
Abstract
Charcot-Marie-Tooth (CMT) disease is the most common disorder in hereditary peripheral nerve neuropathy. Depending on the clinical manifestation, the disease is divided into demyelinating type (CMT1, with autosomal dominant inheritance, and CMT4, with autosomal recessive inheritance), axonal type (CMT2, including both autosomal dominant and autosomal recessive inheritance), and combined type (dominant-intermediate CMT). To date, more than 40 genes have been reported in this field and for some of the causative genes more than 100 mutation sites have been identified. The relationship between genotype and phenotype is variable, with different clinical manifestations resulting from the same mutated gene.
Although our understanding of the pathogenesis of CMT remains limited, accumulating knowledge about the genetic etiology of this disease has provided information about both physiological and pathological myelin formation. In demyelinating CMT, Schwann cell functions are primarily impaired. Various causative mutant proteins are perceived to result in impairment of fundamental cell function during myelination and maintenance; myelin production, degradation of myelin protein (physiological or excessive), and endocytosis. These findings suggest that myelination requires precise regulation of a large amount of myelin proteins. This understanding of the underlying molecular mechanisms is expected to contribute to development of novel therapies for this hereditary neuropathy. Therapeutic approaches are being attempted in which the physiological function of Schwann cells is supported, impaired cell function is modified, and gene expressions are modified.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Endoplasmic reticulum
- Endosome
- Genetic analyses
- Intracellular trafficking
- Lysosome
- Protein misfolding
- Rodent models
- Ubiquitin–proteasome pathway
6.1 Introduction
Charcot-Marie-Tooth syndrome was originally described as a single disorder, in which patients manifest a progressive type of familial peroneal type of progressive muscular atrophy, by J.M. Charcot, P. Marie, and H.H. Tooth in 1886. Later, it was shown that there are several disorders involving peroneal muscle atrophy. The term hereditary motor and sensory neuropathy (HMSN) was introduced to encompass a broad group of inherited neuropathy syndromes that lack known metabolic abnormalities (Dyck and Lambert 1968). Now, inherited peripheral neuropathy encompasses HMSN (CMT), hereditary neuropathy with liability to pressure palsy (HNPP), hereditary motor neuropathy (HMN), and hereditary sensory and autonomic neuropathy (HSAN). Among those disorders, CMT disease is the most common clinical entity.
Classically, CMT is diagnosed and classified according to clinical symptoms, electrophysiological examination, and pathological features. Pathological findings enable the classification of the pathophysiology of CMT into either axonal damage dominant (CMT2), or demyelination dominant (CMT1 and CMT4) (Braathen 2012). Currently, CMT is distinguished according to the responsive genetic mutations, which have revealed the role of Schwann cells in the pathogenesis of the disease. In this chapter, we discuss the pathophysiology of CMT disease, focusing on Schwann cell functional deficits.
6.2 Clinical Features of Charcot-Marie-Tooth Disease
Patients with CMT begin to manifest symptoms in their teens to thirties, typically complaining about difficulties in locomotion. The distribution of peripheral nerve disturbance is indicative of polyneuropathy. Motor function is disturbed symmetrically and distally. Sensory disturbance indicates a distal glove and stocking pattern. Because of the conduction deficit in peripheral nerves in affected limbs, the deep tendon reflexes of the limbs are reduced.
6.3 Clinical Diagnosis
The clinical diagnosis of CMT starts with the history of symptoms, family history, and physical examination. When any type of hereditary peripheral neuropathy is suspected, electrodiagnostic tests are required. In examination of peripheral nerve conduction, a delayed conduction time implies the presence of demyelinating lesions and reduced compound muscle action potential (CMAP) implies axonal damages. The median motor nerve in the forearm is commonly used for examination of nerve conduction velocity (NCV), with a cutoff value of 38 m/s for distinguishing demyelinating types and axonal types (Dyck and Lambert 1968). When the affected patient’s family shows variable NCVs ranging from 10 to 45 m/s, with dominant inheritance, the condition is defined as dominant-intermediate CMT (DI-CMT) (Davis et al. 1978). Together with the family history, autosomal dominant cases with demyelinating findings on a nerve conduction study are diagnosed as CMT1, whereas those with an autosomal recessive family history are diagnosed as CMT4. When males in the patients’ family have severe clinical symptoms, this implies that the genetic abnormality is linked to the X chromosome, and the condition is termed CMTX. On the other hand, CMT2, an axonal type, comprises both autosomal dominant and recessive family histories. When these diagnostic procedures are not conclusive, further analysis, such as nerve biopsy or genetic investigation, should be performed.
Even after all available examinations have been performed, discrimination between axonal types and demyelinating types is not always clear. Axon–Schwann cell interaction is thought to be important, not only in myelin formation but also in maintenance of both cell types. Therefore, severe demyelinating pathology can lead to axonal damage and vice versa, leading to a mixture of both demyelinating lesions and axonal degeneration.
As for CMT1, the most common category, the typical histological findings comprise hypertrophic nerves with an onion bulb shape. Several clinical symptomatic entities are considered CMT-related diseases. Dejerine–Sottas disease (DSD, also called CMT3) is a clinical diagnosis in which patients show a severe and disabling neuropathy in infancy, starting at 3 years of age (Plante-Bordeneuve and Said 2002). Historically, DSD was first recognized as autosomal recessive inheritance, but later an autosomal dominant type was also reported. Congenital hypomyelination (CH or CHM) is also a clinical subtype of HMSN, in which patients have a developmental failure in myelination, manifesting delays in walking and swallowing, or even respiratory difficulties, as early as the first year of life. It is sometimes difficult to distinguish between DSD and CH, because both show markedly slow NCV in physiological examination and similar demyelinating histology in their sural nerve biopsies (Phillips et al. 1999).
Genetic testing is now performed for clinical purposes as well as for research. The precise genetic diagnosis may provide more information about the prognosis of each patient. Moreover, in the attempt to develop novel therapies for CMT, it is important to obtain a homogeneous population for clinical studies. In general, genetic analyses are useful for distinguishing subcategories of CMT1 (A–E) or CMT4 (A–J). However, it should be noted that the genetic background of CMT is not yet fully understood. Although more than 40 genes have been reported to be involved in CMT patients, mutations in those genes account for the disease in only approximately 70 % of CMT patients (Murphy et al. 2012). Therefore, a negative finding on genetic examination cannot exclude the possibility of CMT disease. In addition, several genes in CMT have been reported to be involved in more than two types of this disease. For example, mutations in NEFL have been reported in patients with CMT1E and CMT2E. Additionally, EGR2 is known to be involved in CMT1D and CMT4E. Thus, genetic information has brought greater insights into this disorder; however, much work remains to be done to elucidate the link between genetic changes and disease manifestation.
6.4 Genes Reported in CMT
In demyelinating CMT disease, that is CMT1, CMT4, and CMT1X, 24 genes have been listed in the database: http://www.molgen.ua.ac.be/CMTMutations/Home/IPN.cfm (accessed in June 2013). Table 6.1 shows information about the genes reported in each of the subclasses of CMT1, CMT4, and CMT1X. In several subclasses, such as CMT4G, the responsible genes have not yet been identified and only limited information from linkage analyses is available.
6.5 Prevalence of CMT Diseases
Several studies have revealed the prevalence of CMT disease among inherited peripheral neuropathies. Additionally, recent genetic testing has facilitated data gathering on the frequency of mutations found in CMT disease (Braathen 2012; Murphy et al. 2012). Although there are differences in the populations studied in each report, there are some consistent findings. CMT is the most common disease in inherited peripheral neuropathy. The prevalence of CMT1 is equal to or slightly higher than that of CMT2, whereas these two categories account for 70 % of all CMT disease. As for genetic testing results, abnormalities in four genes, PMP22, MPZ, Cx32, and MFN2 (CMT2), explain 70–90 % of all CMT cases (Saporta et al. 2011). Among them, duplication of PMP22 (CMT1A) is by far more common than other mutations, three- to fivefold more common than mutation of Cx32, the second most common genetic abnormality found in the CMT population. On the other hand, CMT4, a recessively inherited type of demyelinating CMT, is a rare form of CMT, and CMT4 as a whole accounts for less than 10 % of CMT diseases in European countries. However, the prevalence of CMT4 varies depending on area. In Mediterranean countries, where consanguineous marriages are prevalent, autosomal recessive inheritance accounts for 30–50 % of CMT cases (Dubourg et al. 2006). Within CMT4, CMT4A is the most common, and only a very limited number of families have been reported in some CMT4 subclasses, such as CMT4J.
6.6 Pathogenesis of CMT
The identification of specific gene mutations in CMT disease has yielded much insight into the mechanisms underlying CMT pathogenesis and Schwann cell functions during demyelination (Berger et al. 2006b). In some cases, the findings in human genetic testing were used to establish genetic mutant mice. Such animal models allow the use of various experimental methods to reveal molecular functions in the disease. It should be noted that although the causal link between specific gene mutations and CMT pathogenesis is quite clear, current knowledge is insufficient to explain clinical symptoms comprehensively, such as the age of onset and disease severity in each cases.
Hereafter, pathogenesis of CMT is discussed based on causal mutations in genes functioning in Schwann cells.
6.6.1 PMP22
Peripheral myelin protein 22 (PMP22) is a component of compact myelin that makes up 5 % of the peripheral myelin protein. PMP22 is located on chromosome 17, which is the most common causative locus for CMT1 (autosomal dominant inheritance). This subgroup, termed CMT1A, is caused by several types of mutations in PMP22, via various mechanisms, resulting in diverse clinical manifestations.
6.6.1.1 Duplication
A large group of CMT1A patients has a 1.4-Mb chromosome 17 duplication, although some patients have been reported to have smaller duplications (Raeymaekers et al. 1989; Vance et al. 1989). Several animal studies have reported that rodent models overexpressing PMP22 showed demyelination, the severity of which is related to the level of PMP22 expression (Magyar et al. 1996; Sereda et al. 1996). Another transgenic mouse study reported that the demyelination in adult mice is reversible when excessive PMP22 expression is switched off (Perea et al. 2001). Therefore, the gene dosage of PMP22 is critical in the pathogenesis of CMT phenotype.
Although the precise mechanism by which excessive PMP22 protein leads to autosomal dominant demyelination is not yet fully clear, two possibilities have been proposed. First, PMP22 interacts with other myelin proteins, such as P0/MPZ, to form the proper myelin structure (Suter and Snipes 1995). Excess PMP22 may disturb the balance among myelin proteins, leading to destruction of myelin. The other possibility is that excessive PMP22 protein would be degraded by ubiquitination and proteasome pathways, which also maintain the appropriate expression level of PMP22 in healthy, myelinating Schwann cells. When the proteasome system is overwhelmed, the remaining PMP22 protein forms aggregates, which, in turn, exert toxic effects on Schwann cells (Fortun et al. 2005).
6.6.1.2 Point Mutation
Point mutations in PMP22 also cause autosomal dominant CMT, such as CMT1A. Among numerous mutation sites reported, the Leu16Pro mutation is common and is known as the Trembler-J mutation, which is the mutation that occurs in the spontaneous mutant mouse line (Suter et al. 1992a). Another mutation, Gly150Asp, Trembler mutation in mice, has been reported in DSD patients (Suter et al. 1992b; Ionasescu et al. 1997). Besides these missense mutations, other types of mutations, such as nonsense, frameshift, and splice site mutations, are rare and have been reported only in HNPP (Zephir et al. 2005). Because the majority of point mutations result in dominant inheritance of the disease, the mutated gene products likely cause a toxic gain of function.
At present, mutant PMP22 is thought to have a deficit in trafficking to the appropriate position in the plasma membrane. Some mutated proteins, such as Trembler mutation PMP22, can form heterodimers with the wild-type PMP22 protein and interfere with the trafficking of the normal protein (Tobler et al. 1999). Such unsorted proteins may form aggregates, as with duplication of PMP22 gene, leading to overloading of the ubiquitin–proteasome pathway.
Alternatively, the mutated gene may produce unfolded or misfolded proteins. Such proteins, when arriving at the endoplasmic reticulum (ER), induce unfolded-protein reaction (UPR) (D’Urso et al. 1998). When excessive UPR occurs within the ER, it brings about a state of “ER stress,” initiating apoptosis.
6.6.2 P0/MPZ
Myelin protein zero (P0/MPZ) is one of the myelin structural proteins and is also a member of the immunoglobulin superfamily. It is the most abundant protein constituting myelin (50 % of peripheral myelin protein) and also forms complexes with other myelin proteins such as PMP22. The corresponding gene, MPZ, is a causative gene for CMT1B, and more than 100 mutations, most of which are missense mutations, have been reported in this gene (Shy et al. 2004). These mutations predominantly occur in the extracellular domain, where extensive posttranslational modifications are added during transit through the ER and Golgi apparatus. The altered structure of MPZ induces destruction of the myelin structure, leading to early-onset demyelinating neuropathy (Shy 2006). It has also been suggested that protein misfolding and/or inadequate intracellular trafficking may induce ER stress and cell damage (Khajavi et al. 2005). From a clinical perspective, a distinct subgroup of MPZ mutations shows late-onset neuropathy with both axonal degeneration and mild demyelination. The pathogenesis of this subgroup may differ from the early-onset type and may include altered Schwann cell–axon interaction (Shy 2006).
It should be noted that there are similarities in clinical manifestations between PMP22 and MPZ mutations (D’Urso et al. 1999). This similarity could be explained by the fact that both proteins play a similar physiological function in maintaining myelin structure and forming heterocomplexes to some extent. Furthermore, misfolding of these proteins has a dominant toxic effect on intracellular systems, such as the ER or ubiquitin–proteasome pathway.
6.6.3 SIMPLE
The small integral membrane protein of lysosome/late endosome (SIMPLE) is encoded on chromosome 16p13.1, a locus that is linked to CMT1C. Physiologically, SIMPLE has been reported to be involved in the ubiquitin–proteasome pathway. Although the physiological functions of the protein are not yet fully understood, SIMPLE interacts with the E3 ubiquitin ligase, NEDD4, mediating degradation of membrane proteins via the lysosomes (Shirk et al. 2005). Therefore, dysfunction of SIMPLE may lead to failure in turnover of myelin proteins, such as PMP22. Additionally, mutated SIMPLE itself participates in aggregate formation, which, in turn, places a load on the proteasome pathway. To date, six SIMPLE mutations have been reported, five of which have been reported in autosomal dominant CMT1C patients and one in autosomal dominant axonal type CMT (CMT2) (Houlden and Reilly 2006). Because the ubiquitin–proteasome pathway is involved in the pathogenesis of mutated myelin structural proteins (PMP22 and MPZ), the mechanisms by which SIMPLE mutations cause CMT disease may overlap with that of mutations involved in CMT1A and -1B. There has also been a report of a patient with both a SIMPLE mutation and a duplication of PMP22 who manifested with early-onset neuropathy, whereas the parents, with single mutations, had minimal symptoms (Meggouh et al. 2005).
6.6.4 EGR2
Early growth response 2 (EGR2) is a zinc finger transcription factor with the mouse orthologue Krox20. Krox20 plays a pivotal role in myelin formation by Schwann cells, by activating transcription of various myelin proteins, such as MPZ and myelin basic protein. Although Krox20-knockout mice show myelin formation failure, as expected, the heterozygous mice with half the normal dosage of Krox20 are phenotypically normal (Schneider-Maunoury et al. 1993). This, together with the fact that EGR2 mutations are mainly linked to a dominantly inherited disease, CMT1D, indicates that the pathogenesis of EGR2 mutations in CMT is not caused by the absence of this transcriptional factor but rather its dominant-negative function. Among nine mutation sites reported in EGR, all CMT1D patients have mutations in the zinc finger domain, which usually functions as a DNA-binding domain (Houlden and Reilly 2006). It has been reported that the presence of dominant mutations in Krox20 reduces transcription of MPZ. More specifically, dominant mutant Krox20 interferes with DNA binding of Sox10, another crucial transcription factor that binds adjacent to Krox20 in the MPZ regulatory region (LeBlanc et al. 2007). To date, it is not yet clear whether the same dominant-negative effects take place in the regulatory regions of other molecules regulated by Krox20.
There are also cases of recessive inheritance caused by mutations in EGR2, specifically in congenital hypomyelinating neuropathy (CMT4E). A mutation was found in the R1 domain (Ile268Asn), where NAB co-repressors bind. A deficit in the transcriptional complex formed between EGR2 (Krox20) and NAB proteins (NAB1 and NAB2) is thought to be responsible for failure in promoting myelin protein transcription, which leads to Schwann cell dysfunction in myelin formation (Le et al. 2005). Consistent with this proposal, NAB1 and NAB2 double-knockout mice present with severe congenital hypomyelination.
6.6.5 NEFL
Neurofilament protein, light filament (NEFL), is an intermediate neurofilament found in axons. It is the smallest member of three neurofilaments, known as NEFL (68 kDa), NF medium (125 kDa), and NF heavy (200 kDa). As expected, mutations in NEFL result in axonal CMT, such as CMT2E. Such mutations are also found in autosomal dominant CMT1 patients, such as CMT1F. It seems that the dominant pathogenic effect of mutant NEFL affects axons but not Schwann cells. Because NEFL plays a part in maintaining neurofilament assembly, point mutations cause dysfunction in axonal transport and mitochondria localization (Jordanova et al. 2003). These perturbations in axons may interfere with physiological axon–Schwann cell interaction, resulting in a demyelinating phenotype.
6.6.6 Connexin 32
In contrast to PMP22 and MPZ, which are localized in compact myelin, connexin 32 (Cx32/GJB1) is present in paranodal loops, internodal zones, and Schmidt–Lanterman incisures. Cx32/GJB1 is a member of gap junction-forming proteins and mediates the exchange of ions and metabolites. In the case of myelin-forming Schwann cells, the molecule connects different layers of myelin, allowing ions to move between the innermost and outermost myelin (Balice-Gordon et al. 1998). Although Cx32/GJB1-deficient mice can form functional gap junctions, the mice show demyelinating lesions and axonal loss, consistent with the symptoms found in CMTX1 (Scherer et al. 1998). Such phenotypes can be rescued by introducing Cx32/GJB1 expression specifically in Schwann cells, indicating that the pathogenesis is Schwann cell dependent and axonal loss is a secondary effect of Schwann cell dysfunction (Scherer et al. 2005).
More than 400 mutations related to Cx32/GJB1 have been reported in CMTX1 patients. These mutations are distributed, throughout all regions of the molecule, include nonsense, frameshift, and deletion mutations (Kleopa et al. 2012). As the background of various mutations shows a relatively similar degree of clinical symptoms, the pathomechanism of CMTX1 is caused by the loss of function of Cx32/GJB1. Indeed, several mutant proteins have been shown to form nonfunctional or abnormally functioning gap junction formation. Abnormal trafficking is also observed when mutations occur in the C-terminal region of the protein. Such abnormalities in trafficking lead to both changes in channel properties and accumulation of proteins in the ER and Golgi. Unlike PMP22 or MPZ, accumulation of Cx32/GJB1 has not been shown to overwhelm degradation pathways. Therefore, most of the mutations cause loss-of-function of proper channel function in Schwann cells. In rare cases, mutant proteins, such as those involving S85C and F235C, exert gain-of-function effects, by forming dysfunctional channels, resulting in female CTMX1 cases (Liang et al. 2005).
6.6.7 GDAP1
Mutations in the gene encoding ganglioside-induced differentiation-associated protein 1 (GDAP1) is most frequently found in recessively inherited CMT, named CMT4A, while mutations in the same gene have also been reported in other types of CMT (recessive-intermediate CMT A, CMT2K) (Cuesta et al. 2002). GDAP1 is located in the outer mitochondrial membrane, where it regulates mitochondrial function. More than 40 mutations in this gene have been reported, causing truncation of functional domains or mutations within the GST domains (Cassereau et al. 2011). Overexpression of wild-type GDAP1, but not GDAP1 carrying a recessive mutation, increases the total cellular level of the antioxidant glutathione and increases the mitochondrial membrane potential (Noack et al. 2012). These findings suggest that loss of GDAP1 function leads to overproduction of reactive oxygen species (ROS) and activation of the related pathways, including apoptosis.
6.6.8 MTMR2 and MTMR13
Myotubularin-related protein (MTMR) constitutes a large family of phosphoinositide lipid 3-phosphatases that regulate the endosomal pathway. In CMT disease, mutations in the corresponding genes are found in autosomal recessive demyelinating cases, named CMT4B1 for MTMR2 mutations and CMT4B2 for MTMR13 mutations. MTMR2 and MTMR13 form heterocomplexes in which MTMR13 exerts a regulatory function on MTMR2 (Berger et al. 2006a). The synthesis of PI(3)P in the early endosome and PI(3,5)P2 in the late endosome is responsible for the formation of the endosome and associated lysosomal activities. MTMR family molecules are phosphatases for these phosphoinositides and are expected to function as regulators of the endosome system (Ng et al. 2013). A recent study showed that MTMR2 binds to various molecules, such as Dlg1, thereby regulating protein trafficking and membrane addition (Bolis et al. 2009). In spite of the fact that MTMR2 is not only expressed in Schwann cells, but also in neurons, Schwann cell-specific MTMR2 ablation in mutant mice confirmed that lack of MTMR2 in Schwann cells alone is sufficient to cause the demyelinating disease (Bolis et al. 2005). MTMR2-deficient mice are characterized by excessive redundant myelin, known as myelin outfoldings in CMT4B patients. Because the interaction of MTMR2 with Dlg1 negatively regulates membrane formation, a lack of MTMR2 induces excessive amounts of membrane.
6.6.9 SH3TC2/KIAA1985
The locus for CMT4C was assigned to chromosome 5q23–33 and mutations were identified in the SH3 domain and tetratricopeptide repeat domain 2 of SH3TC2, of which the product is expressed in the plasma membrane and perinuclear endocytic recycling compartment (Arnaud et al. 2009). Recently, molecular interaction was found between SH3TC2 and Rab11, a pivotal regulatory molecule involved in recycling of internalized membranes and receptors back to the cell surface in the endosome trafficking pathway. Mutations in SH3TC2 disturb SH3TC2/Rab11 complex formation and impair endocytic recycling (Stendel et al. 2010). Although the dominant-negative form of Rab11 has been reported to impair myelin formation, it remains unknown which constituent of myelin protein is specifically transported by SH3TC2/Rab11-dependent trafficking.
6.6.10 NDRG1
N-myc downstream-regulated gene 1 (NDRG1) is ubiquitously expressed and plays a role in growth arrest and cell differentiation (Melotte et al. 2010). A lack of NDRG1 impairs trafficking in the cytoplasm and nucleus, similar to that seen with SH3TC2 (CMT4C) mutation (Kalaydjieva et al. 2000; King et al. 2011). The mutation in NDRG1 was initially reported in Lom-type HMNS (HMNSL), and the clinical subclass is named CMT4D. In the clinical specimen, demyelination occurred together with severe axonal loss, suggesting impairment in axon–glial interaction.
6.6.11 PRX
Periaxin (PRX) is a member of the PDZ-domain proteins, having two isoforms, L-periaxin and S-periaxin. The PDZ domain facilitates interaction with other molecules, forming dystroglycan–dystrophin-related protein 2 (DRP2) complexes at the cell membrane of myelinating Schwann cells. This complex links the Schwann cell cytoskeleton to the extracellular matrix, and laminin in particular, and plays a crucial role in maintaining myelin structure (Sherman et al. 2001). Sixteen nonsense or frameshift mutations have been reported in PRX among autosomal recessive CMT patients (CMT4F) and Dejerine–Sottas disease patients. Because the phenotype of PRX-deficient mice recaptures the clinical symptoms of human patients, the demyelinating lesion is likely a loss of function of PRX. In some clinical cases, a truncated form of PRX is found complexed with DRP2, suggesting that pathogenesis of the recessive phenotype is independent of the interaction between PRX and DRP2 (Takashima et al. 2002). There is also a possibility that the truncated form of PRX is resistant to degradation, which produces gain-of-function effects on myelin stability.
6.6.12 Frabin/FGD4
Frabin, known as a causative gene for CMT4H, is a member of the GDP/GTP nucleotide exchange factors (GEF) and is a specific activator of Cdc42, a member of the Rho GTPase family, which regulate assembly of the actin cytoskeleton and microtubules (Delague et al. 2007; Horn et al. 2012). Cdc42 is reported to be involved in cell migration, polarization, division, and membrane trafficking. Besides a Dbl homology (DH) domain, the region responsible for regulating Cdc42 function, Frabin also contains other domains, such as a pleckstrin homology (PH) domain and cysteine-rich FYVE domain. These domains are known to be necessary for activation of Cdc42. The presence of PH and FYVE domains also is important for activating c-Jun N-terminal kinase (JNK), as well as for facilitating the binding of Frabin to phosphoinositide phosphates (PIPs), substrates of the myotubularin-related proteins MTMR2 and MTMR13, which are phosphatases for PIPs (Stendel et al. 2007). As mutations in MTMR2 and MTMR13 impair endosomal regulation of myelin proteins, leading to CMT4B1 and CMTB2, respectively, association between Frabin and MTMRs may characterize the importance of Frabin in membrane transport in myelinating Schwann cells. It is noteworthy that pathological findings between CMT4B and CMT4H are similar; that is, both conditions are characterized by myelin outfoldings, irregular folding, and redundant loops of myelin (Tazir et al. 2013).
6.6.13 FIG4
FIG4 is a phosphatase acting on the 5-phosphate from phosphoinositide PI (3,5)P2 on the surface of vesicles in the endosome/lysosome pathway. FIG4 acts on the same target as MTMR2, which is causative for CMT4B, but removes a different phosphate. Mutations in FIG4 have been reported in a severely recessive subgroup of CMT, CMT4J (Chow et al. 2007). The majority of the patients are compound heterozygotes carrying the missense allele I41T, in combination with a null allele of FIG4. The I41T mutant can exert partial functions of FIG4 but is unstable because of impairment of its interaction with the scaffold protein, VAC14. The loss of function of FIG4 results in accumulation of endosomal/lysosomal vesicles, affecting proper myelin maintenance (Lenk et al. 2011).
6.6.14 DNM2
Dynamin 2 (DNM2) is a member of the large GTPase family, regulating membrane trafficking from the trans-Golgi network, actin cytoskeletal dynamics, and membrane fusion (Praefcke and McMahon 2004). Dynamin is recruited to the site of endocytosis and its GTPase activity provides energy for the endocytosis processes. DNM2 is expressed ubiquitously and is reportedly linked to a dominant-intermediate CMT (DI-CMTB). Most of the reported mutation sites among DI-CMTB patients are located around the PH domain that mediates interaction with phosphoinositides, recruiting DNM2 to the vesicles. The mutated proteins have reduced capacity to bind to vesicles, resulting in impairment of endocytosis (Zuchner et al. 2005).
6.7 General Aspects of CMT Pathogenesis
As listed here, there are many genes and mutations reported to be involved in the pathogenesis of demyelinating CMT diseases. In each subgroup, further detail in our understanding of the function of the affected genes in physiology and pathology is required. However, to develop therapeutic strategies for CMT diseases, it would be advantageous to perceive the pathogenesis of the disease from the point of view of generalized pathways involved in demyelinating CMT.
In a simplified summary, the mechanisms underlying demyelinating CMT involve disruption of the membrane structure or accumulation of abnormal or overexpressed proteins. Furthermore, disruption of the membrane can be caused by a lack of components, which usually is prominent in recessively inherited conditions, or the presence of abnormal (or excessive) proteins, by which the physiological molecular interaction in the membrane is disturbed in a dominantly inherited manner.
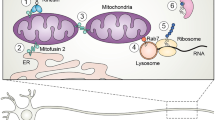
Accumulation of myelin or membrane proteins may exert harmful effects upon Schwann cells through various pathways. To understand those pathways, it is important to understand the physiological protein quality control mechanisms within Schwann cells. It has been reported that up to 80 % of the newly synthesized PMP22 is degraded within 30 min via the proteasome, because of misfolding of the protein or excessive production (Ryan et al. 2002). Such high turnover maintenance of myelin protein is one of the reasons that dysfunction of the degradative pathways can lead to accumulation of the protein. During the process of myelination and remyelination, transactivation of myelin proteins is upregulated, so that heterogeneous mutations can result in excessive levels of myelin proteins, which cannot be integrated into the myelin membrane and are destined to be degraded. Figure 6.1 summarizes the intracellular systems that handle misfolded or excessive proteins.

Pathways for protein degradation in Schwann cells. The production of myelin proteins is tightly regulated with several pathways to degrade undesired proteins. Many causative genes in Charcot-Marie-Tooth (CMT) diseases are related to these pathways, indicating their importance. ER endoplasmic reticulum, ERAD ER-associated degradation, Ub ubiquitination, ROS reactive oxygen species
6.7.1 UPR at ER
Misfolded myelin proteins are retained in the ER and their accumulation activates the unfolded protein response (UPR), characterized by activation of Ire-1, Perk, and ATF6 pathways (Ron and Walter 2007). As an adaptive response, UPR induces specific chaperone expression and at the same time reduces translation, to prevent further accumulation of the unfolded proteins. Transgenic mice models overexpressing wild or mutant forms of MPZ showed that although the S63del mutant is retained in the ER and induces the UPR response, normal MPZ, another MPZ mutant (S63C), and the Trembler-J mutant of PMP22 do not activate the UPR (Pennuto et al. 2008). Although the UPR itself is a physiological component of cell maintenance, severe ER stress can also induce the pro-apoptotic molecule, CHOP. In general, induction of CHOP expression after ER stress triggers the cell death program to eliminate the affected cells. The involvement of CHOP in the pathogenesis of the MPZ S63del is proven by the fact that ablation of Chop from S63del-transgenic mice results in a reduction of demyelinating lesions. Interestingly, the number of Schwann cells is not altered significantly by the absence of CHOP; it is now understood that activation of the CHOP pathway induces dysfunction of Schwann cells, which is independent of cell death (Pennuto et al. 2008).
6.7.2 Ubiquitin–Proteasome Pathway
When misfolded proteins are not fully refolded in the ER, these proteins are released into the cytosol, where specific E3 ligases target the molecule and add several ubiquitin molecules. The poly-ubiquitinated protein is then recognized by proteasome systems, including the lysosome where the protein is finally degraded. In CMT disease, excessive PMP22, mutant PMP22, and mutant MPZ have been reported to be processed by the proteasome via the ER, whereas mutant SIMPLE (CMT1C) is degraded by the proteasome in ER-independent mechanisms (Lee et al. 2012). The function of proteasome is inhibited by oxidative stress, age, anti-cancer drugs, the presence of mutant PMP22 (Trembler-J mutant), or the presence of other protein aggregates. Impaired function of the proteasome leads to a further increase of cytoplasmic protein aggregates (Fortun et al. 2006). The presence of aggregates is thought to be a causative event, not only in CMT, but also in various degenerative diseases, such as Huntington disease. One of the possible mechanisms is that the aggregates entrap other molecules to form nonfunctional complexes. In the case of Trembler-J mice, MBP is mislocalized to PMP22 protein aggregates, reducing the proper sorting of MBP into the compact myelin (Fortun et al. 2005). Although protein aggregates exert cytotoxic effects in other degenerative diseases, whether myelin protein aggregates induce cell death in Schwann cells is not yet clear.
6.7.3 Aggresome–Autophagy Pathway
Another protein quality control system involved in degrading misfolded and aggregated proteins is the aggresome–autophagy pathway. Aggregated proteins that are not handled by the proteasome pathway are sequestered to the perinuclear aggresome through microtubule-dependent transport. The aggresomes are then surrounded by the autophagosome and degraded via autophagy. Because autophagy is an alternative way to handle abnormal protein aggregates, some attempts have been made to enhance autophagy activity in CMT model mice to reduce the amount of aggregates (Fortun et al. 2007).
6.7.4 Inflammation
Although the majority of studies of demyelinating CMT diseases focused on intrinsic pathological events in Schwann cells, the secondary involvement of extrinsic events may be crucial in determining disease progression and severity. The presence of low-grade inflammation is reported both in specimens of CMT patients and in mice models (Malandrini et al. 1999; Kohl et al. 2010). In the mouse model, the number of macrophages infiltrated into the peripheral nerves correlates with axonal damage. Among numerous inflammatory mediators, MCP-1/CCL2 is thought to be a crucial molecule in PMP22tg mice, because ablation of the gene encoding this chemokine can prevent accumulation of macrophages (Kohl et al. 2010). The involvement of inflammation in CMT disease is also suggested from the study of crossbreeding of Cx32-deficient mice (a model of CMTX1) with colony-stimulating factor-1 (CSF-1)-deficient mice. The lack of CSF-1, a cytokine that recruits and activates macrophages, leads to reduction of demyelinating lesions in the mouse model (Groh et al. 2012). The source of these cytokines may not be restricted to Schwann cells, but may also involve other structural cells in the peripheral nerves, such as endoneural fibroblasts.
6.8 Therapeutic Approaches for CMT Diseases
It has been described that there is no established disease-modifying therapy for CMT disease, although supportive care, including use of orthotics and pain management, have been well developed. Nevertheless, the accumulation of knowledge about the cellular and molecular mechanisms of myelination and demyelination provides several possibilities to approach the disease (Herrmann 2008).
6.8.1 Ascorbic Acid
The necessity of ascorbic acid for myelin formation by Schwann cells has been well documented in Schwann cell–dorsal root ganglion co-culture studies. Ascorbic acid is required to form the basement membrane before the initiation of myelination. These in vitro studies provide the rationale for an animal study in which ascorbic acid was given to a mouse model of CMT1A (Passage et al. 2004). In these experiments, ascorbic acid reduced PMP22 mRNA at the transcriptional level. Based on these preclinical data, two clinical trials of ascorbic acid in CMT1A patients are currently underway (Lewis et al. 2013; Pareyson et al. 2006).
6.8.2 Progesterone Antagonist
Progesterone has been shown to promote expression of myelin-related genes, such as PMP22 and MPZ. Therefore, antagonizing its function may convey beneficial effects in the case of excessive myelin protein production, as occurs with duplication of PMP22 (CMT1A). A progesterone receptor antagonist, onapristone, improved the pathological phenotype of CMT1A model mice when the chemical was applied in neonates (Sereda et al. 2003). However, the further clinical application of onapristone is not promising, as this drug is unsafe for use in humans.
6.8.3 Neurotrophin-3
Schwann cell myelination is influenced by various extrinsic molecules, including nerve growth factors. Among these, neurotrophin-3 (NT-3) is a promising molecule for promoting myelin formation. Elevated myelin formation has been shown in both in vitro and in vivo experiments with NT-3 treatment. A pilot study using NT-3 in CMT1A patients reported some beneficial effects of this treatment (Sahenk et al. 2005).
6.8.4 RNA and Gene-Based Therapy
It is assumed that the development of gene-based technologies will provide a better opportunity for establishing therapeutic approaches by directly targeting mRNA or genes. One of the most promising methods would be RNA interference methods. By using short interfering RNA, it is possible to downregulate the expression of undesired mRNA specifically (Bhindi et al. 2007). Therefore, the gain-of-function mutations in CMT, mainly CMT1, would be an ideal target for this approach. However, because CMT disease is a congenital and lifelong disorder, undesired gene expression should be regulated throughout life. Considering that the lesion occurs within the peripheral nerve, which is more difficult to access as compared to muscles, these drug delivery issues should be resolved in future.
6.9 Conclusion
To understand the pathogenesis of CMT, a focus on Schwann cell functions has given us the opportunity to understand how myelination is regulated and maintained in peripheral nerves. The fact that several causative gene products in CMT are expressed ubiquitously, but result specifically in peripheral neuropathy, indicates that achieving proper localization of adequate amounts of myelin proteins requires precise performance of various cell functions. Therefore, Schwann cell myelination would be an informative experimental model for studying cell functions, such as membrane trafficking and degradation. With respect to future challenges for novel therapeutic approaches, it should be kept in mind that the majority of CMT patients bear mutations in PMP22 or MPZ (CMT1A and -B), with a minor population bearing mutations in other genes. In the case of recessive CMT4, it is not realistic to organize a clinical study including more than 100 patients because the patient population for each subtype is too small. Therefore, basic research into CMT should explore mechanisms of general myelination pathways further to contribute to CMT-related diseases as a whole.
References
Arnaud E, Zenker J, de Preux Charles AS, Stendel C, Roos A, Medard JJ, Tricaud N, Kleine H, Luscher B, Weis J, Suter U, Senderek J, Chrast R (2009) SH3TC2/KIAA1985 protein is required for proper myelination and the integrity of the node of Ranvier in the peripheral nervous system. Proc Natl Acad Sci USA 106:17528–17533
Balice-Gordon RJ, Bone LJ, Scherer SS (1998) Functional gap junctions in the Schwann cell myelin sheath. J Cell Biol 142:1095–1104
Berger P, Berger I, Schaffitzel C, Tersar K, Volkmer B, Suter U (2006a) Multi-level regulation of myotubularin-related protein-2 phosphatase activity by myotubularin-related protein-13/set-binding factor-2. Hum Mol Genet 15:569–579
Berger P, Niemann A, Suter U (2006b) Schwann cells and the pathogenesis of inherited motor and sensory neuropathies (Charcot-Marie-Tooth disease). Glia 54:243–257
Bhindi R, Fahmy RG, Lowe HC, Chesterman CN, Dass CR, Cairns MJ, Saravolac EG, Sun LQ, Khachigian LM (2007) Brothers in arms: DNA enzymes, short interfering RNA, and the emerging wave of small-molecule nucleic acid-based gene-silencing strategies. Am J Pathol 171:1079–1088
Bolis A, Coviello S, Bussini S, Dina G, Pardini C, Previtali SC, Malaguti M, Morana P, Del Carro U, Feltri ML, Quattrini A, Wrabetz L, Bolino A (2005) Loss of Mtmr2 phosphatase in Schwann cells but not in motor neurons causes Charcot-Marie-Tooth type 4B1 neuropathy with myelin outfoldings. J Neurosci 25:8567–8577
Bolis A, Coviello S, Visigalli I, Taveggia C, Bachi A, Chishti AH, Hanada T, Quattrini A, Previtali SC, Biffi A, Bolino A (2009) Dlg1, Sec8, and Mtmr2 regulate membrane homeostasis in Schwann cell myelination. J Neurosci 29:8858–8870
Braathen GJ (2012) Genetic epidemiology of Charcot-Marie-Tooth disease. Acta Neurol Scand 126(Suppl 193):1–22
Cassereau J, Chevrollier A, Gueguen N, Desquiret V, Verny C, Nicolas G, Dubas F, Amati-Bonneau P, Reynier P, Bonneau D, Procaccio V (2011) Mitochondrial dysfunction and pathophysiology of Charcot-Marie-Tooth disease involving GDAP1 mutations. Exp Neurol 227:31–41
Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, Szigeti K, Shy ME, Li J, Zhang X, Lupski JR, Weisman LS, Meisler MH (2007) Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature (Lond) 448:68–72
Cuesta A, Pedrola L, Sevilla T, Garcia-Planells J, Chumillas MJ, Mayordomo F, Leguern E, Marin I, Vilchez JJ, Palau F (2002) The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet 30:22–25
D’Urso D, Prior R, Greiner-Petter R, Gabreels-Festen AA, Muller HW (1998) Overloaded endoplasmic reticulum-Golgi compartments, a possible pathomechanism of peripheral neuropathies caused by mutations of the peripheral myelin protein PMP22. J Neurosci 18:731–740
D’Urso D, Ehrhardt P, Muller HW (1999) Peripheral myelin protein 22 and protein zero: a novel association in peripheral nervous system myelin. J Neurosci 19:3396–3403
Davis CJ, Bradley WG, Madrid R (1978) The peroneal muscular atrophy syndrome: clinical, genetic, electrophysiological and nerve biopsy studies. I. Clinical, genetic and electrophysiological findings and classification. J Hum Genet 26:311–349
Delague V, Jacquier A, Hamadouche T, Poitelon Y, Baudot C, Boccaccio I, Chouery E, Chaouch M, Kassouri N, Jabbour R, Grid D, Megarbane A, Haase G, Levy N (2007) Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive Charcot-Marie-Tooth type 4H. Am J Hum Genet 81:1–16
Dubourg O, Azzedine H, Verny C, Durosier G, Birouk N, Gouider R, Salih M, Bouhouche A, Thiam A, Grid D, Mayer M, Ruberg M, Tazir M, Brice A, Leguern E (2006) Autosomal-recessive forms of demyelinating Charcot-Marie-Tooth disease. Neuromolecular Med 8:75–86
Dyck PJ, Lambert EH (1968) Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch Neurol 18:603–618
Fortun J, Li J, Go J, Fenstermaker A, Fletcher BS, Notterpek L (2005) Impaired proteasome activity and accumulation of ubiquitinated substrates in a hereditary neuropathy model. J Neurochem 92:1531–1541
Fortun J, Go JC, Li J, Amici SA, Dunn WA Jr, Notterpek L (2006) Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiol Dis 22:153–164
Fortun J, Verrier JD, Go JC, Madorsky I, Dunn WA, Notterpek L (2007) The formation of peripheral myelin protein 22 aggregates is hindered by the enhancement of autophagy and expression of cytoplasmic chaperones. Neurobiol Dis 25:252–265
Groh J, Weis J, Zieger H, Stanley ER, Heuer H, Martini R (2012) Colony-stimulating factor-1 mediates macrophage-related neural damage in a model for Charcot-Marie-Tooth disease type 1X. Brain 135:88–104
Herrmann DN (2008) Experimental therapeutics in hereditary neuropathies: the past, the present, and the future. Neurotherapeutics 5:507–515
Horn M, Baumann R, Pereira JA, Sidiropoulos PN, Somandin C, Welzl H, Stendel C, Luhmann T, Wessig C, Toyka KV, Relvas JB, Senderek J, Suter U (2012) Myelin is dependent on the Charcot-Marie-Tooth Type 4H disease culprit protein FRABIN/FGD4 in Schwann cells. Brain 135:3567–3583
Houlden H, Reilly MM (2006) Molecular genetics of autosomal-dominant demyelinating Charcot-Marie-Tooth disease. Neuromolecular Med 8:43–62
Ionasescu VV, Searby CC, Ionasescu R, Chatkupt S, Patel N, Koenigsberger R (1997) Dejerine-Sottas neuropathy in mother and son with same point mutation of PMP22 gene. Muscle Nerve 20:97–99
Jordanova A, De Jonghe P, Boerkoel CF, Takashima H, De Vriendt E, Ceuterick C, Martin JJ, Butler IJ, Mancias P, Papasozomenos S, Terespolsky D, Potocki L, Brown CW, Shy M, Rita DA, Tournev I, Kremensky I, Lupski JR, Timmerman V (2003) Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain 126:590–597
Kalaydjieva L, Gresham D, Gooding R, Heather L, Baas F, De Jonge R, Blechschmidt K, Angelicheva D, Chandler D, Worsley P, Rosenthal A, King RH, Thomas PK (2000) N-myc downstream-regulated gene 1 is mutated in hereditary motor and sensory neuropathy-Lom. Am J Hum Genet 67:47–58
Khajavi M, Inoue K, Wiszniewski W, Ohyama T, Snipes GJ, Lupski JR (2005) Curcumin treatment abrogates endoplasmic reticulum retention and aggregation-induced apoptosis associated with neuropathy-causing myelin protein zero-truncating mutants. Am J Hum Genet 77:841–850
King RH, Chandler D, Lopaticki S, Huang D, Blake J, Muddle JR, Kilpatrick T, Nourallah M, Miyata T, Okuda T, Carter KW, Hunter M, Angelicheva D, Morahan G, Kalaydjieva L (2011) Ndrg1 in development and maintenance of the myelin sheath. Neurobiol Dis 42:368–380
Kleopa KA, Abrams CK, Scherer SS (2012) How do mutations in GJB1 cause X-linked Charcot-Marie-Tooth disease? Brain Res 1487:198–205
Kohl B, Fischer S, Groh J, Wessig C, Martini R (2010) MCP-1/CCL2 modifies axon properties in a PMP22-overexpressing mouse model for Charcot-Marie-tooth 1A neuropathy. Am J Pathol 176:1390–1399
Le N, Nagarajan R, Wang JY, Svaren J, Lapash C, Araki T, Schmidt RE, Milbrandt J (2005) Nab proteins are essential for peripheral nervous system myelination. Nat Neurosci 8:932–940
LeBlanc SE, Ward RM, Svaren J (2007) Neuropathy-associated Egr2 mutants disrupt cooperative activation of myelin protein zero by Egr2 and Sox10. Mol Cell Biol 27:3521–3529
Lee SM, Chin LS, Li L (2012) Protein misfolding and clearance in demyelinating peripheral neuropathies: therapeutic implications. Commun Integr Biol 5:107–110
Lenk GM, Ferguson CJ, Chow CY, Jin N, Jones JM, Grant AE, Zolov SN, Winters JJ, Giger RJ, Dowling JJ, Weisman LS, Meisler MH (2011) Pathogenic mechanism of the FIG4 mutation responsible for Charcot-Marie-Tooth disease CMT4J. PLoS Genet 7:e1002104
Lewis RA, McDermott MP, Herrmann DN, Hoke A, Clawson LL, Siskind C, Feely SM, Miller LJ, Barohn RJ, Smith P, Luebbe E, Wu X, Shy ME (2013) High-dosage ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A: results of a randomized, double-masked, controlled trial. JAMA Neurol 70(8):981–987
Liang GS, de Miguel M, Gomez-Hernandez JM, Glass JD, Scherer SS, Mintz M, Barrio LC, Fischbeck KH (2005) Severe neuropathy with leaky connexin 32 hemichannels. Ann Neurol 57:749–754
Magyar JP, Martini R, Ruelicke T, Aguzzi A, Adlkofer K, Dembic Z, Zielasek J, Toyka KV, Suter U (1996) Impaired differentiation of Schwann cells in transgenic mice with increased PMP22 gene dosage. J Neurosci 16:5351–5360
Malandrini A, Villanova M, Dotti MT, Federico A (1999) Acute inflammatory neuropathy in Charcot-Marie-Tooth disease. Neurology 52:859–861
Meggouh F, de Visser M, Arts WF, De Coo RI, van Schaik IN, Baas F (2005) Early onset neuropathy in a compound form of Charcot-Marie-Tooth disease. Ann Neurol 57:589–591
Melotte V, Qu X, Ongenaert M, van Criekinge W, de Bruine AP, Baldwin HS, van Engeland M (2010) The N-myc downstream regulated gene (NDRG) family: diverse functions, multiple applications. FASEB J 24:4153–4166
Murphy SM, Laura M, Fawcett K, Pandraud A, Liu YT, Davidson GL, Rossor AM, Polke JM, Castleman V, Manji H, Lunn MP, Bull K, Ramdharry G, Davis M, Blake JC, Houlden H, Reilly MM (2012) Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry 83:706–710
Ng AA, Logan AM, Schmidt EJ, Robinson FL (2013) The CMT4B disease-causing phosphatases Mtmr2 and Mtmr13 localize to the Schwann cell cytoplasm and endomembrane compartments, where they depend upon each other to achieve wild-type levels of protein expression. Hum Mol Genet 22:1493–1506
Noack R, Frede S, Albrecht P, Henke N, Pfeiffer A, Knoll K, Dehmel T, Meyer Zu Horste G, Stettner M, Kieseier BC, Summer H, Golz S, Kochanski A, Wiedau-Pazos M, Arnold S, Lewerenz J, Methner A (2012) Charcot-Marie-Tooth disease CMT4A: GDAP1 increases cellular glutathione and the mitochondrial membrane potential. Hum Mol Genet 21:150–162
Pareyson D, Schenone A, Fabrizi GM, Santoro L, Padua L, Quattrone A, Vita G, Gemignani F, Visioli F, Solari A (2006) A multicenter, randomized, double-blind, placebo-controlled trial of long-term ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL): the study protocol [EudraCT no.: 2006-000032-27]. Pharmacol Res 54:436–441
Passage E, Norreel JC, Noack-Fraissignes P, Sanguedolce V, Pizant J, Thirion X, Robaglia-Schlupp A, Pellissier JF, Fontes M (2004) Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med 10:396–401
Pennuto M, Tinelli E, Malaguti M, Del Carro U, D’Antonio M, Ron D, Quattrini A, Feltri ML, Wrabetz L (2008) Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron 57:393–405
Perea J, Robertson A, Tolmachova T, Muddle J, King RH, Ponsford S, Thomas PK, Huxley C (2001) Induced myelination and demyelination in a conditional mouse model of Charcot-Marie-Tooth disease type 1A. Hum Mol Genet 10:1007–1018
Phillips JP, Warner LE, Lupski JR, Garg BP (1999) Congenital hypomyelinating neuropathy: two patients with long-term follow-up. Pediatr Neurol 20:226–232
Plante-Bordeneuve V, Said G (2002) Dejerine-Sottas disease and hereditary demyelinating polyneuropathy of infancy. Muscle Nerve 26:608–621
Praefcke GJ, McMahon HT (2004) The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol 5:133–147
Raeymaekers P, Timmerman V, De Jonghe P, Swerts L, Gheuens J, Martin JJ, Muylle L, De Winter G, Vandenberghe A, Van Broeckhoven C (1989) Localization of the mutation in an extended family with Charcot-Marie-Tooth neuropathy (HMSN I). Am J Hum Genet 45:953–958
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529
Ryan MC, Shooter EM, Notterpek L (2002) Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol Dis 10:109–118
Sahenk Z, Nagaraja HN, McCracken BS, King WM, Freimer ML, Cedarbaum JM, Mendell JR (2005) NT-3 promotes nerve regeneration and sensory improvement in CMT1A mouse models and in patients. Neurology 65:681–689
Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME (2011) Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol 69:22–33
Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K, Bone LJ (1998) Connexin32-null mice develop demyelinating peripheral neuropathy. Glia 24:8–20
Scherer SS, Xu YT, Messing A, Willecke K, Fischbeck KH, Jeng LJ (2005) Transgenic expression of human connexin32 in myelinating Schwann cells prevents demyelination in connexin32-null mice. J Neurosci 25:1550–1559
Schneider-Maunoury S, Topilko P, Seitandou T, Levi G, Cohen-Tannoudji M, Pournin S, Babinet C, Charnay P (1993) Disruption of Krox-20 results in alteration of rhombomeres 3 and 5 in the developing hindbrain. Cell 75:1199–1214
Sereda M, Griffiths I, Puhlhofer A, Stewart H, Rossner MJ, Zimmerman F, Magyar JP, Schneider A, Hund E, Meinck HM, Suter U, Nave KA (1996) A transgenic rat model of Charcot-Marie-Tooth disease. Neuron 16:1049–1060
Sereda MW, Meyer Zu Horste G, Suter U, Uzma N, Nave KA (2003) Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat Med 9:1533–1537
Sherman DL, Fabrizi C, Gillespie CS, Brophy PJ (2001) Specific disruption of a Schwann cell dystrophin-related protein complex in a demyelinating neuropathy. Neuron 30:677–687
Shirk AJ, Anderson SK, Hashemi SH, Chance PF, Bennett CL (2005) SIMPLE interacts with NEDD4 and TSG101: evidence for a role in lysosomal sorting and implications for Charcot-Marie-Tooth disease. J Neurosci Res 82:43–50
Shy ME (2006) Peripheral neuropathies caused by mutations in the myelin protein zero. J Neurol Sci 242:55–66
Shy ME, Jani A, Krajewski K, Grandis M, Lewis RA, Li J, Shy RR, Balsamo J, Lilien J, Garbern JY, Kamholz J (2004) Phenotypic clustering in MPZ mutations. Brain 127:371–384
Stendel C, Roos A, Deconinck T, Pereira J, Castagner F, Niemann A, Kirschner J, Korinthenberg R, Ketelsen UP, Battaloglu E, Parman Y, Nicholson G, Ouvrier R, Seeger J, De Jonghe P, Weis J, Kruttgen A, Rudnik-Schoneborn S, Bergmann C, Suter U, Zerres K, Timmerman V, Relvas JB, Senderek J (2007) Peripheral nerve demyelination caused by a mutant Rho GTPase guanine nucleotide exchange factor, frabin/FGD4. Am J Hum Genet 81:158–164
Stendel C, Roos A, Kleine H, Arnaud E, Ozcelik M, Sidiropoulos PN, Zenker J, Schupfer F, Lehmann U, Sobota RM, Litchfield DW, Luscher B, Chrast R, Suter U, Senderek J (2010) SH3TC2, a protein mutant in Charcot-Marie-Tooth neuropathy, links peripheral nerve myelination to endosomal recycling. Brain 133:2462–2474
Suter U, Snipes GJ (1995) Biology and genetics of hereditary motor and sensory neuropathies. Annu Rev Neurosci 18:45–75
Suter U, Moskow JJ, Welcher AA, Snipes GJ, Kosaras B, Sidman RL, Buchberg AM, Shooter EM (1992a) A leucine-to-proline mutation in the putative first transmembrane domain of the 22-kDa peripheral myelin protein in the trembler-J mouse. Proc Natl Acad Sci USA 89:4382–4386
Suter U, Welcher AA, Ozcelik T, Snipes GJ, Kosaras B, Francke U, Billings-Gagliardi S, Sidman RL, Shooter EM (1992b) Trembler mouse carries a point mutation in a myelin gene. Nature (Lond) 356:241–244
Takashima H, Boerkoel CF, De Jonghe P, Ceuterick C, Martin JJ, Voit T, Schroder JM, Williams A, Brophy PJ, Timmerman V, Lupski JR (2002) Periaxin mutations cause a broad spectrum of demyelinating neuropathies. Ann Neurol 51:709–715
Tazir M, Bellatache M, Nouioua S, Vallat JM (2013) Autosomal recessive Charcot-Marie-Tooth disease: from genes to phenotypes. J Peripher Nerv Syst 18:113–129
Tobler AR, Notterpek L, Naef R, Taylor V, Suter U, Shooter EM (1999) Transport of Trembler-J mutant peripheral myelin protein 22 is blocked in the intermediate compartment and affects the transport of the wild-type protein by direct interaction. J Neurosci 19:2027–2036
Vance JM, Nicholson GA, Yamaoka LH, Stajich J, Stewart CS, Speer MC, Hung WY, Roses AD, Barker D, Pericak-Vance MA (1989) Linkage of Charcot-Marie-Tooth neuropathy type 1a to chromosome 17. Exp Neurol 104:186–189
Zephir H, Stojkovic T, Latour P, Hurtevent JF, Blankaert F, Vermersch P (2005) A family with a novel frameshift mutation in the PMP22 gene (c.433_434insC) causing a phenotype of hereditary neuropathy with liability to pressure palsies. Neuromuscul Disord 15:493–497
Zuchner S, Noureddine M, Kennerson M, Verhoeven K, Claeys K, De Jonghe P, Merory J, Oliveira SA, Speer MC, Stenger JE, Walizada G, Zhu D, Pericak-Vance MA, Nicholson G, Timmerman V, Vance JM (2005) Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet 37:289–294
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Japan
About this chapter
Cite this chapter
Ogata, T. (2014). Charcot-Marie-Tooth Disease. In: Sango, K., Yamauchi, J. (eds) Schwann Cell Development and Pathology. Springer, Tokyo. https://doi.org/10.1007/978-4-431-54764-8_6
Download citation
DOI: https://doi.org/10.1007/978-4-431-54764-8_6
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-54763-1
Online ISBN: 978-4-431-54764-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)