
Abstract
At least 150 different human proteins are anchored to the plasma membrane via glycosylphosphatidylinositol (GPI). GPI anchor and precursor proteins are separately synthesized in the endoplasmic reticulum, and GPI anchor is attached to the proteins’ carboxyl-terminus as a posttranslational modification by GPI transamidase. Twenty-two PIG (for phosphatidylinositol glycan) genes are involved in the biosynthesis and protein attachment of GPI. After attachment to proteins, both lipid and glycan moieties of GPI are remodeled in the endoplasmic reticulum and Golgi apparatus. Four PGAP (for post-GPI attachment to proteins) genes are involved in the remodeling of GPI. An acquired GPI deficiency, paroxysmal nocturnal hemoglobinuria, is a hematopoietic disease, which is caused by the somatic mutation of PIGA gene in the hematopoietic stem cell.
Recently, many inherited GPI deficiencies caused by germline mutations in the PIG and PGAP genes have been found. They are characterized by intellectual disability often accompanied by epilepsy and hyperphosphatasia. The characteristics of the 26 PIG and PGAP genes and the GPI deficiencies caused by mutations in these genes are discussed.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others

Keywords
- Glycosylphosphatidylinositol
- Somatic mutation
- Germline mutation
- Inherited GPI deficiency
- Epilepsy
- Hyperphosphatasia
- Intellectual disability
Introduction
At least 150 different human proteins are posttranslationally modified by glycosylphosphatidylinositol (GPI) at the carboxyl-terminus. These proteins are termed GPI-anchored proteins (GPI-APs) expressing on the cell surface by being anchored to the outer leaflet of the plasma membrane via the phosphatidylinositol (PI) and share similar characteristics based on this anchor structure. GPI-APs are typical raft-associated proteins and can be released from the cell after cleavage by GPI-cleaving enzymes or GPIases. These characteristics are critical for the functions of individual GPI-APs and for embryogenesis, neurogenesis, fertilization, and the immune system. Complete GPI deficiency causes embryonic death because of total loss of GPI-APs from the cell surface, as GPI-APs include various important proteins, such as hydrolytic enzymes, adhesion molecules, receptors, protease inhibitors, and complement regulatory proteins (Kinoshita et al. 2008).
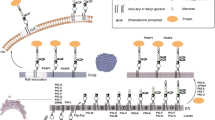
Protein-bound mammalian GPI anchors have a core backbone, which can be modified by side branches (Fig. 1). The structure of the core backbone is EtNP-6Manα1-2Manα1-6Manα1-4GlcNα1-6myoinositol-phospholipid (where EtNP is ethanolamine phosphate, Man is mannose, and GlcN is glucosamine) and is highly conserved among various species. GPI precursors are synthesized in the endoplasmic reticulum (ER) and transferred en bloc to proteins that bear a GPI-attachment signal sequence at the carboxyl-terminus (Fig. 2). After attachment to proteins, both lipid and glycan moieties of GPI are remodeled in the ER and Golgi apparatus. There are at least 22 genes, termed PIG (for phosphatidylinositol glycan) genes, involved in the biosynthesis and protein attachment of GPI, and four genes, termed PGAP (for post-GPI attachment to proteins) genes, are involved in the remodeling of GPI (Kinoshita et al. 2008).

Structure of mammalian GPI-APs. The structure of core backbone is EtNP-6Manα1-2Manα1-6Manα1-4GlcNα1-6myoinositol-phospholipid. The EtNP linked to Man3, the α1-2linked Man, makes an amide bond with the C-terminus

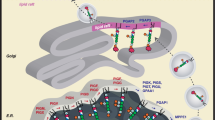
Biosynthesis and remodeling of GPI-AP. GPI precursor is biosynthesized in the ER by at least 11 stepwise reactions and en bloc transferred to the carboxyl-terminus of proteins. After attachment of GPI to proteins in the ER, the inositol-linked acyl chain and Man1-linked EtNP side branch are removed by PGAP1 and PGAP5, respectively. In the Golgi, an unsaturated fatty acid at the sn-2 position is removed by PGAP3, generating lyso-GPI-AP, which is reacylated with a saturated fatty acid by PGAP2. After this fatty acid remodeling, GPI-APs are expressed on the outer leaflet of the plasma membrane (PM)
Several human diseases are caused by the mutations in these genes. Somatic mutation in PIGA, which encodes the first enzyme of GPI biosynthesis, results in paroxysmal nocturnal hemoglobinuria (PNH), an acquired hemolytic disease. Additionally, autosomal or X-linked mutations of various PIG and PGAP genes have been reported to cause autosomal or X-linked recessive inherited GPI deficiency (IGD) , which is characterized by intellectual disability and epilepsy. This review mainly focused on this recently found disease, inherited GPI deficiency.
Biosynthesis and Remodeling of GPI-Anchored Proteins (GPI-APs) and Their Deficiencies
PIG Genes Involved in Biosynthesis of GPI-APs
GPI is synthesized in the ER and is transferred en bloc to proteins by GPI transamidase (Fig. 2). The synthesis of GPI is initiated by the addition of an N-acetylglucosamine (GlcNAc) to free PI on the cytosolic face of the ER. This reaction is catalyzed by the enzyme complex composed of seven components, such as PIG-A (a catalytic subunit), PIG-Q, PIG-C, PIG-H, PIG-P, DPM2, and PIG-Y. Deacetylation of GlcNAc catalyzed by PIG-L results in GlcN-PI. GlcN-PI most likely flips across the ER membrane into the luminal side. The flippase that catalyzes the flip of GPIs has not been identified. An acyl chain (generally palmitate) is transferred from an acyl-CoA to the 2-position of the inositol in GlcN-PI by PIG-W, resulting in GlcN-(acyl)PI. Mature GPI is synthesized in the lumen by the addition of three mannoses from a donor substrate, dolichol-phosphate-mannose, by PIG-M/PIG-X complex, PIG-V, and PIG-B, and by the addition of three EtNPs from phosphatidylethanolamine by PIG-N, PIG-O/PIG-F complex, and PIG-G/PIG-F complex. Finally the assembled GPI anchor is transferred to a protein by a GPI transamidase complex that is composed of PIG-K (a catalytic subunit), PIG-S, PIG-T, PIG-U, and GPAA1. An EtNP attached to Man3 bridges the GPI anchor via an amide bond with the processed carboxyl-terminus of the precursor protein whose GPI-attachment signal peptide is recognized and removed by the GPI transamidase complex. The GPI transamidase forms transient intermediates with the precursor proteins, and their GPI-attachment signal peptides have particular conserved structural features. When GPI anchor is limited because of hypomorphic defect in one of PIG genes, the precursor proteins, which have strong GPI-attachment signal, are preferentially anchored.
GPI Deficiencies Caused by Somatic and Germline Mutations in PIG Genes
Paroxysmal Nocturnal Hemoglobinuria (PNH)
PNH is an acquired hematopoietic disease, which is caused by the somatic mutation of PIGA gene in a hematopoietic stem cell. A major clinical symptom is intravascular hemolysis of the affected erythrocytes attacked by their own complement proteins because of defect in GPI-anchored complement regulatory proteins, such as CD59 and DAF on the cell surface. As PIGA is an only X-linked gene among the PIG genes, it has been the only gene responsible for PNH because one hit is sufficient to inactivate the gene. However, it was reported recently that the somatic mutation of PIGT gene in combination with germline mutation of another allele caused PNH. One big question to be solved in PNH is that what causes the clonal expansion of GPI-negative hematopoietic stem cells. Survival advantage against the cytotoxic cells or benign tumorlike phenotype obtained by additional mutations might cause the clonal expansion, which is not completely proven.
Inherited GPI Deficiencies (IGDs) Caused by Germline Mutation of PIG Genes
In case of germline mutations, only the hypomorphic mutations are compatible with live birth. The first reported case was PIGM deficiency caused by the point mutation in promoter region (Almeida et al. 2006). The homozygous mutation disrupted an Sp1-binding site and caused reduced PIGM mRNA levels and partial reduction of GPI-APs on the surface of granulocytes, B-lymphoblastoid cells, and skin fibroblasts. The affected individuals suffered from seizures and thrombosis in hepatic and/or portal veins. From 2010, using the second-generation sequencer, responsible genes for many familial diseases are determined. Many IGDs are reported, such as PIGA (Johnston et al. 2012; Kato et al. 2014), PIGQ (Martin et al. 2014), PIGL (Ng et al. 2012), PIGW (Chiyonobu et al. 2014), PIGN (Maydan et al. 2011), PIGV (Krawitz et al. 2010), PIGO (Krawitz et al. 2012), and PIGT (Kvarnung et al. 2013) deficiencies. X-linked PIGA deficiency affects only males and frequency should be rather high compared to other IGDs. Among them, PIGW, PIGV, and PIGO deficiencies caused hyperphosphatasia and mental retardation syndrome (HPMRS), also termed Mabry syndrome. Alkaline phosphatase (ALP), a GPI-AP, is processed by the GPI transamidase complex in the ER and secreted without GPI anchoring because of its shortage. On the contrary, when a GPI transamidase component PIGT is defective, ALP is completely degraded leading to hypophosphatasia and bone abnormalities (Kvarnung et al. 2013). The main symptoms of IGD are intellectual disability , often accompanied by epilepsy and hyperphosphatasia, sometimes with dysmorphic facial features, brachytelephalangy, progressive central nervous system abnormalities, cutaneous abnormalities, and multiple organ anomalies, the severity of which is dependent on how much reduction in GPI-anchor biosynthesis occurs.
PGAP Genes Involved in Remodeling of GPI-APs
Soon after GPI attachment to proteins, inositol-linked acyl chain is removed by PGAP1, an inositol deacylase (Fig. 2). Bacterial PI-specific phospholipase C (PI-PLC) cleaves GPI and releases proteins from the cell surface. However, inositol-acylated GPI-APs are resistant to PI-PLC because PI-PLC requires a 2-position hydroxyl group for cleavage. In addition, ER-to-Golgi transport of GPI-APs was threefold slower than in wild-type cells in PGAP1 mutant Chinese hamster ovary cells, although the cell surface expression levels of GPI-APs were normal. Then the Man2-linked EtNP side branch is removed by PGAP5/MPPE1, which is necessary for efficient GPI-AP recruitment into the ER exit sites and transport to the Golgi. In the Golgi, fatty acids of GPI-APs are further remodeled by sequential actions of PGAP3 and PGAP2 (Fig. 2). This reaction is essential for GPI-APs’ localization within lipid rafts. PGAP3, involved in the elimination of the unsaturated fatty acid (mainly C20:4, arachidonic acid) in sn-2 position, belongs to a hydrolase superfamily and is most likely a GPI-AP-specific phospholipase A2, although the enzyme activity has yet to be demonstrated in vitro. PGAP2 is involved in the second step of GPI-AP fatty acid remodeling, in that the lyso-GPI-AP intermediate is reacylated by stearic acid (C18:0). There is no significant sequence homology between PGAP2 and known acyltransferases, and the issue of whether PGAP2 is the acyltransferase itself or a regulatory protein remains to be determined.
GPI Deficiencies Caused by Germline Mutations in PGAP Genes
PGAP1 deficiency showed severe mental retardation with seizures (Murakami et al. 2014). GPI-APs in PGAP1-deficient cells are expressed on the cell surface at normal level, but structure of GPI is abnormal. GPI-APs on PGAP1-deficient cells have inositol-linked acyl chain unremoved and an unsaturated fatty acid at sn-2 position because fatty acid remodeling requires removal of the inositol-linked acyl chain. Pgap1 knockout mice showed otocephaly or holoprosencephaly. Abnormal Wnt or nodal signaling was reported to cause these phenotypes.
PGAP2 deficiency also causes HPMRS similarly to PIGO or PIGV deficiencies but with a different mechanism (Krawitz et al. 2013). Without PGAP2, GPI-APs are transported to the cell surface as a lyso-form, which is rapidly released into serum leading to decrease in the surface expression and hyperphosphatasia.
PGAP3 deficiency also causes HPMRS (Howard et al. 2014). Without PGAP3, GPI-APs are transported to the cell surface without fatty acid remodeling leading to failure to raft association, which might cause the increased release of GPI-APs from the cell surface and slight reduction of the surface expression.
These facts clearly indicate that not only the surface expression level of GPI-APs but also the structure of GPI anchor is important for normal neuronal development. Only the hypomorphic mutations are found in PIG gene deficiencies, whereas even null mutations are found in PGAP gene deficiencies. All these deficiencies are now included in congenital disorders of glycosylation (CDG) and are described as CDG (name of the gene responsible in non-italicized letters), such as CDG-PIGA and CDG-PIGM.
Concluding Remarks
To date, IGDs caused by mutations in twelve PIG and PGAP genes have been reported. Loss-of-function mutations in any one of the 26 PIG and PGAP genes described in this chapter would cause IGDs. Moreover, it is possible that other genes, which affect the transport of the GPI-APs or regulate its expression, might also cause IGDs and spectrum of this new disease will be broader. With the accumulation of more genetic, biochemical, cell biological, and clinical information from many more cases of IGDs, we will achieve better understanding of the mechanistic bases of various symptoms, improved ways of diagnosis, and hopefully useful treatment measures.
References
Almeida AM, Murakami Y, Layton DM et al (2006) Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med 12:846–851
Chiyonobu T, Inoue N, Morimoto M et al (2014) Glycosylphosphatidylinositol (GPI) anchor deficiency caused by mutations in PIGW is associated with West syndrome and hyperphosphatasia with mental retardation syndrome. J Med Genet 51:203–207
Howard MF, Murakami Y, Pagnamenta AT et al (2014) Mutations in PGAP3 impair GPI-anchor maturation and lead to intellectual disability with hyperphosphatasia and additional phenotypic features. Am J Hum Genet 94:278–287
Johnston JJ, Gropman AL, Sapp JC et al (2012) The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet 90:295–300
Kato M, Saitsu H, Murakami Y et al (2014) PIGA mutations cause early-onset epileptic encephalopathies and distinctive features. Neurol 82:1587–1596
Kinoshita T, Fujita M, Maeda Y (2008) Biosynthesis, remodelling and functions of mammalian GPI-anchored proteins: recent progress. J Biochem 144:287–294
Krawitz PM, Murakami Y, Hecht J et al (2012) Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am J Hum Genet 91:146–151
Krawitz PM, Murakami Y, Riess A et al (2013) PGAP2 mutations, affecting the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation syndrome. Am J Hum Genet 92:584–589
Krawitz PM, Schweiger MR, Rodelsperger C et al (2010) Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet 42:827–829
Kvarnung M, Nilsson D, Lindstrand A et al (2013) A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet 50:521–528
Martin HC, Kim GE, Pagnamenta AT et al (2014) Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet in press
Maydan G, Noyman I, Har-Zahav A et al (2011) Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 48:383–389
Murakami Y, Tawamie H, Maeda Y et al (2014) Null mutation in PGAP1 impairing GPI-anchor maturation in patients with intellectual disability and encephalopathy. PLOS Genet 10: e1004320
Ng BG, Hackmann K, Jones MA et al (2012) Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am J Hum Genet 90:685–688
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Japan
About this entry
Cite this entry
Murakami, Y., Kinoshita, T. (2015). Congenital Disorders of Glycosylation: Glycosylphosphatidylinositol (GPI)-Related. In: Taniguchi, N., Endo, T., Hart, G., Seeberger, P., Wong, CH. (eds) Glycoscience: Biology and Medicine. Springer, Tokyo. https://doi.org/10.1007/978-4-431-54841-6_171
Download citation
DOI: https://doi.org/10.1007/978-4-431-54841-6_171
Received:
Accepted:
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-54840-9
Online ISBN: 978-4-431-54841-6
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences