John SA, Weiss JN (2002) Spermine block of the strong inward rectifier potassium channel Kir2.1: dual roles of surface charge screening and pore block. J Gen Physiol 120:53–66
Xu Y, Shin HG, Szep S, Lu Z (2009) Physical determinants of strong voltage sensitivity of K(+) channel block. Nat Struct Mol Biol 16:1252–1258
Yellen G (2002) The voltage-gated potassium channels and their relatives. Nature (Lond) 419:35–42
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Japan
About this chapter
Cite this chapter
Kurata, H.T. (2015). Polyamine Block of Inwardly Rectifying Potassium (Kir) Channels. In: Kusano, T., Suzuki, H. (eds) Polyamines. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55212-3_18
Download citation
DOI: https://doi.org/10.1007/978-4-431-55212-3_18
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55211-6
Online ISBN: 978-4-431-55212-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)