
Abstract
Eukaryotic cells depend upon oxygen (O2) for their survival and elaborate mechanisms have evolved in multicellular animals, especially vertebrates, to monitor the availability of environmental O2, the efficiency of O2 extraction from the environment, ensure adequate O2 delivery to tissues and even to regulate cellular metabolism when O2 availability is compromised. In vertebrates, specialized O2 “sensing” cells have developed to carry out many of these processes. Although all O2 sensing cells ultimately couple low Po 2 (hypoxia) to physiological responses, how these cells actually detect hypoxia, i.e., the “O2 sensor” remains controversial. We have recently proposed that hydrogen sulfide (H2S) through its O2-dependent metabolism is a universal and phylogenetically ancient O2 sensing mechanism. This hypothesis is based on a variety of experimental evidence including; (1) the effects of exogenous H2S mimic hypoxia, (2) H2S production and/or metabolism is biochemically coupled to O2, (3) tissue H2S concentration is inversely related to Po 2 at physiologically relevant Po 2s, (4) compounds that inhibit or augment H2S production inhibit and augment hypoxic responses, (5) H2S acts upon effector mechanisms known to mediate hypoxic responses, (6) H2S was key to the origin of life and the advent of eukaryotic cells and the reciprocal relationship between O2 and H2S has been inexorably intertwined throughout evolution. The evidence for H2S-mediated O2 sensing is critically examined in this review.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
2.1 Introduction
Specialized O2 sensing tissues in vertebrates are strategically placed to monitor ambient O2, O2 transport in blood, and to match blood flow with ventilation or tissue demand. Neuroepithelial cells (NEC) are present on the external surfaces of fish gills and monitor water Po 2 (Milsom and Burleson 2007). This is especially important for these aquatic vertebrates because, compared to air, water has considerably less O2 (1/30), slower rates of O2 diffusion (Krogh’s diffusion coefficients 1/200,000) and higher viscosity (60 times). Even more problematic, aquatic O2 levels can vary both temporally (minutes to seasons) and spatially within meters (Bickler and Buck 2007). In lungs of newborn mammals, cells similar to neuroepithelial-like cells are found in clusters (called neuroepithelial bodies, NEB) near airway bifurcations and here they may be important in the transition from the relatively hypoxic uterine environment during and shortly after birth (Kemp et al. 2002). External O2 sensors other than NEB are relatively uncommon in terrestrial vertebrates. Presumably this is because atmospheric O2 is relatively constant (21 %) and internal O2 sensors appear to be able to accommodate changes in O2 availability (such as in borrows or with increasing altitude) if needed. Arterial O2 sensors that monitor blood Po 2 and O2 delivery are found in fish as vascular-facing NEC and in higher vertebrates as the type I glomus cells of carotid and aortic bodies. (It is perhaps no coincidence that the first gill arch of fish is the homolog of the mammalian carotid body.) Mammalian adrenal medullary cells and homologous chromaffin cells that line systemic veins in fish secrete catecholamines in response to hypoxemia (Nurse et al. 2006; Perry et al. 2000) and may monitor tissue O2 extraction. Perhaps the best characterized O2 sensing tissues are the blood vessels themselves. It has generally been accepted that hypoxia relaxes systemic vessels thereby matching tissue perfusion to metabolic demand, whereas hypoxia contracts pulmonary vessels to match ventilation to perfusion (Sylvester et al. 2012). However, recent studies have shown that many systemic vessels in nonmammalian vertebrates are contracted by hypoxia (Russell et al. 2008) and hypoxia relaxes pulmonary vessels in diving mammals to prevent pulmonary hypertension that would otherwise occur during a prolonged dive (Olson et al. 2010). Although the vascular response to hypoxia is intrinsic to vascular smooth muscle (Madden et al. 1992) there appears to be considerable plasticity in the functional organization of this response. These atypical responses have been key to evaluating H2S-mediated mechanisms for acute O2 sensing as described in this chapter.
There is considerable controversy concerning the actual mechanism with which these cells detect O2 levels or availability and then transduce this into physiologically relevant signals and of the numerous proposed O2 sensing mechanisms none have received unanimous support. Our work has suggested that the O2 dependent metabolism of hydrogen sulfide (H2S) is an effective and efficient mechanism of H2S sensing. This review presents the evidence supporting our hypothesis.
2.2 Mechanism(s) of O2 Sensing
Despite the apparent ubiquity of O2 sensing tissues in vertebrates there has been little consensus on the mechanism that specifically measures Po 2 or O2 concentration. Potassium channels have long been a likely candidate (Weir and Archer 1995) although it is now believed that they operate downstream of the actual sensing mechanism. The various theories of O2 sensing mechanisms have been extensive reviewed Sylvester et al. (2012). Because mitochondria account for most of a cell’s O2 consumption they are central to most O2 sensing theories and implicit in the three most prevalent theories, the redox hypothesis, the reactive oxygen species (ROS) hypothesis and the energy state/AMPK (AMP-activated protein kinase) hypothesis. In the redox hypothesis, hypoxia suppresses mitochondrial oxidative phosphorylation which further reduces the cytosol and decreases ROS production. Voltage-gated potassium (Kv) channels that were tonically kept open during normoxia by ROS now close and the resulting cell depolarization opens voltage-gated calcium channels and the influx of calcium produces contraction. Essentially the opposite occurs in the ROS hypothesis where hypoxia increases mitochondrial production of ROS, namely superoxide (O2 −) and probably more importantly hydrogen peroxide (H2O2). The ROS thus produced activate a variety of intracellular signaling cascades that also increase intracellular calcium concentration. In the energy state/AMPK hypothesis, hypoxia decreases mitochondrial ATP production which increases the AMP to ATP ratio and activates AMP kinase. The resulting production of cyclic ADP ribose then brings about an increase in intracellular calcium and contraction. It should be noted, however, that vascular smooth muscle and endothelial cells can and do derive their energy from glycolysis and even though they respond to hypoxia, their ATP levels do not appear to be affected (Dromparis and Michelakis 2013). Other O2 sensing mechanisms such as heme oxygenase (which generates the gasotransmitter carbon monoxide, CO), plasma membrane bound NADPH oxidase or a yet identified hemoprotein or mitochondrial complex III and nitric oxide (NO) have also been described for various tissues (Evans et al. 2011; Gonzalez et al. 2010; Haldar and Stamler 2013; Waypa and Schumacker 2010; Wolin et al. 2010).
2.3 H2S Oxidation as an O2 Sensing Mechanism
We (Olson et al. 2006) originally proposed that the O2-dependent metabolism of endogenously generated, and biologically active H2S functioned as an efficient O2 sensing mechanism that initiated hypoxic pulmonary vasoconstriction (HPC) and hypoxic systemic vasodilation (HSD). This hypothesis appears to fulfill the criteria for an O2 sensing mechanism (Olson 2011) in that; (1) the effects of exogenous H2S mimic hypoxia, (2) H2S production and/or metabolism is biochemically coupled to O2, (3) tissue H2S concentration is inversely related to Po 2 at physiologically relevant Po 2s, (4) compounds that inhibit or augment H2S production inhibit and augment hypoxic responses, (5) H2S acts upon effector mechanisms known to mediate hypoxic responses, (6) H2S-mediated O2 sensing has an evolutionary precedent and a phylogenetic history. The following sections describe how these criteria have been met.
2.3.1 The Effects of Exogenous H2S Mimic Hypoxia
2.3.1.1 Cardiovascular System
Mechanical responses to hypoxia are identical to those produced by H2S in all vessels (Fig. 2.1). Relaxation of rat thoracic aorta and portal vein was one of the first “physiological” effects of H2S identified (Hosoki et al. 1997) and this mimics the well-known hypoxic vasodilation of mammalian systemic vessels. Similar hypoxic and H2S vasodilations have been observed in all classes of vertebrates from the most primitive hagfish and lamprey to mammals (cf Russell et al. 2008; Dombkowski et al. 2005). This response has now been observed in over 20 mammalian studies. H2S also dilates the mouse ductus arteriosus (Baragatti et al. 2013) which would be expected to keep this vessel patent in the relative hypoxic intrauterine environment. H2S vasodilation of mammalian systemic vessels has also been observed in perfused organs (Cheng et al. 2004) and in vivo (Derwall et al. 2011; Leffler et al. 2010; Mustafa et al. 2009; Yan et al. 2004; Yang et al. 2008). Consistent with hypoxic pulmonary vasoconstriction, H2S constricts isolated bovine pulmonary arteries (Olson et al. 2006, 2010), increases vascular resistance in perfused fish gills (Skovgaard and Olson 2012) and perfused rat lungs (Madden et al. 2012) and increases pulmonary arterial blood pressure in vivo (Derwall et al. 2011; Sowmya et al. 2010).

Hypoxia (95 % N2/5 % CO2 a, b, d, e; or 100 % N2, c) and H2S (300 μmol/l) produce identical responses in conductance (>500 μm diameter) vessels from rat thoracic aorta (a), rat pulmonary artery (b), lamprey dorsal aorta (c) and bovine pulmonary artery (d). Vessels pre-contracted with 1 μmol/l norepinephrine (NE) or U-46619 (thromboxane A2 mimetic; 0.1 μmol/l); air aeration with room air; w wash; 1, 2, 3, tri-phasic response. Horizontal time bar in a–d = 10 min, vertical tension scale = 0.5 g. (e), hypoxia (Po 2 ~ 50 mmHg) and H2S (3 × 10−4 M) produce identical contractions of cow resistance (<500 μm diameter) pulmonary arteries while both stimuli relax sea lion resistance pulmonary arteries. Mean ± SE; N (animals,vessels) = cow (9,9), sea lion (3,5). (f, g), H2S has two dose-dependent effects on pre-contracted (0.1 μmol/l U-46619) bovine pulmonary arteries. H2S appears to produce a dose-dependent relaxation between 10−8 and 10−5 mol/l, whereas above 10−5 mol/l it produces a dose-dependent constriction. The EC50 for relaxation (5.5 ± 1.8 × 10−7 M) is significantly (p < 0.05) different from the EC50 for contraction (3.7 ± 1.5 × 10−4 M). (f) single trace of cumulative doses, arrows indicate log M [H2S]; w wash; (g) average (+SE) of eight vessels, filled circles denote relaxation and open squares contraction (values extrapolated where curves overlap ~ 10−5 M) (a–d, f, g Adapted from Olson et al. 2006, with permission; e adapted from Olson et al. 2010, with permission)
Hypoxic responses of non-mammalian vertebrates are considerably more variable in that systemic vessels can be either dilated or contracted by hypoxia and often these effects are multiphasic. Nevertheless, in over 30 animals from all vertebrate classes, the hypoxic responses of both systemic and respiratory vessels are consistently mimicked by H2S (summarized in Olson and Whitfield 2010). This includes the signature multiphasic contraction-relaxation-contraction of rat pulmonary arteries (Fig. 2.1b) and the unique hypoxic and H2S-mediated dilation of sea lion pulmonary resistance vessels (Fig. 2.1e). It is also evident that many of these multiphasic responses such as the initial relaxation followed by constriction in bovine pulmonary arteries are the results of separate, but dose-dependent effects of H2S (Fig. 2.1f), a hint of which can also be seen during the onset of hypoxia (Fig. 2.1d). Hypoxia and H2S also relax non-vascular smooth muscle of fish urinary bladder and the gastrointestinal tract and in the gastrointestinal tract both of these stimuli produce a unique and transient increase in spontaneous contraction frequency and amplitude prior to the onset of the inhibitory effects (Dombkowski et al. 2006, 2011). H2S also relaxes human corpus cavernosum and urinary bladder smooth muscle (d’Emmanuele di Villa Bianca et al. 2009; Fusco et al. 2012).
Involvement of H2S has also been shown in other hypoxia-related responses. In the cardiovascular system this includes angiogenesis, ischemia reperfusion injury (RI) and pre- and post-conditioning against RI (Bian et al. 2006; Cai et al. 2007; Calvert et al. 2010; Lavu et al. 2010; Liu et al. 2010; Pan et al. 2006; Papapetropoulos et al. 2009; Szabo and Papapetropoulos 2011; Wang et al. 2010a, b; Yong et al. 2008). H2S also contributes to hypoxia-induced radiation resistance (Zhang et al. 2011) and it is a cryogenic mediator of hypoxia-induced anapyrexia (Kwiatkoski et al. 2012).
2.3.1.2 Respiration
Intravascular injection or inhalation of H2S at low concentrations has long been known to mimic hypoxemia by stimulating respiration in many mammals (Beauchamp et al. 1984; Reiffenstein et al. 1992; Haggard and Henderson 1922; Haouzi et al. 2009 [in mice but not rats], 2011; Haouzi 2012; Van de Louw and Haouzi 2012) and birds (Klentz and Fedde 1978). This may have both a central and peripheral component mediated through the carotid bodies. Intracerebroventricular injection of H2S produces a KATP channel-mediated dose dependent bradycardia and hypotension (Liu et al. 2011) akin to the hypoxic diving reflex. In more specific studies it was observed that H2S increases discharge frequency from the pre-Bötzinger (pB) dorsal inspiratory respiratory group and it may initially produce transient inhibition of the pB by stimulating the nearby parafacial respiratory group (Chen et al. 2013; Hu et al. 2008). H2S stimulates peripheral chemoreceptors (neuroepithelial cells) in the fish gill (Olson et al. 2008) and mammalian carotid body (Li et al. 2010; Makarenko et al. 2012; Peng et al. 2010) and it stimulates the mammalian adrenal medulla (Peng et al. 2010; Zhu et al. 2012) and homologous fish chromaffin cells (Perry et al. 2009). H2S may also contribute to the sequela of events in which heart failure, hypertension and renal failure activate the carotid body leading to breathing instability and increased sympathetic nerve activity (Schultz et al. 2012).
2.3.2 H2S Production and/or Metabolism is Biochemically Coupled to O2
2.3.2.1 Biosynthesis
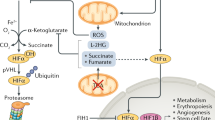
Pathways for H2S synthesis and metabolism are shown in Fig. 2.2. l-cysteine and l-homocysteine account for most H2S production through the activity of four enzymes, cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE aka CGL) and sequential catalysis by cysteine aminotransferase (CAT) and 3-mercaptopyruvate sulfurtransferase (3-MST). H2S may be synthesized in the cytosol as CBS and CSE and CAT/3-MST are cytosolic enzymes, or it may be synthesized in the mitochondria as CAT/3-MST are present there as well (Kamoun 2004). 3-MST is especially abundant in the mitochondrial matrix (Mikami et al. 2011a) where it can take advantage of threefold higher cysteine concentration than in the cytosol (Fu et al. 2012). CSE can also be translocated to the mitochondria by a variety of stress-related stimuli (Fu et al. 2012). Initially it was believed that CBS was predominantly found in the brain and CSE in the cardiovascular system (reviewed in Kimura 2010), although a broader distribution is becoming evident, i.e., CBS has been identified in vascular endothelium, CAT and 3-MST in vascular endothelium and brain and MST, but not CAT, in vascular smooth muscle (Kimura 2010; Olson et al. 2010). CBS and CSE have also been identified in human plasma (Bearden et al. 2010). H2S can be generated from d-cysteine, however, this pathway appears limited to the brain and kidney where it protects the former from oxidative stress and the latter from re-perfusion injury; it may be of limited function elsewhere (Shibuya et al. 2013). CBS, CSE and CAT are pyridoxal 5′phosphate (PLP)-dependent, enzymes. S-adenosylmethionine allosterically activates CBS (Stipanuk 2004). CBS contains a heme group that can be inhibited by presumably physiological levels of carbon monoxide (CO; inhibition constant, Ki = 5.6 μM) and although it is also inhibited by nitric oxide (NO), the Ki is so high (320 μM) that it is of questionable physiological significance; O2 does not affect CBS activity (Banerjee and Zou 2005). CSE and both cytosolic and mitochondrial CAT activity are inhibited by calcium, independent of calmodulin (Mikami et al. 2011b, 2013).

Pathways for H2S production and degradation. H2S is synthesized from homocysteine or cysteine by the cytosolic enzymes cystathionine β-synthase (CBS), cystathionine γ-lyase, or the tandem action of cysteine aminotransferase (CAT) and 3-mercaptopyruvate sulfur transferase (3-MST) in both the cytosol and mitochondria. Other potential mechanisms for H2S biosynthesis have been described in invertebrates (dotted enclosure; Julian et al. 2002) but have yet to be confirmed in mammals. 3-mercaptopyruvate can also be produced from d-cysteine in the peroxisome by d-amino acid oxidase (DAO). Oxidation of H2S in the mitochondria (blue letters) is initiated by interaction with the enzyme sulfide:quinone oxidoreductase (SQR) producing two SQR persulfides that are then transferred to a mobile sulfide carrier (RSSH), one of which is oxidized to sulfite (SO3 2−) by mitochondrial sulfur dioxygenase (ETHE1). Sulfur transferase (ST) transfers the other persulfide to form thiosulfate (S2O3 2−). H2S may be regenerated from thiosulfate by 3-MST or rhodanase (Rde), or the thiosulfate may be oxidized to sulfate (SO4 2−) by the sequential actions of thioredoxin reductase (TRD) in combination with glutathione (GSH) and sulfur oxidase (SO). Oxidation of H2S donates electrons to the respiratory chain (Q, III, IV) that has been shown in invertebrates and mammals to result in ATP production and O2 consumption. An alternative oxidase (AOX) that oxidizes H2S without concomitant ATP production has also been observed in invertebrates. H2S can also be regenerated from thiosulfate by 3-mercaptopyruvate sulfur transferase (3-MST) or rhodanase (Rde) in the presence of other reducing disulfides such as thioredoxin (Trx) or dihydrolipoic acid (DHLA). Circled numbers indicate actual or potential hypoxia-sensitive sites (see text for details) (Modified from Olson et al. (2012c), with permission)
2.3.2.2 Metabolism (Inactivation)
Mathematical models of H2S diffusion out of cells versus intracellular metabolism suggest that most H2S is inactivated intracellularly (Olson 2013), and this is supported by the considerable body of evidence that mitochondria efficiently oxidize H2S (reviewed in Olson 2012c). Mitochondrial enzymes, sulfide:quinone oxidoreductase (SQR), 3-MST, rhodanase (Rde), thiosulfate reductase (TR), sulfur dioxygenase (ETHE1) and sulfite oxidase (SO) ultimately oxidize H2S to sulfate (SO4 2−) which is then excreted. Sulfite (SO3 2−) and thiosulfate (S2O3 2−) are intermediates. SQR is bound to the inner mitochondrial membrane and it is closely associated with the respiratory chain “supercomplex”(Hildebrandt 2011) which provides a hint at its O2 sensing function as the mitochondrion is a lead candidate for the site of O2 sensing (Sylvester et al. 2012). Not surprisingly, 3-MST, rhodanase and thiosulfate reductase are abundant in the mitochondrial matrix and to a lesser extent the intermembrane space (Koj et al. 1975). H2S oxidation begins with H2S binding to the highly conserved Cys-Cys disulfide bridge of SQR. The sulfide is oxidized to elemental sulfur forming SQR persulfide (sulfane sulfur) with the now-reduced SQR cysteine (SQR-SSH). Two H2S and two SQR are involved, one persulfide sulfur is transferred to sulfur dioxygenase (SDO) where it is oxidized to sulfite while sulfur from the second persulfide is transferred from the SQR to sulfite by sulfur transferase (ST) producing thiosulfate. One electron from each of the two H2S are fed via the quinone pool (Q) into the respiratory chain (Marcia et al. 2010) and they ultimately reduce O2 at complex IV. SQR is bound to the inner mitochondrial membrane and it is believed that sulfur is shuttled from SQR by a mobile carrier such as glutathione (GSH), dihydrolipoate, thioredoxin or even sulfite (Hildebrandt and Grieshaber 2008; Jackson et al. 2012; Theissen and Martin 2008). One of the mobile persulfides is oxidized to sulfite by ETHE1 which consumes molecular O2 and water in the process. Metabolism of H2S through SQR appears ubiquitous in tissues although the brain may be an exception (Hildebrandt 2011; Lagoutte et al. 2010; Linden et al. 2011; but see Ackermann et al. 2011). The capacity of cells to oxidize sulfide appears to be greater than the estimated rate of sulfide production (Bouillaud and Blachier 2011; Furne et al. 2008). Thus it is expected that intracellular H2S concentrations are very low under normoxic conditions.
Under normoxic conditions, most thiosulfate is further metabolized to sulfate by thiosulfate reductase (TR) and sulfite oxidase (SO). Elimination of one sulfur atom as sulfate is accompanied by four atoms of O2. Even though sulfur excretion as thiosulfate would conserve O2, apparently there is little need for this when is O2 plentiful and thiosulfate excretion in vertebrates is generally low. In fact, it is not clear why most vertebrates, especially terrestrial ones, would bother with this pathway at all. However, as described below, it may be an important avenue for regeneration of H2S during hypoxia. Details of H2S biosynthesis and metabolism can be found in (Kabil and Banerjee 2010; Kimura 2010; Olson 2012c; Stipanuk 2004; Stipanuk et al. 2009).
2.3.2.3 H2S Oxidation via Electron Transport
In our original mechanism of H2S-mediated O2 sensing (Olson et al. 2006) we proposed that H2S was constitutively produced in the cytosol through transsulfurration and oxidized in the mitochondria. As the amount of O2 available to the mitochondria determined the rate of H2S oxidation, and hence H2S concentration, this was the “O2 sensor.” Although this is still likely an integral mechanism, it is now evident that there are a number of other mechanisms with which O2 can influence H2S concentration and thereby contribute to O2 sensing. These mechanisms, some of which are expected to rapidly respond to O2 while others may provide a longer bias of H2S concentration, are described in the following paragraphs and shown numerically in Fig. 2.2. While many of these pathways remain to be verified in the context of O2 sensing, there is accumulating evidence, largely through studies on enzyme deficiencies, that their dysfunction will impact sulfide metabolism and either directly or indirectly increase H2S concentration.
2.3.2.3.1 Rapid Effectors of H2S Concentration
Electron transport: Disruption of electron flow down the respiratory chain by insufficient O2 delivery to the mitochondrion at complex IV still remains a very likely and highly effective mechanism to regulate H2S as it directly couples O2 availability to H2S inactivation. This will prevent any further oxidation of H2S that was derived from transsulfurration. This pathway of H2S oxidation has been well-established in the context of ATP synthesis (Goubern et al. 2007; Lagoutte et al. 2010; Modis et al. 2013).
ETHE1: The mitochondrial dioxygenase, ETHE1, uses molecular O2 and water to oxidize the mobile persulfide from SQR to form sulfite. Inhibition of this pathway will prevent H2S binding to SQR. Although the effects of O2 on this pathway have not been examined in detail, they would presumably be similar to the well-characterized ETHE1 deficiencies in experimental animals (ETHE1−/−mice) and humans, the pathology of which is characterized by greatly elevated tissue H2S and thiosulfate (Di Meo et al. 2011; Drousiotou et al. 2011; Giordano et al. 2011; Tiranti et al. 2009).
Sulfite oxidase: Sulfite oxidase (SO) in the mitochondrial innermembrane space catalyzes the oxidation of sulfite to sulfate by transferring an atom of O2 from water to sulfite and in the process the enzyme undergoes a 2-electron reduction (Rajapakshe et al. 2012). These electrons are then transferred from SO to cytochrome c and shuttled into the electron transport chain with molecular oxygen as the terminal acceptor. This couples sulfite concentration to O2 availability and suggests that a hypoxia-induced increase in sulfite would increase thiosulfate concentration and ultimately H2S production. SO deficiency in a human was first demonstrated in 1967 and, as might be expected, this patient presented with elevated urinary thiosulfate (Mudd et al. 1967).
Thiosulfate reduction: As described above, inhibition of either ETHE1 or SO will prevent further oxidation of thiosulfate and this thiosulfate can now directly produce H2S under appropriate conditions by reduction. The sulfur atoms on thiosulfate have different oxidation states, the inner sulfur is +5 and the outer (persulfide) sulfur is −1. In the presence of a reducing reagent such as the endogenous mitochondrial reductant, dihydrolipoic acid (DHLA), 3-MST or thiosulfate reductase (TR; aka rhodanase, Rde) can catalyze the removal of the persulfide and generate H2S (Mikami et al. 2011; Villarejo and Westley 1963). H2S generation from thiosulfate under these conditions has been demonstrated in a variety of mammalian and non-mammalian tissues and it is enhanced by hypoxia (Olson et al. 2013). Thiosulfate can also be generated from d-cysteine by d-amino acid oxidase through formation of 3-mercaptopyruvate (Huang et al. 1998) although the biological significance of this pathway is unknown. H2S generation from thiosulfate will be greatest where thiosulfate concentrations are the highest and when the immediate environment becomes more reduced and the relevant reducing molecules become more available. Thiosulfate concentrations are probably highest in or near the inner mitochondrial membrane, the site of SQR. This is also likely to experience the greatest increase in hypoxia-induced reducing equivalents such as DHLA or thioredoxin because as cells become hypoxic, reactive oxygen species (ROS) increase in both the cytosol and mitochondrial intermembrane space, whereas ROS decrease in the mitochondrial matrix (Waypa et al. 2010). Thus, thiosulfate reduction could significantly contribute to the initial increase in H2S concentration that activates the O2 sensing cascade and by recycling sulfur it conserves biologically relevant thiols.
2.3.2.3.2 Long-Term Effectors of H2S Concentration
Cysteine dioxygenase: Cysteine dioxygenase (CDO), a cytosolic enzyme, effectively eliminates sulfur from entering the H2S pool by irreversibly catalyzing the oxidation of cysteine to cysteinesulfinate which can be further metabolized to hypotaurine, then taurine or sulfite and then sulfate for excretion (Stipanuk et al. 2009). One of the primary functions of CDO is believed to be the detoxification of excess dietary or metabolic cysteine as CDO activity is dynamically regulated by cysteine (as much as 450-fold) whereas cysteine desulfuration, the H2S-forming transsulfuration pathway is not regulated (Stipanuk et al. 2009). CDO also contributes to degradation of methionine and homocysteine after their conversion to cysteine (Stipanuk and Ueki 2011). Impairment of this pathway, which has been demonstrated in CDO knockout (CDO−/−) mice, redirects sulfur through the desulfuration pathway and increases thiosulfate and H2S production (Ueki et al. 2011; Roman et al. 2013). As molecular O2 is the only other substrate in CDO-mediated cysteine oxidation it is likely that hypoxia will impair cysteine oxidation and favor H2S production. It is not likely that this would contribute to the rapid on/off signaling observed acute hypoxia, but it could place a long-term bias on chronic H2S-mediated H2S sensing. An inability of this pathway to handle a large transient cysteine load may partially explain how hypoxic responses are augmented by exogenous cysteine (see below). Ueki et al. (2011) also noted the striking similarities between CDO−/−and ETHE1−/−pathologies and suggested that sulfide/H2S was the common factor.
CSE translocation to mitochondria: In vascular smooth muscle cells, hypoxia stimulates CSE translocation from the cytosol to the mitochondria where it utilizes the threefold increase in cysteine concentration to generate H2S which can be subsequently used in ATP synthesis (Fu et al. 2012). It has been proposed that this H2S improves mitochondrial ATP production and it is protective during hypoxia (Fu et al. 2012). There are, however, several problems with this hypothesis. First, vascular smooth muscle can derive sufficient energy from anaerobic metabolism and does not need oxidative phosphorylation to supply energy, even during hypoxia (Dromparis and Michelakis 2013). Second, because ATP generated from H2S is ultimately dependent on O2, it is unclear what the electron acceptor will be in the absence of O2. However, the H2S formed by CSE translocation could clearly contribute to O2 sensing and hypoxic vasodilation.
2.3.2.3.3 Indirect O2 Effects
3-MST and thioredoxin catalytic-site cysteines: Many enzymes contain cysteine in the catalytic site and because these cysteines generally have a low pKa, they are redox active (Nagahara 2011). The catalytic cysteine in 3-MST (Cys247; rat) is one example. The exposed, Cys247 sulfur is readily oxidized to a sulfenyl (R-SO) by O2, peroxide (H2O2) or other oxidants under mild oxidizing conditions; the sulfenyl is also reduced (to R-SH) by reduced thioredoxin (Trx) but not GSH (Nagahara 2012). Monomeric Rat 3-MST can also dimerize by mild oxidation of two other exposed cysteines, Cys154 and Cys263; both oxidization of Cys247 and dimerization inactivate the enzyme. A defect in 3-MST activity, presented clinically as mercaptolactate-cysteine disulfidia, is believed to be associated with deficient H2S production (Nagahara 2012). While the three external cysteines of 3-MST may well allow it to serve as an effective antioxidant (Nagahara 2012) it could also be a key component of the H2S-mediated O2 sensing mechanism. Furthermore, as 3-MST is found in both the cytosol and mitochondrial matrix and during hypoxia ROS in the cytosol increase while ROS in the matrix decrease, in both pulmonary and systemic arterial smooth muscle cells (Waypa et al. 2010), this will favor mitochondrial H2S production and inhibit it in the cytosol. Parenthetically, the now-oxidized Trx can be reduced by thioredoxin reductase (TRD) using NADPH; which may be an overlooked, but key explanation for why NADPH has been central in many O2-sensing theories (Gupte and Wolin 2008).
2.3.3 Tissue H2S Concentration Is Inversely Related to Po 2 at Physiologically Relevant Po 2s
If H2S and O2 coexist in the environment or in cells it is only transient. In fact, hypoxia, or more often anoxia, is generally (although rarely appreciated) a requisite for all measurements of tissue H2S production, other than those using polarographic H2S sensors (Olson 2012a). Using the polarographic sulfide sensor, originally developed by Jeroschewski et al. (1996), Kraus and Doeller (2004) observed that excised gills and gill mitochondria from sulfide-adapted mussel, Geukensia demissa, rapidly consumed H2S in the presence of O2 and that the rate of H2S consumption was reduced 50-fold in anoxia and 75 % inhibited by cyanide. The authors estimated the P50 (the partial pressure of O2 at half maximal rate) for H2S oxidation in mitochondria of approximately 7.5 mmHg. Both Furne et al. (2008) and Doeller et al. (2005) measured the relationship between O2 and H2S production in rat tissues and observed that H2S production was suppressed at normoxic Po 2 and Furne et al. (2008) observed a switch from H2S production in hypoxia to H2S consumption in normoxia in mouse liver and brain. Similar observations have been made in a variety of other tissues (Fig. 2.3; Dombkowski et al. 2011; Linden et al. 2011; Madden et al. 2012; Olson et al. 2008, 2010; Olson and Whitfield 2010; Whitfield et al. 2008). A compelling argument for H2S-mediated O2 sensing can be made by comparing bovine and sea lion lungs (Olson et al. 2010); while both tissues clearly demonstrate an identical and reciprocal relationship between H2S production/consumption and O2 (Fig. 2.3a, b), both hypoxia and H2S constrict the former and dilate the latter (Fig. 2.1e).

Reciprocal relationship between O2 and H2S in tissues. (a–c) Realtime measurements of O2 and H2S in homogenized cow lung (a), homogenized sea lion lung (b) and homogenized trout intestine (c) using gas-specific polarographic electrodes. H2S production increases after addition of 1 mM cysteine (Cys) and α-ketoglutarate (α-Kg) or Cys alone but only in the absence of O2. Injection of a small air bubble (air) which increases O2 concentration immediately reduces H2S concentration all tissues. When the O2 is consumed, H2S concentration again increases. These results show that H2S production is inversely related to O2 in a variety of tissues, even though bovine pulmonary arteries are constricted by hypoxia whereas sea lion pulmonary arteries and trout intestines are relaxed. (d) H2S consumption as a function of O2 availability (Po 2) by bovine mitochondria (mito), homogenized bovine lung tissue (bovine lung) and bovine pulmonary artery smooth muscle cells (PASMC). H2S is rapidly metabolized (100 % inactivation rate) until Po 2 falls to ~ 15 mmHg (1–2 mmHg for mito); H2S metabolism rapidly fails as Po 2 continues to fall. The Po 2 values at which H2S metabolism is impaired are at the low end of cytosolic and mitochondrial Po 2 and would be expected during hypoxia. The Po 2 at which H2S metabolism is reduced to half (P50) in lung tissue and PASMC is essentially the same Po 2 that produces half-maximal hypoxic vasoconstriction of bovine pulmonary arteries (vessel). (a, b, d, Modified from Olson et al. 2010, c modified from Dombkowski et al. 2011, with permission)
To date, there has only been one study in which the rate of H2S consumption by tissue has been measured at carefully controlled Po 2 (Olson et al. 2010). As shown in Fig. 2.3d, the efficiency of H2S oxidation by bovine lung homogenate, bovine pulmonary arterial smooth muscle cells, or purified bovine heart mitochondria begins to fail at physiologically relevant Po 2s and at Po 2s routinely encountered during hypoxia H2S metabolism becomes highly sensitive to O2 availability. Further demonstration of the physiological relevancy of this process is the observation that the Po 2 at which the ability of pulmonary arterial smooth muscle cells to oxidize H2S is halved (P50) is identical to the P50 of hypoxic pulmonary vasoconstriction. As might be expected, mitochondria function at a Po 2 below the cytosolic Po 2 and it is evident in Fig. 2.3d that the H2S oxidation curve is left-shifted accordingly. These studies clearly show that the metabolism of H2S is O2 dependent, that the ability of tissues to metabolize H2S fails at physiologically relevant Po 2s, and this provides a sensitive and efficient mechanism for O2 sensing.
2.3.4 Compounds that Augment or Inhibit H2S Production Augment or Inhibit Hypoxic Responses
The ability of sulfur donors especially cysteine to augment hypoxic responses has been well documented (Fig. 2.4). Cysteine increases the magnitude of hypoxic vasoconstriction of isolated lamprey aortas (Olson et al. 2006), bovine pulmonary arteries (Olson et al. 2006, 2010) and it increases vascular resistance in the perfused rat lung (Madden et al. 2012). Both reduced and oxidized glutathione augment hypoxic vasoconstriction in pulmonary arteries and the perfused rat lung and cysteine plus α-ketoglutarate (presumably utilizing the CAT/3-MST pathway) increases hypoxic vasoconstriction in bovine pulmonary arteries (Madden et al. 2012; Olson et al. 2010). Continuous utilization of cysteine to sustain a hypoxic vasoconstriction is evident in Fig. 2.4d where it clearly sustains the hypoxic response. Exogenous cysteine also augments hypoxic relaxation of rat aortas (Bucci et al. 2010), the relaxation component of the perfused trout gill (Skovgaard and Olson 2012) as well as hypoxic relaxation of trout urinary bladder (Dombkowski et al. 2006) and salmon intestine (Dombkowski et al. 2011).

Substrates for H2S biosynthesis augment hypoxic responses. (a) In the lamprey aorta cysteine (Cys, 1 mM) nearly doubles a hypoxic contraction but does not affect a KCl (80 mM) contraction. Both Cys and glycine (Gly, 1 mM) had a slight effect but Gly does not affect the hypoxic response. (b) Cys (1 mM), reduced glutathione (GSH, 1 mM) and oxidized glutathione (GSSG, 1 mM) all significantly increase perfusion pressure in the perfused rat lung. (c) Cys (1 mM), GSH (1 mM), GSSG (1 mM) and Cys plus α-ketoglutarate (Cys + αKg, 1 mM) enhance consecutive hypoxic contractions of bovine pulmonary arteries. (d) Representative myograph traces illustrating the ability of Cys (1 mM) to prolong a hypoxic contraction in an isolated bovine pulmonary artery (Modified from Olson 2012b, with permission)
Hypoxic responses of lamprey aorta, bovine pulmonary arteries, rat aorta and the perfused trout gill and rat lung are also inhibited by inhibitors of H2S synthesis (Fig. 2.5; Olson et al. 2006; Madden et al. 2012; Skovgaard and Olson 2012). Although inhibitors of H2S are notoriously nonspecific and often poorly absorbed by tissues (Szabó 2007), their application can provide some information on the biosynthetic pathways that are being used to produce H2S. Not surprisingly, CSE appears to be the major pathway for H2S production by systemic vessels (Fig. 2.5c). However, in bovine pulmonary arteries inhibition of CBS, but not CSE, reduces the hypoxic response, whereas hydroxyl amine, which inhibits all pyridoxal phosphate dependent enzymes, including CBS, CSE and CAT, completely inhibits hypoxic vasoconstriction (Fig. 2.5b). This suggests that both the CBS and CAT/3-MST contribute to H2S production in bovine pulmonary vessels. The CAT/3-MST pathway can also be utilized in the rat lung as the competitive inhibitor, aspartate, prevents the augmented hypoxic response produced by exogenous α-ketoglutarate (Fig. 2.5c). There appears to be some species variation in the enzymatic pathways employed as CSE may be a major component of H2S production mediating hypoxic responses of the perfused rat lung (Madden et al. 2012). Inhibitors of H2S biosynthesis have also been shown to inhibit hypoxic relaxation of rainbow trout urinary bladder (Dombkowski et al. 2006) and rainbow trout and Coho salmon intestine (Dombkowski et al. 2011).

Inhibitors of H2S biosynthesis inhibit hypoxic vasoconstriction in the lamprey aorta (a), and bovine pulmonary artery (b) and hypoxic vasodilation of the norepinephrine (NE, 1 μM) precontracted rat aorta. Inhibiting H2S biosynthesis also inhibits hypoxic vasoconstriction in the perfused rat lung (c) and perfused trout gill (d). CBS cystathionine β-synthase, CSE cystathionine γ-lyase, A amino-oxyacetate a CBS inhibitor (1 mM), P propargyl glycine a CSE inhibitor (10 mM), HA, CBS and CSE inhibitor hydroxylamine (1 mM), α-Kg α-ketoglutarate a substrate for mercaptopyruvate sulfur transferase (MST), Asp aspartic acid, an inhibitor of MST (10 mM) (From Olson 2012b, with permission)
Inhibiting CBS in the mouse carotid body decreases hypoxia-stimulated afferent nerve activity in vitro and blunts the hypoxic hyperventilation in vivo (Li et al. 2010). Conversely, Peng et al. (2010) observed an inverse Po 2-dependent increase in H2S production by rat carotid bodies and both H2S production and sinus nerve activity could be blocked by inhibiting CSE; in a mouse CSE knockout (CSE−/−) hypoxic responses of glomus cells were significantly reduced. Hypoxia-evoked catecholamine secretion from adrenal glands was also inhibited in CSE−/−mice or by inhibiting CSE in rats Peng et al. (2010). Clearly additional studies are needed so sort out the specific metabolic pathways for H2S production in the glomus cells, but nevertheless, a strong case can be made for the involvement of H2S in hypoxic signal transduction.
2.3.5 H2S Acts Upon Effector Mechanisms Known to Mediate Hypoxic Responses
The recent identification of H2S signaling through sulfhydration of protein cysteine molecules (Mustafa et al. 2009) has not only contributed to our overall understanding of H2S signaling pathways, but it has shed some light on the mechanisms of H2S signaling in hypoxia. Because many proteins such as enzymes and structural proteins are regulated through active-site cysteines (Nagahara 2011) is also evident that H2S signaling is most likely an autocrine activity and that even within the cell it must be highly spatially regulated. This is supported by the models predicting that hypoxic signaling proceeds in the immediate mitochondrial environment (Olson 2013). As would be expected, the mechanisms with which inhibits or activates H2S cells is commensurate with the intended outcome of H2S signaling.
It is well-known that hypoxic vasodilation is mediated in part by KATP channels (Weir and Archer 1995) and these channels were one of the first targets identified for H2S signaling (Zhao et al. 2001). Since then a variety of channels in vascular smooth muscle and endothelial cells have been shown to be affected by H2S leading to vasodilation. These include KATP, intermediate conductance (IKCa) and Kv7 potassium channels (Jiang et al. 2010; Liang et al. 2011; Martelli et al. 2013; Mustafa et al. 2011). H2S also relaxes newborn piglet cerebral arteries by increasing [Ca2+] in the sarcoplasmic reticulum. This stimulates Ca2+ sparks, increases current through KCa channels and hyperpolarizes the cells thereby lowering global intracellular [Ca2+] (Liang et al. 2012). H2S did not directly affect KCa channels in these studies. H2S activates the Kir 6.1 subunit of KATP channel through sulfhydration of specific cysteine residues, especially Cys-34, this decreases the inhibitory effect of ATP on these channels while increasing binding of the activator phosphatidylinositol (4,5)-bisphosphate (PIP2)28 to Kir 6.1; other channels such as the endothelial intermediate conductance (IKCa) channel and other cysteine residues such as Cys6 and Cys26 may also be sulfhydrated and contribute to H2S relaxation (Jiang et al. 2010; Mustafa et al. 2011). Intermittent hypoxia down regulates CSE and increases vascular tone via the loss of H2S activation of BKCa channels (Jackson-Weaver et al. 2011) suggesting a longer time-scale of vascular regulation.
H2S can also activate cells. We (Sudhahar et al. 2013) have recently shown that H2S induces membrane trafficking of protein kinase Cε (PKCε) through specific sulfhydration of Cys-13 and Cys-74 in the C2 domain. This is consistent with the well known role of PKCε activation in hypoxic pulmonary vasoconstriction (Sylvester et al. 2012). H2S also activates the carotid body through inhibition of large-conductance calcium activated potassium (BKCa) channels (Li et al. 2010; Telezhkin et al. 2009, 2010) and/or inhibition of background (TASK) potassium channels (Buckler 2012); both lead to membrane depolarization and voltage-gated Ca2+ entry. Although specific channels were not identified, inhibition of potassium channels is consistent with hypoxia- and H2S-mediated depolarization of zebrafish neuroepithelial cells (Olson et al. 2008), bovine pulmonary arteries (Olson et al. 2006) and lamprey aortas (Madden and Olson, unpublished). H2S stimulates catecholamine release from rat adrenal cells via H2S inhibition of I K(Ca) current (Zhu et al. 2012). In other cells, H2S directly increases BK channel activity in rat GH(3) pituitary tumor cells through its reducing action on sulfhydryl groups of the channel protein (Sitdikova et al. 2010). A direct link between H2S signaling and hypoxia has been shown by Tao et al. (2012) where the Cys1045-Cys1024 disulfide bond of VEGFR2 is targeted by H2S and serves as a specific molecular switch for hypoxia mediated migration of vascular endothelial cells.
It is not too surprising that the acute hypoxia signal, H2S, interacts with the long-term hypoxia signaling hypoxia-inducible factors (HIFs). In general, H2S decreases HIF-1α expression in a variety of mammalian tissues (Kai et al. 2012; Si et al. 2013; Wu et al. 2012). H2S inhibits HIF-1α protein accumulation during hypoxia (1 % O2) or hypoxia-mimetic conditions by enhancing HIF2α phosphorylation independent of protein synthesis or ubiquitin-proteasomal degradation (Kai et al. 2012; Wu et al. 2012). Hypoxic pre- (and post-)conditioning, which appears to involve H2S signaling (Bian et al. 2006; Lavu et al. 2010; Pan et al. 2006; Yong et al. 2008) is also associated with a decrease in HIF1-α expression (Sims et al. 2012). Interestingly, H2S has the opposite effect on the nematode, Caenorhabditis elegans, where it increases HIF-1 activity, although this effect appears to be independent of hypoxia-mediated HIF-1 expression (Budde and Roth 2010). Other effectors of H2S-mediated cellular protection from hypoxia include heat shock protein 90 (Meng et al. 2011), inhibition of ROS-activated pathways such as NF-κB/COX-2 (Yang et al. 2011) and ERK1/2 and p38MAPK (Lan et al. 2011).
Direct competition between hypoxia and H2S for the same effector response, seen as the inability of one stimulus to produce a response when the tissue is activated by the other, has been demonstrated in a variety of blood vessels (Olson et al. 2006; Skovgaard and Olson 2012). This provides additional, albeit indirect, evidence that H2S is involved in hypoxic signaling.
2.3.6 H2S-Mediated O2 Sensing Has an Evolutionary Precedent and a Phylogenetic History
H2S was likely an important energy source and structural entity in the origin of life and for the first 500 million years after the origin of eukaryotes, the latter occurring in sulfidic and anoxic environments (reviewed in Olson 2012c). Thus, there is a long evolutionary history of H2S in intracellular energy trafficking and signaling. Some of these facets are retained today as SQR, the initial enzyme in H2S oxidation, is an integral component of the mitochondrial electron transport chain, as is SO (Hildebrandt 2011). It is evident in the present day, however, that H2S and O2 are not mutually compatible either in the environment, or in cells. Thus, as the Earth’s O2 levels began to rise some 800 million years ago, environmental H2S fell and H2S was no longer available as a substrate for energy production. However, it is clear that cells retained much of their metabolic capabilities and with an ironic, but well established metabolic twist, they now use H2S as a reporter of O2 availability.
2.4 Conclusions
There is now considerable evidence that the reciprocal relationship between H2S and O2 provides cells with an effective and accurate mechanism with which to couple O2 availability to a variety of effector responses in O2 sensing and perhaps all cells. This hypothesis is supported by the ubiquitous similarity between the effects of hypoxia and H2S on a variety of tissues, that net tissue H2S is exquisitely controlled by O2 availability at physiologically relevant O2 levels, that factors that augment or inhibit H2S production have a similar effect on hypoxic responses and that the downstream effectors for hypoxia and H2S appear to be identical. This mechanism appears to have originated early on in evolution and it is likely widespread in the animal kingdom. The next step in evaluating this signaling mechanism will be to determine how, and where, this mechanism operates at the sub-cellular level.
Abbreviations
- 3MP:
-
3-Mercaptopyruvate
- 3-MST:
-
3-Mercaptopyruvate sulfur transferase
- AMPK:
-
AMP-activated protein kinase
- AOA:
-
Amino-oxyacetate
- AOX:
-
Alternative oxidase
- Asp:
-
Aspartic acid
- CAT:
-
Cysteine aminotransferase
- CBS:
-
Cystathionine β-synthase
- CDO:
-
Cysteine dioxygenase
- CO:
-
Carbon monoxide
- CSE:
-
Cystathionine γ-lyase
- Cys:
-
Cysteine
- DAO:
-
d-amino acid oxidase
- DHLA:
-
Dihydrolipoic acid
- EC50 :
-
Effective concentration for half-maximal activity
- ETHE1:
-
Mitochondrial sulfur dioxygenase
- Gly:
-
Glycine
- GSH:
-
Reduced glutathione
- GSSG:
-
Oxidized glutathione
- H2O2 :
-
Hydrogen peroxide
- H2S:
-
Hydrogen sulfide
- HA:
-
Hydroxylamine
- HIF:
-
Hypoxia-inducible factor
- HPC:
-
Hypoxic pulmonary vasoconstriction
- HSD:
-
Hypoxic systemic vasodilation
- IKCa :
-
Intermediate conductance potassium channel
- KATP :
-
Adenosine triphosphate sensitive potassium channel
- KCl:
-
Potassium chloride
- Ki :
-
Inhibition constant
- Kv :
-
Voltage-gated potassium channels
- NEB:
-
Neuroepithelial bodies
- NEC:
-
Neuroepithelial cells
- NO:
-
Nitric oxide
- O2 − :
-
Superoxide
- PASMC:
-
Pulmonary artery smooth muscle cells
- pB:
-
Pre-Bötzinger respiratory group
- PKCε:
-
Protein kinase C epsilon
- PLP:
-
Pyridoxal 5′phosphate
- Po 2 :
-
Partial pressure of oxygen
- PPG:
-
Propargyl glycine
- Rde:
-
Rhodanase
- RI:
-
Ischemia reperfusion injury
- ROS:
-
Reactive oxygen species
- R-SO:
-
Sulfenyl
- S2O3 2− :
-
Thiosulfate
- SO:
-
Sulfur oxidase
- SO3 2− :
-
Sulfite
- SO4 2− :
-
Sulfate
- SQR:
-
Sulfide:quinone oxidoreductase
- ST:
-
Sulfur transferase
- TASK:
-
Acid-sensitive potassium channel
- TR:
-
Thiosulfate reductase, a.k.a. rhodanase
- TRD:
-
Thioredoxin reductase
- Trx:
-
Thioredoxin
- α-Kg:
-
α-ketoglutarate
References
Ackermann M, Kubitza M, Maier K, Brawanski A, Hauska G, Pina AL (2011) The vertebrate homolog of sulfide-quinone reductase is expressed in mitochondria of neuronal tissues. Neuroscience 199:1–12
Banerjee R, Zou CG (2005) Redox regulation and reaction mechanism of human cystathionine-beta-synthase: a PLP-dependent hemesensor protein. Arch Biochem Biophys 433:144–156
Baragatti B, Ciofini E, Sodini D, Luin S, Scebba F, Coceani F (2013) Hydrogen sulfide in the mouse ductus arteriosus: a naturally occurring relaxant with potential EDHF function. Am J Physiol Heart Circ Physiol 304:H927–H934
Bearden SE, Beard RS Jr, Pfau JC (2010) Extracellular transsulfuration generates hydrogen sulfide from homocysteine and protects endothelium from redox stress. Am J Physiol Heart Circ Physiol 299:H1568–H1576
Beauchamp RO Jr, Bus JS, Popp JA, Boreiko CJ, Andjelkovich DA (1984) A critical review of the literature on hydrogen sulfide toxicity. Crit Rev Toxicol 13:25–97
Bian JS, Yong QC, Pan TT, Feng ZN, Ali MY, Zhou S, Moore PK (2006) Role of hydrogen sulfide in the cardioprotection caused by ischemic preconditioning in the rat heart and cardiac myocytes. J Pharmacol Exp Ther 316:670–678
Bickler PE, Buck LT (2007) Hypoxia tolerance in reptiles, amphibians, and fishes: life with variable oxygen availability. Annu Rev Physiol 69:145–170
Bouillaud F, Blachier F (2011) Mitochondria and sulfide: a very old story of poisoning, feeding and signaling? Antioxid Redox Signal 15:379–391
Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Pyriochou A, Roussos C, Roviezzo F, Brancaleone V, Cirino G (2010) Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler Thromb Vasc Biol 30:1998–2004
Buckler KJ (2012) Effects of exogenous hydrogen sulphide on calcium signalling, background (TASK) K channel activity and mitochondrial function in chemoreceptor cells. Pflugers Arch 463:743–754
Budde MW, Roth MB (2010) Hydrogen sulfide increases hypoxia-inducible factor-1 activity independently of von Hippel-Lindau tumor suppressor-1 in C. elegans. Mol Biol Cell 21:212–217
Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC (2007) The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res 76:29–40
Calvert JW, Elston M, Nicholson CK, Gundewar S, Jha S, Elrod JW, Ramachandran A, Lefer DJ (2010) Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation 122:11–19
Chen L, Zhang J, Ding Y, Li H, Nie L, Zhou H, Tang Y, Zheng Y (2013) Site-specific hydrogen sulfide-mediated central regulation of respiratory rhythm in medullary slices of neonatal rats. Neuroscience 233:118–126
Cheng Y, Ndisang JF, Tang G, Cao K, Wang R (2004) Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol 287:H2316–H2323
Derwall M, Francis RC, Kida K, Bougaki M, Crimi E, Adrie C, Zapol WM, Ichinose F (2011) Administration of hydrogen sulfide via extracorporeal membrane lung ventilation in sheep with partial cardiopulmonary bypass perfusion: a proof of concept study on metabolic and vasomotor effects. Crit Care 15:R51
di d’Emmanuele Villa Bianca, Sorrentino R, Maffia P, Mirone V, Imbimbo C, Fusco F, De PR, Ignarro LJ, Cirino G (2009) Hydrogen sulfide as a mediator of human corpus cavernosum smooth-muscle relaxation. Proc Natl Acad Sci USA 106:4513–4518
Di Meo I, Fagiolari G, Prelle A, Viscomi C, Zeviani M, Tiranti V (2011) Chronic exposure to sulfide causes accelerated degradation of cytochrome C oxidase in ethylmalonic encephalopathy. Antioxid Redox Signal 15:353–362
Doeller JE, Isbell TS, Benavides G, Koenitzer J, Patel H, Patel RP, Lancaster JR Jr, Drley-Usmar VM, Kraus DW (2005) Polarographic measurement of hydrogen sulfide production and consumption by mammalian tissues. Anal Biochem 341:40–51
Dombkowski RA, Russell MJ, Schulman AA, Doellman MM, Olson KR (2005) Vertebrate phylogeny of hydrogen sulfide vasoactivity. Am J Physiol Regul Integr Comp Physiol 288:R243–R252
Dombkowski RA, Doellman MM, Head SK, Olson KR (2006) Hydrogen sulfide mediates hypoxia-induced relaxation of trout urinary bladder smooth muscle. J Exp Biol 209:3234–3240
Dombkowski RA, Naylor MG, Shoemaker E, Smith M, DeLeon ER, Stoy GF, Gao Y, Olson KR (2011) Hydrogen sulfide (HS) and hypoxia inhibit salmonid gastrointestinal motility: evidence for HS as an oxygen sensor. J Exp Biol 214:4030–4040
Dromparis P, Michelakis ED (2013) Mitochondria in vascular health and disease. Annu Rev Physiol 75:95–126
Drousiotou A, Di Meo I, Mineri R, Georgiou T, Stylianidou G, Tiranti V (2011) Ethylmalonic encephalopathy: application of improved biochemical and molecular diagnostic approaches. Clin Genet 79:385–390
Evans AM, Hardie DG, Peers C, Mahmoud A (2011) Hypoxic pulmonary vasoconstriction: mechanisms of oxygen-sensing. Curr Opin Anaesthesiol 24:13–20
Fu M, Zhang W, Wu L, Yang G, Li H, Wang R (2012) Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc Natl Acad Sci USA 109:2943–2948
Furne J, Saeed A, Levitt MD (2008) Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am J Physiol Regul Integr Comp Physiol 295:R1479–R1485
Fusco F, di d’Emmanuele V, Mitidieri E, Cirino G, Sorrentino R, Mirone V (2012) Sildenafil effect on the human bladder involves the L-cysteine/hydrogen sulfide pathway: a novel mechanism of action of phosphodiesterase type 5 inhibitors. Eur Urol 62:1174–1180
Giordano C, Viscomi C, Orlandi M, Papoff P, Spalice A, Burlina A, Di Meo I, Tiranti V, Leuzzi V, di Amati G, Zeviani M (2011) Morphologic evidence of diffuse vascular damage in human and in the experimental model of ethylmalonic encephalopathy. J Inherit Metab Dis 35(3):451–458
Gonzalez C, Agapito MT, Rocher A, Gomez-Niño A, Rigual R, Castañeda J, Conde SV, Obeso A (2010) A revisit to O2 sensing and transduction in the carotid body chemoreceptors in the context of reactive oxygen species biology. Respir Physiol Neurobiol 174:317–330
Goubern M, Andriamihaja M, Nubel T, Blachier F, Bouillaud F (2007) Sulfide, the first inorganic substrate for human cells. FASEB J 21:1699–1706
Gupte SA, Wolin MS (2008) Oxidant and redox signaling in vascular oxygen sensing: implications for systemic and pulmonary hypertension. Antioxid Redox Signal 10:1137–1152
Haggard HW, Henderson Y (1922) The influence of hydrogensulphide upon respiration. Am J Physiol 61:289–297
Haldar SM, Stamler JS (2013) S-nitrosylation: integrator of cardiovascular performance and oxygen delivery. J Clin Invest 123:101–110
Haouzi P (2012) Ventilatory and metabolic effects of exogenous hydrogen sulfide. Respir Physiol Neurobiol 184:170–177
Haouzi P, Bell HJ, Notet V, Bihain B (2009) Comparison of the metabolic and ventilatory response to hypoxia and H2S in unsedated mice and rats. Respir Physiol Neurobiol 167:316–322
Haouzi P, Bell H, Philmon M (2011) Hydrogen sulfide oxidation and the arterial chemoreflex: effect of methemoglobin. Respir Physiol Neurobiol 177:273–283
Hildebrandt TM (2011) Modulation of sulfide oxidation and toxicity in rat mitochondria by dehydroascorbic acid. Biochim Biophys Acta 1807:1206–1213
Hildebrandt TM, Grieshaber MK (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J 275:3352–3361
Hosoki R, Matsuki N, Kimura H (1997) The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun 237:527–531
Hu H, Shi Y, Chen Q, Yang W, Zhou H, Chen L, Tang Y, Zheng Y (2008) Endogenous hydrogen sulfide is involved in regulation of respiration in medullary slice of neonatal rats. Neuroscience 156:1074–1082
Huang J, Khan S, O’Brien PJ (1998) The glutathione dependence of inorganic sulfate formation from L- or D-cysteine in isolated rat hepatocytes. Chem Biol Interact 110:189–202
Jackson MR, Melideo SL, Jorns MS (2012) Human sulfide: quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 51:6804–6815
Jackson-Weaver O, Paredes DA, Gonzalez Bosc LV, Walker BR, Kanagy NL (2011) Intermittent hypoxia in rats increases myogenic tone through loss of hydrogen sulfide activation of large-conductance Ca(2+)-activated potassium channels. Circ Res 108:1439–1447
Jeroschewski P, Steuckart C, Kuhl M (1996) An amperometric microsensor for the determination of H2S in aquatic environments. Anal Chem 68:4351–4357
Jiang B, Tang G, Cao K, Wu L, Wang R (2010) Molecular mechanism for H(2)S-induced activation of K(ATP) channels. Antioxid Redox Signal 12:1167–1178
Julian D, Statile JL, Wohlgemuth SE, Arp AJ (2002) Enzymatic hydrogen sulfide production in marine invertebrate tissues. Comp Biochem Physiol A Mol Integr Physiol 133:105–115
Kabil O, Banerjee R (2010) Redox biochemistry of hydrogen sulfide. J Biol Chem 285:21903–21907
Kai S, Tanaka T, Daijo H, Harada H, Kishimoto S, Suzuki K, Takabuchi S, Takenaga K, Fukuda K, Hirota K (2012) Hydrogen sulfide inhibits hypoxia- but not anoxia-induced hypoxia-inducible factor 1 activation in a von hippel-lindau- and mitochondria-dependent manner. Antioxid Redox Signal 16:203–216
Kamoun P (2004) Endogenous production of hydrogen sulfide in mammals. Amino Acids 26:243–254
Kemp PJ, Lewis A, Hartness ME, Searle GJ, Miller P, O’Kelly I, Peers C (2002) Airway chemotransduction: from oxygen sensor to cellular effector. Am J Respir Crit Care Med 166:S17–S24
Kimura H (2010) Hydrogen sulfide: its production, release and functions. Amino Acids 41:113–121
Klentz RD, Fedde MR (1978) Hydrogen sulfide: effects on avian respiratory control and intrapulmonary CO2 receptors. Respir Physiol 32:355–367
Koj A, Frendo J, Wojtczak L (1975) Subcellular distribution and intramitochondrial localization of three sulfurtransferases in rat liver. FEBS Lett 57:42–46
Kraus DW, Doeller JE (2004) Sulfide consumption by mussel gill mitochondria is not strictly tied to oxygen reduction: measurements using a novel polargraphic sulfide sensor. J Exp Biol 207:3667–3679
Kwiatkoski M, Soriano RN, Francescato HD, Batalhao ME, Coimbra TM, Carnio EC, Branco LG (2012) Hydrogen sulfide as a cryogenic mediator of hypoxia-induced anapyrexia. Neuroscience 201:146–156
Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F (2010) Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta 1797:1500–1511
Lan A, Liao X, Mo L, Yang C, Yang Z, Wang X, Hu F, Chen P, Feng J, Zheng D, Xiao L (2011) Hydrogen sulfide protects against chemical hypoxia-induced injury by inhibiting ROS-activated ERK1/2 and p38MAPK signaling pathways in PC12 cells. PLoS One 6:e25921
Lavu M, Bhushan S, Lefer DJ (2010) Hydrogen sulfide-mediated cardioprotection: mechanisms and therapeutic potential. Clin Sci (Lond) 120:219–229
Leffler CW, Parfenova H, Basuroy S, Jaggar JH, Umstot ES, Fedinec AL (2010) Hydrogen sulfide and cerebral microvascular tone in newborn pigs. Am J Physiol Heart Circ Physiol 300:H440–H447
Li Q, Sun B, Wang X, Jin Z, Zhou Y, Dong L, Jiang LH, Rong W (2010) A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxid Redox Signal 12:1179–1189
Liang GH, Adebiyi A, Leo MD, McNally EM, Leffler CW, Jaggar JH (2011) Hydrogen sulfide dilates cerebral arterioles by activating smooth muscle cell plasma membrane KATP channels. Am J Physiol Heart Circ Physiol 300:H2088–H2095
Liang GH, Xi Q, Leffler CW, Jaggar JH (2012) Hydrogen sulfide activates Ca(2)(+) sparks to induce cerebral arteriole dilatation. J Physiol 590:2709–2720
Linden DR, Furne J, Stoltz GJ, Ddel-Rehim MS, Levitt MD, Szurszewski JH (2011) Sulfide quinone reductase contributes to hydrogen sulfide metabolism in murine peripheral tissues but not in the central nervous system. Br J Pharmacol 165:2178–2190
Liu X, Pan L, Zhuo Y, Gong Q, Rose P, Zhu Y (2010) Hypoxia-inducible factor-1 alpha is involved in the pro-angiogenic effect of hydrogen sulfide under hypoxic stress. Biol Pharm Bull 33:1550–1554
Liu WQ, Chai C, Li XY, Yuan WJ, Wang WZ, Lu Y (2011) The cardiovascular effects of central hydrogen sulphide are related to K(ATP) channels activation. Physiol Res 60:729–738
Madden JA, Vadula MS, Kurup VP (1992) Effects of hypoxia and other vasoactive agents on pulmonary and cerebral artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 263:L384–L393
Madden JA, Ahlf SB, Dantuma MW, Olson KR, Roerig DL (2012) Precursors and inhibitors of hydrogen sulfide synthesis affect acute hypoxic pulmonary vasoconstriction in the intact lung. J Appl Physiol 112:411–418
Makarenko VV, Nanduri J, Raghuraman G, Fox AP, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR (2012) Endogenous H2S is required for hypoxic sensing by carotid body glomus cells. Am J Physiol Cell Physiol 303:C916–C923
Marcia M, Ermler U, Peng G, Michel H (2010) A new structure-based classification of sulfide: quinone oxidoreductases. Proteins 78:1073–1083
Martelli A, Testai L, Breschi MC, Lawson K, McKay NG, Miceli F, Taglialatela M, Calderone V (2013) Vasorelaxation by hydrogen sulphide involves activation of Kv7 potassium channels. Pharmacol Res 70:27–34
Meng JL, Mei WY, Dong YF, Wang JH, Zhao CM, Lan AP, Yang CT, Chen PX, Feng JQ, Hu CH (2011) Heat shock protein 90 mediates cytoprotection by H2S against chemical hypoxia-induced injury in PC12 cells. Clin Exp Pharmacol Physiol 38:42–49
Mikami Y, Shibuya N, Kimura Y, Nagahara N, Ogasawara Y, Kimura H (2011a) Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem J 439:479–485
Mikami Y, Shibuya N, Kimura Y, Nagahara N, Yamada M, Kimura H (2011b) Hydrogen sulfide protects the retina from light-induced degeneration by the modulation of Ca2+ influx. J Biol Chem 286:39379–39386
Mikami Y, Shibuya N, Ogasawara Y, Kimura H (2013) Hydrogen sulfide is produced by cystathionine gamma-lyase at the steady-state low intracellular Ca(2+) concentrations. Biochem Biophys Res Commun 431:131–135
Milsom WK, Burleson ML (2007) Peripheral arterial chemoreceptors and the evolution of the carotid body. Respir Physiol Neurobiol 157:4–11
Modis K, Coletta C, Erdelyi K, Papapetropoulos A, Szabo C (2013) Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J 27:601–611
Mudd SH, Irreverre F, Laster L (1967) Sulfite oxidase deficiency in man: demonstration of the enzymatic defect. Science 156:1599–1602
Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH (2009) H2S signals through protein S-sulfhydration. Sci Signal 2:ra72
Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, Amzel LM, Berkowitz DE, Snyder SH (2011) Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res 109:1259–1268
Nagahara N (2011) Catalytic site cysteines of thiol enzyme: sulfurtransferases. J Amino Acids 2011:1–7
Nagahara N (2012) Regulation of mercaptopyruvate sulfurtransferase activity via intrasubunit and intersubunit redox-sensing switches. Antioxid Redox Signal
Nurse CA, Buttigieg J, Thompson R, Zhang M, Cutz E (2006) Oxygen sensing in neuroepithelial and adrenal chromaffin cells. Novartis Found Symp 272:106–114
Olson KR (2011) Hydrogen sulfide is an oxygen sensor in the carotid body. Respir Physiol Neurobiol 179:103–110
Olson KR (2012a) A practical look at the chemistry and biology of hydrogen sulfide. Antioxid Redox Signal 17:32–44
Olson KR (2012b) Hydrogen sulfide as an oxygen sensor. Clin Chem Lab Med 51:623–632
Olson KR (2012c) Mitochondrial adaptations to utilize hydrogen sulfide for energy and signaling. J Comp Physiol B 182:881–897
Olson KR (2013) A theoretical examination of hydrogen sulfide metabolism and its potential in autocrine/paracrine oxygen sensing. Respir Physiol Neurobiol 186:173–179
Olson KR, Whitfield NL (2010) Hydrogen sulfide and oxygen sensing in the cardiovascular system. Antioxid Redox Signal 12:1219–1234
Olson KR, Dombkowski RA, Russell MJ, Doellman MM, Head SK, Whitfield NL, Madden JA (2006) Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J Exp Biol 209:4011–4023
Olson KR, Healy M, Qin J, Skovgaard N, Vulesevic B, Duff DW, Whitfield NL, Yang G, Wang R, Perry SF (2008) Hydrogen sulfide as an oxygen sensor in trout gill chemoreceptors. Am J Physiol Regul Integr Comp Physiol 295:R669–R680
Olson KR, Whitfield NL, Bearden SE, St. Leger J, Nilson E, Gao Y, Madden JA (2010) Hypoxic pulmonary vasodilation: a paradigm shift with a hydrogen sulfide mechanism. Am J Physiol Regul Integr Comp Physiol 298:R51–R60
Olson KR, DeLeon ER, Gao Y, Hurley K, Sadauskas V, Batz C (2013) Thiosulfate: a readily accessible source of hydrogen sulfide in oxygen sensing. Am J Physiol Regul Integr Comp Physiol (in press)
Pan TT, Feng ZN, Lee SW, Moore PK, Bian JS (2006) Endogenous hydrogen sulfide contributes to the cardioprotection by metabolic inhibition preconditioning in the rat ventricular myocytes. J Mol Cell Cardiol 40:119–130
Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, Jeschke MG, Branski LK, Herndon DN, Wang R, Szabo C (2009) Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci USA 106:21972–21977
Peng YJ, Nanduri J, Raghuraman G, Souvannakitti D, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR (2010) H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci USA 107:10719–10724
Perry SF, Montpetit CJ, Borowska M (2000) The effects of acute hypoxia on chemically or neuronally induced catecholamine secretion in rainbow trout (Oncorhynchus mykiss) in situ and in vivo. J Exp Biol 203:1487–1495
Perry SF, McNeill B, Elia E, Nagpal A, Vulesevic B (2009) Hydrogen sulfide stimulates catecholamine secretion in rainbow trout (Oncorhynchus mykiss). Am J Physiol Regul Integr Comp Physiol 296:R133–R140
Rajapakshe A, Tollin G, Enemark JH (2012) Kinetic and thermodynamic effects of mutations of human sulfite oxidase. Chem Biodivers 9:1621–1634
Reiffenstein RJ, Hulbert WC, Roth SH (1992) Toxicology of hydrogen sulfide. Annu Rev Pharmacol Toxicol 32:109–134
Roman HB, Hirschberger LL, Krijt J, Valli A, Kozich V, Stipanuk MH (2013) The cysteine dioxygenase knockout mouse: altered cysteine metabolism in nonhepatic tissues leads to excess H2S/HS production and evidence of pancreatic and lung toxicity. Antioxid Redox Signal
Russell MJ, Dombkowski RA, Olson KR (2008) Effects of hypoxia on vertebrate blood vessels. J Exp Zool A Ecol Genet Physiol 309:55–63
Schultz HD, Del RR, Ding Y, Marcus NJ (2012) Role of neurotransmitter gases in the control of the carotid body in heart failure. Respir Physiol Neurobiol 184:197–203
Shibuya N, Koike S, Tanaka M, Ishigami-Yuasa M, Kimura Y, Ogasawara Y, Fukui K, Nagahara N, Kimura H (2013) A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat Commun 4:1366
Si YF, Wang J, Guan J, Zhou L, Sheng Y, Zhao J (2013) Treatment with hydrogen sulfide alleviates streptozotocin-induced diabetic retinopathy in rats. Br J Pharmacol 169:619–631
Sims B, Clarke M, Francillion L, Kindred E, Hopkins ES, Sontheimer H (2012) Hypoxic preconditioning involves system Xc- regulation in mouse neural stem cells. Stem Cell Res 8:285–291
Sitdikova GF, Weiger TM, Hermann A (2010) Hydrogen sulfide increases calcium-activated potassium (BK) channel activity of rat pituitary tumor cells. Pflugers Arch 459:389–397
Skovgaard N, Olson KR (2012) Hydrogen sulfide mediates hypoxic vasoconstriction through a production of mitochondrial ROS in trout gills. Am J Physiol Regul Integr Comp Physiol 303:R487–R494
Sowmya S, Swathi Y, Yeo AL, Shoon ML, Moore PK, Bhatia M (2010) Hydrogen sulfide: regulatory role on blood pressure in hyperhomocysteinemia. Vascul Pharmacol 53:138–143
Stipanuk MH (2004) Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annu Rev Nutr 24:539–577
Stipanuk MH, Ueki I (2011) Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J Inherit Metab Dis 34:17–32
Stipanuk MH, Ueki I, Dominy JE Jr, Simmons CR, Hirschberger LL (2009) Cysteine dioxygenase: a robust system for regulation of cellular cysteine levels. Amino Acids 37:55–63
Sudhahar CG, Lee J, Buck LE, Olson KR, Stahelin RV (2013) Hydrogen sulfide induces the membrane translocation of protein kinase Cε through sulfhydration of the C2 domain. Antioxid Redox Signal (in press)
Sylvester JT, Shimoda LA, Aaronson PI, Ward JP (2012) Hypoxic pulmonary vasoconstriction. Physiol Rev 92:367–520
Szabó C (2007) Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov 6:917–935
Szabo C, Papapetropoulos A (2011) Hydrogen sulfide and angiogenesis: mechanisms and applications. Br J Pharmacol 164:853–865
Tao BB, Liu SY, Zhang CC, Fu W, Cai WJ, Wang Y, Shen Q, Wang MJ, Chen Y, Zhu Y, Zhu YC (2012) VEGFR2 functions as an H2S-targeting receptor protein kinase with its novel Cys1045-Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells. Antioxid Redox Signal. doi:10.1089/ars.2012.4565
Telezhkin V, Brazier SP, Cayzac S, Muller CT, Riccardi D, Kemp PJ (2009) Hydrogen sulfide inhibits human BK(Ca) channels. Adv Exp Med Biol 648:65–72
Telezhkin V, Brazier SP, Cayzac SH, Wilkinson WJ, Riccardi D, Kemp PJ (2010) Mechanism of inhibition by hydrogen sulfide of native and recombinant BKCa channels. Respir Physiol Neurobiol 172:169–178
Theissen U, Martin W (2008) Sulfide: quinone oxidoreductase (SQR) from the lugworm Arenicola marina shows cyanide- and thioredoxin-dependent activity. FEBS J 275:1131–1139
Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, Levitt MD, Prelle A, Fagiolari G, Rimoldi M, Zeviani M (2009) Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med 15:200–205
Ueki I, Roman HB, Valli A, Fieselmann K, Lam J, Peters R, Hirschberger LL, Stipanuk MH (2011) Knockout of the murine cysteine dioxygenase gene results in severe impairment in ability to synthesize taurine and an increased catabolism of cysteine to hydrogen sulfide. Am J Physiol Endocrinol Metab 301:E668–E684
Van de Louw A, Haouzi P (2012) Inhibitory effects of hyperoxia and methemoglobinemia on H(2)S induced ventilatory stimulation in the rat. Respir Physiol Neurobiol 181:326–334
Villarejo M, Westley J (1963) Mechanism of rhodanese catalysis of thiosulfate-lipoate oxidation-reduction. J Biol Chem 238:4016–4020
Wang MJ, Cai WJ, Li N, Ding YJ, Chen Y, Zhu YC (2010a) The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxid Redox Signal 12:1065–1077
Wang MJ, Cai WJ, Zhu YC (2010b) Mechanisms of angiogenesis: role of hydrogen sulphide. Clin Exp Pharmacol Physiol 37:764–771
Waypa GB, Schumacker PT (2010) Hypoxia-induced changes in pulmonary and systemic vascular resistance: where is the O2 sensor? Respir Physiol Neurobiol 174:201–211
Waypa GB, Marks JD, Guzy R, Mungai PT, Schriewer J, Dokic D, Schumacker PT (2010) Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res 106:526–535
Weir EK, Archer SL (1995) The mechanism of acute hypoxic pulmonary vasoconstriction: the tale of two channels. FASEB J 9:183–189
Whitfield NL, Kreimier EL, Verdial FC, Skovgaard N, Olson KR (2008) Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am J Physiol Regul Integr Comp Physiol 294:R1930–R1937
Wolin MS, Gupte SA, Neo BH, Gao Q, Ahmad M (2010) Oxidant-redox regulation of pulmonary vascular responses to hypoxia and nitric oxide-cGMP signaling. Cardiol Rev 18:89–93
Wu B, Teng H, Yang G, Wu L, Wang R (2012) Hydrogen sulfide inhibits the translational expression of hypoxia-inducible factor-1alpha. Br J Pharmacol 167:1492–1505
Yan H, Do J, Tang C (2004) The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun 313:22–27
Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R (2008) H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322:587–590
Yang C, Yang Z, Zhang M, Dong Q, Wang X, Lan A, Zeng F, Chen P, Wang C, Feng J (2011) Hydrogen Sulfide Protects against Chemical Hypoxia-Induced Cytotoxicity and Inflammation in HaCaT Cells through Inhibition of ROS/NF-kappaB/COX-2 Pathway. PLoS One 6:e21971
Yong QC, Lee SW, Foo CS, Neo KL, Chen X, Bian JS (2008) Endogenous hydrogen sulphide mediates the cardioprotection induced by ischemic postconditioning. Am J Physiol Heart Circ Physiol 295:H1330–H1340
Zhang J, Xie Y, Xu Y, Pan Y, Shao C (2011) Hydrogen sulfide contributes to hypoxia-induced radioresistance on hepatoma cells. J Radiat Res 52:622–628
Zhao W, Zhang J, Lu Y, Wang R (2001) The vasorelaxant effect of H2S as a novel endogenous KATP channel opener. EMBO J 20:6008–6016
Zhu D, Yu X, Sun J, Li J, Ma X, Yao W (2012) H2S induces catecholamine secretion in rat adrenal chromaffin cells. Toxicology 302:40–43
Acknowledgments
The author wishes to acknowledge the numerous colleagues that contributed to this research. The author’s work has been supported by National Science Foundation Grants, IBN 0235223, IOS 0641436 and IOS 1051627.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Wien
About this chapter
Cite this chapter
Olson, K.R. (2013). Hydrogen Sulfide as an Oxygen Sensor. In: Kimura, H. (eds) Hydrogen Sulfide and its Therapeutic Applications. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1550-3_2
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1550-3_2
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1549-7
Online ISBN: 978-3-7091-1550-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)