
Abstract
Bioconjugations with synthetic polymers have been a versatile way to add new value, advanced features and unique properties to biomolecules. Smart polymer-protein conjugates have been investigated over the past 30 years. Since the conjugation of smart polymer to single molecule can generate a nano-scale switch, many researchers have conjugated smart polymers to proteins for a great variety of applications in affinity separations, enzyme bioprocesses, drug delivery, diagnostics and biosensors, cell culture processes including tissue engineering, and DNA motors. Biomolecules that can be conjugated with smart polymers include not only proteins but also peptides, polysaccharides, and DNA, and lipids etc. This chapter reviews different types of smart polymer-biomolecule conjugates that have been developed in the last decades.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
6.1 Introduction
This chapter focuses on the synthesis, characterization, and applications of bioconjugates of smart polymers and bioactive molecules such as drugs, peptides, enzymes, antibodies, viruses and cells. Bioconjugations with synthetic polymers have been a versatile way to add new value, advanced features and unique properties to inert biomolecules. Polymer-protein conjugates, for example, have been extensively investigated over the past 35 years. In 1970s, Ringsdorf [1] proposed a general scheme of designing a drug delivery system using a synthetic polymer backbone as a carrier for low-molecular weight drugs. The proposed system contains solubilizing groups or targeting moieties that render water solubility and targeting properties on the carrier. In 1980s, Hiroshi Maeda laid the foundations for Ringsdorf’s visionary model by showing efficient accumulation of polymer-antitumor protein conjugates within tumors, so-call EPR (enhanced permeability and retention) effect [2]. Covalent attachment of polymer chains to bioactive molecules also offers the possibility of exerting control over their biological activity. This principle is now being widely exploited in pharmaceutical development, as covalent attachment of polyethylene glycol (PEG) to therapeutic proteins, called “PEGylation”, has been shown to improve the safety and efficiency without losing their biological functions [3]. Many PEGylated pharmaceuticals are currently on the market. This technology can be also used as a means to improve the in vivo pharmacokinetics of the viral vectors [4, 5] or to silence the antigenic response of red blood cells (RBCs) toward the development of universal blood transfusion [6–8].
Although polymer-conjugation studies are already well advanced, controlling the specific functions of these molecules in biological systems under changing conditions is difficult. In this context, smart polymer-protein conjugates have been also investigated over the past 30 years. The conjugation of smart polymer to single molecule can generate a nano-scale switch (Fig. 6.1). The early publications on smart-polymer conjugates were appeared in 1970s, where proteins were conjugated to carboxylated polymers that phase-separated either at low pH or by the addition of calcium ions [9, 10]. Until today, pioneering work in this area has been carried out by Hoffman and co-workers [11, 12]. They first demonstrated the selective separation of certain proteins out of blood serum using “monomer conjugation plus copolymerization” method. They conjugated vinyl groups to a protein and then copolymerized the monomer-derivatized protein with hydroxyethyl methacrylate (HEMA), causing it to phase separation because HEMA polymer was not water soluble while HEMA monomer was water soluble. In 1980s, Hoffman and co-workers [13, 14] have further developed the thermally-induced phase separation immunoassay (TIPSIA) using temperature-responsive polymers, such as poly(N-isopropylacrylamide) (PNIPAAm). PNIPAAm undergoes a sharp coil-globule transition in water at 32 °C (lower critical solution temperature: LCST), being hydrophilic below this temperature and hydrophobic above it. The PNIPAAm-monoclonal antibody conjugates thus enabled the partition and separation of target antigen in aqueous environment on the basis of solubility. Since then, many researchers have conjugated smart polymers to proteins for a great variety of applications in affinity separations [15, 16], enzyme bioprocesses [17–19], drug delivery [20–23], diagnostics and biosensors [24, 25], cell culture processes including tissue engineering [26–28], and DNA motors [29, 30]. Biomolecules that can be conjugated with smart polymers include not only proteins but also peptides [31, 32], polysaccharides [33–35], and DNA [25, 36], and lipids [37, 38] etc. Smart polymers may be conjugated randomly or site-specifically to biomolecules to produce a new hybrid type of molecules that can synergistically combine the individual properties of the two components to yield a large family of polymer-biomolecule systems that can respond to biological as well as physical and chemical stimuli. The activity of the overall conjugate can be regulated by altering the response stimulus, and how and where it is attached. There are many different chemical reactions that can be used to derivatize polymer-reactive groups for subsequent conjugation to biomolecules. Polymers with pendant reactive groups can be synthesized using random copolymerization techniques with functional comonomers. One of the most attractive advantages of random copolymers is that they can be designed to react with more than one protein or at multiple sites. Polymer with only one reactive end group can be also synthesized using chain transfer polymerization techniques with chain transfer agents. Usually, the lysine amino groups are the easiest and preferred reactive sites for conjugation of polymers to proteins, but other possible sites include -COOH groups of aspartic or glutamic acid, -OH groups of serine or tyrosine and, especially, -SH groups of cysteine residues. Because the conjugations usually are designed to occur randomly at lysine sites, the polymer may react with and conjugate to several different lysine sites on the same protein surface. If the amino acid sequence of a protein is known, on the other hand, site-specific mutagenesis of the protein may be used to substitute one amino acid at a specific site with another. For example, a cysteine residue can be introduced by such techniques to yield a mutant protein with an exposed thiol group. Then, polymers with terminal or pendant maleimide, vinyl, or vinyl sulfone groups can be conjugated to the protein. Stayton et al. [39] were the first to demonstrate the versatility of site-specific conjugation by conjugating PNIPAAm far away from and close to the active site of streptavidin. In another approach, non-natural amino acids may be inserted into a protein structure by genetic engineering of the protein expression process within cells [40]. Although coupling polymers to reactive sites on proteins allows the synthesis of conjugates from a library of preformed polymers (commonly called the “grafting-to” approach), it is also possible to derivatize the thiol group with a polymerization initiator group via a disulfide linkage, and then polymerize the smart polymer directly from that particular site. This “grafting-from” approach is particularly well suited to the preparation of conjugates with high molecular weight homopolymers or block copolymers. This approach also offers the ease purification because unreacted monomer is readily removed from the final polymer-protein conjugate.

Schematic illustration of a smart-polymer-conjugated enzyme. The smart polymer serves as a molecular antenna and actuator to reversibly turn the enzyme activity on and off in response to external stimuli
In the following sections of this chapter, we review different types of smart polymer-biomolecule conjugates that have been developed in the last few decades. In Sect. 6.2, we describe the classifications of smart bioconjugates on the basis of conjugation routes and types of biomolecule that may be conjugated to a smart polymer. The characterization methods are discussed in Sect. 6.3. In Sect. 6.4, certain applications of the smart bioconjugates are discussed. The chapter ends with an overview of some of the future trends in applications in biotechnology and biomedicine.
6.2 Classification on the Basis of Conjugation Routes
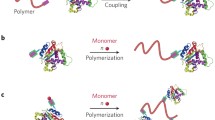
Nowadays, a variety of bioconjugation techniques are known for both in vitro and in vivo studies. In general, conjugation of biomolecules can be broadly separated into two categories: (1) covalent and (2) noncovalent conjugations. The covalent conjugation is divided into three sub-categories: (a) random conjugation (b) site-specific conjugation, and (c) grafting from approach using protein-reactive initiators. The noncovalent conjugation is divided into two subcategories: (d) receptor-ligand and (e) electrostatic conjugations (see Fig. 6.2).

Schematic illustration of polymer-protein conjugation methods by (1) covalent and (2) noncovalent modification. The covalent conjugation is divided into three subcategories: a random conjugation, b site-specific conjugation, and c grafting-from protein-reactive initiators. The noncovalent conjugation is divided into two subcategories: d receptor-ligand and e electrostatic conjugation
6.2.1 Random Conjugation
The random conjugation has been achieved through pendant groups along the polymer backbone or one end of a polymer (Fig. 6.2a). Common types of random conjugation are amine coupling of lysine amino acid residues through amine-reactive succinimidyl esters, sulfhydryl coupling of cysteine residues with either other sulfhydryl groups or in a Michael addition via a sulfhydryl-reactive maleimide. The use of N-hydroxyl-succinimidyl ester (NHS) is one of the most common methods for condensation reaction between a carboxylic acid and an amino group of membrane proteins. Other possible sites include -COOH groups of aspartic or glutamic acid, -OH groups of serine or tyrosine, and -SH groups of cysteine residues. For example, free-radical polymerization of NIPAAm initiated by 2,2’-azobisisobutyronitrile (AIBN) with β-mercaptoethanol as the chain-transfer agent resulted in the α-hydroxyl-functionalized PNIPAAm. The α-hydroxyl group was then converted to the vinyl sulfone through coupling with divinyl sulfone in the presence of a base. Maleimides react selectively with the thiols of cysteine residues in the pH range of 6.5–7.5 via Michael addition. Multiple attachments are especially likely to occur if a polymer chain has multiple pendant amino-reactive groups and the protein has multiple lysine sites. Conjugations via end groups, especially where there is only one reactive end group, usually yield conjugates that are the most clearly defined in structure and composition. Chen et al. [41] have compared the enzyme activity of α-amylase covalently immobilized with NIPAAm by single-point attachment and multiple-point attachment. α-Amylase immobilized to the polymer with a single functional end group had a higher activity than that immobilized to the polymer with multiple functional groups, and the specific activity can be higher than that of the free enzyme. However, the enzyme immobilized to the latter polymer is more thermally stable and the recovery is better than with the former one. Matsukata et al. [42] have also compared the enzyme activity of trypsin covalently immobilized with NIPAAm by single-point attachment and multiple- point attachment. The single-point attachment was achieved by conjugating a semitelechelic copolymer of NIPAAm with N,N-dimethylacrylamide (DMAAm) with a single carboxyl end group to trypsin. The multiple-point attachment was achieved by conjugating a copolymer of NIPAAm with DMAAm and acrylic acid (AAc) to trypsin. The trypsin conjugated to the polymer with multiple points showed higher stability than that immobilized to the polymer with a single point. Polymers with only one reactive end group may be synthesized using traditional chain transfer polymerization techniques with mercaptyl amines, carboxylic acids, or alcohols as chain transfer agents.
PNIPAAm with one carboxyl group at the end of the chain has been conjugated to β-d-glucosidase [43] and trypsin [44]. The carboxyl groups were activated using N,N’-dicyclohexylcarbodiimide (DCC) and N-hydroxysuccinimide (NHS). More than 95 % of the conjugate can be recovered by thermally induced precipitation. Takei et al. have polymerized a carboxyl-terminated PNIPAAm by polymerization with 3-mercaptopropionic acid, and it was used for conjugation to IgG via a coupling reaction of an activated ester with a protein amino group. The IgG-PNPIPAm conjugates reduced the original specific antigen binding activity by approximately 60 % [16]. More recently, “living” free radical polymerization methods, such as reversible addition-fragmentation chain transfer (RAFT) polymerization [45] and atom transfer radical polymerization (ATRP) [46], have been used to yield polymers with controlled MWs, narrow MW distributions, and one reactive end group. Amine-reactive polymers have been synthesized with NHS- and acetal-functionalized initiators for ATPR in order to target lysine side chains in proteins. The NHS-functionalized initiator was synthesized in one step by coupling NHS with 2-bromopropionic acid to form the 2-bromopropionate NHS ATRP initiator [47]. Acetal-functionalized initiators are employed because the resulting polymer can be hydrolyzed, exposing aldehyde groups for reaction with amines via reductive amination [48]. This functionalized initiator for ATRP was synthesized by reaction of 2-chloro-1,1-dimethoxyethane, followed by esterification with 2-bromoisobutyryl bromide. One special feature of these controlled, living free radical polymerizations is that block copolymers may also be synthesized where one or both of the blocks are smart polymers. In the latter case, the two blocks can have different responses to different stimuli [35, 49].
In the case of random conjugation, however, problems in controlling the conjugation chemistry frequently arise because the number and location of lysine residues vary greatly in native proteins and because the location of reactive groups on proteins is random. Thus, the stoichiometry of the protein-polymer conjugate and the attachment sites of the polymer to the protein cannot be precisely controlled, although they are factors that might have important implications for protein stability and function. These limitations can be circumvented by the appropriate design of polymer-protein conjugates.
6.2.2 Site-Specific Conjugation
Although random conjugation strategies offer facile procedures, the conjugated products are heterogeneously linked at different sites with distinct activity, binding efficacy, or in vivo pharmacokinetics. Therefore, site-specific conjugation strategies have been developed (Fig. 6.2b). If the amino acid sequence of a protein is known, site-specific mutagenesis (“genetic engineering” of the protein) may be used to substitute one amino acid at a specific site with another. For example, a cysteine residue can be introduced by such techniques to yield a mutant protein with an exposed thiol group. Then, polymers with terminal or pendant maleimide, vinyl, or vinyl sulfone groups can be conjugated to the protein (Fig. 6.3). These groups react preferentially with thiol groups rather than with lysine amino groups [39, 50]. Furthermore, pendant or terminal thiol or disulfide groups on smart polymers may be used to form disulfide linkages of the polymer to the protein [51]. Such site-specific conjugation of smart polymers to proteins can be carried out either near or far away from the active site. In the former case, the protein activity can be directly affected. Thus, by stimulating the polymer first to collapse and then to rehydrate, the collapsed smart polymer coil can first “block” (turn “off’”) the protein active site, and then “unblock”’ (turn back “on”) the active site [52]. However, it is expected that both the location of the conjugation site and the volume (or MW) of the smart polymer coil, either before or after collapse, can control protein activity. Krishnamurthy et al. [53] have reported dissociation constants and “effective molarities” (M eff ) for the intramolecular binding of a ligand covalently attached to the surface of a protein by oligo(ethylene glycol) (oEG) linkers of different lengths (n = 0, 2, 5, 10, and 20) and compared these experimental values with theoretical estimates from polymer theory. They found a strong influence on enzyme inhibition of both the conjugation site and the MW of a PEG-inhibitor molecule conjugated at various sites on an enzyme (see Fig. 6.4). The value of M eff is lowest when the linker is too short (n = 0) to allow the ligand to bind noncovalently at the active site of the protein without strain. On the other hand, the value of M eff is highest when the linker has the optimal length (n = 2) to allow such binding to occur, and decreases monotonically as the length increases past this optimal value. These experimental results are not compatible with a model in which the single bonds of the linker are quantitatively compatible with a model that treats the linker as a random-coil polymer.

Thiol-reactive end-groups. a Vinyl sulfone end-group, b maleimide end-group, c a,w-pyridyl disulfide end-group, and d methoxy carbonyl end-group

Effective molarity (M eff) with linker length (n; related to the root-mean-square distance between the ends of the linker) for an intramolecular protein-ligand binding event [53]
Chilkoti et al. [54] were the first to demonstrate the versatility of site-specific conjugation by conjugating PNIPAAm far away from the active site of cytochrome-b 5 . A genetically engineered mutant of cytochrome-b 5 , incorporating a unique cysteine residue, was conjugated to maleimide end-functionalized PNIPAAm. Since the native cytochrome-b 5 does not contain any cysteine residues, this substitution provided a unique attachment point for the polymer. The resultant polymer-protein conjugate displayed LCST behavior and could be reversibly precipitated from solution by varying the temperature. This approach has proved to be very versatile, and a large number of polymer-biopolymer conjugates incorporating biological components as diverse as antibodies, protein A, streptavidin, proteases, and hydrolases have now been prepared. The biological functions or activities of these conjugate systems were all similar to their native counterparts, but were switched on or off as a result of thermally induced polymer phase transitions. Both temperature and photochemically switchable enzymes, which display varying and opposite activities depending on temperature or UV/Vis illumination, have also been demonstrated [19, 55]. Pyridyl disulfide- and maleimide-functionalized initiators for ATRP have also been synthesized to enable conjugation to the free cysteines of proteins. Unlike targeting amines, this approach allows for the formation of well-defined conjugates. The pyridyl disulfide-functionalized 2-bromoisobutyrate initiator for ATRP was synthesized in two steps by the reaction of 2,2’-dithiopyridine with 3-mercapto-1-propanol to form pyridyl disulfide propanol [56]. Subsequent esterification yielded the ATRP initiator. An advantage of this approach is that no post polymerization modification of the chains was necessary prior to conjugate formation and that the conjugate formation was reversible. The maleimide initiator for ATRP was synthesized in three steps beginning with a Diels–Alder reaction between maleic anhydride and furan to form 3,6-epoxy-1,2,3,6-tetrahydrophthalic anhydride [57]. Subsequent reaction with ethanolamine formed the protected maleimide alcohol, which was then esterified with 2-bromoisobutyryl bromide to afford the initiator.
Another approach for a site-specific conjugation is to take advantage of the presence of an N-terminal serine or threonine, which can be converted by very mild periodate oxidation to a glyoxylyl derivative [58], and to functionalize the polymer with a complementary reactive function, the aminooxy group. An analogous method was shown to be straightforward for incorporating synthetic peptides into a protein backbone by introducing a hydrazide function at the C-terminus of one fragment or peptide to be recoupled [59] and a glyoxylyl group at the N-terminus of the other. In the same way, the aminooxy function will specifically react with the generated aldehyde group at the N-terminus of the polypeptide to form an oxime bond, which is more stable than the hydrazone bond. This approach has already been exploited for the site-specific labeling of IL-8 at the N-terminus with an aminooxy-functionalized fluorescent probe [60]. A convenient method for the construction of site-specifically modified polymer-protein conjugates by generating a reactive carbonyl group in place of the terminal amino group has also been reported [61]. If the protein has N-terminal serine or threonine, this can be achieved by very mild periodate oxidation and generates a glyoxylyl group. A method less restricted by the nature of the N-terminal residue, but which requires somewhat harsher conditions, is metal-catalyzed transamination, which gives a keto group. The N-terminal-introduced reactive carbonyl group specifically reacts, under mild acidic conditions, with an aminooxy-functionalized polymer to form a stable oxime bond.
6.2.3 Grafting from Protein-Reactive Initiators
Generating polymers directly from proteins at defined initiation sites provides the opportunity to evade all post polymerization modification strategies and protein-polymer coupling reactions (Fig. 6.2c). An additional advantage of polymerizing directly from proteins is that the purification of the final bioconjugate from an unreacted monomer or catalyst is simplified. Also, the precise number and placement of polymer chains is predetermined, thereby facilitating the synthesis and characterization of well-defined conjugates. RAFT and ATRP have been proven most effective for the synthesis of polymer-protein conjugates by the grafting-from approach. ATRP is well suited for the preparation of bioconjugates by a grafting-from approach (Fig. 6.5). Early reports described the preparation of “protein-reactive initiators” of bovine serum albumin (BSA) [62] and chymotrypsin [46].

Protein-reactive initiators. a NHS, b acetal, c pyridyl disulfide, d protected maleimide, e azide, and f biotinylated initiators for ATRP. g Biotinylated RAFT chain transfer agent. h Biotinylated-initiator for cyanoxyl-mediated polymerization
ATRP is well suited for the preparation of bioconjugates by a grafting-from approach owing to its applicability to a wide range of monomers commonly polymerized in aqueous environments, such as (meth)acrylates and (meth)acrylamides. Heredia et al. [62] have developed a technique to produce well-defined conjugates via ATPR directly from a protein. Free cysteines, Cys-34 of bovine serum albumin (BSA) and Cys-131 of T4 lysozyme V131C, were modified with initiators for ATRP either through a reversible disulfide linkage or irreversible bond by reaction with pyridyl disulfide- and maleimide-functionalized initiators, respectively (Fig. 6.5c and d). Polymerization of NIPAAm from the protein macroinitiators resulted in thermosensitive BSA-PNIPAAm and lysozyme-PNIPAAm in greater than 65 % yield. Bontempo and Maynard have also conducted polymerization directly from an SA macroinitiator [63]. The protein initiator was formed by coupling a biotinylated ATRP initiator with SA in phosphate-buffered saline (PBS)-methanol to obtain an SA macroinitiator (Fig. 6.5f). Polymerization of NIPAAm in water at ambient temperature with copper bromide and 2,2’-bipyridine (bipy) formed the SA-PNIPAAm conjugates. Polymerization was conducted in the presence of 2-bromoisobutyrate-functionalized Wang resin as the sacrificial initiator in order to increase the concentration of initiation sites. Le Droumaguet and Velonia [64] reported on the facile and high-yielding ATRP-mediated preparation of a giant amphiphile of BSA-polystyrene. In this work, a maleimide-capped ATRP initiator was synthesized and coupled to C34 of BSA under a mild condition. Depp et al. [65] have reported a comparison of the in vitro serum stability and enzyme activity retention for PEGylated chymotrypsin along with biocompatible chymotrypsin conjugates prepared by polymerizing from chymotrypsin covalently modified with an ATRP initiator. The chymotrypsin-initiated ATRP conjugates had higher catalytic activity than PEGylated chymotrypsin. These exciting results suggest that polymerizing from proteins is promising for the development of long-lasting biocompatible pharmaceuticals. Magnusson et al. [66] have also demonstrated improved pharmacokinetic properties upon polymerization from recombinant human growth hormone (rhGH) via ATRP. Gao et al. [67] have reported PEG conjugates via site-specific polymerization from the N-terminus of myoglobin and the C-terminus of green fluorescent protein (GFP) [68].
Despite the success of ATRP for the synthesis of polymer-protein conjugates, the reliance on transition metal catalysts has often been cited as a potential concern. In this respect, RAFT becomes a highly useful method for the synthesis of polymer conjugates. Polymer conjugates have been prepared by polymerization from protein modified with suitable RAFT chain transfer agents. The RAFT agent (Z-C(=S)S-R) is immobilized to the biological substrate via its R-group such that the thiocarbonylthio moiety is distal to the protein and readily accessible for chain transfer with propagating chains in solution, a key step of the RAFT mechanisms responsible for molecular weight control. Moreover, this “R-group approach” leads to the relatively labile thiocarbonylthio group residing on the free end of the immobilized polymer such that it is not responsible for the conjugation linkage. Therefore, the conjugates prepared in this way may demonstrate increased stability and the potential for transformation into thiol groups for subsequent surface immobilization, labeling, or chain extension. For example, a RAFT agent containing an activated ester was conjugated to amines on lysozyme, and NIPAAm was polymerized by grafting directly from the modified protein in aqueous buffer [69]. Retention of the active thiocarbonylthio moieties on the ω end of the conjugated chains allowed chain extension via polymerization of N,N-dimethylacrylamide (DMAAm) to yield lysozyme-PNIPAAm-b-PDMAAm block copolymer conjugates. De et al. [70] have prepared PNIPAAm-BSA conjugates by RAFT polymerization from a macro-RAFT agent prepared by modifying the BSA at its single reduced cysteine residue. This was accomplished in aqueous media. Hong and Pan [71] synthesized PNIPAAm-b-poly(N-(2-hydroxylpropyl)methacrylamide) (PHPMA) via one-step RAFT polymerization using biotinylated trithiocarbonate as the RAFT agent (Fig. 6.5h). An alternative approach for the formation of well-defined polymer-protein conjugates involves immobilization of the RAFT agent to the protein via its Z-group. While not strictly a grafting-from process, this highly efficient “transfer-to” method was the first reported to lead to polymer-protein conjugates directly during RAFT polymerization [72]. A benefit of this approach is that having the polymer linked to the protein via its Z-group ensures that only dormant “living” chains are conjugated, because all termination products remain in solution. Additionally, to allow separate characterization of the polymer or for in vivo applications in which triggered chain cleavage is beneficial, having the polymer and protein linked via the relatively labile thiocarbonylthio group can be advantageous.
In another approach, nonnatural amino acids may be inserted into a protein structure by genetic engineering of the protein expression process within cells, combined with appropriate cell culture conditions [40, 73–76]. Figure 6.6 shows a variety of nonnatural amino acids bearing different reactive groups. Recently, the synthesis of a nonnatural amino acid bearing an ATRP initiating group, 4-(2’-bromoisobutyramido) phenylalanine, and its genetic expression into the GFP have been reported [77, 78]. This was accomplished using an engineered M. jannaschii tyrosyl tRNA/aminoacyl-tRNA-synthetase pair vector in E. coli. By using genetic engineering, the specific placement of the initiating amino acid was selected to be expressed at the 134 position of GFP, thereby protecting the protein’s active sites and structurally weak regions. Furthermore, genetic engineering allows precise control over the number of chains attached (i.e., the number of initiation sites) to the protein, overcoming a primary drawback of traditional protein-polymer conjugates. Thus, genetically engineered proteins have unique capabilities to solve some of the traditional limitations of smart conjugates.

Structures of non-natural amino acids carrying a variety of specialty side groups
6.2.4 Receptor-Ligand Conjugation
An alternate route to prepare well-defined protein reactive polymers involves using polymers containing ligand end-groups (Fig. 6.2d). The near-covalent bond between SA and biotin has generated much interest in the synthesis of polymers with biotin end-groups for applications in biotechnology owing to the high-affinity interaction with K a ≈1015 M−1. The naturally occurring carboxylic acid of biotin presents an easy handle for modification. Biotinylated smart polymers have been synthesized for conjugation to both SAs. Kulkarni et al. [79] synthesized a biotinylated PNIPAAm via RAFT polymerization of NIPAAm. Hydrolysis of the dithioester end-group in a methanol-aqueous sodium hydroxide solution resulted in a thiol-terminated PNIPAAm. Subsequent coupling with a maleimide-functionalized biotin formed the biotinylated PNIPAAm. Aggregation of mesoscale SA-PNIPAAm particles prepared from these conjugates was investigated for potential use in microfluidic devices for the capture and release of biomolecules [80]. Giant amphiphiles comprised of an SA-polystyrene conjugate were synthesized through the use of a biotinylated polystyrene [81]. The biotin functionality was introduced via coupling of an amine-functionalized biotin derivative with a carboxylic acid-terminated polystyrene. Cofactor reconstitution of horseradish peroxidase (HRP) was also demonstrated to prepare amphiphilic, bioactive protein-polymer conjugates [82]. A cofactor-terminated polymer was synthesized via amidation between a carboxylic-acid-terminated polystyrene and a monoprotected diamine. Subsequent deprotection to form the free amine, coupling with one carboxylic acid group of protoporphyrin IX, and addition of ferrous chloride tetrahydrate resulted in a polystyrene cofactor. HRP was reconstituted with the polymeric cofactor, resulting in vesicular aggregates that displayed enzymatic activity. This approach has great potential for producing controlled nanostructures of bioactive enzymes and proteins for biotechnology applications. Sun and colleagues [83, 84] have employed a biotinylated initiator for cyanoxyl-mediated free-radical polymerization of glycomonomers. The arylamine initiator was synthesized in two steps after coupling an NHS-activated biotin with p-nitrobenzylamine and reducing the nitro group with hydrogen/palladium. The formation of the diazonium salt followed by the addition of sodium cyanate and glycomonomer generated the biotinylated polymer. SA-glycopolymer conjugates were readily achieved by the interaction of the two species. The biotinylated ATRP initiators were also synthesized by activating the carboxyl group of biotin with N,N’-disuccinimidyl carbonate, followed by the addition of 2-(2-aminoethoxy)ethanol. Esterification of the resulting alcohol with 2-chloropropionic acid or 2-bromoisobutyric acid formed the biotin ATRP initiators. Biotinylated PNIPAAm was synthesized from these initiators [85].
6.2.5 Smart Conjugations with Multiple Proteins
The majority of protein-polymer conjugates contain one protein and one to several polymer chains. Recently, homo- and heterotelechelic bioreactive polymers have been synthesized. The incorporation of the same protein onto each end of a polymer generates a triblock material with a wide range of possible applications. The polymer should provide the same benefits as described above, and in addition, exhibit higher binding affinities than mono-functionalized conjugates with improved pharmacokinetics over dimers synthesized using small molecules.
ATRP telechelic polymers have been prepared by employing bis-functional initiators, with subsequent transformation of the halogen chain ends (Fig. 6.7a). Kopping et al. [86] reported the synthesis of aminooxy-end functionalized polystyrene using ATRP followed by ATR coupling. A 1-bromoethyl ATRP initiator with an NHS group was used to polymerize styrene by copper-mediated ATRP with a high initiator efficiency. Polymerization kinetics showed that polymers were produced with the targeted molecular weights and low PDIs. Subsequent ATR coupling and deprotection with hydrazine yielded the telechelic aminooxy end-functionalized polymers. End-group reactivity was demonstrated using 4-bromobenzaldehyde as a model to confirm efficient and complete oxime bond formation. In a similar manner, cysteine reactive telechelic polystyrene was also synthesized [87]. In this example, a dimethylfulvene-protected maleimide-functionalized ATRP initiator was used to polymerize styrene. The authors have also reported the straightforward synthesis of homodimeric protein polymer conjugates by RAFT polymerization that exploits the inherent reactivity of trithiocarbonate end-groups [88]. A telechelic maleimide-end-functionalized PNIPAAm was synthesized by RAFT polymerization. Subsequent radical addition of the protected maleimide azo-initiator to the chain ends and retro-Diels Alder afforded the telechelic maleimide-end-functionalized PNIPAAm. Conjugation to a V131C mutant T4 lysozyme with only one reactive cysteine gave the protein dimer. Four-armed PNIPAAm was also synthesized by RAFT polymerization in the presence of a tetrafunctionalized trithiocarbonate chain transfer agent (CTA) (Fig. 6.7b) [89]. Maleimide functional groups were introduced at the chain ends by heating the polymers in the presence of a furan-protected azo-initiator. This allowed for site-specific conjugation of V131C T4 lysozyme to the polymers to generate multimeric protein-polymer conjugates. This simple strategy provides ready access to star protein-polymer conjugates for application in the fields of drug discovery, drug delivery, and nanotechnology.

Heterodimeric conjugates offer other interesting possibilities. A variety of strategies have been extended toward the synthesis of heterodimeric protein-polymer conjugates (Fig. 6.7c). An α-azide, ω-pyridyl disulfide CTA was used to produce well-defined PNIPAAm [90]. The azide was utilized for a Huisgen cycloaddition with an alkyne-functionalized biotin, allowing for conjugation to SA. The pyridyl disulfide was used for the reversible conjugation of glutathione or BSA. Heredia et al. [91] have demonstrated the ability to synthesize heterodimeric protein-polymer conjugates via radical coupling with a furan-protected azo-initiator to biotinylated PNIPAAm synthesized by RAFT. NIPAAm was polymerized in the presence of a biotinylated CTA and biotinylated CTA with a disulfide between biotin and the site of polymer growth [92]. BSA was then conjugated to the maleimide end-group of both polymers. The ability to tether two biomolecules would allow for surface immobilization or attachment of various tags, fluorophores, peptides, antibodies, and other proteins to be linked to the biomolecule-polymer conjugate of interest.
6.2.6 Electrostatic Conjugation
Electrostatic interactions also represent an interesting class of noncovalent conjugations useful for stabilizing proteins via multiple-point attachment (Fig. 6.2e). Although the resulting conjugates are less well-defined than their analogs prepared by single-point attachment, the multi-point attachment of enzymes to polymers has been widely studied and is considered to be the most general approach to stabilize these proteins against different denaturing conditions [93]. Indeed, multi-point attachment has increased the thermal stability and enzymatic activity of various enzymes. It has also been reported that ionic interactions between the polymer/enzyme played a role in the pH-dependent activity profiles of the conjugate [94]. Several groups have explored the interactions of cationic polymers and DNA [95]. Both electrostatic and hydrophobic interactions contribute to the formation of complexes between cationic polymer/DNA. Because many cationic polymers can condense DNA spontaneously, a series of chemically different cationic polymers are currently being investigated for gene delivery applications. The cationic polymer/DNA binding has also been utilized in a microarray format for the sensitive detection of DNA without the need for target labeling. Electrostatic conjugation has also been employed for the surface coating of cells or viruses represented by a layer-by-layer (LbL) technique of anionic and cationic polymers. The versatility of this technique allows for the construction of a thin polymer layer on the surface. In addition, the surface property can be controlled by the outermost layer of the polymer [96]. When the LbL method is applied on the erythrocyte surface, a camouflage shell can prevent antibody recognition [7, 8]. The multilayered film consisting of a protective shell and a camouflage shell was successfully created on RBCs through the LBL technique by Mansouri et al. [8]. A drawback of this technique is cytotoxicity when polycations are used as the first layer, which directly interacts with the cell surface. In addition to mammalian cells, electrostatic conjugation of surface proteins has evolved into a powerful tool for engineering the surfaces of viruses, bacteria, and yeast cells, for example. The viral surface can potentially be modified to provide a better means of preparation, purification, concentration, detection, tracking, imaging, and targeting for diagnostics, vaccine development, and drug/gene therapy. We have successfully created a nanoscale layer of hyaluronic acid (HA) on inactivated Hemagglutinating Virus of Japan Envelope (HVJ-E) via an LbL assembly technique for CD-44 targeted delivery [97].
6.2.7 Elastin-Like Polypeptides
Recombinant artificial elastin-like polypeptides (ELPs), which are composed of Val-Pro-Gly-Xaa-Gly amino acid repeat units (Xaa is a ‘guest residue’ except proline), were reported to replace synthetic smart polymers as smart bioconjugates. Interestingly, ELPs are water soluble below their transition temperature, but they precipitate owing to their aggregation caused by hydrophobic interactions above the transition temperature. The phase transition temperature can be precisely controlled by varying the chain length and peptide sequence. Various ELP versions responsive to pH, light, and other stimuli, such as an electrochemical potential or analyte concentration, can be found in the literature. Rodríguez-Cabello et al. [98] have developed the ELPs that presented modulated pH and T sensitivities covering the most interesting range of biomedical applications. ELPs have also been modified with photoresponsive molecules such as azobenzenes and spiropyranes. A series of experiments have been performed by Urry [99] for the study of designed ELPs for more than a decade. The genetically engineered protein-based polymers can provide many advantages. First, they will, in principle, be able to show the same simple or complex properties present in natural proteins. In this sense, this method offers an opportunity to exploit the huge functional resources that have been hoarded and refined to the extreme by biology during the long process of natural selection. Second, we can design and produce materials exhibiting desired functions of particular technological interest because we can construct a coding gene. Third, they are characterized as being strictly monodisperse and can range from a few hundred daltons to more than 200 kDa. Fourth, the number of different combinations attainable by combining the 20 natural amino acids is practically infinite. In a simple calculation, the number of possible different combinations to obtain a small protein consisting of 100 amino acids is as high as 1.3 × 10130. The smart response of ELPs has already found applications in different fields. For example, Chilkoti and colleagues have designed temperature- and pH-responsive ELPs for targeted drug delivery [23]. They have obtained responsive ELPs that conjugate to drugs and enable thermally targeted drug delivery to solid tumors with their transition temperature between body temperature and the temperature in a locally heated region [100]. In another example, Kostal et al. [101] have designed tunable ELPs for heavy metal removal. The presence of the histidine clusters enabled Cd2C to bind strongly to the biopolymers. Recovery of biopolymer-Cd2C complexes was easily achieved by triggering aggregation upon increasing the temperature above the transition temperature.
6.3 Characterization Methods
A variety of techniques for characterizing bioconjugates have been reported in the literature. The most commonly used methods include microscopy, spectroscopy, scattering, mass spectroscopy, and thermal technique. Some of the most recently used methods for conjugate characterization are described in the following subsections.
6.3.1 Determination of Molecular Sizes
Owing to an important influence of polymer architecture on the grafting process, the determination of the molecular weight is necessary. Gel permeation chromatography (GPC) is an established method of determining the molecular mass of polymers. The detectors used are either refractive index (RI) or ultraviolet (UV) detectors. Compared with other methods of analysis, such as osmometry and static light scattering, it has the advantage that it determines not merely average values but the complete distribution of molecular weights. In GPC, molecules are separated according to their hydrodynamic volume. Their molecular weights (MWs) and molecular weight distributions can be determined from the measured retention volume (RV) using a calibration curve (log MW against RV), which must be set up with the aid of a number of standards of known molecular weight. However, as the relationship between molecular weight and size depends on the type of polymer, the calibration curve depends on the polymer used, with the result that true molecular masses can only be obtained if the calibration standards and the sample are of the same type. In all other instances, the results are only relative. Large deviations from the true molecular weight occur in branched samples in particular, because the molecular density in such a case is substantially higher than in linear chains.
To overcome this limitation and meet the growing demand for the characterization of increasingly complex polymers, detectors sensitive to molecular weight, such as light scattering, can be used in GPC. The signal of light-scattering detectors is directly proportional to the molecular weights of the polymers. The concentration and refractive index increment (dn/dc) must be known to determine the molecular mass. The advantage of light scattering in GPC is that the molecular weight can be directly determined without a calibration curve, provided that the signal-to-noise ratio is adequate for this. When it comes to determining the structure, on the other hand, viscosity detectors are more suitable, because they measure structural differences directly and can be used over a substantially wider range of molecular weights. Therefore, triple detection combines these detection capabilities into a single system to give true molecular weight distributions as well as size distributions and further structural information, such as the degree of polymer branching.
Mass spectrometry (MS) also deals with accurate mass measurement by producing charged molecular species in vacuum and their separation by magnetic and electric fields on the basis of the mass-to-charge ratio. Because MS requires the ability to deconvolute molecular fragments, it is more appropriate for low-molecular-weight derivatized proteins and conjugates. The development of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) allows the ionization of fragile macromolecules such as proteins of 105 molecular weight without promoting decomposition. Therefore, MALDI-TOF-MS is an attractive approach for the determination of the accurate molecular weight of intact proteins [88, 102]. Tao et al. used MALDI-TOF-MS to determine the architecture of multimeric lysozyme/PNIPAAm conjugates. They prepared polymer samples by mixing trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene] malononitrile (DTCB) matrix, potassium trifluoroacetate, and polymer in a ratio of 5:1:5 v/v/v [89]. The MALDI-TOF spectra gives an MW of 5470 Da for four-armed PNIPAAm, which was close to the theoretical value (6,000 Da) (Fig. 6.8a). Aminolysis with butylamine was utilized to cleave the arms from the core to confirm that the polymer contained four arms of similar molecular weight. The mass of the cleaved arms was 1,435 Da (theoretical value, 1,351 Da) (Fig. 6.8b). This result suggested that the desired four-armed star had been synthesized. The choice of the matrix depends on the type of analyte to be analysed.

MALDI-TOF spectra of four-armed PNIPAAm before a and after b aminolysis with butylamine [89]
6.3.2 Determination of Conjugation
A number of methods have demonstrated wide utility in the characterization of polymer/bioconjugates. Gel electrophoresis (GE) and GPC can be used to compare conjugate molecular weight to native protein and provide a semiquantitative measure of conjugate polydispersity. In GE, the presence of more than one conjugate band for a given conjugation protocol indicates a polydisperse conjugate population. The free (unconjugated) polymer usually results in a blurry band. Therefore, an ion exchange column or thermal precipitation may be needed to remove the conjugates from the free polymer. When protein is reacted with polymer to form 1:1 protein:polymer conjugates, several distinct bands are usually visualized. Each band corresponds to an integer multiple of polymer, indicating that there is a significant population of conjugates with more than one polymer per protein. This can be rationalized using the polymer micelle results. Tao and coworkers have performed sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis to determine the resulting structure of homodimeric lysozyme/PNIPAAm conjugates [88]. After Coomassie staining, the gel revealed two new spots with higher molecular weight than that of unmodified lysozyme. These new spots were attributed to the monomer and dimer protein/polymer conjugate adducts (Fig. 6.9). They have also performed SDS-PAGE analysis to determine the structure of heterotelechelic SA-BSA/PNPAAm conjugates [91]. Blue fluorescent SA was used to enable visualization by UV light and Coomassie Blue staining. SDS-PAGE of the SA-BSA heterodimer was observed first under UV light and then with Coomassie Blue stain. Using both visualization techniques, it was clarified that the SA was present in the conjugate and that the SA-BSA heterodimer was formed.

When biotinylated PNIPAAm is conjugated to SA, 2-(4’-hydroxyazobenzene) benzoic acid (HABA) assay can be used to characterize the binding stoichiometry of PNIPAAm to SA. In the HABA assay, displacement of HABA from SA by biotinylated polymer results in a loss of absorbance by HABA at 500 nm. This change in absorbance can be compared with a standard curve for the measurement of the average protein:polymer ratio. For molecular weights lower than 20 kDa, no more than four polymers may be added to a single SA. When the polymer molecular weight is above 20 kDa, only one polymer may be added to a given SA face, resulting in a maximum of two polymers per protein. By mixing SA:30 kDa PNIPAAm in a 1:1.5 mol ratio, conjugates with an average of 1:1 polymer:protein were created. These conjugates should provide one open face for subsequent biotin binding and are useful for further conjugation reactions.
6.3.3 Structural Analysis
Circular dichroic (CD) spectroscopy has also been applied to examine the changes in the tertiary structure of the enzyme before and after conjugation. CD has proved to be extremely useful in understanding the various structural elements in proteins, and structural transitions from order to disorder can be well documented by CD. Yan et al. [103] measured the CD spectra of the native trypsin and the conjugated trypsin with poly(3-dimethyl(methacryloyloxyethyl) ammonium propane sulfonate) (PDMAPS). The native trypsin has 8.6 % α-helix and 32.3 % β-sheet, while the conjugated trypsin has 7.17 % α-helix and 24.8 % β-sheet. The conjugation with PDMAPS had a weak effect on the content of the α-helix but a slightly stronger effect on the content of the β-sheet, leading to a reduction from 30.3 to 24.8 %. Sharma et al. [104] conjugated α-chymotrypsin with Eudragit S-100. The CD showed that there are definite changes in the secondary structure upon immobilization. The small α-helical content is totally gone and there is a decrease in the content of the β-sheet/β-turn structure. The randomness in the structure increased by about 17 %.
6.3.4 Determination of Transition-Point
The phase separation behavior of smart bioconjugate aqueous solutions has been investigated in the literature by a wide variety of experimental techniques, including IR spectroscopy, viscometry, light scattering, 1H NMR spectroscopy, fluorescence, calorimetry, Raman spectroscopy, and FTIR spectroscopy. These experimental techniques can provide different information during phase separation of smart polymers. The simplest method of detecting a point of phase separation in aqueous polymer solutions is the cloud-point method. It is based on a visual observation of the macroscopic phase separation at a specific temperature, pH, or salt concentration. The strong increase in the scattering intensity results from the grown concentration fluctuations near the critical point when the size of those fluctuations exceeds the wavelength of the scattered light. Usually, a standard UV-Vis spectrophotometer is used to determine the cloud point. One of the major advantages of this method is rapid and easy measurement. However, the cloud-point method only measures the relative changes in scattering intensity in the course of nearing the transition point. In addition, the solution may remain transparent, and it may be difficult to determine the phase transition point based on visual observation or even with the use of a UV-Vis apparatus.
Thermal techniques can aid in determining the thermal stability of the biomolecules. Differential scanning calorimetry (DSC) is used to study various material transitions including melting, crystallization, glass transition, and decomposition. DSC is an inexpensive and rapid method of measuring heat capacities of condensed phases. From these measurements, enthalpy changes for phase transitions can easily be determined. DSC has been applied to a wide variety of problems, from coal combustion to protein denaturation. Subsequent analysis can indicate the state of the bioconjugate, including the stability of the biomolecules, and structural information on both the polymer and biomolecule.
The light scattering (LS) technique is the most precise method of observing the process of phase separation not only in the polymer solutions, but also in binary mixtures of organic solvents. Static light scattering (SLS) allows one to follow the growing fluctuations of concentration by recording the scattering intensity at the scattering angles suitable for the hydrodynamic regime. DLS allows one to detect changes in the hydrodynamic size and shape of macromolecules and their aggregation following their precipitation in the course of transition. Kulkarni et al. [79] observed reversible particle formation and dissolution kinetics of mesoscale PNIPAAm/streptavidin conjugates by DLS. The scattering intensity in counts per second (CPS) was used as an index of nanoparticle formation, since sizing measurements cannot be performed within short time intervals. The transition from soluble conjugates to particles was found to be rapid (within 20 s) and to occur in a narrow temperature range at the critical point. The time lapse of ca. 100 s was observed after the temperature of the solution increased because there was an induction period with early nucleation events. After reversal of the temperature stimulus, the scattering intensity decreased by more than 90 % within 2 min. The particle formation and dissolution kinetics of smart bioconjugates also depend on the concentration. The particle size increases with the concentration of the bioconjugate. Interestingly, the particles were stable once formed (>16 h) and further dilution at the elevated temperatures did not significantly affect the size of the particles. Particle sizes were comparable for the 25.9, 14.9, and 4.8 kDa conjugates, with no significant change in size with a change in molecular weight (Fig. 6.10). The particle sizes also depend on the molecular weight of the PNIPAm used for conjugation. Conjugates of higher molecular weight polymers form more uniform particles. The heating rate is also important for particle formation of smart bioconjugates. Smaller particles were formed at higher heating rates, and nanoparticles of fixed sizes could be formed by varying the heating rate. For example, the size of the 14.9 kDa PNIPAAm-streptavidin conjugate could be tightly controlled to 675 nm at a concentration of 1 μM by choosing a heating rate of 0.12 °C/s. The particles had a wider distribution when formed at lower heating rates. On the other hand, the final temperature did not affect the particle size significantly. These results indicate that the particle size is largely governed by the kinetics of aggregation at defined concentrations and polymer molecular weights.

DLS analysis of the particle formation kinetics of mesoscale PNIPAAm/SA conjugates above the LCST. The particles formed by the PNIPAAm/SA conjugates are stable in size for over 16 h. In contrast, aggregates of biotinylated PNIPAAm aggregated over time into larger particles [79]
6.3.5 Determination of Catalytic or Binding Activity Assay
One of the most important characterizations for polymer/protein conjugates to exploit is the effect of polymer conjugation on the protein activity. Heterogeneity in the structure is generally reflected in the biological properties of the conjugate and often results in decreased protein activity. Enzyme activities are determined by measuring the amount of product that is formed when an enzyme acts upon a specific substrate. The speed at which the enzyme acts upon a substrate and converts it to a product is affected by several factors, including the specific substrate that is used in the reagent, the concentration of the substrate, the pH of the reagent containing the substrate, and the presence or absence of certain compounds in the reagent that modulate the rate at which an enzyme can work. Heredia et al. [62] compared the lytic ability of the PNIPAAm/lysozyme with respect to the lyophilized substrate Micrococcus lysodeikticus. Lysozyme, lysozyme initiators, and isolated lysozyme conjugates were prepared with equal protein concentrations, and then the enzyme solution was mixed with M. lysodeikticus solution. Upon cell wall lysis, the solution became less turbid, and this decrease in absorbance was monitored at 450 nm. Activity was expressed in activity units (AU). One AU is defined as a change in absorbance of 0.001 per min. The lytic activity assay was also performed on the lysozyme conjugates using fluorescein-labeled M. lysodeikticus. When protein/polymer conjugates were prepared by polymerization from the initiator-modified proteins, the enzymatic activity was fully retained. Therefore, creating well-defined conjugates is important, and site-specific modification of the protein is a better approach for preparing such biomolecules.
Site-specific modification of proteins is also important for directed immobilization onto surfaces and ensures that biorecognition sites are accessible. For smart polymer switches, placement of the polymer chain near the protein or enzyme active site is critical for reversible activity control. Shimoboji et al. [19, 55] have successfully demonstrated thermally or photoinduced switching of enzyme activity on the basis of the site-directed conjugation of end-reactive smart polymers to a unique cysteine residue positioned near the enzyme active site. The polymer was conjugated to the endoglucanase 12 A (EG 12A) site-directed mutant N55C, directly adjacent to the cellulose binding cleft, and to the S25C mutant, where the conjugation site is more distant. The N55C conjugate displayed a greater activity shutoff efficiency in the collapsed polymer state than the S25C conjugate. Increasing the polymer molecular weight was also shown to increase the shutoff efficiency of the switch. Related to these effects of conjugation site and polymer size, the switching efficiency was found to be strongly dependent on substrate size. With a small substrate, o-nitrophenyl-β-D-cellobioside (ONPC), there was minimal blocking of enzyme activity when the polymer was in the expanded state. With a large substrate, hydroxyethyl cellulose (HEC), there was a large reduction of enzyme activity in the polymer expanded state, even with relatively small polymer chains, and a further reduction when the polymer was collapsed. Similar general trends for the interactive effects of conjugation site, polymer size, and substrate size were observed for immobilized conjugates. To elucidate the mechanism of the stimuli-response switches, the kinetic parameters K m and k cat were determined by Lineweaver-Burk analysis assuming Michaelis-Menten conditions. Kinetic studies demonstrated that the switching activity was due to the blocking of substrate association by the collapsed polymers. These investigations provided mechanistic insight that can be utilized to design molecular switches for a variety of stimuli-responsive polymer-protein conjugates. When polymers are conjugated to an antibody, a competition assay and an ELISA can be used to characterize the binding activity through the measurement of analyte depletion.
6.3.6 Cytotoxicity Assay
Since many of the polymer bioconjugates are intended for use as biomedical and/or therapeutic polymers, in vitro cytotoxicity assays are essential for determining the responses of cells to bioconjugates. Cell viability, cell proliferation and many important live-cell functions, including apoptosis, cell adhesion, chemotaxis, multidrug resistance, endocytosis, secretion, and signal transduction, can be stimulated or monitored with various chemical and biological reagents. Many of these processes lead to changes in intracellular radicals, free-ion concentrations, or membrane potential that can be followed with appropriately responsive fluorescent indicators. Proliferation assays are primarily designed to monitor the growth rate of a cell population or to detect daughter cells in a growing population. Fluorometric assays of cell viability and cytotoxicity are easy to perform with the use of a fluorescence microscope, fluorometer, fluorescence microplate reader, or flow cytometer, and they offer many advantages over traditional colorimetric and radioactivity-based assays. The LIVE/DEAD Viability/Cytotoxicity kit provides an exceptionally easy fluorescence-based method for determining the viability of adherent or nonadherent cells and for assaying cytotoxicity. The kit comprises two probes: calcein AM and ethidium homodimer-1. Calcein AM is a fluorogenic esterase substrate that is hydrolyzed to a green-fluorescent product; thus, green fluorescence is an indicator of cells that have esterase activity as well as an intact membrane to retain the esterase products. Ethidium homodimer-1 is a high-affinity, red-fluorescent nucleic acid stain that is only able to pass through the compromised membranes of dead cells.
Reagents for counting cells and quantitating cell proliferation are also valuable research and diagnostic tools. Most cell proliferation assays estimate the number of cells either by incorporating 5-bromo-2-deoxyuridine (BrdU) into cells during proliferation or by measuring the total nucleic acid or protein content of lysed cells. BrDU is a synthetic nucleoside that is an analogue of thymidine. BrDU is commonly used in the detection of proliferating cells in living tissues. BrDU can be incorporated into the newly synthesized DNA of replicating cells (during the S phase of the cell cycle), substituting for thymidine during DNA replication.
The MTT calorimetric assay determines the ability of viable cells to convert a soluble tetrazolium salt [3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT) into an insoluble formazan precipitate. Tetrazolium salts accept electrons from oxidized substrates or appropriate enzymes, such as NADH and NADPH. In particular, MTT is reduced at the ubiquinone and cytochrome b and c sites of the mitochondrial electron transport system and is the result of succinate dehydrogenase activity. This reaction converts the yellow salts to blue-colored formazan crystals that can be dissolved in an organic solvent whose concentration can be spectrophotometrically determined. Owing to the many advantages of the assay, it is considered today a significant advance over traditional techniques. In fact, it is rapid, versatile, quantitative, and highly reproducible with a low intratest variation between data points. Moreover, the test can also be used for floating cells, such as those of leukemia and small cell lung carcinoma, and always allows sufficient time for cell replication, drug-induced cell death, and loss of enzymatic activity, which generates the formazan product from the MTT substrate.
6.4 Applications of Smart Bioconjugates
There have been several successful applications in medicine and biotechnology for such smart polymer biomolecule systems, and as such, they represent an important extension of polymeric biomaterials. The smart polymers can provide switchable-solubilization/precipitation for biomolecules such as enzymes, liposomes, and plasmid vectors, when these polymers are conjugated to specific sites within these biomolecules. Some of the most successfully demonstrated applications are described in the following subsections.
6.4.1 Bioseparations and Immunoassays
Smart conjugate systems have been used for many years in bioseparations and immunoassays. Hoffman and coworkers were the first to demonstrate a phase separation immunoassay [11–13]. The thermally induced precipitation of a PNIPAAm-protein bioconjugate from a complex solution can simultaneously and selectively remove only the protein that is conjugated to the PNIPAAm from the solution (Fig. 6.11). They have used this phenomenon for the separation of an enzyme from its reaction solution, to enable both recovery of the product from the supernatant and recycling of the enzyme. If the conjugated protein forms a complex with another biomolecule, for example, by affinity recognition, then the complex will also be selectively precipitated from solution. This phenomenon can be used to selectively remove IgG from solution as a PNIPAAm-protein A/IgG complex, in a fashion similar to affinity chromatography, but in this case, it is carried out by reversible phase separation from a solution, instead of flowing through and eluting from a packed column. This thermally induced affinity precipitation process may be extended to stimuli-induced phase separation of a biotinylated target molecule that is complexed to avidin or streptavidin. In this case, a biotinylated target molecule is first complexed with an excess of avidin or streptavidin molecules in solution, such that at least one of the four (on average) biotin binding sites remains free. Then, an end-linked biotin smart polymer conjugate is permitted to bind to the free site on the avidin or streptavidin molecules. Following this, the bioconjugate/affinity complex can be phase-separated by raising the temperature above the LCST of the smart polymer, which selectively removes the biotinylated target molecule from solution. Hoffman and coworkers have extended the affinity phase separation concept to the selective isolation and assay of an analyte from a complex mixture such as a serum sample. This is done by conjugating a first antibody to the polymer, complexing the analyte by affinity to the first antibody, and then introducing a second, labeled antibody, which then binds to the analyte by affinity to a similar or different site (epitope) on the analyte. This yields a temperature-sensitive polymer conjugated to an immune complex sandwich, which can then be selectively removed by thermally induced precipitation. This is an especially important separation step, because an excess of the labeled, second antibody is usually added to the sample. Washing and dissolution in cold buffer permit easy assay of the analyte. This immunoassay resembles an enzyme-linked immunosorbent assay (ELISA) carried out in solution. This concept has been extended to the assay of two different analytes in the same test sample. If NIPAAm is copolymerized with a more hydrophilic or a more hydrophobic comonomer, then copolymers with higher or lower LCSTs can be obtained, respectively. If one of each of these two different LCST copolymers is conjugated to a different antibody, then two different analytes may be assayed in the same serum sample by sequentially raising the temperature of the system to sequentially phase-separate the two different polymer-conjugated immune complex sandwiches. One could also carry out such a dual affinity separation or dual immunoassay using combinations of two different pH-sensitive smart polymers, or one temperature- and one pH-sensitive smart polymer. Another approach to controlling biological reactions using smart polymers is to prepare recombinant proteins with built-in polymer binding sites close to ligand or cell binding sites. This technique has been used to control ligand and cell binding activity, based on a variety of triggers including temperature and light. The time and costs involved in purifying proteins might be reduced significantly by using these smart polymer systems.

Schematic illustration of the thermally induced phase separation of a smart polymer and a ligand or a receptor, such as enzymes, antibodies, cell membrane receptors, and many others. This general process should be useful for removing any specific molecule from a complex mixture for the purpose of recovery and possible recycling of that molecule
Concanavalin A (ConA)- or wheat germ lectin (WGL)-conjugated PNIPAAm have also been used in the purification of various polysaccharides or polysaccharide-containing compounds such as glucan [105]. The thermally reversible soluble-insoluble PNIPAAm-dextran derivative conjugate has been synthesized by conjugating amino-terminated PNIPAAm to a dextran derivative via ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC), and the conjugate was used as a tool to purify polyclonal antibodies in serum samples from rabbits subcutaneously immunized with the derivatized dextran [106]. Chang et al. reported an effective and rapid method of purifying glutathione S-transferase (GST) using glutathione (GSH)-modified PNIPAAm and mild thermal conditions. They employed a chain transfer agent modified with pyridyl disulfide in the RAFT polymerization of NIPAAm [107]. Conjugation of GSH to the pyridyl disulfide-PNIPAAm reached 95 % within 30 min, as determined by UV-Vis monitoring of the release of pyridine-2-thione. GST was successfully thermoprecipitated upon heating the GSH-PNIPAAm above the LCST. Owing to its simplicity and high efficiency, this method holds great potential for large-scale purification of GST-tagged proteins. A thermally induced phase separation system has also been used for the specific separation of animal cells [28]. Monoclonal antibodies were modified with itaconic anhydride and copolymerized with NIPAAm, and ligand-conjugated carriers were added to the PEG 800-dextran T500 aqueous two-phase systems. CD34-positive human acute myeloid leukemia cells were specifically separated from human T lymphoma cells by applying anti-CD34 conjugated with PNIPAAm in the aqueous two-phase system.
Extensive efforts by Mattiasson and colleagues [108–110] have also been made to establish effective metal-chelate affinity precipitation using smart polymers. In metal chelating affinity precipitation, metal ligands such as imidazole are covalently coupled to the reversible soluble–insoluble smart polymers. The copolymers carrying metal-chelating ligands are charged with metal ions and the target protein binds the metal-loaded polymer in solution via the interaction between the histidine on the protein and the metal ion. Many proteins both containing natural metal ion binding residues and recombinant proteins containing His-tag residues have been purified using metal chelate affinity precipitation. Therefore, His-tagged proteins or cells or bioparticles can be purified through the precipitation of a target molecule–metal-loaded polymer complex from the mixture. The precipitated complex is solubilized by reversing the precipitation conditions, and the target molecule is dissociated from the precipitated polymer by using imidazole or EDTA as an eluting agent. The biomolecule is recovered from the copolymer by precipitating the latter at elevated temperature in the presence of NaCl. In a recent study, purification of extracellularly expressed six histidine-tagged single-chain Fv-antibody fragments (His6-scFv fragments) from a recombinant Escherichia coli cell culture broth was performed. The precipitation efficiency was lower with Ni(II)-poly(VI-NIPAAm) than with Cu(II)-poly(VI-NIPAAm), but the selectivity was better in the former case. The bound His6-scFv fragments were recovered almost completely (>95 %) by elution with 50 mM EDTA buffer, pH 8.0 [111].
Besides protein purification, the metal-ion charged copolymer of poly(VI-NIPAAm) can also be applied to the separation of single-stranded nucleic acids, such as RNA from double-stranded linear and plasmid DNA, by affinity precipitation [112]. The separation method utilizes the interaction of metal ions to the aromatic nitrogens in exposed purines in single-stranded nucleic acids [113].
6.4.2 Molecular Switching
Since an important aspect of many technologies that utilize biomolecular components is the control of their recognition properties, there is considerable interest in developing molecular switches that control recognition processes with mild signals. Smart polymers have been utilized to control protein activity as molecular switches. The ability to reversibly control protein and enzyme activities using external stimuli could provide new opportunities for the development of molecular diagnostics, bioprocesses, affinity separations, lab assays, BioMEMS, bioelectronics, and biosensor technologies. Chilkoti et al. have been demonstrating a new approach to molecular switches, in which site-specific conjugation of smart polymers is utilized. The site-specific conjugation is designed to assure minimal loss in the activity of the protein after conjugation with the smart polymer. They first reported the site-specific attachment of maleimide-terminated PN1PAAm to a genetically engineered mutant of cytochrome b 5, incorporating a unique cysteine residue by site-directed mutagenesis techniques [54]. This conjugation technique permits the site-specific and stoichiometric conjugation of the polymer with the protein. A genetically engineered SA mutant, which has only one cysteine residue, was also conjugated site specifically via sulfhydryl groups with a PNIPAAm that has pendant sulfhydryl-reactive vinyl sulfone groups [39]. Cys was substituted for Asn at position 49, which is near the outer edge of the biotin-binding pocket. Normal binding of biotin to the modified SA occurred below the LCST, whereas above the LCST, the polymer collapsed and blocked binding. Site-directed mutagenesis techniques were also used to replace the native glutamate at position 116 with cysteine [114]. Since the conjugation site is near the tryptophan 120 residue, which forms a van der Waals contact with biotin, which is important in generating the large binding free energy, the temperature-induced conformational change of the polymer at position 116 may lead to structural changes in the region of tryptophan 120, which are responsible for the reversible binding between biotin and the conjugated SA. The conjugate repeatedly bound and released biotin as temperature was cycled through the LCST. With the incorporation of pH-sensitive units such as acrylic acid (AAc) in a random copolymer with NIPAAm, a copolymer becomes completely soluble at 37 °C and pH 7.4 and insoluble at 37 °C and pH 4.0. This copolymer has been conjugated to a specific cysteine thiol site inserted by genetic engineering near the recognition site of SA [115]. The biotin binding at 37 °C is significantly reduced at pH 4.0, compared with pH 7.4. This is most likely due to the more compact copolymer coil at pH 4.0 and 37 °C compared with that at pH 7.4 and 37 °C. The bound biotin was able to be released by changing the conditions to pH 4.0.
To investigate the detailed mechanism of the smart polymer activity switches, the roles of the conjugation site and the free polymer were determined using a new end-reactive polymer, N,N-dimethyl acrylamide-co-4-phenylazophenylacrylate (DMAA). The use of this polymer and a control SA mutant has allowed an accurate determination of the conjugation ratio. The site selectivity of the switch was first investigated with the S139C SA mutant that contains a unique cysteine at the C-terminus. The purified S139C conjugate, where the DMAA is distant from the binding site, did not display switching activity. The previously characterized E116C streptavidin mutant displayed switching activity when the DMAA was conjugated at this cysteine near the biotin binding site [116]. Because the release of biotin was shown to proceed with the same kinetics as the biotin off-rate for wild-type streptavidin, the mechanism for release was shown to be the blocking of biotin reassociation by the collapsed DMAA. The reversible temperature-induced collapse of DMAA has also been used as a molecular switch to control the catalytic activity of endoglucanase 12A (EG 12A) (Fig. 6.12) [117]. The polymer was conjugated to the EG 12A site-directed mutant N55C, directly adjacent to the cellulose binding cleft, and to the S25C mutant, where the conjugation site is more distant. The N55C conjugate displayed a larger activity shutoff efficiency in the collapsed polymer state than the S25C conjugate. Increasing the polymer molecular weight was also shown to increase the shutoff efficiency of the switch. Related to these effects of conjugation site and polymer size, the switching efficiency was found to be strongly dependent on substrate size. With a small substrate, there was minimal blocking of enzyme activity when the polymer was in the expanded state. With a large substrate, on the other hand, there was a large reduction in enzyme activity in the polymer expanded state, even with relatively small polymer chains, and a further reduction when the polymer was collapsed. Similar general trends for the interactive effects of the conjugation site, polymer size, and substrate size were observed for immobilized conjugates.

(a) Schematic model of site-directed mutant endoglucanase 12A (EG 12A). The red residues represent the catalytic glutamic acid side chains at the active site of EG 12A, the green residue is the Asn 55 position, the purple residue is the Ser 25 position, and the N-terminus is represented as the blue circle. The polymer is conjugated to the EG 12A site-directed mutant N55C, directly adjacent to the active site (b), and to the S25C mutant, where the conjugation site is more distant (c). The N55C conjugate displays a larger activity shutoff efficiency in the collapsed polymer state than the S25C conjugate [117]
Temperature-responsive poly(N, N-diethylacrylamide) (PDEAAm) was attached to the SA approximately 20 Å from the binding site for biotinylated proteins. Below the LCST, the polymer is in its extended state and acts as a ‘shield’ to block the binding of large biotinylated proteins. Above the LCST, it collapses and exposes the binding site, thereby allowing binding. The degree of shielding depends on both the size of the biotinylated protein and the size of PDEAAm (Fig. 6.13) [50]. The biotinylated IgG (MW 150 kD), for example, was unable to bind to the SA conjugate whether the polymer was above or below its LCST, whereas the biotinylated Protein G (MW 6.2 kD) was able to bind whether the polymer was hydrated or collapsed. An intermediate-size biotinylated bovine serum albumin (BSA) (MW 67 kD) exhibited increased binding as the temperature was raised through the LCST. These results suggest that smart polymer shields could be tailored to achieve a wide range of size-dependent ligand discrimination for use in affinity separations, biosensors, and diagnostics technologies.

Size-dependent control of the binding of biotinylated proteins to SA using a polymer shield. The biotinylated IgG is unable to bind to the SA conjugate whether the polymer was above or below its LCST, whereas the biotinylated Protein G is able to bind whether the polymer was hydrated or collapsed. An intermediate-size biotinylated BSA exhibited increased binding as the temperature was raised through the LCST [50]
Light-regulated molecular switches that reversibly control biomolecular function could also provide new opportunities for controlling activity in diagnostics, affinity separations, bioprocessing, therapeutics, and bioelectronics applications. Hohsaka et al. [118] have prepared a monoclonal antibody against a non-natural amino acid carrying an azobenzene group, L-p-(phenylazo)phenylalanine. The antibody binds an azobenzene group when it is in the trans form, but releases it when the latter is photoisomerized to the cis form. Ueda et al. have replaced Trp3 of phospholipase A2(PLA2) by non-natural aromatic amino acids, 3-(2-naphthyl)-L-alanine (Nap), 3-(9-anthryl)-DL-alanine (Ant), and p-phenylazo-L-phenylalanine (AzoF). UV irradiation during the hydrolysis reduced the activities of Nap-AMPA and Ant-AMPA. However, AzoF-AMPA with a cis(Z) configuration of the AzoF unit showed, upon UV irradiation, hydrolytic activity. The change in enzymatic activity induced by UV irradiation is ascribed to a conformational change in the mutant proteins. Shimoboji et al. [55] have successfully shown that the photoinduced changes in the size and hydration of a smart polymer chain coil can be used to regulate substrate access and enzyme activity when conjugated to the enzyme at a specific point just outside the active site (Fig. 6.14). In order to enable photocontrol of enzyme activity, two different copolymers that exhibit combined temperature sensitivity and photosensitivity were prepared by copolymerizing N,N-dimethyl acrylamide (DMA) with two different light-sensitive comonomers, 4-phenylazophenyl acrylate (AZAA) and N-4-phenylazophenyl acrylamide (AZAAm). Under the isothermal conditions at their photoresponsive temperatures, the AZAAm copolymer precipitated under UV light irradiation (350 nm), whereas the AZAA copolymer dissolved under the same UV light irradiation. The copolymers were conjugated to the E116C SA mutant via thiol coupling. One of these copolymer-E116C SA conjugates exhibited blocking of free biotin and triggered the release of bound biotin under VIS irradiation; the other demonstrated the same phenomena under UV irradiation [19]. These opposite photoresponsive biotin-blocking or biotin-releasing responses corresponded to the original photoinduced phase transition properties of the copolymers. The photoresponsive switch was not observed when a 4-phenylazoaleinanil monomer was conjugated directly to the protein.

a Schematic model of the photoresponsive enzyme switch. b Sequential photoswitching of the activity of the DMAA-VS and DMAAm-VS conjugates [55]
Smart polymer conjugation techniques have also been utilized for the control of gene expression. Murata et al. [119] have prepared a smart antisense reagent comprised of phosphodiester-linked oligodeoxynucleotides (ODNs) and PNIPAAm, and evaluated the antisense activity and nuclease stability of the conjugate. The antisense ODN-PNIPAAm conjugate demonstrated a thermoresponsive regulation of the hybridization between the conjugate and the target RNAs [36]. In this strategy, antisense ODN can be used without chemical modification at the phosphodiester linkage of the ODN, which means that the conjugate may retain its binding affinity for the target RNA. These characteristics of the conjugate are promising for its use as an antisense ODN carrier in gene therapy. Pennadam et al. [29] have successfully designed protein-polymer nanomachines to control the EcoR124I motor function (Fig. 6.15). PNIPAAm with reactive end-groups was attached to the motor subunit of EcoR124I via coupling of a maleimide-tipped linker. The protein-polymer conjugates were stable to extensive purification and, when combined with the M2S complex, the activity of this conjugate motor system was similar to that of its native counterpart, but can be switched on or off as a result of thermally induced polymer phase transitions [30]. Thus, the conjugation of the responsive polymer to the molecular motor generates a nanoscale switchable device, which can translocate DNA under one set of conditions.

Schematic illustration of the switchable control of molecular motor function by a temperature-responsive polymer switch. M, R, and S denote the specific motor subunit. Chain extension of the polymer below LCST provides a steric shield blocking the active site. Chain collapse (above LCST) enables access to the active site and restoration of enzyme function [29]
6.4.3 Drug/Gene Delivery
There are many peptide-, protein-, antibody-, and DNA-based therapeutics under development in the biotechnology and pharmaceutical industries. Intracellular delivery, for example, represents a promising approach for the treatment of a wide variety of diseases. DNA delivery systems based on attenuated viruses have shown the greatest successes to date, but numerous concerns over the safety of these vectors have led to the development of nonviral vectors for use in gene therapy. The efficacy achieved with these nonviral vectors, however, is significantly lower than that achieved with viral vectors. The low transfection efficiencies are due to the multiple biological barriers encountered during delivery from extracellular locations into the cell nucleus. A number of biological organisms have evolved surface proteins to solve similar trafficking problems, and a variety of viral-based, fusogenic peptides have been studied as pH-dependent membrane-disruptive components in gene delivery systems to enhance transport from the endosome to the cytoplasm. Lackey et al. [120] have investigated whether synthetic polymers could provide an alternative approach to pH-dependent membrane destabilization in drug delivery systems. They focused on pH-responsive polymers containing hydrophobic moieties inspired by the amino acid side chains of leucine and isoleucine, for example, and demonstrated that poly(propyl acrylic acid) (PPAAc) displays interesting improvements in the pH dependence of red blood cell hemolysis. PPAAc is not hemolytic at pH 7.4 and reaches maximum hemolysis at approximately pH 6.0 and below. This pH-sensitive, membrane-disruptive PPAAc can be used to enhance the release of drugs from the acidic endosomal compartment to the cytoplasm. Jones et al. [121] compared the hemolytic ability of three poly(2-alkylacrylic acid)s, PPAAc, poly(2-ethylacrylic acid) (PEAAc), and poly(2-methylacrylic acid) (PMAAc). PMAAc showed little hemolytic activity at all pHs tested, reaching a maximum of about 2 % at pH 5.0. By contrast, both PEAAc and PPAAc showed significant levels of haemolysis. As the pH decreased, the hemolytic activity of PEAAc increased, reaching a peak at pH 5.4. In contrast, the haemolytic activity of PPAAc increased with pH to a maximum at pH 5.8–6.2. Kyriakides et al. [122] have found that one PPAAc significantly enhances in vitro transfections of lipoplex formulations in cell culture, and does so in the presence of as much as 50 % serum. They have also extended the cell culture studies to an in vivo murine excisional wound healing model. A pilot study with a green fluorescent protein (GFP)-encoding plasmid indicated that injection of formulations containing PPAAc into healing wounds resulted in increased GFP expression. Subsequently, by administering sense and antisense DNA for the angiogenesis inhibitor thrombospondin-2 (TSP2), they were able to alter the wound healing response in TSP2-null and wild-type mice, respectively. These results suggest that PPAAc can provide significant improvements in the in vivo efficacy of drugs such as DNA-controlled medications.
Cell membrane ligands may also be conjugated to the polymer molecule when it has two reactive end groups or multiple reactive pendant groups, for targeting a conjugated protein drug to a specific cell. Antibody-targeting ligands have been linked to a pH-sensitive smart polymer by biotinylating both the antibody and the smart polymer and using SA to link the two together [22]. A biotinylated monoclonal antibody to a CD-3 lymphoma cell receptor and a biotinylated pH-sensitive, lipid-membrane-disruptive smart polymer, PPAAc, were linked together by SA. The PPAAc phase separates sharply as the pH in the endosome drops, and in so doing, PPAAc disrupts the endosomal vesicle membrane. Thus, when the antibody-SA-PPAA conjugate was endocytosed after binding to the CD-3 receptor in Jurkat lymphoma cells, the conjugate was observed to escape from the endosome to the cytosol. This is a desirable event for a protein drug since, otherwise, it would be trafficked to the lysosome where lysosomal enzymes would degrade it. Berguig et al. [123] used the antibody-SA-polymer conjugate to study the intracellular trafficking dynamics of an anti-CD22-internalizing HD39 monoclonal antibody (mAb) with PPAAc (Fig. 6.16). The PPAAc conjugate was shown to alter the intracellular trafficking kinetics strongly relative to HD39/SA alone or HD39/SA conjugates with a control polymer, PMAAc. Subcellular trafficking studies revealed that after 6 h, only 11 % of the HD39/SA–PPAAc conjugates had been trafficked to acidic lysosomal compartments with values at or below pH 5.6. In contrast, the average intracellular pH of HD39/SA alone dropped from 6.7 ± 0.2 at 1 h to 5.6 ± 0.5 after 3 h and 4.7 ± 0.6 after 6 h. Conjugation of the control polymer PMAA to HD39/SA showed an average pH drop similar to that of HD39/SA. Subcellular fractionation studies with tritium-labeled HD39/SA demonstrated that after 6 h, 89 % of HD39/SA was associated with endosomes and lysosomes, while 45 % of HD39/SA–PPAAc was translocated to the cytosol. These results demonstrate the endosomal-releasing properties of PPAAc with antibody-polymer conjugates and detail their intracellular trafficking dynamics and subcellular compartmental distributions over time. Dubé et al. [124] have demonstrated that folate-targeting conjugates can facilitate tumor cell uptake of the conjugates. They designed a family of NIPAAm copolymers carrying a small number of octadecyl groups and folic acid residues grafted along the chain via a short ethylenedioxy chain. The folate conjugates were obtained by amidation of an aminated precursor copolymer. Cytotoxicity assays confirmed that the folate-NIPAAm conjugates bind specifically to KB cells overexpressing the folate receptor. The cellular uptake of the copolymer was found to be temperature dependent. Benoit et al. [125] also reported a facile approach to provide folate-receptor-specific delivery of polymer therapeutics by employing a de novo synthesized folate-functionalized CTA. Folate functionalization provided specific polymer therapeutic-folate receptor interactions and elicited gene knockdown through the delivery of siRNA. Many polymer therapeutics can be functionalized by this strategy, and this approach is highly versatile for functionalizing RAFT polymers.

Intracellular trafficking of the HD39/SA-PPAAc conjugate. Ligation of the anti-CD22 monoclonal antibody (HD39) to CD22 leads to receptor-mediated endocytosis. A portion of the conjugate is trafficked from endosomes to lysosomes while a second fraction is released into the cytosol via endosomal escape mediated by PPAAc [123]
The PPAAc-based membrane-disruptive system has also been used for antigen delivery systems. While many infectious diseases are controlled by vaccine strategies, important limitations continue to motivate the development of better antigen delivery systems. Flanary et al. [126] developed a pH-sensitive polymeric carrier based on PPAAc to address the need for more potent CD8 cytotoxic T-cell (CTL) responses. Ovalbumin, as the protein antigen, was conjugated to poly(propylacrylic acid-co-pyridyldisulfide acrylate) (PPAA-PDSA) by disulfide exchange to form reversible conjugates that could be reduced by the glutathione redox system in the cytosol of antigen-presenting cells. The PPAA-PDSA ovalbumin conjugates displayed the pH-sensitive membrane disruptive properties of the parent polymer as determined by their hemolysis activities. The polymer-ovalbumin conjugates exhibited strong 22-fold increases in the MHC-1 presentation and ovalbumin-specific CTL activation compared with free ovalbumin. No CTL activation was observed with control conjugates of ovalbumin and PMAAc. This system was further evaluated to test whether improved cytosolic delivery of a protein antigen could enhance CD8+ cytotoxic lymphocyte generation and prophylactic tumor vaccine responses. PPAA was directly conjugated to the ovalbumin antigen via reducible disulfide linkages and was also tested in a particulate formulation after condensation with cationic poly(dimethylaminoethyl methacrylate) (PDMAEMA) [127]. In an EG.7-OVA mouse tumor protection model, PPAA-containing carriers robustly inhibited tumor growth and led to an approximately 3.5-fold increase in the longevity of tumor-free survival relative to controls. Mechanistically, this response was attributed to the 8-fold increase in the production of ovalbumin-specific CD8+ T-lymphocytes and an 11-fold increase in the production of anti-ovalbumin IgG. This is one of the first demonstrated examples of in vivo immunotherapeutic efficacy using soluble protein-polymer conjugates. This system shows promise for protein vaccine strategies against cancer and viruses and is also applicable to any technique requiring improved delivery of a protein cargo to the cytoplasm of a cell. Albarran et al. [128] showed that PPAAc can strongly enhance target cell killing through the intracellular delivery of a functional proapoptotic peptide. The Bak BH3 peptide induces apoptosis via antagonization of suppressor targets such as Bcl-2 and Bcl-xL. A genetically engineered streptavidin that contains an N-terminal TAT peptide sequence was used to optimize the pinocytotic cell uptake of biotinylated BH3 peptide and end-biotinylated PPAAc. Approximately 30 % of cells treated with TAT-SA:BH3 complexes revealed morphologically distinct nuclear condensation, a hallmark of apoptosis. Together with the PPAA, the TAT-SA adaptor complex could prove useful as a carrier of peptide/protein cargo to cultured cells. Fujimoto et al. [32] reported a novel drug delivery system for apoptosis induction by a smart polymer vehicle possessing temperature responsivity and bioaffinity. The cell-adhesive RGDS peptide was conjugated with the NIPAAm copolymer as a ligand model for bioaffinity. Dolichyl phosphate (dol-p), which is an apoptotic inducer, was added to the copolymer at around the precipitation temperature for incorporation. Aggregates incorporating dol-p were added to a human promonocytic leukemia U937 cell suspension at 37 °C. When the temperature was lowered to 25 °C, cells underwent apoptosis in the presence of Ca2+ because copolymer vehicles were concentrated on the cell surface through the binding of RGDS and integrin, and lipid inducers were released by the disruption of vehicles in response to temperature. This system would also be expected to provide a therapeutic application for targeting a drug to the cell and triggering its release in the body by cooling.
In addition to the pH-responsive system, reduction-oxidation-sensitive polymers have also been utilized for intracellular delivery, because the reduction-oxidation state of intracellular compartments changes: the endosome is rendered reductive, whereas the lysosomal compartment is substantially oxidative, compared with the mildly oxidative extracellular environment. Oxidation-sensitive smart polymers can be engineered by designing block copolymers containing a hydrophobic polypropylene sulphide (PPS). On exposure to oxidative conditions, the block is converted to hydrophilic polypropylene sulphoxide and, ultimately, to the more hydrophilic polypropylene sulphone [129]. On the other hand, earlier in the processes of endolysosomal processing, endocytosed compounds encounter a reductive environment. Smart bioconjugates with a reducible disulphide connection have been shown to destabilize, releasing the pendant drugs or DNA. El-Sayed et al. [130] reported membrane-destabilizing and glutathione-reactive polymers. ODN was ionically complexed to cationic peptides grafted onto the polymer backbone via disulfide linkages to the pyridyl disulfide acrylate (PDSA) units. This unique design allowed for the release of the disulfide-conjugated cationic peptides with the complexed ODN into the cytoplasm by the reducing action of the glutathione enzyme, which is commonly present in the cytoplasm. Manickam and Oupicky [131] synthesized high-molecular-weight polypeptides containing disulfide bonds in the backbone by oxidative copolymerization of a histidine-rich peptide (HRP) and a nuclear localization sequence (NLS) peptide. Cytotoxicity and transfection activity of DNA polyplexes were evaluated in vitro. In comparison with control polyethylenimine (PEI), only minimum toxic effects were observed on the metabolic activity and membrane integrity of human endothelial cells.
Furgeson et al. [23] have developed a smart doxorubicin-polypeptide conjugate for thermally targeted delivery to solid tumors. A temperature-responsive, genetically engineered ELP was conjugated to doxorubicin (Dox) molecules through pH-sensitive, maleimide-activated, hydrazone linkers. The highest release of the ELP–Dox conjugate by cleavage of the hydrazone bond at pH 4 was nearly 80 % over 72 h and was exhibited by the conjugate with the shortest linker. The endocytotic uptake of a thermally responsive ELP was also observed to be significantly enhanced by the thermally triggered phase transition of the polypeptide in cell culture for three different tumor cell lines [132]. ELPs conjugated to drugs also enable thermally targeted drug delivery to solid tumors if their transition temperatures are between body temperature and the temperature in a locally heated region. In vivo studies of ELP delivery to human tumors (SKOV-3 ovarian carcinoma and D-54MG glioma) implanted in nude mice demonstrated that hyperthermic targeting of the thermally responsive ELP for 1 h provides a twofold increase in tumor localization compared with the same polypeptide without hyperthermia. By exploiting the phase-transition-induced aggregation of these polypeptides, this method provides a new way of thermally targeting polymer-drug conjugates to solid tumors [100].
6.4.4 Diagnostic Technologies
The ability to sequentially control biomolecular recognition and activity could open new opportunities or improve existing applications in the diagnostic fields. Since the smart polymers serve as both antennae and actuators, to sense signals and respond to them, leading to the control of biorecognition events, smart conjugates with protein and DNA have also drawn considerable attention in the diagnostic fields. Their characteristic ‘on-off’ responses to small changes in pH, temperature, and/or UV-visible light permits rapid and precise control of molecular events. Agasti et al. [133] for example, developed a photocleavable DNA barcode-antibody conjugate for rapid, quantitative, and multiplexed detection of protein expression in single live cells (Fig. 6.17). Irradiation of the labeled cells with light (∼365 nm) cleaves the linker between the antibodies and the barcodes, causing the barcodes to be released into the solution for easy isolation. Barcode amplification by polymerase chain reaction (PCR) and subsequent gel electrophoresis analysis of the amplified barcodes allows simultaneous detection and quantification of multiple protein analytes from single cells. Maeda and coworker [25, 134–136] have developed a single-nucleotide polymorphism (SNP)-responsive diagnostic using DNA-PNIPAAm conjugates. The graft copolymer consisting of PNIPAAm and single-stranded DNA (ssDNA) forms nanoparticles above physiological temperature. Non-crosslinking aggregation of the DNA-PNIPAAm nanoparticles is induced by the hybridization of the surface DNA with the full-match complementary DNA (Fig. 6.18). This aggregation mechanism was applicable for the target 24mer DNA corresponding to k-ras (oncogene) codons 10–17, as well as for SNP sites of CYP2C9. Each nanoparticle was aggregated by the hybridization with its full-match complementary DNA fragment, but not with one-base mismatch. These results demonstrated that the non-crosslinking aggregation of DNA-PNIPAAm nanoparticles is useful for analyzing various SNPs.

Schematic illustration of the light-mediated cellular barcoding strategy. Protein targets are labeled with DNA-barcode-conjugated antibodies and then photocleaved to release the DNA barcodes. Amplified barcodes are analyzed using gel electrophoresis for multiplexed detection of protein biomarkers from single cells [133]

The switchability of smart polymer conjugates also opens the door to potential uses in microfluidic formats where the differential diffusive and physical properties might be exploited for separation, analyte concentration, and signal generation. Microfluidic platforms have shown promise for conducting diagnostic measurements in both clinical and point-of-care settings [137, 138]. While great strides have been made, the potential of this technology has not yet been fully realized and several challenges remain. One important outstanding need is the handling of dilute antigens and biomarkers, particularly their purification and enrichment from complex biological fluids. Several chromatographic strategies have been demonstrated to address analyte purification and enrichment issues in microfluidic systems. Affinity moieties have been conjugated directly to microchannel walls, attached to particles and packed into microchannels and immobilized within porous monolithic slabs. These techniques suffer from several shortcomings, including complex packing and modification steps, high flow resistance and limitations in scaling-up and manufacturability. These strategies also usually allow the separation of only a single analyte and cannot be modified once introduced. Kulkarni et al. [49, 79] have developed a series of PNIPAAm-protein conjugates that separate and enrich analytes from solution and enable detection. Because smart conjugates bind analytes in solution prior to separation, conjugate-analyte binding avoids steric and mass transport limitations associated with surface-based techniques [139, 140]. A reversible microchannel surface capture system has been further developed for bioanalytical samples [141–143]. The capture/release efficiency and enrichment of PNIPAAm-antibody conjugates in PNIPAAm-grafted PDMS microchannels have been investigated using a helical flow, circular microreactor. The conjugate’s immobilization and release were limited by mass transport to and from the functionalized PNIPAAm surface. Transport and adsorption efficiencies were dependent on the aggregate size of the PNIPAAm–streptavidin conjugate above the LCST as well as on whether the conjugates were heated in the presence of the stimuli-responsive surface or pre-aggregated and then flowed across the surface. Mixing and recirculation substantially increase the conjugate release rate and sharpness once the temperature has dropped below the phase transition temperature. The concentration of protein-polymer conjugates could be achieved by a continuous conjugate flow into the heated recirculator, allowing nearly linear enrichment of the conjugate reagent from larger volumes. This capability was shown with anti-p24 HIV monoclonal antibody reagents that were enriched over fivefold by using this protocol. pH-responsive surface traps have also been constructed in the channel wall by the same methods. Magnetic nanoparticles have also been developed for diagnostic target isolation because smaller particles display better association and binding properties to the target analytes [144, 145]. A reversible surface capture system for smart bioconjugates has also been demonstrated on porous membrane filters for the detection of the malaria antigen Plasmodium falciparum histidine-rich protein 2 (PfHRP2) (Fig. 6.19) [24]. The carboxyl end-groups of semi-telechelic PNIPAAm synthesized by RAFT polymerization were modified with tetrafluorophenol to yield amine-reactive ester groups for conjugation to amine groups of anti-streptavidin and anti-PfHRP2 antibodies. Stimuli-responsive membranes were constructed from 1.2-μm-pore-size, hydroxylated, nylon-6,6 filters. The surface hydroxyl groups on the filters were conjugated to a 2-ethylsulfanylthiocarbonylsulfanyl-2-methyl propionic acid (EMP) RAFT chain transfer agent, and the surface-grafted PNIPAAm was obtained by subsequent polymerization. The PNIPAAm grafted membranes showed greater than 80 % anti-streptavidin capture efficiency. The PfHRP2 antigen could be processed and detected at clinically relevant concentrations of this malaria biomarker. These studies provide insight into the mechanism of smart polymer–protein conjugate capture and release in grafted channels and show the potential of this purification and enrichment module for processing diagnostic samples.

Smart fluidic system for purifying and concentrating diagnostic biomarkers using temperature-responsive antibody conjugates and membranes. PNIPAAm-conjugated antibodies are captured at the membrane above the LCST, while unconjugated plasma components flow through (a). When the membrane region is cooled, the conjugates are released back into the flow stream (b) [24]
6.5 Conclusions and Future Trends
Smart bioconjugate-based technologies are becoming the premier tool for a wide range of applications in the areas of medicine and biotechnology. Conjugating proteins, peptides, DNA, and cells with smart polymers has been a versatile way to add new value, advanced features and unique properties to inert polymers. For example, creating nanoscale antennae and actuators to sense signals and respond to them significantly improves biological properties as well as introduces new unique properties, leading to the control of biorecognition events. This chapter mainly addresses the developments of smart bioconjugates specifically applied in bioseparation, molecular switching, diagnosis, sensors, and drug/gene delivery. An ongoing challenge is to synthesize well-defined conjugates. Traditionally, the reactive polymers were prepared by the modification of preformed chains. However, with the advent of new controlled/‘living’ polymerization techniques that are tolerant to a wide range of functional groups, the use of protein-reactive initiators to form well-defined polymers is now possible. This strategy is less time-consuming and results in polymers amenable to coupling to proteins without any further modification. Native chemical ligation, tRNA engineering methods, and other advances in protein engineering also allow for the synthesis of proteins containing non-natural amino acids. So far, the incorporation of functional groups that react orthogonally to natural amino acids has been underexploited in the preparation of polymer bioconjugates. The combination of controlled radical polymerization with non-natural protein engineering could result in unprecedented control over polymer conjugation, resulting in precise bioconjugates for a variety of applications. Therefore, novel strategies for the design of smart bioconjugates including proteins, DNA, mammalian cells, bacteria, yeast cells, and viruses will facilitate the creation of a new class of biomaterials in the future.
References
Ringsdorf H (1975) Structure and properties of pharmacologically active polymers. J Polym Sci: Polym Symp 51:135–153. doi:10.1002/polc.5070510111
Matsumura Y, Maeda H (1986) A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 46:6387–6392
Abuchowski A, McCoy JR, Palczuk NC, van Es T, Davis FF (1977) Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J Biol Chem 252:3582–3586
Mok H, Palmer DJ, Ng P, Barry MA (2005) Evaluation of polyethylene glycol modification of first-generation and helper-dependent adenoviral vectors to reduce innate immune responses. Mol Ther 11:66–79
O’Riordan CR, Song A (2008) PEGylated adenovirus for targeted gene therapy. In: Doux J (ed) Gene therapy protocols, vol 434. Methods in molecular biology™. Humana Press, pp 133–160. doi:10.1007/978-1-60327-248-3_9
Scott MD, Murad KL, Koumpouras F, Talbot M, Eaton JW (1997) Chemical camouflage of antigenic determinants: Stealth erythrocytes. In: Proceedings of the national academy of sciences, vol 94. U.S.A., pp 7566–7571
Hashemi-Najafabadi S, Vasheghani-Farahani E, Shojaosadati SA, Rasaee MJ, Armstrong JK, Moin M, Pourpak Z (2006) A method to optimize PEG-coating of red blood cells. Bioconjug Chem 17:1288–1293. doi:10.1021/bc060057w
Mansouri S, Merhi Y, Winnik FM, Tabrizian M (2011) Investigation of layer-by-layer assembly of polyelectrolytes on fully functional human red blood cells in suspension for attenuated immune response. Biomacromolecules 12:585–592. doi:10.1021/bm101200c
Charles M, Coughlin RW, Hasselberger FX (1974) Soluble–insoluble enzyme catalysts. Biotechnol Bioeng 16:1553–1556. doi:10.1002/bit.260161113
Van Leemputten E, Horisberger M (1976) Soluble–insoluble complex of trypsin immobilized on acrolein–acrylic acid copolymer. Biotechnol Bioeng 18:587–590. doi:10.1002/bit.260180410
Shoemaker SG, Hoffman AS, Priest JH (1987) Synthesis and properties of vinyl monomer/enzyme conjugates. Appl Biochem Biotechnol 15:11–24. doi:10.1007/bf02798503
Auditore-Hargreaves K, Houghton RL, Monji N, Priest JH, Hoffman AS, Nowinski RC (1987) Phase-separation immunoassays. Clin Chem 33:1509–1516
Hoffman AS (1987) Applications of thermally reversible polymers and hydrogels in therapeutics and diagnostics. J Controlled Release 6:297–305. http://dx.doi.org/10.1016/0168-3659(87)90083-6
Monji N, Hoffman A (1987) A novel immunoassay system and bioseparation process based on thermal phase separating polymers. Appl Biochem Biotechnol 14:107–120. doi:10.1007/bf02798429
Luong JHT, Nguyen A-L (1990) Affinity partitioning of bioproducts. Nat Biotech 8:306–307
Takei YG, Aoki T, Sanui K, Ogata N, Sakurai Y, Okano T, Matsukata M, Kikuchi A (1994) Temperature-responsive bioconjugates. 3. Antibody-poly(N-isopropylacrylamide) conjugates for temperature-modulated precipitations and affinity bioseparations. Bioconjug Chem 5:577–582. doi:10.1021/bc00030a013
Fujimura M, Mori T, Tosa T (1987) Preparation and properties of soluble–insoluble immobilized proteases. Biotechnol Bioeng 29:747–752. doi:10.1002/bit.260290612
Ito Y, Kotoura M, Chung DJ, Imanishi Y (1993) Trypsin modification by vinyl polymers with variable solubilities in response to external signals. Bioconjug Chem 4:358–361. doi:10.1021/bc00023a009
Shimoboji T, Ding ZL, Stayton PS, Hoffman AS (2002) Photoswitching of ligand association with a photoresponsive polymer-protein conjugate. Bioconjug Chem 13:915–919. doi:10.1021/bc010057q
Dainiak MB, Izumrudov VA, Muronetz VI, Galaev IY, Mattiasson B (1998) Conjugates of monoclonal antibodies with polyelectrolyte complexes—an attempt to make an artificial chaperone. Biochim Biophys Acta 1381:279–285. http://dx.doi.org/10.1016/S0304-4165(98)00035-X
Muronetz VI, Kazakov SV, Dainiak MB, Izumrudov VA, Galaev IY, Mattiasson B (2000) Interaction of antibodies and antigens conjugated with synthetic polyanions: on the way of creating an artificial chaperone. Biochim Biophys Acta 1475:141–150. http://dx.doi.org/10.1016/S0304-4165(00)00060-X
Lackey CA, Press OW, Hoffman AS, Stayton PS (2002) A biomimetic pH-responsive polymer directs endosomal release and intracellular delivery of an endocytosed antibody complex. Bioconjug Chem 13:996–1001. doi:10.1021/bc010053l
Furgeson DY, Dreher MR, Chilkoti A (2006) Structural optimization of a “smart” doxorubicin–polypeptide conjugate for thermally targeted delivery to solid tumors. J Controlled Release 110:362–369. http://dx.doi.org/10.1016/j.jconrel.2005.10.006
Golden AL, Battrell CF, Pennell S, Hoffman AS, J. Lai J, Stayton PS (2010) Simple fluidic system for purifying and concentrating diagnostic biomarkers using stimuli-responsive antibody conjugates and membranes. Bioconjug Chem 21:1820–1826. doi:10.1021/bc100169y
Mori T, Maeda M (2003) Temperature-Responsive formation of colloidal nanoparticles from poly(N-isopropylacrylamide) grafted with single-stranded DNA. Langmuir 20:313–319. doi:10.1021/la0356194
Ebara M, Uto K, Idota N, Hoffman JM, Aoyagi T (2012) Shape-memory surface with dynamically tunable nano-geometry activated by body heat. Adv Mater 24:273–278. doi:10.1002/adma.201102181
Kim Y-J, Ebara M, Aoyagi T (2012) A smart nanofiber web that captures and releases cells. Angew Chem Int Ed 51:10537–10541. doi:10.1002/anie.201204139
Kumar A, Kamihira M, Galaev IY, Mattiasson B, Iijima S (2001) Type-specific separation of animal cells in aqueous two-phase systems using antibody conjugates with temperature-sensitive polymers. Biotechnol Bioeng 75:570–580. doi:10.1002/bit.10080
Pennadam S, Firman K, Alexander C, Gorecki D (2004) Protein-polymer nano-machines. Towards synthetic control of biological processes. J Nanobiotechnol 2:8
Pennadam SS, Lavigne MD, Dutta CF, Firman K, Mernagh D, Górecki DC, Alexander C (2004) Control of a multisubunit DNA Motor by a thermoresponsive Polymer Switch. J Am Chem Soc 126:13208–13209. doi:10.1021/ja045275j
Stoica F, Alexander C, Tirelli N, Miller AF, Saiani A (2008) Selective synthesis of double temperature-sensitive polymer-peptide conjugates. Chem Commun 37:4433–4435
Fujimoto K, Iwasaki C, Arai C, Kuwako M, Yasugi E (2000) Control of cell death by the smart polymeric vehicle. Biomacromolecules 1:515–518. doi:10.1021/bm000082j
Vázquez-Dorbatt V, Tolstyka ZP, Maynard HD (2009) Synthesis of aminooxy end-functionalized PNIPAAm by RAFT polymerization for protein and polysaccharide conjugation. Macromolecules 42:7650–7656. doi:10.1021/ma9013803
Lima A, Song W, Blanco-Fernandez B, Alvarez-Lorenzo C, Mano J (2011) Synthesis of temperature-responsive dextran-MA/PNIPAAm particles for controlled drug delivery using superhydrophobic surfaces. Pharm Res 28:1294–1305. doi:10.1007/s11095-011-0380-2
Kotsuchibashi Y, Ebara M, Idota N, Narain R, Aoyagi T (2012) A ‘smart’ approach towards the formation of multifunctional nano-assemblies by simple mixing of block copolymers having a common temperature sensitive segment. Polym Chem-Uk 3:1150–1157
Murata M, Kaku W, Anada T, Sato Y, Kano T, Maeda M, Katayama Y (2003) Novel DNA/polymer conjugate for intelligent antisense reagent with improved nuclease resistance. Bioorg Med Chem Lett 13:3967–3970. http://dx.doi.org/10.1016/j.bmcl.2003.08.062
Ta T, Convertine AJ, Reyes CR, Stayton PS, Porter TM (2010) Thermosensitive liposomes modified with poly(N-isopropylacrylamide-co-propylacrylic acid) copolymers for triggered release of doxorubicin. Biomacromolecules 11:1915–1920. doi:10.1021/bm1004993
Kono K, Nakai R, Morimoto K, Takagishi T (1999) Thermosensitive polymer-modified liposomes that release contents around physiological temperature. Biochim Biophys Acta 1416:239–250. http://dx.doi.org/10.1016/S0005-2736(98)00226-0
Stayton PS, Shimoboji T, Long C, Chilkoti A, Ghen G, Harris JM, Hoffman AS (1995) Control of protein-ligand recognition using a stimuli-responsive polymer. Nature 378:472–474
Kiick KL, Tirrell DA (2000) Protein engineering by in vivo incorporation of non-natural amino acids: control of incorporation of methionine analogues by methionyl-trna synthetase. Tetrahedron 56:9487-9493. http://dx.doi.org/10.1016/S0040-4020(00)00833-4
Chen J-P, Chu D-H, Sun Y-M (1997) Immobilization of α-amylase to temperature-responsive polymers by single or multiple point attachments. J Chem Technol Biotechnol 69:421–428. doi:10.1002/(sici)1097-4660(199708)69:4<421:aid-jctb730>3.0.co;2-3
Matsukata M, Aoki T, Sanui K, Ogata N, Kikuchi A, Sakurai Y, Okano T (1996) Effect of molecular architecture of poly(N-isopropylacrylamide)–trypsin conjugates on their solution and enzymatic properties. Bioconjug Chem 7:96–101. doi:10.1021/bc950082u
Chen G, Hoffman AS (1993) Preparation and properties of thermoreversible, phase-separating enzyme-oligo (N-isopropylacrylamide) conjugates. Bioconjug Chem 4:509–514. doi:10.1021/bc00024a013
Plourde R, Phillips AT, Wu CH, Hays RM, Chowdhury JR, Chowdhury NR, Wu GY (1996) A hepatocyte-targeted conjugate capable of delivering biologically active colchicine in vitro. Bioconjug Chem 7:131–137. doi:10.1021/bc950083m
Chiefari J, Chong YK, Ercole F, Kristina J, Jeffery J, Le TPT, Mayadunne RTA, Meijs GF, Moad CL, Moad G, Rizzardo E, Thang SH (1998) Living free-radical polymerization by reversible addition—fragmentation chain transfer: the RAFT process. Macromolecules 31:5559–5562. doi:10.1021/ma9804951
Lele BS, Murata H, Matyjaszewski K, Russell AJ (2005) Synthesis of uniform protein-polymer conjugates. Biomacromolecules 6:3380–3387. doi:10.1021/bm050428w
Lecolley F, Tao L, Mantovani G, Durkin I, Lautru S, Haddleton DM (2004) A new approach to bioconjugates for proteins and peptides (“pegylation”) utilising living radical polymerisation. Chem Commun 0:2026–2027
Tao L, Mantovani G, Lecolley F, Haddleton DM (2004) α-Aldehyde terminally functional methacrylic polymers from living radical polymerization: application in protein conjugation “pegylation”. J Am Chem Soc 126:13220–13221. doi:10.1021/ja0456454
Kulkarni S, Schilli C, Grin B, Müller AHE, Hoffman AS, Stayton PS (2006) Controlling the aggregation of conjugates of streptavidin with smart block copolymers prepared via the RAFT copolymerization technique. Biomacromolecules 7:2736–2741. doi:10.1021/bm060186f
Ding Z, Fong RB, Long CJ, Stayton PS, Hoffman AS (2001) Size-dependent control of the binding of biotinylated proteins to streptavidin using a polymer shield. Nature 411:59–62
Bulmus V, Woodward M, Lin L, Murthy N, Stayton P, Hoffman A (2003) A new pH-responsive and glutathione-reactive, endosomal membrane-disruptive polymeric carrier for intracellular delivery of biomolecular drugs. J Controlled Release 93:105–120. http://dx.doi.org/10.1016/j.jconrel.2003.06.001
Stayton PS, Hoffman AS, El-Sayed M, Kulkarni S, Shimoboji T, Murthy N, Bulmus V, Lackey C (2005) Intelligent biohybrid materials for therapeutic and imaging agent delivery. Proc IEEE 93:726–736. doi:10.1109/jproc.2005.844619
Krishnamurthy VM, Semetey V, Bracher PJ, Shen N, Whitesides GM (2007) Dependence of effective molarity on linker length for an intramolecular protein–ligand system. J Am Chem Soc 129:1312–1320. doi:10.1021/ja066780e
Chilkoti A, Chen G, Stayton PS, Hoffman AS (1994) Site-specific conjugation of a temperature-sensitive polymer to a genetically engineered protein. Bioconjug Chem 5:504–507. doi:10.1021/bc00030a004
Shimoboji T, Larenas E, Fowler T, Kulkarni S, Hoffman AS, Stayton PS (2002) Photoresponsive polymer–enzyme switches. Proc Natl Acad Sci 99:16592–16596. doi:10.1073/pnas.262427799
Bontempo D, Heredia KL, Fish BA, Maynard HD (2004) Cysteine-reactive polymers synthesized by atom transfer radical polymerization for conjugation to proteins. J Am Chem Soc 126:15372–15373. doi:10.1021/ja045063m
Mantovani G, Lecolley F, Tao L, Haddleton DM, Clerx J, Cornelissen JJLM, Velonia K (2005) Design and synthesis of N-maleimido-functionalized hydrophilic polymers via copper-mediated living radical polymerization: a suitable alternative to PEGylation chemistry. J Am Chem Soc 127:2966–2973. doi:10.1021/ja0430999
Geoghegan KF, Stroh JG (1992) Site-directed conjugation of nonpeptide groups to peptides and proteins via periodate oxidation of a 2-amino alcohol. Application to modification at N-terminal serine. Bioconjug Chem 3:138–146. doi:10.1021/bc00014a008
Gaertner HF, Offord RE, Cotton R, Timms D, Camble R, Rose K (1994) Chemo-enzymic backbone engineering of proteins. Site-specific incorporation of synthetic peptides that mimic the 64-74 disulfide loop of granulocyte colony-stimulating factor. J Biol Chem 269:7224–7230
Alouani S, Gaertner HF, Mermod J-J, Power CA, Bacon KB, Wells TNC, Proudfoot AEI (1995) A fluorescent interleukin-8 receptor probe produced by targetted labelling at the amino terminus. Eur J Biochem 227:328–334. doi:10.1111/j.1432-1033.1995.tb20393.x
Gaertner HF, Offord RE (1996) Site-specific attachment of functionalized poly (ethylene glycol) to the amino terminus of proteins. Bioconjug Chem 7:38–44. doi:10.1021/bc950074d
Heredia KL, Bontempo D, Ly T, Byers JT, Halstenberg S, Maynard HD (2005) In situ preparation of protein—“smart” polymer conjugates with retention of bioactivity. J Am Chem Soc 127:16955–16960. doi:10.1021/ja054482w
Bontempo D, Maynard HD (2005) Streptavidin as a macroinitiator for polymerization. In Situ protein-polymer conjugate formation. J Am Chem Soc 127:6508–6509. doi:10.1021/ja042230+
Le Droumaguet B, Velonia K (2008) In Situ ATRP-mediated hierarchical formation of giant amphiphile bionanoreactors. Angew Chem Int Ed 47:6263–6266. doi:10.1002/anie.200801007
Depp V, Alikhani A, Grammer V, Lele BS (2009) Native protein-initiated ATRP: a viable and potentially superior alternative to PEGylation for stabilizing biologics. Acta Biomater 5:560–569. http://dx.doi.org/10.1016/j.actbio.2008.08.010
Magnusson JP, Bersani S, Salmaso S, Alexander C, Caliceti P (2010) In situ growth of side-chain PEG polymers from functionalized human growth hormone—a new technique for preparation of enhanced protein-polymer conjugates. Bioconjugate Chem 21:671–678. doi:10.1021/bc900468v
Gao W, Liu W, Mackay JA, Zalutsky MR, Toone EJ, Chilkoti A (2009) In situ growth of a stoichiometric PEG-like conjugate at a protein’s N-terminus with significantly improved pharmacokinetics. Proc Natl Acad Sci 106:15231–15236. doi:10.1073/pnas.0904378106
Gao W, Liu W, Christensen T, Zalutsky MR, Chilkoti A (2010) In situ growth of a PEG-like polymer from the C terminus of an intein fusion protein improves pharmacokinetics and tumor accumulation. Proc Natl Acad Sci. doi:10.1073/pnas.1006044107
Li H, Li M, Yu X, Bapat AP, Sumerlin BS (2011) Block copolymer conjugates prepared by sequentially grafting from proteins via RAFT. Polym Chem Uk 2:1531–1535
De P, Li M, Gondi SR, Sumerlin BS (2008) Temperature-regulated activity of responsive polymer-protein conjugates prepared by grafting-from via RAFT polymerization. J Am Chem Soc 130:11288–11289. doi:10.1021/ja804495v
Hong C-Y, Pan C-Y (2006) Direct synthesis of biotinylated stimuli-responsive polymer and diblock copolymer by RAFT polymerization using biotinylated trithiocarbonate as RAFT agent. Macromolecules 39:3517–3524. doi:10.1021/ma052593+
Liu J, Bulmus V, Herlambang DL, Barner-Kowollik C, Stenzel MH, Davis TP (2007) In situ formation of protein-polymer conjugates through reversible addition fragmentation chain transfer polymerization. Angew Chem Int Ed 46:3099–3103. doi:10.1002/anie.200604922
Hohsaka T, Sisido M (2002) Incorporation of non-natural amino acids into proteins. Curr Opin Chem Biol 6:809–815. http://dx.doi.org/10.1016/S1367-5931(02)00376-9
Katti KS, Ambre AH, Peterka N, Katti DR (2010) Use of unnatural amino acids for design of novel organomodified clays as components of nanocomposite biomaterials. Philoso Trans R Soc A 368:1963–1980. doi:10.1098/rsta.2010.0008
Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM (2006) Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc Natl Acad Sci 103:9482–9487. doi:10.1073/pnas.0601637103
Kolb HC, Finn MG, Sharpless KB (2001) Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed 40:2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004:aid-anie2004>3.0.co;2-5
Peeler JC, Woodman BF, Averick S, Miyake-Stoner SJ, Stokes AL, Hess KR, Matyjaszewski K, Mehl RA (2010) Genetically encoded initiator for polymer growth from proteins. J Am Chem Soc 132:13575–13577. doi:10.1021/ja104493d
Averick SE, Magenau AJD, Simakova A, Woodman BF, Seong A, Mehl RA, Matyjaszewski K (2011) Covalently incorporated protein-nanogels using AGET ATRP in an inverse miniemulsion. Polym Chem-Uk 2:1476–1478
Kulkarni S, Schilli C, Müller AHE, Hoffman AS, Stayton PS (2004) Reversible meso-scale smart polymer-protein particles of controlled sizes. Bioconjugate Chem 15:747–753. doi:10.1021/bc034215k
Hoffman JM, Ebara M, Lai JJ, Hoffman AS, Folch A, Stayton PS (2010) A helical flow, circular microreactor for separating and enriching “smart” polymer-antibody capture reagents. Lab Chip 10:3130–3138
Hannink JM, Cornelissen JJLM, Farrera JA, Foubert P, De Schryver FC, Sommerdijk NAJM, Nolte RJM (2001) Protein-polymer hybrid amphiphiles. Angew Chem Int Ed 40:4732–4734. doi:10.1002/1521-3773(20011217)40:24<4732:aid-anie4732>3.0.co;2-p
Boerakker MJ, Hannink JM, Bomans PHH, Frederik PM, Nolte RJM, Meijer EM, Sommerdijk NAJM (2002) Giant amphiphiles by cofactor reconstitution. Angew Chem Int Ed 41:4239–4241. doi:10.1002/1521-3773(20021115)41:22<4239:aid-anie4239>3.0.co;2-e
Sun X-L, Faucher KM, Houston M, Grande D, Chaikof EL (2002) Design and synthesis of biotin chain-terminated glycopolymers for surface glycoengineering. J Am Chem Soc 124:7258–7259. doi:10.1021/ja025788v
Hou S, Sun X-L, Dong C-M, Chaikof EL (2004) Facile synthesis of chain-end functionalized glycopolymers for site-specific bioconjugation. Bioconjug Chem 15:954–959. doi:10.1021/bc0499275
Bontempo D, Li RC, Ly T, Brubaker CE, Maynard HD (2005) One-step synthesis of low polydispersity, biotinylated poly(N-isopropylacrylamide) by ATRP. Chem Commun 0:4702–4704
Kopping JT, Tolstyka ZP, Maynard HD (2007) Telechelic aminooxy polystyrene synthesized by ATRP and ATR coupling. Macromolecules 40:8593–8599. doi:10.1021/ma071606b
Tolstyka ZP, Kopping JT, Maynard HD (2007) Straightforward synthesis of cysteine-reactive telechelic polystyrene. Macromolecules 41:599–606. doi:10.1021/ma702161q
Tao L, Kaddis CS, Ogorzalek Loo RR, Grover GN, Loo JA, Maynard HD (2009) Synthetic approach to homodimeric protein–polymer conjugates. Chem Commun 0:2148–2150
Tao L, Kaddis CS, Loo RRO, Grover GN, Loo JA, Maynard HD (2009) Synthesis of maleimide-end-functionalized star polymers and multimeric protein-polymer conjugates. Macromolecules 42:8028–8033. doi:10.1021/ma901540p
Boyer C, Liu J, Bulmus V, Davis TP, Barner-Kowollik C, Stenzel MH (2008) Direct synthesis of well-defined heterotelechelic polymers for bioconjugations. Macromolecules 41:5641–5650. doi:10.1021/ma800289u
Heredia KL, Grover GN, Tao L, Maynard HD (2009) Synthesis of heterotelechelic polymers for conjugation of two different proteins. Macromolecules 42:2360–2367. doi:10.1021/ma8022712
Heredia KL, Tao L, Grover GN, Maynard HD (2010) Heterotelechelic polymers for capture and release of protein-polymer conjugates. Polym Chem-Uk 1:168–170
Yoshimoto T, Takahashi K, Ajima A, Matsushima A, Saito Y, Tamaura Y, Inada Y (1985) Activation and stabilization of asparaginase by anti-asparaginase IgG and its Fab. FEBS Lett 183:170–172. http://dx.doi.org/10.1016/0014-5793(85)80978-9
Zhang Y-Q, Tao M-L, Shen W-D, Mao J-P, Chen Y-h (2006) Synthesis of silk sericin peptides–L-asparaginase bioconjugates and their characterization. J Chem Technol Biotechnol 81:136–145. doi:10.1002/jctb.1370
Xia F, Zuo X, Yang R, Xiao Y, Kang D, Vallée-Bélisle A, Gong X, Heeger AJ, Plaxco KW (2010) On the binding of cationic, water-soluble conjugated polymers to DNA: electrostatic and hydrophobic interactions. J Am Chem Soc 132:1252–1254. doi:10.1021/ja908890q
Decher G (1997) Fuzzy nanoassemblies: toward layered polymeric multicomposites. Science 277:1232–1237. doi:10.1126/science.277.5330.1232
Okada T, Uto K, Sasai M, Lee CM, Ebara M, Aoyagi T (2013) Nano-decoration of Hemagglutinating Virus of Japan Envelope (HVJ-E) using Layer-by-Layer Assembly Technique. Langmuir 29:7384–7392. doi:10.1021/la304572s
Carlos Rodríguez-Cabello J, Reguera J, Girotti A, Alonso M, Testera AM (2005) Developing functionality in elastin-like polymers by increasing their molecular complexity: the power of the genetic engineering approach. Prog Polym Sci 30:1119–1145. http://dx.doi.org/10.1016/j.progpolymsci.2005.07.004
Urry DW (2004) The change in Gibbs free energy for hydrophobic association: derivation and evaluation by means of inverse temperature transitions. Chem Phys Lett 399:177–183. http://dx.doi.org/10.1016/j.cplett.2004.09.137
Meyer DE, Kong GA, Dewhirst MW, Zalutsky MR, Chilkoti A (2001) Targeting a genetically engineered elastin-like polypeptide to solid tumors by local hyperthermia. Cancer Res 61:1548–1554
Kostal J, Mulchandani A, Chen W (2001) Tunable biopolymers for heavy metal removal. Macromolecules 34:2257–2261. doi:10.1021/ma001973m
Wan X, Zhang G, Ge Z, Narain R, Liu S (2011) Construction of polymer-protein bioconjugates with varying chain topologies: polymer molecular weight and steric hindrance effects. Chem Asian J 6:2835-2845. doi:10.1002/asia.201100489
Yan M, Ge J, Dong W, Liu Z, Ouyang P (2006) Preparation and characterization of a temperature-sensitive sulfobetaine polymer–trypsin conjugate. Biochem Eng J 30:48–54. http://dx.doi.org/10.1016/j.bej.2006.02.001
Sharma S, Kaur P, Jain A, Rajeswari MR, Gupta MN (2003) A smart bioconjugate of chymotrypsin. Biomacromolecules 4:330–336. doi:10.1021/bm0256799
Pan LC, Chien CC (2003) A novel application of thermo-responsive polymer to affinity precipitation of polysaccharide. J Biochem Bioph Methods 55:87–94. http://dx.doi.org/10.1016/S0165-022X(02)00180-X
Anastase-Ravion S, Ding Z, Pellé A, Hoffman AS, Letourneur D (2001) New antibody purification procedure using a thermally responsive poly(N-isopropylacrylamide)–dextran derivative conjugate. J Chromatogr B: Biomed Sci Appl 761:247–254. http://dx.doi.org/10.1016/S0378-4347(01)00336-X
Chang C-W, Nguyen TH, Maynard HD (2010) Thermoprecipitation of glutathione S-transferase by glutathione–poly(N-isopropylacrylamide) Prepared by RAFT Polymerization. Macromol Rapid Commun 31:1691–1695. doi:10.1002/marc.201000333
Mattiasson B, Kumar A, Galaev IY (1998) Affinity precipitation of proteins: design criteria for an efficient polymer. J Mol Recognit 11:211–216. doi:10.1002/(sici)1099-1352(199812)11:1/6<211:aid-jmr425>3.0.co;2-y
Galaev IY, Mattiasson B (1993) Affinity thermoprecipitation: contribution of the efficiency of ligand–protein interaction and access of the ligand. Biotechnol Bioeng 41:1101–1106. doi:10.1002/bit.260411113
Kumar A, Srivastava A, Galaev IY, Mattiasson B (2007) Smart polymers: physical forms and bioengineering applications. Prog Polym Sci 32:1205–1237. http://dx.doi.org/10.1016/j.progpolymsci.2007.05.003
Kumar A, Wahlund P-O, Kepka C, Galaev IY, Mattiasson B (2003) Purification of histidine-tagged single-chain Fv-antibody fragments by metal chelate affinity precipitation using thermoresponsive copolymers. Biotechnol Bioeng 84:494–503. doi:10.1002/bit.10810
Balan S, Murphy J, Galaev I, Kumar A, Fox G, Mattiasson B, Willson R (2003) Metal chelate affinity precipitation of RNA and purification of plasmid DNA. Biotechnol Lett 25:1111–1116. doi:10.1023/a:1024148316322
Murphy JC, Jewell DL, White KI, Fox GE, Willson RC (2003) Nucleic Acid Separations Utilizing Immobilized Metal Affinity Chromatography. Biotechnol Progr 19:982–986. doi:10.1021/bp025563o
Ding Z, Long CJ, Hayashi Y, Bulmus EV, Hoffman AS, Stayton PS (1999) Temperature control of biotin binding and release with a streptavidin-poly(N-isopropylacrylamide) site-specific conjugate. Bioconjug Chem 10:395–400. doi:10.1021/bc980108s
Bulmus V, Ding Z, Long CJ, Stayton PS, Hoffman AS (1999) Site-specific polymer–streptavidin bioconjugate for ph-controlled binding and triggered release of biotin. Bioconjug Chem 11:78–83. doi:10.1021/bc9901043
Shimoboji T, Ding Z, Stayton PS, Hoffman AS (2001) Mechanistic investigation of smart polymer-protein conjugates. Bioconjug Chem 12:314–319. doi:10.1021/bc000107b
Shimoboji T, Larenas E, Fowler T, Hoffman AS, Stayton PS (2003) Temperature-induced switching of enzyme activity with smart polymer–enzyme conjugates. Bioconjug Chem 14:517–525. doi:10.1021/bc025615v
Hohsaka T, Kawashima K, Sisido M (1994) Photoswitching of NAD+-mediated enzyme reaction through photoreversible antigen-antibody reaction. J Am Chem Soc 116:413–414. doi:10.1021/ja00080a064
Murata M, Kaku W, Anada T, Soh N, Katayama Y, Maeda M (2003) Thermoresponsive DNA/polymer conjugate for intelligent antisense strategy. Chem Lett 32:266–267
Lackey CA, Murthy N, Press OW, Tirrell DA, Hoffman AS, Stayton PS (1999) Hemolytic activity of pH-responsive polymer-streptavidin bioconjugates. Bioconjugate Chem 10:401–405. doi:10.1021/bc980109k
Jones RA, Cheung CY, Black FE, Zia JK, Stayton PS, Hoffman AS, Wilson MR (2003) Poly(2-alkylacrylic acid) polymers deliver molecules to the cytosol by pH-sensitive disruption of endosomal vesicles. Biochem J 372:65–75. doi:10.1042/bj20021945
Kyriakides TR, Cheung CY, Murthy N, Bornstein P, Stayton PS, Hoffman AS (2002) pH-Sensitive polymers that enhance intracellular drug delivery in vivo. J Controlled Release 78:295–303. http://dx.doi.org/10.1016/S0168-3659(01)00504-1
Berguig GY, Convertine AJ, Shi J, Palanca-Wessels MC, Duvall CL, Pun SH, Press OW, Stayton PS (2012) Intracellular delivery and trafficking dynamics of a lymphoma-targeting antibody-polymer conjugate. Mol Pharm 9:3506–3514. doi:10.1021/mp300338s
Dubé D, Francis M, Leroux J-C, Winnik FM (2002) Preparation and Tumor Cell Uptake of Poly(N-isopropylacrylamide) Folate Conjugates. Bioconjugate Chem 13:685–692. doi:10.1021/bc010084g
Benoit DSW, Srinivasan S, Shubin AD, Stayton PS (2011) Synthesis of Folate-Functionalized RAFT Polymers for Targeted siRNA Delivery. Biomacromolecules 12:2708–2714. doi:10.1021/bm200485b
Flanary S, Hoffman AS, Stayton PS (2009) Antigen delivery with poly(Propylacrylic acid) conjugation enhances MHC-1 presentation and t-cell activation. Bioconjug Chem 20:241–248. doi:10.1021/bc800317a
Foster S, Duvall CL, Crownover EF, Hoffman AS, Stayton PS (2010) Intracellular delivery of a protein antigen with an endosomal-releasing polymer enhances cd8 t-cell production and prophylactic vaccine efficacy. Bioconjug Chem 21:2205–2212. doi:10.1021/bc100204m
Albarran B, Hoffman AS, Stayton PS (2011) Efficient intracellular delivery of a pro-apoptotic peptide with a pH-responsive carrier. React Funct Polym 71:261–265. http://dx.doi.org/10.1016/j.reactfunctpolym.2010.09.008
Napoli A, Valentini M, Tirelli N, Muller M, Hubbell JA (2004) Oxidation-responsive polymeric vesicles. Nat Mater 3:183–189. http://www.nature.com/nmat/journal/v3/n3/suppinfo/nmat1081_S1.html
El-Sayed MEH, Hoffman AS, Stayton PS (2005) Rational design of composition and activity correlations for pH-sensitive and glutathione-reactive polymer therapeutics. J Controlled Release 101:47–58. http://dx.doi.org/10.1016/j.jconrel.2004.08.032
Manickam DS, Oupický D (2006) Multiblock reducible copolypeptides containing histidine-rich and nuclear localization sequences for gene delivery. Bioconjug Chem 17:1395–1403. doi:10.1021/bc060104k
Raucher D, Chilkoti A (2001) Enhanced Uptake of a Thermally Responsive Polypeptide by Tumor Cells in Response to Its Hyperthermia-mediated Phase Transition. Cancer Res 61:7163–7170
Agasti SS, Liong M, Peterson VM, Lee H, Weissleder R (2012) Photocleavable DNA barcode-antibody conjugates allow sensitive and multiplexed protein analysis in single cells. J Am Chem Soc 134:18499–18502. doi:10.1021/ja307689w
Maeda M (2006) Sequence-specific aggregation behavior of DNA-carrying colloidal nanoparticles prepared from poly(N-isopropylacrylamide)-graft-oligodeoxyribonucleotide. Polym J 38:1099–1104
Mori T, Maeda M (2001) Formation of DNA-carrying colloidal particle from poly(N-isopropylacrylamide)-graft-DNA copolymer and its assembly through hybridization. Polym J 33:830
Mori T, Maeda M (2002) Stability change of DNA-carrying colloidal particle induced by hybridization with target DNA. Polym J 34:624
Marie Dupuy A, Lehmann S, Paul Cristol J (2005) Protein biochip systems for the clinical laboratory. Clin Chem Lab Med 43:1291–1302. doi:10.1515/cclm.2005.223
Toner M, Irimia D (2005) Blood-on-a-Chip. Annu Rev Biomed Eng 7:77–103. doi:10.1146/annurev.bioeng.7.011205.135108
Malmstadt N, Hoffman AS, Stayton PS (2004) “Smart” mobile affinity matrix for microfluidic immunoassays. Lab Chip 4:412–415
Malmstadt N, Yager P, Hoffman AS, Stayton PS (2003) A smart microfluidic affinity chromatography matrix composed of poly(N-isopropylacrylamide)-coated beads. Anal Chem 75:2943–2949. doi:10.1021/ac034274r
Ebara M, Hoffman JM, Hoffman AS, Stayton PS (2006) Switchable surface traps for injectable bead-based chromatography in PDMS microfluidic channels. Lab Chip 6:843–848
Ebara M, Hoffman JM, Stayton PS, Hoffman AS (2007) Surface modification of microfluidic channels by UV-mediated graft polymerization of non-fouling and ‘smart’ polymers. Radiat Phys Chem 76:1409–1413. http://dx.doi.org/10.1016/j.radphyschem.2007.02.072
Ebara M, Hoffman AS, Stayton PS, Lai JJ (2013) A photo-induced nanoparticle separation in microchannels via pH-sensitive surface traps. Langmuir 29:5388–5393. doi:10.1021/la400347r
Lai JJ, Hoffman JM, Ebara M, Hoffman AS, Estournès C, Wattiaux A, Stayton PS (2007) Dual magnetic-/temperature-responsive nanoparticles for microfluidic separations and assays. Langmuir 23:7385–7391. doi:10.1021/la062527g
Lai JJ, Nelson KE, Nash MA, Hoffman AS, Yager P, Stayton PS (2009) Dynamic bioprocessing and microfluidic transport control with smart magnetic nanoparticles in laminar-flow devices. Lab Chip 9:1997–2002
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Copyright information
© 2014 National Institute for Materials Science, Japan. Published by Springer Japan
About this chapter
Cite this chapter
Ebara, M. et al. (2014). Smart Bioconjugates. In: Smart Biomaterials. NIMS Monographs. Springer, Tokyo. https://doi.org/10.1007/978-4-431-54400-5_6
Download citation
DOI: https://doi.org/10.1007/978-4-431-54400-5_6
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-54399-2
Online ISBN: 978-4-431-54400-5
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)