
Abstract
Rheumatoid arthritis (RA) is a chronic inflammatory disease, associated with inflammation of the synovial tissue lining joints and tendons, which leads to degradation of underlying cartilage and bone. Extra-articular manifestations of RA, including depression and anaemia, combine with inflammation and joint destruction to impact significantly on patients’ quality of life. RA is also associated with co-morbidities such as increased cardiovascular disease. Successful development of therapies to treat RA requires an understanding of the cellular and molecular events underlying the disease. Angiogenesis is now understood to play an important role. Inadequate oxygenation (hypoxia) is believed to drive the increase in synovial angiogenesis which occurs in RA, through expression of hypoxia-inducible molecules, including vascular endothelial growth factor (VEGF). This allows further infiltration of inflammatory cells and production of inflammatory mediators, perpetuating synovitis. In parallel, inflammatory molecules and cells, particularly activated macrophages, are observed in synovial tissue and can also directly affect angiogenesis.
The current chapter describes the importance of angiogenesis in RA and discusses whether angiogenesis may be a potential therapeutic target in RA. Furthermore, we will review how angiogenesis and inflammation may interact to promote, maintain and resolve synovitis in RA, with a particular focus on the functional responses of macrophages in the context of RA. Successful treatment of RA is associated with reduced levels of pro-angiogenic factors such as VEGF, supporting the concept that modulation of blockade could be of therapeutic benefit in RA.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disease, which affects approximately 1 % of the population worldwide, and is primarily characterised by inflammation of the synovial membrane which lines the joint spaces, leading to the localised invasion and destruction of underlying cartilage and bone. RA may begin at any time from the first few weeks of life until the ninth decade, although the peak time of presentation is 35–45 years of age. Early diagnosis of RA is challenging, due the heterogeneity of its clinical presentation and lack of sufficiently specific and sensitive laboratory tests. Patients present with painful, stiff and swollen joints, predominantly the small joints of the hands and wrists, as well as the metatarsophalangeal joints, ankles, knees and cervical spine. In most patients, symptoms appear over weeks to months, and peri-articular structures such as tendon sheaths may be inflamed. RA is associated with a range of other symptoms such as fatigue, anaemia, weight loss and vasculitis. Depressive symptoms occur in many patients, and there is some evidence that depression may exacerbate pain and disease activity [1, 2]. Indeed more recent clinical trials of new therapeutics in RA include at least one generic patient-reported outcome (PRO) instrument, usually the Health Assessment Questionnaire-Disability Index, to assess function, and/or the so-called ‘SF-36 survey’, to assess Health Related Quality of Life (HRQoL). These instruments have shown validity and sensitivity for assessment of changes in clinical trials of disease modifying anti-rheumatic drugs (DMARD) and biological therapies. Further instruments have been designed to address problems specific to the RA population. The Arthritis Impact Measurement Scale (AIMS) was one of the first PRO measures developed to evaluate HRQoL in RA [3]. In particular, RA Impact of Disease (RAID) is a new PRO instrument recently developed by EULAR (European League Against Rheumatism) for use in clinical trials, as a measure of the impact of RA on HRQoL, and takes into account pain, functional capacity, fatigue and physical and emotional well-being, together with outcomes such as quality of sleep [4].
Up to 30 % of people with RA become permanently work-disabled within 3 years of diagnosis if they do not have medical treatment [5]. For example, it has been documented in a recent study that at the time of first symptoms of RA, 86 % of men and 64 % of women below 65 years of age were working. More than a third (37 %) of these patients reported subsequent work disability, and the probabilities of continuing to work were 80 % and 68 % at 2 and 5 years, respectively [6]. Furthermore, the standardised mortality ratio for patients with RA is more than 1.5–2.5-fold higher than the ratio for the general population [7]. Although RA patients are at increased risk of dying of urogenital, gastrointestinal, respiratory infections and cancer [8, 9], the major cause of mortality (more than 40 % of deaths) is cardiovascular disease, including ischemic heart disease and heart failure [10]. The odds ratio for RA patients for the risk of all-category stroke was 1.64, with an odds ratio of 2.66 for ischemic stroke [11]. Another study in the USA found that subjects with RA had higher odds ratio for congestive heart failure (3.59) [12]. In a prospective cohort study, which comprised more than 100,000 women free of RA and cardiovascular disease at baseline, the adjusted relative risks of myocardial infarction and stroke in women subsequently diagnosed with RA were 2.00 and 1.48, respectively, when compared to women without RA [13]. A high 10-year risk of cardiovascular disease in newly diagnosed RA patients has been reported, with the absolute cardiovascular risk in RA patients similar to that in non-RA subjects who were 5–10 years older [14]. RA patients also have an increased risk of fatality following myocardial infarction (assessed as the 30-day mortality rates following a first acute cardiovascular event) [15]. A recent study of cardiovascular autopsy findings found that RA patients had more frequent myocardial abnormalities (21 %) than those without RA (12 %) [16].
In terms of disease pathogenesis, interplay between environmental and genetic factors, sex hormones and perhaps an infectious agent or other immune-activating factor is thought to initiate an autoimmune response that culminates in a disease with inflammatory and destructive features [17]. The most common genetic risk factors associated with RA comprise the shared epitope (SE) alleles of the human leukocyte antigen (HLA) gene and a polymorphism of protein tyrosine phosphatase N22 (PTPN22) [18, 19]. Over 80 % of Caucasian RA patients have SE conserved across the HLA-DR1 and HLA-DR4 haplotypes (0101, 0401, 0404 and 1402) [20]. This genetic predisposition has been additionally linked to environmental factors such as smoking [21] and certain autoantibodies. Smoking history, when combined with the presence of HLA-DR SE, increases the risk of RA 21-fold compared with non-smokers carrying no SE genes [20]. Epidemiological and genetic studies of RA have also demonstrated significant differences in subsets of patients with and without the presence of antibodies to citrullinated protein antigens (ACPA). ACPA are found in approximately 60 % of RA patients but only in 2 % of the normal population, making them highly specific for RA [22–24]. An association between smoking, HLA-DRB1 SE and antibodies to cyclic citrullinated peptides (CCP) has been demonstrated [20]. Furthermore, in anti-CCP-positive individuals, antibodies to the immunodominant citrullinated α-enolase-1 epitope were detected in approximately 40–60 % of RA patients, and this was linked to HLA-DRB1*04, suggesting that citrullinated α-enolase may be an autoantigen linking smoking to genetic risk factors in the development of RA [25, 26].
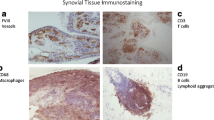
At the tissue and cellular level, RA is characterised by inflammation, hyperplasia of the synovial lining layer and marked infiltration by blood-derived cells, particularly lymphocytes and macrophages (Fig. 16.1). The normal synovium is generally one to three cell layers thick and is composed of loosely associated macrophage- and fibroblast-like cells, as well as vascular endothelial cells. In RA, the synovium is altered to a thickened tissue several cell layers thick, which covers and erodes the adjacent cartilage, bone and tendon. Histologically, the inflamed synovium shows pronounced angiogenesis, cellular hyperplasia and influx of cells, in particular T-cells, macrophages and dendritic cells. The invasive and destructive synovium is responsible for the erosions observed in RA, in that differentiation of monocyte/macrophage cells leads to formation of osteoclasts, which resorb bone matrix. Progressive destruction of the cartilage and bone produces the deformities characteristic of long-standing RA and results in functional deterioration and disability.

Mechanisms of RA pathogenesis. The normal synovium is generally one to three cell layers thick but in RA becomes thickened (due to fibroblast proliferation) and infiltrated by blood-derived cells, especially lymphocytes and macrophages. Neutrophils accumulate in the synovial fluid within the joint space. This inflamed synovial tissue invades underlying bone and cartilage, particularly as a result of osteoclast activation, leading to joint destruction
In the past, treatment of RA used a pyramidal approach starting with non-steroidal anti-inflammatory drugs at the base of the pyramid and progressing to DMARD such as gold and methotrexate (MTX). However, in spite of such pharmacological interventions, up to 90 % of patients with aggressive synovitis exhibited radiological evidence of bone erosion within 2 years of diagnosis. Major advances in the understanding of the pathogenesis of RA, based on bench-bedside studies of human tissue and animal models of disease, have led to the identification of a number of new biological targets for intervention, the first of which was tumour necrosis factor α (TNFα), which mediates many inflammatory and immunoregulatory activities relevant to RA. Other biologicals used in RA include rituximab (which targets B-cells), abatacept (cytotoxic T-lymphocyte antigen 4 fusion protein which prevents T-cell activation) and anti-interleukin (IL)-6 receptor antibody tocilizumab.
2 Role of Angiogenesis in RA
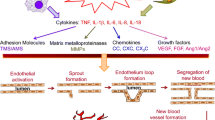
RA synovium is a site of active angiogenesis, due to the expression of numerous angiogenic factors, leading to the formation of new blood vessels. Blood vessels fulfil an important role in RA, fuelling synovial expansion and infiltration by cells from the blood, by supplying oxygen and nutrients necessary for cell metabolism and division, as well as by bringing in leukocytes and signalling mediators such as cytokines and growth factors [27–33]. As the synovium expands, more blood vessels are needed to supply poorly perfused and hypoxic areas distant from the pre-existing blood vessels with oxygen and nutrients (Fig. 16.2). The number of synovial blood vessels has been found to correlate with synovial cell hyperplasia and indices of joint tenderness [34]. When affected joints of RA patients were examined using power Doppler ultrasonography, increased synovial blood flow was observed in 81 % of RA patients, but only in less than 10 % of healthy controls, with a correlation between intra-articular microvascular power Doppler flow and clinical synovitis in RA [35]. A number of subsequent studies have confirmed these findings and have further shown that ultrasonographic measures correlate with disease severity in RA and may be useful as a marker of response to therapy [36–39]. Endothelial cells lining blood vessels within RA synovium have been shown to express cell cycle-associated antigens [40], and endothelial proliferation was shown to be increased in synovium from patients with RA [41]. This results in altered synovial blood vessel density [42, 43], with blood vessels of different sizes observed both in areas of diffuse synovitis and in regions of large leukocytic infiltrates with germinal centre-like structures [42, 44]. Despite the formation of blood vessels, their morphology appears similar to that seen in post-capillary venules of lymphoid tissue, and it is thought that there may be a failure to form functional mature vessels. RA synovium contains a significant fraction of neoangiogenic, immature and leaky blood vessels which may be observed from early stages of RA. Comparison of the staining patterns for CD31 and the pericyte marker α-smooth muscle cell actin revealed a significant fraction of CD31-positive but α-smooth muscle cell actin-negative cells in RA tissue when compared to osteoarthritis (OA) or control tissue [45, 46]. The presence or density of immature vessels is increased in patients with longer disease duration, higher activity and greater cell infiltration [45].

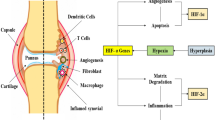
Central role of angiogenesis in RA. Angiogenesis plays a key role in perpetuating disease in RA, by fuelling synovial hyperplasia. As cells such as FLS proliferate and become increasingly distant from pre-existing blood vessels, the synovium becomes hypoxic. Reduced oxygen tension and activation of HIF signalling drives expression of angiogenic factors such as VEGF, ANGPTL4, leptin and EFNA3, further promoting angiogenesis. Hypoxia also promotes expression of inflammatory cytokines, chemokines, molecules involved in metabolism (such as the glucose transporter GLUT-1) and MMPs and thus maintains inflammation and synovial destruction. Potential interactions between HIF and NFκB signalling, in part mediated by inhibition of PHD/FIH-1 leading to downstream NFκB activation, also contributes to the destructive and inflammatory synovial milieu
2.1 Angiogenic Factors Expressed In RA: A Key Role for VEGF
A range of different factors can promote angiogenesis, directly or indirectly, and many of these factors have been reported to be expressed in RA [47, 48]. Over 30 years ago it was reported that synovial fluids from patients with RA, as well as from those with OA, contained a low molecular weight angiogenesis factor, now termed ESAF (endothelial cell stimulating angiogenesis factor) [49]. In 1994, the groups of Koch and Fava almost simultaneously reported vascular endothelial growth factor (VEGF) expression in RA synovial fluids and tissue [50, 51]. VEGF expression is localised to synovial macrophages, neutrophils and fibroblasts [52, 53], and stimuli for VEGF release include pro-inflammatory cytokines expressed in RA such as IL-1 [54], TNFα [55] and transforming growth factor β [56], as well as hypoxia, which will be discussed in more detail later. VEGF isoforms VEGF-165 and VEGF-121 appear to be the predominant forms expressed [57]. VEGF receptors (VEGFR) VEGFR1 and VEGFR2 are expressed by RA synovial microvascular endothelial cells [51], and conditioned medium from synovial tissue explants was shown to be mitogenic for endothelial cells, an activity reduced by anti-VEGF antibody [50]. Another study utilised an antibody that selectively recognises VEGFR2 when complexed with VEGF and found that vessel density as assessed using this antibody expression was elevated in RA synovium compared to OA and normal synovium [58]. In addition to synovial expression of VEGF, circulating (serum) levels of VEGF are increased and correlate with inflammatory response markers [54, 59–62]. VEGF levels are increased even in RA patients with a disease duration of less than 2 years [63, 64] and are higher in patients with extra-articular manifestations of RA [65]. Treatment of RA with TNFα inhibitors (alone or with MTX) or anti-IL-6 receptor antibody significantly reduced serum VEGF concentrations [54, 66–70]. Single nucleotide polymorphisms in the VEGF may alter circulating VEGF levels and have been suggested to correlate in RA with both disease activity [71, 72] and onset age [73], although contradictory effects of VEGF polymorphisms on the risk of cardiovascular disease in RA have been reported [74, 75]. In addition to members of the VEGF family, expression of angiopoietin (Ang)-1 and Ang-2 [76, 77] and angiopoietin receptors Tie (Tyrosine kinase with immunoglobulin-like and epidermal growth factor-like domains)-1 and Tie-2 [78–80] in RA has been described. Additionally, fibroblast growth factor (FGF)-1 and FGF-2 have been detected in RA synovial tissue [81, 82], together with platelet-derived growth factor (PDGF) [83, 84] and hepatocyte growth factor (HGF) [85].
While VEGF is traditionally considered for its role in angiogenesis, there may be another important function for this growth factor in terms of blood vessel development in RA. In RA, CD133/CD34/VEGFR2-positive endothelial progenitor cells were found close to RA synovial blood vessels [86]. These cells were generated at a higher rate from bone marrow samples taken from RA patients, compared to normal subjects. Furthermore, the capacity of bone marrow-derived cells from RA patients to progress into endothelial cells correlated with synovial microvessel density [87]. Circulating endothelial progenitor cells in patients with active RA have been reported to be lower than in individuals with inactive disease or in healthy controls [88]. Interestingly, a recent study assessed late-outgrowth endothelial progenitor cells and actually found these to be enhanced in RA and suggested that these cells contribute to synovitis perpetuation by promoting blood vessel formation through vasculogenesis [89]. It therefore seems likely that increased synovial vessel density in RA is not just due to angiogenesis but also results from post-natal vasculogenesis, due to increased mobilisation of endothelial progenitors from the bone marrow driven by VEGF and increased endothelial progenitor cell homing to the synovium.
Finally, there is evidence that angiogenesis may be a feature of other joint diseases. In OA, expression of VEGF has been reported, together with chemokines, HGF and members of the Ang-Tie family [90–93]. Expression of Ang-2 and VEGF is higher in synovium of patients with psoriatic arthritis, relative to RA, whereas Ang-1 levels were more comparable. Psoriatic arthritis and RA exhibited different features in terms of vascular morphology, in that blood vessels in psoriatic synovium were highly tortuous in appearance, compared to the straight and branching vessels seen in RA, suggesting that the balance between Ang and VEGF may affect vessel growth and maturation in arthritic synovium [44].
In summary, a strong pro-angiogenic drive appears to exist in RA. However, increased blood-vessel formation might not necessarily result in improved perfusion and oxygenation, due to the formation of immature vessels with abnormal morphology, and might explain the apparent paradox that although synovial blood vessel density is increased, hypoxia is nevertheless a feature of RA. VEGF is a key pro-angiogenic factor expressed in RA, although its function as a vascular permeability factor may contribute to the enhanced vessel leakiness which is a feature of RA. VEGF may also contribute to synovial vasculogenesis in RA, which may impact detrimentally on vasculogenesis in other organs and could be linked to the increased cardiovascular disease seen in RA patients.
2.2 Regulation of Angiogenesis by Hypoxia in RA
Oxygen is a key molecular ingredient in life, and hence hypoxia—disruption in O2 homeostasis caused when oxygen demand exceeds oxygen supply—leads to a number of adaptive changes in cellular responses. The alterations in gene expression induced by hypoxia underlie various physiological processes, including normal embryonic development [94–97] and adaptation to exercise and/or high altitudes [98]. Hypoxia is a feature of certain diseases, in particular solid tumours, but also, with relevance to this chapter, hypoxia plays a key role in RA [33]. The objective of this section is to describe the role of hypoxia in the setting of RA, particularly in terms of angiogenesis, and the potential crosstalk between hypoxia-mediated pathways and inflammatory signalling cascades. As macrophages represent important cellular players in RA, their specific responses to the hypoxic synovial microenvironment will also be considered.
The molecular basis for cellular sensing and adaptation to oxygen-depleted conditions has been extensively described, revealing key roles for members of the family of transcription factors termed hypoxia-inducible factors (HIFs), frequently termed the ‘master regulators’ of the response to alterations in oxygen tension [99]. HIF is a heterodimeric transcription factor composed of two subunits, namely HIF-α (regulated by oxygen levels and post-translational modifications that are sensitive to oxygen levels) and HIF-β (expressed constitutively in the nucleus) [99–101]. HIF-α accumulates in the cytoplasm, followed by translocation into the nucleus, where it dimerises with HIF-β and binds HIF co-activators, before binding hypoxia-response elements (HRE) in target genes to initiate transcription. The main regulators of HIF-α post-translational modifications are dioxygenases requiring O2, 2-oxoglutarate, ferrous iron and ascorbic acid, named HIF prolyl hydroxylase domain-containing enzymes (PHDs) and factor inhibiting HIF-1 (FIH-1). The PHD enzymes hydroxylate proline residues within HIF-α, thus making HIF-α recognisable by the von Hippel Lindau tumour suppressor, which leads to polyubiquitination and proteasomal degradation [102]. In contrast, hydroxylation of asparagine residues by FIH-1 prevents recruitment of co-activators p300/CBP (CREB-binding protein) [103]. Under conditions of oxygen deprivation, PHD and FIH-1 are inactive, allowing HIF nuclear translocation and induction of transcription of HRE-containing genes.
As mentioned above, hypoxia is a feature of some diseases, particularly when tissue expansion and cellular metabolism result in oxygen demand exceeding supply, as is the case in RA synovium. Both HIF-α isoforms (HIF-1α, HIF-2α) are reported to be expressed in the setting of human RA synovium. In particular, HIF-1α was expressed abundantly by macrophages in most rheumatoid synovia, predominantly close to the intimal layer but also in sub-intimal zone. Of note there was markedly lower expression of HIF-1α in OA synovia, and HIF-1α was absent from healthy synovium [104, 105]. In a subsequent study, using a microelectrode technique in patients having elective hand surgery for RA tendon disease, synovial hypoxia and expression of HIF-2α were shown in inflammatory infiltrates [106]. Synovial hypoxia is probably the main driving force behind angiogenesis in RA. HIF-α expression in human RA synovium correlates with angiogenesis onset [105] and has been proved to control the expression of a wide range of genes with various roles. VEGF is one of the best characterised HIF-regulated genes, and increased levels of VEGF and VEGFR in RA have been reported in many studies, as mentioned earlier [50, 51, 54, 107]. Importantly, hypoxia increased the angiogenic potential of RA synovium-derived cells, as demonstrated by enhanced blood vessel formation using in vitro angiogenesis assay, together with enhanced synovial cell invasiveness [108]. Hypoxia also drives the expression of other pro-angiogenic players including the chemokines IL-8 (also known as CXC chemokine ligand 8 or CXCL8) [109], CC chemokine ligand 20 [110] and SDF-1 (stromal cell-derived factor 1, CXCL12) [111–113]. We and others have also reported that hypoxia increases expression of angiogenic genes other than VEGF, including angiopoietin-like 4 (ANGPTL4), ephrin A3 (EFNA3) and leptin [114, 115]. Increased levels of pro-inflammatory cytokines such as IL-6 and matrix metalloproteinases (MMP) MMP-1 and MMP-3 [109] have also been reported in response to hypoxia. Synovial hypoxia is, therefore, likely to contribute to RA by promoting inflammation, angiogenesis, cellular infiltration and cartilage degradation.
2.3 Interplay Between Hypoxia, Angiogenesis and Inflammation
As previously mentioned, low oxygen levels are associated with accumulation of inflammatory cells, enhanced expression of HIF and hypoxia-responsive genes and increased levels of pro-inflammatory cytokines, formally establishing a link between hypoxia and inflammation in the context of many diseases including RA (Fig. 16.2) [116]. Indications that HIF-α could have an important role in inflammation came from seminal studies utilising mice bearing myeloid-specific deletion of the Hif-α gene showing that HIF-1α allows myeloid cells to generate ATP in oxygen-deprived inflamed tissues, thereby operating energy-requiring processes including aggregation, motility, invasiveness and bactericidal activity [117, 118]. Indeed, the fact that hypoxia and the HIF pathway influence many inflammatory and immune responses relevant to synovitis, including monocyte–macrophage responses, is now well recognised. Pro- and anti-inflammatory cytokines are capable of directly affecting the HIF pathway, thus highlighting the convergence between inflammatory and oxygen-dependent signalling pathways. For instance, studies on gingival fibroblasts and RA fibroblast-like synoviocytes (FLS) have demonstrated that IL-1β and TNFα are capable of activating HIF in normoxia, an effect requiring both mitogen-activated protein kinase and phosphoinositide 3-kinase (PI3K) pathways [119, 120]. In contrast to HIF-α protein stabilisation that occurs under hypoxic conditions due to inhibition of PHDs and FIH-1, cytokines have been reported to stimulate HIF-α gene transcription in macrophages and RA FLS [120, 121]. Importantly, it has been demonstrated that T helper cell-type (TH) 1 cytokines can synergise with hypoxia to induce HIF-1 in various cells, including RA FLS [120, 122, 123]. We have also shown that TH1 cytokines in combination with hypoxia are not sufficient to induce angiogenic activity by RA FLS despite inducing HIF-1 and VEGF. In contrast, TH2 cytokines induce pro-angiogenic activity in normoxia and hypoxia, despite their inability to activate HIF-1 in FLS, highlighting the complex relationships between hypoxia, angiogenesis and inflammation in RA [114]. These observations might go some way to explain the apparent paradox of the concurrent presence of hypoxia and angiogenesis in RA synovium, since TH1 cytokines and hypoxia may not lead to formation of fully functional blood vessels, which have been shown to be present in RA [45, 46].
As well as FLS, monocytes and/or macrophages are considered key players within the context of RA, and thus hypoxia inevitably influences critical aspects of their behaviour, namely the transcriptional programme, polarisation status and metabolism, which might determine the perpetuation or resolution of the inflammatory disease. HIF-1 and HIF-2 were shown to be transcriptional effectors regulating macrophage responses to hypoxia [121, 124–126], and hypoxia upregulates a number of HRE-containing genes, including VEGF and ANGPTL4 [125, 127]. With regard to polarisation status, Mantovani and colleagues [128] presented a classification system for macrophage activation in which macrophages are divided into two groups: M1-type macrophages, characterised by enhanced ability of killing intracellular microorganisms and exhibiting pro-inflammatory properties, and M2-type macrophages, which exert anti-inflammatory effects [128]. So-called ‘classically activated’ M1 macrophages stimulate delayed-type hypersensitivity responses and increase surface levels of major histocompatibility complex (MHC) class II molecules, expression of markers such as inducible nitric oxide synthase (iNOS) and secretion of pro-inflammatory cytokines (IL-1β, TNFα, IL-6, IL-12, IL-15). M2 (‘alternatively activated’) macrophages are activated in response to IL-4 or IL-13, synthesise anti-inflammatory cytokines (such as IL-10) and facilitate TH2 responses. The relevance of HIF factors to macrophage polarisation ability comes from studies involving a specific macrophage subset, primarily expressed in tumour regions, called tumour associated macrophages (TAMs) and exhibiting a M1/M2 mixed phenotype with M2 being slightly more prominent. Werno et al., utilising a tumour-spheroid model, showed that HIF-1α-deficient macrophages developed a more prominent TAM marker profile (M2-skewed) together with reduced cytotoxicity and also displayed reduced angiogenic potential [129]. The precise role of HIF-2α during macrophage-mediated inflammatory responses has also been investigated. It was shown that mice lacking HIF-2α in myeloid cells are resistant to lipopolysaccharide (LPS)-induced endotoxemia and display an inability to mount inflammatory responses to cutaneous and peritoneal irritants. This phenotype was also associated with reduced TAM infiltration in murine hepatocellular and colitis-associated colon carcinoma models, and reduced tumour cell proliferation and progression, probably due to reduced expression of the macrophage-colony stimulating factor receptor and the CXC chemokine receptor 4 [126]. Chemical stabilisation of HIF following treatment with the HIF hydroxylase inhibitor dimethyloxaloylglycine (DMOG) has also been described to promote LPS-induced tolerance via expansion of the M2 macrophage population and activation of nuclear factor κ B (NFκB) signalling [130]. Hypoxia-mediated angiogenesis via enhanced blood vessel formation facilitates tissue oxygen delivery, thus contributing to repair of injured tissues, which is a characteristic function of M2 macrophages. On the other hand, hypoxia/HIF induces the expression of pro-angiogenic molecules such as VEGF. A recently published study investigated the differential roles of macrophage-released HIF-1α and HIF-2α with regard to angiogenesis progression. In this setting, it was shown that HIF-1α exhibits pro-angiogenic behaviour via its effects on VEGF, but that HIF-2α displays anti-angiogenic behaviour through production of the angiogenesis inhibitor soluble VEGFR1 [131]. HIF hydroxylases have also been ascribed different roles in macrophage polarisation. PHD-2 haplodeficient (Phd-2+/−) mice displayed preformed collateral arteries that preserved limb perfusion and prevented tissue necrosis in ischemia. Improved arteriogenesis in Phd-2+/− mice was due to expansion of tissue-resident, M2-like macrophages, with higher expression of M2-type genes. Conversely, several pro-inflammatory or anti-angiogenic (M1-type) molecules were downregulated, including IL-6, iNOS and IL-12 [132]. On the other hand, PHD-3 was found to be preferentially expressed by M1 macrophages [133]. Modulation of the HIF-PHD-NFκB axis might thus represent a novel approach for targeting inflammatory diseases where a macrophage polarisation switch may be a contributory factor.
The presence of hypoxia and HIF expression against a background of inflammation has prompted the question of whether hypoxia can activate signalling pathways other than the HIF cascade. Of particular relevance in inflammation is the dynamic interplay between the HIF and NFκB pathways. The NFκB pathway is activated in many different cells, including macrophages, dendritic cells, fibroblasts and endothelial cells, important cellular components in the establishment and progression of RA. Activation of this key transcription factor can be induced by a large number of stimuli, including bacterial products, viral molecules and pro-inflammatory cytokines. Hypoxia has been shown to drive NFκB activation and thereby regulate the release of critical pro-inflammatory mediators [134, 135]. The very first indication that HIF induces NFκB activation came from Walmsley et al. providing data showing that hypoxia promotes human neutrophil survival, an effect requiring HIFα-dependent regulation of inhibitor of NF-κB (IκB) α and NFκB [136]. Furthermore, the participation of PHD HIF hydroxylases in NFκB activation has been proposed [103, 137–140]. In the canonical pathway of NFκB activation, activity is controlled by IκB kinases (IKK), which mediate serine phosphorylation and degradation of IκBα and allow nuclear accumulation of NFκB. IKK-2 contains an evolutionarily conserved amino acid consensus motif which can potentially be hydroxylated by PHD. Mimicking hypoxia by using small interfering RNA against PHD-1 or PHD-2 resulted in NFκB activation in HeLa cells [137]. Similar studies in other cells also suggest that PHD may negatively regulate IKK via prolyl hydroxylation [138, 139]. Potential candidates for NFκB activation in the setting of RA include pro-inflammatory cytokines, which are likely to cause a vicious cycle of signals that result in chronic inflammation [141]. Activation of NFκB and HIF by converging hypoxic and inflammatory signalling pathways thus probably leads to pathological changes associated with RA, such as inflammation, angiogenesis and bone or cartilage destruction.
3 Angiogenesis Inhibition as a Therapeutic Option in RA
The principle underlying angiogenesis blockade for treatment of cancer is to ‘starve’ the tumour of nutrients and oxygen. There is now increasing appreciation that RA can also be classified as an angiogenesis-dependent disease, due to the many parallels with solid tumours, such as hypoxia, inflammation and altered vascularity. We and others have extensively described potential therapeutic applications aiming at inhibiting angiogenesis in RA [27, 29, 31–33, 142]. Therefore in this chapter we will focus on VEGF as a target in RA and review more extensively new potential angiogenesis inhibitors for treatment of RA (Table 16.1).
Over the last decade anti-VEGF biologicals have been approved for the treatment of angiogenesis-related diseases. The best examples are anti-VEGF antibody bevacizumab, used in different forms of cancer including metastatic colon cancer, and ranibizumab and pegaptanib sodium (anti-VEGF antibody and VEGF aptamer respectively) for age-related macular degeneration. The promising clinical outcomes of the use of such biologicals suggested that such treatments may also be an option to treat other angiogenic disorders in which VEGF is over-expressed including RA. Murine collagen-induced arthritis (CIA) is a model widely used for the testing of potential therapeutics for RA and was utilised to develop TNFα inhibitors as a therapeutic modality for RA. In CIA, expression of VEGF and VEGFR has been demonstrated [143–146]. Anti-VEGF antibody in this model delayed disease onset but appeared less effective when administered during the chronic phase of disease [144]. In another study, anti-VEGF inhibited synovitis in CIA, as indicated by a reduction in clinical score and paw swelling relative to untreated mice [147]. A soluble form of VEGFR1 has also been shown to significantly suppress established arthritis [148, 149]. A different strategy to limit angiogenesis via the VEGF pathway was to directly target VEGFR. In a spontaneous model of arthritis, De Bandt et al. observed that treatment with anti-VEGFR1 antibody abrogated bone and cartilage destruction. The antibody delayed the onset of arthritis and attenuated the severity of disease [150]. The group of Carmeliet also demonstrated that treatment with antibody against VEGFR1 reduced the incidence of joint disease, whereas antibody specific for VEGFR2 appeared ineffective [151]. More recently, Kong et al. reported that inhibition of VEGF via neuropilin-1 (NRP-1), an alternative VEGFR, significantly inhibited the survival, adhesion and migration of FLS. The anti-NRP-1 peptide also inhibited the proliferation, capillary tube formation and migration of endothelial cells in vitro, and neovascularisation in vivo, and suppressed experimentally induced arthritis in mice by inhibiting hyperplasia and angiogenesis in the arthritic joints, suggesting that anti-NRP-1 may offer a new approach for the treatment of RA [152]. To provide a more profound understanding of arthritis-associated angiogenesis, we recently evaluated the expression of angiogenesis modulating genes in CIA. One of the important findings of our study was that NRP-1 was a key player in the pathogenesis of CIA. Treatment with anti-NRP-1 antibody significantly reduced disease severity and joint destruction in CIA [145]. Kong et al. further developed a previously investigated hexapeptide which prevents VEGF binding to VEGFR1 by generating a stereochemical D-form of the peptide which was conjugated to PEG to prolong its half-life and found that the new peptide was much more beneficial in the treatment of angiogenesis in RA than the unmodified peptide. The effect was due to increased stability and delivery, suggesting that further development of current strategies could be applied to treat angiogenesis and RA in humans [153].
In addition to VEGF, pro-inflammatory cytokines have been demonstrated to be important in promoting angiogenesis. In 2010 Pickens et al. documented a novel role for IL-17A (also known as IL-17) in mediating angiogenesis, showing that IL-17 concentrations similar to those found in RA joints were capable of inducing endothelial cell migration and tube formation through the PI3K/AKT pathway. Furthermore, local expression of IL-17 in mouse ankles was able to induce joint inflammation and vascularity and demonstrated that IL-17 was angiogenic and promoted blood vessel formation in mice [154]. In continuation of their previous work the same group has further demonstrated that antibody blockade of CXCL5 ameliorates the angiogenic effect of IL-17 [155]. The ability of IL-17 to stimulate endothelial tube formation and invasiveness was also demonstrated by another recent study, highlighting the potential of targeting IL-17 in treatment of RA [156]. In an endothelial and synovial cell co-culture system, it was shown that IL-6 can also induce angiogenesis [157]. A follow-up study reported that IL-6 stimulation induced endothelial cell growth, by decreasing Ang-1 and increasing VEGF and Ang-2, and destabilised angiogenesis in RA, suggesting that targeting IL-6 signalling in RA (e.g. using tocilizumab) could inhibit angiogenesis [158]. Other approaches have targeted the Ang-Tie pathway [159–161] and the methionine aminopeptidase enzyme involved in endothelial proliferation [162–164].
Chemically modified heparins have also been developed as possible candidates for inhibiting angiogenesis; however, heparin therapy is limited by poor oral bioavailability. A newly synthesised low molecular weight heparin and deoxycholic acid conjugate (6ODS-LHbD) has been described to possess high oral bioavailability in rats. In a murine anti-collagen antibody-induced arthritis model this compound inhibited joint neovascularisation and disease development, suggesting that 6ODS-LHbD may be a promising candidate as an orally active angiogenesis inhibitor for treatment of RA [165]. In another recent study Kumagai and colleagues examined the anti-arthritic effect of ICM0301B, an angiogenesis inhibitor first described in 2004 [166]. They found that in the mouse CIA model ICM0301B inhibited disease development with an efficacy comparable to that of a non-steroidal anti-inflammatory drug, indomethacin, and suggested that the observed anti-arthritic effect of ICM0301B might be partially attributed to its anti-angiogenic activity [167]. Another novel inhibitor of angiogenesis in RA was described by Tanaka in 2011, who tested the effect of human plasminogen-related protein B in mouse CIA and found a significant reduction of disease development, VEGF expression and new blood vessel formation [168]. Chang and colleagues examined the role of macrophage CCAAT/enhancer binding protein delta (CEBPD) in CIA using Cebpd −/− mice. In the absence of CEBPD, disease development was significantly inhibited, with a decrease in the number of affected paws and reduced angiogenesis. Activation of CEBPD in macrophages was involved in promoting tube formation by endothelial cells and the migration and proliferation of rat synoviocytes [169]. Finally, another study aimed to examine whether angiogenesis induced by inflammatory agents is mediated via monocyte chemotactic protein-induced protein (MCPIP). Inflammatory agents, including TNFα, IL-1β and IL-8, were found to induce endothelial tube formation via MCPIP, and moreover MCPIP was shown to stimulate angiogenesis [170].
4 Conclusions
Many new therapies have been developed for treatment of RA, directed at biological responses which play a role in disease pathogenesis, such as B-cell and T-cell responses, or cascades driven by inflammatory cytokines, including IL-6, IL-1 and, notably, TNFα. These important advances offered a targeted strategy, unlike conventional but non-specific traditional treatments such as DMARD. However, in addition to side effects such as injection/infusion site reactions, the most significant side-effect of these therapies is an increase in the risk of all types of infections, including tuberculosis. Indeed, there is a significantly increased risk of reactivation of latent tuberculosis when using TNF inhibitors, and while pre-treatment screening has allowed identification of at-risk individuals; nonetheless, there is a drive to seek alternative treatment approaches. In this regard, angiogenesis represents an attractive option. VEGF is expressed in RA and VEGF inhibitors are already in clinical use for oncology applications. However, up to now VEGF blockade has been intensively used only in animal models of arthritis and no clinical trials of VEGF inhibition have been undertaken for RA. In addition adverse effects of VEGF-targeted biologicals, such as hypertension and gastro-intestinal perforation, coupled with the high cost of such therapies, are major concerns. Nevertheless, there is a valid hypothesis that such therapies can be beneficial in treating angiogenesis in RA and some groups are considering other approaches established in cancer for treatment of RA [171]. Moreover, the presence of immature blood vessels in RA synovium suggests that simply inhibiting VEGF may not be the best option. Interestingly, treatment of RA patients with TNF inhibitors selectively depleted immature vessels, without affecting the mature vasculature [45]. Haplodeficiency of PHD-2, using an in vivo tumour model in Phd-2+/− mice, actually resulted in vessel maturation, leading to improved tumour perfusion and oxygenation, and reduced tumour cell invasion, intravasation and metastasis [172]. In a model of colitis, inhibition of HIF hydroxylases in vivo using DMOG reduced disease severity. The possible underlying mechanism may be due to expansion of macrophages [130, 173]. These findings suggest that the best tactic to modulate angiogenesis in RA is at present unclear, and further in vitro and in vivo studies are still needed.
Abbreviations
- ACPA:
-
Antibodies to citrullinated protein antigens
- Ang:
-
Angiopoietin
- ANGPTL4:
-
Angiopoietin-like 4
- CCP:
-
Cyclic citrullinated peptides
- CIA:
-
Collagen-induced arthritis
- CXCL:
-
CXC chemokine ligand
- DMARD:
-
Disease modifying anti-rheumatic drug
- DMOG:
-
Dimethyloxaloylglycine
- EFNA3:
-
Ephrin A3
- FGF:
-
Fibroblast growth factor
- FIH-1:
-
Factor inhibiting HIF-1
- FLS:
-
Fibroblast-like synoviocytes
- HGF:
-
Hepatocyte growth factor
- HIF:
-
Hypoxia-inducible factor
- HLA:
-
Human leukocyte antigen
- HRE:
-
Hypoxia-response elements
- IKK:
-
IκB kinases
- IL:
-
Interleukin
- iNOS:
-
Inducible nitric oxide synthase
- IκB:
-
Inhibitor of NF-κB
- LPS:
-
Lipopolysaccharide
- MHC:
-
Major histocompatibility complex
- MMP:
-
Matrix metalloproteinases
- MTX:
-
Methotrexate
- NFκB:
-
Nuclear factor κ B
- NRP-1:
-
Neuropilin-1
- OA:
-
Osteoarthritis
- PDGF:
-
Platelet-derived growth factor
- PHD:
-
Prolyl hydroxylase domain-containing enzyme
- PI3K:
-
Phosphoinositide 3-kinase
- RA:
-
Rheumatoid arthritis
- SE:
-
Shared epitope
- TAM:
-
Tumour associated macrophages
- TH:
-
T helper cell-type
- Tie:
-
Tyrosine kinase with immunoglobulin-like and epidermal growth factor-like domains
- TNFα:
-
Tumour necrosis factor α
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
VEGF receptor
References
Bruce TO (2008) Comorbid depression in rheumatoid arthritis: pathophysiology and clinical implications. Curr Psychiatry Rep 10(3):258–264
Rathbun AM, Reed GW, Harrold LR (2013) The temporal relationship between depression and rheumatoid arthritis disease activity, treatment persistence and response: a systematic review. Rheumatology (Oxford) 52:1785–1794
Meenan RF, Gertman PM, Mason JH (1980) Measuring health status in arthritis. The arthritis impact measurement scales. Arthritis Rheum 23(2):146–152
Gossec L, Paternotte S, Aanerud GJ, Balanescu A, Boumpas DT, Carmona L, de Wit M, Dijkmans BA, Dougados M, Englbrecht M, Gogus F, Heiberg T, Hernandez C, Kirwan JR, Mola EM, Cerinic MM, Otsa K, Schett G, Scholte-Voshaar M, Sokka T, von Krause G, Wells GA, Kvien TK (2011) Finalisation and validation of the rheumatoid arthritis impact of disease score, a patient-derived composite measure of impact of rheumatoid arthritis: a EULAR initiative. Ann Rheum Dis 70(6):935–942
Barrett EM, Scott DG, Wiles NJ, Symmons DP (2000) The impact of rheumatoid arthritis on employment status in the early years of disease: a UK community-based study. Rheumatology (Oxford) 39(12):1403–1409
Sokka T, Kautiainen H, Pincus T, Verstappen SM, Aggarwal A, Alten R, Andersone D, Badsha H, Baecklund E, Belmonte M, Craig-Muller J, da Mota LM, Dimic A, Fathi NA, Ferraccioli G, Fukuda W, Geher P, Gogus F, Hajjaj-Hassouni N, Hamoud H, Haugeberg G, Henrohn D, Horslev-Petersen K, Ionescu R, Karateew D, Kuuse R, Laurindo IM, Lazovskis J, Luukkainen R, Mofti A, Murphy E, Nakajima A, Oyoo O, Pandya SC, Pohl C, Predeteanu D, Rexhepi M, Rexhepi S, Sharma B, Shono E, Sibilia J, Sierakowski S, Skopouli FN, Stropuviene S, Toloza S, Valter I, Woolf A, Yamanaka H (2010) Work disability remains a major problem in rheumatoid arthritis in the 2000s: data from 32 countries in the QUEST-RA Study. Arthritis Res Ther 12(2):R42
Sihvonen S, Korpela M, Laippala P, Mustonen J, Pasternack A (2004) Death rates and causes of death in patients with rheumatoid arthritis: a population-based study. Scand J Rheumatol 33(4):221–227
Kaplan MJ (2006) Cardiovascular disease in rheumatoid arthritis. Curr Opin Rheumatol 18(3):289–297
Van Doornum S, Brand C, King B, Sundararajan V (2006) Increased case fatality rates following a first acute cardiovascular event in patients with rheumatoid arthritis. Arthritis Rheum 54(7):2061–2068
Gkaliagkousi E, Gavriilaki E, Doumas M, Petidis K, Aslanidis S, Stella D (2012) Cardiovascular risk in rheumatoid arthritis: pathogenesis, diagnosis, and management. J Clin Rheumatol 18(8):422–430
Nadareishvili Z, Michaud K, Hallenbeck JM, Wolfe F (2008) Cardiovascular, rheumatologic, and pharmacologic predictors of stroke in patients with rheumatoid arthritis: a nested, case–control study. Arthritis Rheum 59(8):1090–1096
Ong KL, Wu BJ, Cheung BM, Barter PJ, Rye KA (2013) Arthritis: its prevalence, risk factors, and association with cardiovascular diseases in the United States, 1999 to 2008. Ann Epidemiol 23(2):80–86
Solomon DH, Karlson EW, Rimm EB, Cannuscio CC, Mandl LA, Manson JE, Stampfer MJ, Curhan GC (2003) Cardiovascular morbidity and mortality in women diagnosed with rheumatoid arthritis. Circulation 107(9):1303–1307
Kremers HM, Crowson CS, Therneau TM, Roger VL, Gabriel SE (2008) High ten-year risk of cardiovascular disease in newly diagnosed rheumatoid arthritis patients: a population-based cohort study. Arthritis Rheum 58(8):2268–2274
Solomon DH, Goodson NJ, Katz JN, Weinblatt ME, Avorn J, Setoguchi S, Canning C, Schneeweiss S (2006) Patterns of cardiovascular risk in rheumatoid arthritis. Ann Rheum Dis 65(12):1608–1612
Koivuniemi R, Paimela L, Suomalainen R, Leirisalo-Repo M (2013) Cardiovascular diseases in patients with rheumatoid arthritis. Scand J Rheumatol 42(2):131–135
Firestein GS (2005) Pathogenesis of rheumatoid arthritis: how early is early? Arthritis Res Ther 7(4):157–159
Andersson AK, Li C, Brennan FM (2008) Recent developments in the immunobiology of rheumatoid arthritis. Arthritis Res Ther 10(2):204
Orozco G, Rueda B, Martin J (2006) Genetic basis of rheumatoid arthritis. Biomed Pharmacother 60(10):656–662
Klareskog L, Stolt P, Lundberg K, Kallberg H, Bengtsson C, Grunewald J, Ronnelid J, Harris HE, Ulfgren AK, Rantapaa-Dahlqvist S, Eklund A, Padyukov L, Alfredsson L (2006) A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum 54(1):38–46
Silman AJ, Newman J, MacGregor AJ (1996) Cigarette smoking increases the risk of rheumatoid arthritis. Results from a nationwide study of disease-discordant twins. Arthritis Rheum 39(5):732–735
Schellekens GA, Visser H, de Jong BA, van den Hoogen FH, Hazes JM, Breedveld FC, van Venrooij WJ (2000) The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum 43(1):155–163
Vincent C, Nogueira L, Sebbag M, Chapuy-Regaud S, Arnaud M, Letourneur O, Rolland D, Fournie B, Cantagrel A, Jolivet M, Serre G (2002) Detection of antibodies to deiminated recombinant rat filaggrin by enzyme-linked immunosorbent assay: a highly effective test for the diagnosis of rheumatoid arthritis. Arthritis Rheum 46(8):2051–2058
van Gaalen FA, Linn-Rasker SP, van Venrooij WJ, de Jong BA, Breedveld FC, Verweij CL, Toes RE, Huizinga TW (2004) Autoantibodies to cyclic citrullinated peptides predict progression to rheumatoid arthritis in patients with undifferentiated arthritis: a prospective cohort study. Arthritis Rheum 50(3):709–715
Lundberg K, Kinloch A, Fisher BA, Wegner N, Wait R, Charles P, Mikuls TR, Venables PJ (2008) Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum 58(10):3009–3019
Mahdi H, Fisher BA, Kallberg H, Plant D, Malmstrom V, Ronnelid J, Charles P, Ding B, Alfredsson L, Padyukov L, Symmons DP, Venables PJ, Klareskog L, Lundberg K (2009) Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat Genet 41(12):1319–1324
Szekanecz Z, Koch AE (2008) Targeting angiogenesis in rheumatoid arthritis. Curr Rheumatol Rev 4(4):298–303
Paleolog EM (2009) The vasculature in rheumatoid arthritis. Int J Exp Pathol 90(3):249–261
Szekanecz Z, Besenyei T, Paragh G, Koch AE (2009) Angiogenesis in rheumatoid arthritis. Autoimmunity 42(7):563–573
Paleolog EM (2010) Angiogenesis in joint disease: the need for clinical data. Int J Clin Rheumatol 5(4):439–449
Szekanecz Z, Besenyei T, Szentpetery A, Koch AE (2010) Angiogenesis and vasculogenesis in rheumatoid arthritis. Curr Opin Rheumatol 22(3):299–306
Thairu N, Kiriakidis S, Dawson P, Paleolog E (2011) Angiogenesis as a therapeutic target in arthritis in 2011: learning the lessons of the colorectal cancer experience. Angiogenesis 14(3):223–234
Konisti S, Kiriakidis S, Paleolog EM (2012) Hypoxia: a key regulator of angiogenesis and inflammation in rheumatoid arthritis. Nat Rev Rheumatol 8(3):153–162
Rooney M, Condell D, Quinlan W, Daly L, Whelan A, Feighery C, Bresnihan B (1988) Analysis of the histologic variation of synovitis in rheumatoid arthritis. Arthritis Rheum 31(8):956–963
Strunk J, Heinemann E, Neeck G, Schmidt KL, Lange U (2004) A new approach to studying angiogenesis in rheumatoid arthritis by means of power Doppler ultrasonography and measurement of serum vascular endothelial growth factor. Rheumatology (Oxford) 43(12):1480–1483
Taylor PC, Steuer A, Gruber J, Cosgrove DO, Blomley MJ, Marsters PA, Wagner CL, McClinton C, Maini RN (2004) Comparison of ultrasonographic assessment of synovitis and joint vascularity with radiographic evaluation in a randomized, placebo-controlled study of infliximab therapy in early rheumatoid arthritis. Arthritis Rheum 50(4):1107–1116
Taylor PC, Steuer A, Gruber J, McClinton C, Cosgrove DO, Blomley MJ, Marsters PA, Wagner CL, Maini RN (2006) Ultrasonographic and radiographic results from a two-year controlled trial of immediate or one-year-delayed addition of infliximab to ongoing methotrexate therapy in patients with erosive early rheumatoid arthritis. Arthritis Rheum 54(1):47–53
Strunk J, Klingenberger P, Strube K, Bachmann G, Muller-Ladner U, Kluge A (2006) Three-dimensional Doppler sonographic vascular imaging in regions with increased MR enhancement in inflamed wrists of patients with rheumatoid arthritis. Joint Bone Spine 73(5):518–522
Larche MJ, Seymour M, Lim A, Eckersley RJ, Petavy F, Chiesa F, Rioja I, Lukey PT, Binks M, McClinton C, Dolan K, Taylor PC (2010) Quantitative power Doppler ultrasonography is a sensitive measure of metacarpophalangeal joint synovial vascularity in rheumatoid arthritis and declines significantly following a 2-week course of oral low-dose corticosteroids. J Rheumatol 37(12):2493–2501
Ceponis A, Konttinen YT, Imai S, Tamulaitiene M, Li TF, Xu JW, Hietanen J, Santavirta S, Fassbender HG (1998) Synovial lining, endothelial and inflammatory mononuclear cell proliferation in synovial membranes in psoriatic and reactive arthritis: a comparative quantitative morphometric study. Br J Rheumatol 37(2):170–178
Walsh DA, Wade M, Mapp PI, Blake DR (1998) Focally regulated endothelial proliferation and cell death in human synovium. Am J Pathol 152(3):691–702
FitzGerald O, Soden M, Yanni G, Robinson R, Bresnihan B (1991) Morphometric analysis of blood vessels in synovial membranes obtained from clinically affected and unaffected knee joints of patients with rheumatoid arthritis. Ann Rheum Dis 50(11):792–796
Stevens CR, Blake DR, Merry P, Revell PA, Levick JR (1991) A comparative study by morphometry of the microvasculature in normal and rheumatoid synovium. Arthritis Rheum 34(12):1508–1513
Fearon U, Griosios K, Fraser A, Reece R, Emery P, Jones PF, Veale DJ (2003) Angiopoietins, growth factors, and vascular morphology in early arthritis. J Rheumatol 30(2):260–268
Izquierdo E, Canete JD, Celis R, Santiago B, Usategui A, Sanmarti R, Del Rey MJ, Pablos JL (2009) Immature blood vessels in rheumatoid synovium are selectively depleted in response to anti-TNF therapy. PLoS One 4(12):e8131
Kennedy A, Ng CT, Biniecka M, Saber T, Taylor C, O'Sullivan J, Veale DJ, Fearon U (2010) Angiogenesis and blood vessel stability in inflammatory arthritis. Arthritis Rheum 62(3):711–721
Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L (2006) VEGF receptor signalling – in control of vascular function. Nat Rev Mol Cell Biol 7(5):359–371
Bainbridge J, Sivakumar B, Paleolog E (2006) Angiogenesis as a therapeutic target in arthritis: lessons from oncology. Curr Pharm Des 12(21):2631–2644
Brown RA, Weiss JB, Tomlinson IW, Phillips P, Kumar S (1980) Angiogenic factor from synovial fluid resembling that from tumours. Lancet 1(8170):682–685
Koch AE, Harlow LA, Haines GK, Amento EP, Unemori EN, Wong WL, Pope RM, Ferrara N (1994) Vascular endothelial growth factor. A cytokine modulating endothelial function in rheumatoid arthritis. J Immunol 152(8):4149–4156
Fava RA, Olsen NJ, Spencer-Green G, Yeo KT, Yeo TK, Berse B, Jackman RW, Senger DR, Dvorak HF, Brown LF (1994) Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J Exp Med 180(1):341–346
Kasama T, Kobayashi K, Yajima N, Shiozawa F, Yoda Y, Takeuchi HT, Mori Y, Negishi M, Ide H, Adachi M (2000) Expression of vascular endothelial growth factor by synovial fluid neutrophils in rheumatoid arthritis (RA). Clin Exp Immunol 121(3):533–538
Taichman NS, Young S, Cruchley AT, Taylor P, Paleolog E (1997) Human neutrophils secrete vascular endothelial growth factor. J Leukoc Biol 62(3):397–400
Paleolog EM, Young S, Stark AC, McCloskey RV, Feldmann M, Maini RN (1998) Modulation of angiogenic vascular endothelial growth factor by tumor necrosis factor alpha and interleukin-1 in rheumatoid arthritis. Arthritis Rheum 41(7):1258–1265
Bottomley MJ, Webb NJ, Watson CJ, Holt PJ, Freemont AJ, Brenchley PE (1999) Peripheral blood mononuclear cells from patients with rheumatoid arthritis spontaneously secrete vascular endothelial growth factor (VEGF): specific up-regulation by tumour necrosis factor-alpha (TNF-alpha) in synovial fluid. Clin Exp Immunol 117(1):171–176
Berse B, Hunt JA, Diegel RJ, Morganelli P, Yeo K, Brown F, Fava RA (1999) Hypoxia augments cytokine (transforming growth factor-beta (TGF-beta) and IL-1)-induced vascular endothelial growth factor secretion by human synovial fibroblasts. Clin Exp Immunol 115(1):176–182
Pufe T, Petersen W, Tillmann B, Mentlein R (2001) Splice variants VEGF121 and VEGF165 of the angiogenic peptide vascular endothelial cell growth factor are expressed in the synovial tissue of patients with rheumatoid arthritis. J Rheumatol 28(7):1482–1485
Giatromanolaki A, Sivridis E, Athanassou N, Zois E, Thorpe PE, Brekken RA, Gatter KC, Harris AL, Koukourakis IM, Koukourakis MI (2001) The angiogenic pathway “vascular endothelial growth factor/flk-1(KDR)-receptor” in rheumatoid arthritis and osteoarthritis. J Pathol 194(1):101–108
Harada M, Mitsuyama K, Yoshida H, Sakisaka S, Taniguchi E, Kawaguchi T, Ariyoshi M, Saiki T, Sakamoto M, Nagata K, Sata M, Matsuo K, Tanikawa K (1998) Vascular endothelial growth factor in patients with rheumatoid arthritis. Scand J Rheumatol 27(5):377–380
Kikuchi K, Kubo M, Kadono T, Yazawa N, Ihn H, Tamaki K (1998) Serum concentrations of vascular endothelial growth factor in collagen diseases. Br J Dermatol 139(6):1049–1051
Lee SS, Joo YS, Kim WU, Min DJ, Min JK, Park SH, Cho CS, Kim HY (2001) Vascular endothelial growth factor levels in the serum and synovial fluid of patients with rheumatoid arthritis. Clin Exp Rheumatol 19(3):321–324
Sone H, Sakauchi M, Takahashi A, Suzuki H, Inoue N, Iida K, Shimano H, Toyoshima H, Kawakami Y, Okuda Y, Matsuo K, Yamada N (2001) Elevated levels of vascular endothelial growth factor in the sera of patients with rheumatoid arthritis correlation with disease activity. Life Sci 69(16):1861–1869
Latour F, Zabraniecki L, Dromer C, Brouchet A, Durroux R, Fournie B (2001) Does vascular endothelial growth factor in the rheumatoid synovium predict joint destruction? A clinical, radiological, and pathological study in 12 patients monitored for 10 years. Joint Bone Spine 68(6):493–498
Clavel G, Bessis N, Lemeiter D, Fardellone P, Mejjad O, Menard JF, Pouplin S, Boumier P, Vittecoq O, Le Loet X, Boissier MC (2007) Angiogenesis markers (VEGF, soluble receptor of VEGF and angiopoietin-1) in very early arthritis and their association with inflammation and joint destruction. Clin Immunol 124(2):158–164
Kuryliszyn-Moskal A, Klimiuk PA, Sierakowski S, Ciolkiewicz M (2006) A study on vascular endothelial growth factor and endothelin-1 in patients with extra-articular involvement of rheumatoid arthritis. Clin Rheumatol 25(3):314–319
Ballara SC, Taylor PC, Reusch P, Marmé D, Feldmann M, Maini RN, Paleolog EM (2001) Raised serum vascular endothelial growth factor levels are associated with destructive change in inflammatory arthritis. Arthritis Rheum 44(9):2055–2064
Nakahara H, Song J, Sugimoto M, Hagihara K, Kishimoto T, Yoshizaki K, Nishimoto N (2003) Anti-interleukin-6 receptor antibody therapy reduces vascular endothelial growth factor production in rheumatoid arthritis. Arthritis Rheum 48(6):1521–1529
Aggarwal A, Panda S, Misra R (2004) Effect of etanercept on matrix metalloproteinases and angiogenic vascular endothelial growth factor: a time kinetic study. Ann Rheum Dis 63(7):891–892
Macias I, Garcia-Perez S, Ruiz-Tudela M, Medina F, Chozas N, Giron-Gonzalez JA (2005) Modification of pro- and antiinflammatory cytokines and vascular-related molecules by tumor necrosis factor-a blockade in patients with rheumatoid arthritis. J Rheumatol 32(11):2102–2108
Nagashima M, Wauke K, Hirano D, Ishigami S, Aono H, Takai M, Sasano M, Yoshino S (2000) Effects of combinations of anti-rheumatic drugs on the production of vascular endothelial growth factor and basic fibroblast growth factor in cultured synoviocytes and patients with rheumatoid arthritis. Rheumatology (Oxford) 39(11):1255–1262
Han SW, Kim GW, Seo JS, Kim SJ, Sa KH, Park JY, Lee J, Kim SY, Goronzy JJ, Weyand CM, Kang YM (2004) VEGF gene polymorphisms and susceptibility to rheumatoid arthritis. Rheumatology (Oxford) 43(9):1173–1177
Chen Y, Dawes PT, Mattey DL (2012) Polymorphism in the vascular endothelial growth factor A (VEGFA) gene is associated with serum VEGF-A level and disease activity in rheumatoid arthritis: differential effect of cigarette smoking. Cytokine 58(3):390–397
Chen Y, Mattey DL (2012) Age at onset of rheumatoid arthritis: association with polymorphisms in the vascular endothelial growth factor A(VEGFA) gene and an intergenic locus between matrix metalloproteinase (MMP) 1 and 3 genes. Clin Exp Rheumatol 30(6):894–898
Chen Y, Dawes PT, Packham JC, Mattey DL (2011) Interaction between smoking and polymorphism in the promoter region of the VEGFA gene is associated with ischemic heart disease and myocardial infarction in rheumatoid arthritis. J Rheumatol 38(5):802–809
Rodriguez-Rodriguez L, Garcia-Bermudez M, Gonzalez-Juanatey C, Vazquez-Rodriguez TR, Miranda-Filloy JA, Fernandez-Gutierrez B, Llorca J, Martin J, Gonzalez-Gay MA (2011) Vascular endothelial growth factor A and cardiovascular disease in rheumatoid arthritis patients. Tissue Antigens 77(4):291–297
Scott BB, Zaratin PF, Colombo A, Hansbury MJ, Winkler JD, Jackson JR (2002) Constitutive expression of angiopoietin-1 and -2 and modulation of their expression by inflammatory cytokines in rheumatoid arthritis synovial fibroblasts. J Rheumatol 29(2):230–239
Gravallese EM, Pettit AR, Lee R, Madore R, Manning C, Tsay A, Gaspar J, Goldring MB, Goldring SR, Oettgen P (2003) Angiopoietin-1 is expressed in the synovium of patients with rheumatoid arthritis and is induced by tumour necrosis factor alpha. Ann Rheum Dis 62(2):100–107
DeBusk LM, Chen Y, Nishishita T, Chen J, Thomas JW, Lin PC (2003) Tie2 receptor tyrosine kinase, a major mediator of tumor necrosis factor alpha-induced angiogenesis in rheumatoid arthritis. Arthritis Rheum 48(9):2461–2471
Shahrara S, Volin MV, Connors MA, Haines GK, Koch AE (2002) Differential expression of the angiogenic Tie receptor family in arthritic and normal synovial tissue. Arthritis Res 4(3):201–208
Uchida T, Nakashima M, Hirota Y, Miyazaki Y, Tsukazaki T, Shindo H (2000) Immunohistochemical localisation of protein tyrosine kinase receptors Tie-1 and Tie-2 in synovial tissue of rheumatoid arthritis: correlation with angiogenesis and synovial proliferation. Ann Rheum Dis 59(8):607–614
Sano H, Forough R, Maier JA, Case JP, Jackson A, Engleka K, Maciag T, Wilder RL (1990) Detection of high levels of heparin binding growth factor-1 (acidic fibroblast growth factor) in inflammatory arthritic joints. J Cell Biol 110(4):1417–1426
Nakashima M, Eguchi K, Aoyagi T, Yamashita I, Ida H, Sakai M, Shimada H, Kawabe Y, Nagataki S, Koji T, Nakane PK (1994) Expression of basic fibroblast growth factor in synovial tissues from patients with rheumatoid arthritis: detection by immunohistological staining and in situ hybridisation. Ann Rheum Dis 53(1):45–50
Sano H, Engleka K, Mathern P, Hla T, Crofford LJ, Remmers EF, Jelsema CL, Goldmuntz E, Maciag T, Wilder RL (1993) Coexpression of phosphotyrosine-containing proteins, platelet-derived growth factor-B, and fibroblast growth factor-1 in situ in synovial tissues of patients with rheumatoid arthritis and Lewis rats with adjuvant or streptococcal cell wall arthritis. J Clin Invest 91(2):553–565
Remmers EF, Sano H, Lafyatis R, Case JP, Kumkumian GK, Hla T, Maciag T, Wilder RL (1991) Production of platelet derived growth factor B chain (PDGF-B/c-sis) mRNA and immunoreactive PDGF B-like polypeptide by rheumatoid synovium: coexpression with heparin binding acidic fibroblast growth factor-1. J Rheumatol 18(1):7–13
Koch AE, Halloran MM, Hosaka S, Shah MR, Haskell CJ, Baker SK, Panos RJ, Haines GK, Bennett GL, Pope RM, Ferrara N (1996) Hepatocyte growth factor. A cytokine mediating endothelial migration in inflammatory arthritis. Arthritis Rheum 39(9):1566–1575
Ruger B, Giurea A, Wanivenhaus AH, Zehetgruber H, Hollemann D, Yanagida G, Groger M, Petzelbauer P, Smolen JS, Hoecker P, Fischer MB (2004) Endothelial precursor cells in the synovial tissue of patients with rheumatoid arthritis and osteoarthritis. Arthritis Rheum 50(7):2157–2166
Hirohata S, Yanagida T, Nampei A, Kunugiza Y, Hashimoto H, Tomita T, Yoshikawa H, Ochi T (2004) Enhanced generation of endothelial cells from CD34+ cells of the bone marrow in rheumatoid arthritis: possible role in synovial neovascularization. Arthritis Rheum 50(12):3888–3896
Grisar J, Aletaha D, Steiner CW, Kapral T, Steiner S, Seidinger D, Weigel G, Schwarzinger I, Wolozcszuk W, Steiner G, Smolen JS (2005) Depletion of endothelial progenitor cells in the peripheral blood of patients with rheumatoid arthritis. Circulation 111(2):204–211
Jodon de Villeroche V, Avouac J, Ponceau A, Ruiz B, Kahan A, Boileau C, Uzan G, Allanore Y (2010) Enhanced late-outgrowth circulating endothelial progenitor cell levels in rheumatoid arthritis and correlation with disease activity. Arthritis Res Ther 12(1):R27
Bonnet CS, Walsh DA (2005) Osteoarthritis, angiogenesis and inflammation. Rheumatology (Oxford) 44(1):7–16
Haywood L, McWilliams DF, Pearson CI, Gill SE, Ganesan A, Wilson D, Walsh DA (2003) Inflammation and angiogenesis in osteoarthritis. Arthritis Rheum 48(8):2173–2177
Mapp PI, Avery PS, McWilliams DF, Bowyer J, Day C, Moores S, Webster R, Walsh DA (2008) Angiogenesis in two animal models of osteoarthritis. Osteoarthritis Cartilage 16(1):61–69
Mapp PI, Walsh DA, Bowyer J, Maciewicz RA (2010) Effects of a metalloproteinase inhibitor on osteochondral angiogenesis, chondropathy and pain behavior in a rat model of osteoarthritis. Osteoarthritis Cartilage 18(4):593–600
Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL (1998) Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev 12(2):149–162
Ryan HE, Lo J, Johnson RS (1998) HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J 17(11):3005–3015
Peng J, Zhang L, Drysdale L, Fong GH (2000) The transcription factor EPAS-1/hypoxia-inducible factor 2alpha plays an important role in vascular remodeling. Proc Natl Acad Sci USA 97(15):8386–8391
Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL (1998) The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev 12(21):3320–3324
Pagel H, Engel A, Jelkmann W (1992) Erythropoietin induction by hypoxia. A comparison of in vitro and in vivo experiments. Adv Exp Med Biol 317:515–519
Semenza GL (2009) Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 24:97–106
Rey S, Semenza GL (2010) Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc Res 86(2):236–242
Semenza GL (2010) Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29(5):625–634
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399(6733):271–275
Cockman ME, Lancaster DE, Stolze IP, Hewitson KS, McDonough MA, Coleman ML, Coles CH, Yu X, Hay RT, Ley SC, Pugh CW, Oldham NJ, Masson N, Schofield CJ, Ratcliffe PJ (2006) Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc Natl Acad Sci USA 103(40):14767–14772
Hollander AP, Corke KP, Freemont AJ, Lewis CE (2001) Expression of hypoxia-inducible factor 1alpha by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum 44(7):1540–1544
Brouwer E, Gouw AS, Posthumus MD, van Leeuwen MA, Boerboom AL, Bijzet J, Bos R, Limburg PC, Kallenberg CG, Westra J (2009) Hypoxia inducible factor-1-alpha (HIF-1alpha) is related to both angiogenesis and inflammation in rheumatoid arthritis. Clin Exp Rheumatol 27(6):945–951
Sivakumar B, Akhavani MA, Winlove CP, Taylor PC, Paleolog EM, Kang N (2008) Synovial hypoxia as a cause of tendon rupture in rheumatoid arthritis. J Hand Surg [Am] 33(1):49–58
Kurosaka D, Hirai K, Nishioka M, Miyamoto Y, Yoshida K, Noda K, Ukichi T, Yanagimachi M, Furuya K, Takahashi E, Kingetsu I, Fukuda K, Yamada A (2010) Clinical significance of serum levels of vascular endothelial growth factor, angiopoietin-1, and angiopoietin-2 in patients with rheumatoid arthritis. J Rheumatol 37(6):1121–1128
Akhavani MA, Madden L, Buysschaert I, Sivakumar B, Kang N, Paleolog EM (2009) Hypoxia upregulates angiogenesis and synovial cell migration in rheumatoid arthritis. Arthritis Res Ther 11(3):R64
Ahn JK, Koh EM, Cha HS, Lee YS, Kim J, Bae EK, Ahn KS (2008) Role of hypoxia-inducible factor-1alpha in hypoxia-induced expressions of IL-8, MMP-1 and MMP-3 in rheumatoid fibroblast-like synoviocytes. Rheumatology (Oxford) 47(6):834–839
Bosco MC, Delfino S, Ferlito F, Battaglia F, Puppo M, Gregorio A, Gambini C, Gattorno M, Martini A, Varesio L (2008) Hypoxic synovial environment and expression of macrophage inflammatory protein 3gamma/CCL20 in juvenile idiopathic arthritis. Arthritis Rheum 58(6):1833–1838
Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H (2002) Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum 46(10):2587–2597
del Rey MJ, Izquierdo E, Caja S, Usategui A, Santiago B, Galindo M, Pablos JL (2009) Human inflammatory synovial fibroblasts induce enhanced myeloid cell recruitment and angiogenesis through a hypoxia-inducible transcription factor 1alpha/vascular endothelial growth factor-mediated pathway in immunodeficient mice. Arthritis Rheum 60(10):2926–2934
Santiago B, Calonge E, Del Rey MJ, Gutierrez-Canas I, Izquierdo E, Usategui A, Galindo M, Alcami J, Pablos JL (2011) CXCL12 gene expression is upregulated by hypoxia and growth arrest but not by inflammatory cytokines in rheumatoid synovial fibroblasts. Cytokine 53(2):184–190
Larsen H, Muz B, Khong TL, Feldmann M, Paleolog EM (2012) Differential effects of Th1 versus Th2 cytokines in combination with hypoxia on HIFs and angiogenesis in RA. Arthritis Res Ther 14(4):R180
Muz B, Larsen H, Madden L, Kiriakidis S, Paleolog EM (2012) Prolyl hydroxylase domain enzyme 2 is the major player in regulating hypoxic responses in rheumatoid arthritis. Arthritis Rheum 64(9):2856–2867
Giatromanolaki A, Sivridis E, Maltezos E, Athanassou N, Papazoglou D, Gatter KC, Harris AL, Koukourakis MI (2003) Upregulated hypoxia inducible factor-1alpha and -2alpha pathway in rheumatoid arthritis and osteoarthritis. Arthritis Res Ther 5(4):R193–R201
Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS (2003) HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112(5):645–657
Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS (2005) HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest 115(7):1806–1815
Thornton RD, Lane P, Borghaei RC, Pease EA, Caro J, Mochan E (2000) Interleukin 1 induces hypoxia-inducible factor 1 in human gingival and synovial fibroblasts. Biochem J 350(Pt 1):307–312
Westra J, Brouwer E, Bos R, Posthumus MD, Doornbos-van der Meer B, Kallenberg CG, Limburg PC (2007) Regulation of cytokine-induced HIF-1alpha expression in rheumatoid synovial fibroblasts. Ann N Y Acad Sci 1108:340–348
Takeda N, O'Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, Asagiri M, Simon MC, Hoffmann A, Johnson RS (2010) Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev 24(5):491–501
Hellwig-Burgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W (1999) Interleukin-1beta and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood 94(5):1561–1567
Westra J, Brouwer E, Bouwman E, Doornbos-van der Meer B, Posthumus MD, van Leeuwen MA, Limburg PC, Ueda Y, Kallenberg CG (2009) Role for CaMKII inhibition in rheumatoid arthritis: effects on HIF-1-induced VEGF production by rheumatoid synovial fibroblasts. Ann N Y Acad Sci 1173:706–711
Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL (2000) The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol 157(2):411–421
Fang HY, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, Johnson RS, Imityaz HZ, Simon MC, Fredlund E, Greten FR, Rius J, Lewis CE (2009) Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 114(4):844–859
Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ, Hammond R, Gimotty PA, Keith B, Simon MC (2010) Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest 120(8):2699–2714
White JR, Harris RA, Lee SR, Craigon MH, Binley K, Price T, Beard GL, Mundy CR, Naylor S (2004) Genetic amplification of the transcriptional response to hypoxia as a novel means of identifying regulators of angiogenesis. Genomics 83(1):1–8
Sica A, Schioppa T, Mantovani A, Allavena P (2006) Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer 42(6):717–727
Werno C, Menrad H, Weigert A, Dehne N, Goerdt S, Schledzewski K, Kzhyshkowska J, Brune B (2010) Knockout of HIF-1alpha in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis 31(10):1863–1872
Hams E, Saunders SP, Cummins EP, O’Connor A, Tambuwala MT, Gallagher WM, Byrne A, Campos-Torres A, Moynagh PM, Jobin C, Taylor CT, Fallon PG (2011) The hydroxylase inhibitor dimethyloxallyl glycine attenuates endotoxic shock via alternative activation of macrophages and IL-10 production by B1 cells. Shock 36(3):295–302
Eubank TD, Roda JM, Liu H, O’Neil T, Marsh CB (2011) Opposing roles for HIF-1alpha and HIF-2alpha in the regulation of angiogenesis by mononuclear phagocytes. Blood 117(1):323–332
Takeda Y, Costa S, Delamarre E, Roncal C, Leite de Oliveira R, Squadrito ML, Finisguerra V, Deschoemaeker S, Bruyere F, Wenes M, Hamm A, Serneels J, Magat J, Bhattacharyya T, Anisimov A, Jordan BF, Alitalo K, Maxwell P, Gallez B, Zhuang ZW, Saito Y, Simons M, De Palma M, Mazzone M (2011) Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature 479(7371):122–126
Escribese MM, Sierra-Filardi E, Nieto C, Samaniego R, Sanchez-Torres C, Matsuyama T, Calderon-Gomez E, Vega MA, Salas A, Sanchez-Mateos P, Corbi AL (2012) The prolyl hydroxylase PHD3 identifies proinflammatory macrophages and its expression is regulated by activin A. J Immunol 189(4):1946–1954
Taylor CT (2008) Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J Physiol 586(Pt 17):4055–4059
Oliver KM, Taylor CT, Cummins EP (2009) Hypoxia. Regulation of NFkappaB signalling during inflammation: the role of hydroxylases. Arthritis Res Ther 11(1):215
Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med 201(1):105–115
Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT (2006) Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA 103(48):18154–18159
Fu J, Taubman MB (2010) Prolyl hydroxylase EGLN3 regulates skeletal myoblast differentiation through an NF-kappaB-dependent pathway. J Biol Chem 285(12):8927–8935
Xue J, Li X, Jiao S, Wei Y, Wu G, Fang J (2010) Prolyl hydroxylase-3 is down-regulated in colorectal cancer cells and inhibits IKKbeta independent of hydroxylase activity. Gastroenterology 138(2):606–615
Ferguson JE III, Wu Y, Smith K, Charles P, Powers K, Wang H, Patterson C (2007) ASB4 is a hydroxylation substrate of FIH and promotes vascular differentiation via an oxygen-dependent mechanism. Mol Cell Biol 27(18):6407–6419
Simmonds RE, Foxwell BM (2008) Signalling, inflammation and arthritis: NF-kappaB and its relevance to arthritis and inflammation. Rheumatology (Oxford) 47(5):584–590
Muz B, Khan MN, Kiriakidis S, Paleolog EM (2009) The role of hypoxia and HIF-dependent signalling events in rheumatoid arthritis. Arthritis Res Ther 11(1):201
Hermann LM, Pinkerton M, Jennings K, Yang L, Grom A, Sowders D, Kersten S, Witte DP, Hirsch R, Thornton S (2005) Angiopoietin-like-4 is a potential angiogenic mediator in arthritis. Clin Immunol 115(1):93–101
Lu J, Kasama T, Kobayashi K, Yoda Y, Shiozawa F, Hanyuda M, Negishi M, Ide H, Adachi M (2000) Vascular endothelial growth factor expression and regulation of murine collagen-induced arthritis. J Immunol 164(11):5922–5927
Raatz Y, Ibrahim S, Feldmann M, Paleolog EM (2012) Gene expression profiling and functional analysis of angiogenic markers in murine collagen-induced arthritis. Arthritis Res Ther 14(4):R169
Thornton S, Sowders D, Aronow B, Witte DP, Brunner HI, Giannini EH, Hirsch R (2002) DNA microarray analysis reveals novel gene expression profiles in collagen-induced arthritis. Clin Immunol 105(2):155–168
Sone H, Kawakami Y, Sakauchi M, Nakamura Y, Takahashi A, Shimano H, Okuda Y, Segawa T, Suzuki H, Yamada N (2001) Neutralization of vascular endothelial growth factor prevents collagen-induced arthritis and ameliorates established disease in mice. Biochem Biophys Res Commun 281(2):562–568
Miotla J, Maciewicz R, Kendrew J, Feldmann M, Paleolog E (2000) Treatment with soluble VEGF receptor reduces disease severity in murine collagen-induced arthritis. Lab Invest 80(8):1195–1205
Afuwape AO, Feldmann M, Paleolog EM (2003) Adenoviral delivery of soluble VEGF receptor 1 (sFlt-1) abrogates disease activity in murine collagen-induced arthritis. Gene Ther 10(23):1950–1960
de Bandt M, Ben Mahdi MH, Ollivier V, Grossin M, Dupuis M, Gaudry M, Bohlen P, Lipson KE, Rice A, Wu Y, Gougerot-Pocidalo MA, Pasquier C (2003) Blockade of vascular endothelial growth factor receptor I (VEGF-RI), but not VEGF-RII, suppresses joint destruction in the K/BxN model of rheumatoid arthritis. J Immunol 171(9):4853–4859
Luttun A, Tjwa M, Moons L, Wu Y, Angelillo-Scherrer A, Liao F, Nagy JA, Hooper A, Priller J, De Klerck B, Compernolle V, Daci E, Bohlen P, Dewerchin M, Herbert JM, Fava R, Matthys P, Carmeliet G, Collen D, Dvorak HF, Hicklin DJ, Carmeliet P (2002) Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med 8(8):831–840
Kong JS, Yoo SA, Kim JW, Yang SP, Chae CB, Tarallo V, De Falco S, Ryu SH, Cho CS, Kim WU (2010) Anti-neuropilin-1 peptide inhibition of synoviocyte survival, angiogenesis, and experimental arthritis. Arthritis Rheum 62(1):179–190
Kong JS, Yoo SA, Kang JH, Ko W, Jeon S, Chae CB, Cho CS, Kim WU (2011) Suppression of neovascularization and experimental arthritis by D-form of anti-flt-1 peptide conjugated with mini-PEG™. Angiogenesis 14(4):431–442
Pickens SR, Volin MV, Mandelin AM II, Kolls JK, Pope RM, Shahrara S (2010) IL-17 contributes to angiogenesis in rheumatoid arthritis. J Immunol 184(6):3233–3241
Pickens SR, Chamberlain ND, Volin MV, Gonzalez M, Pope RM, Mandelin AM II, Kolls JK, Shahrara S (2011) Anti-CXCL5 therapy ameliorates IL-17-induced arthritis by decreasing joint vascularization. Angiogenesis 14(4):443–455
Moran EM, Connolly M, Gao W, McCormick J, Fearon U, Veale DJ (2011) Interleukin-17A induction of angiogenesis, cell migration, and cytoskeletal rearrangement. Arthritis Rheum 63(11):3263–3273
Hashizume M, Hayakawa N, Suzuki M, Mihara M (2009) IL-6/sIL-6R trans-signalling, but not TNF-alpha induced angiogenesis in a HUVEC and synovial cell co-culture system. Rheumatol Int 29(12):1449–1454
Kayakabe K, Kuroiwa T, Sakurai N, Ikeuchi H, Kadiombo AT, Sakairi T, Matsumoto T, Maeshima A, Hiromura K, Nojima Y (2012) Interleukin-6 promotes destabilized angiogenesis by modulating angiopoietin expression in rheumatoid arthritis. Rheumatology (Oxford) 51(9):1571–1579
Chen Y, Donnelly E, Kobayashi H, Debusk LM, Lin PC (2005) Gene therapy targeting the Tie2 function ameliorates collagen-induced arthritis and protects against bone destruction. Arthritis Rheum 52(5):1585–1594
Jin P, Zhang J, Sumariwalla PF, Ni I, Jorgensen B, Crawford D, Phillips S, Feldmann M, Shepard HM, Paleolog EM (2008) Novel splice variants derived from the receptor tyrosine kinase superfamily are potential therapeutics for rheumatoid arthritis. Arthritis Res Ther 10(4):R73
Malik NM, Jin P, Raatz Y, Sumariwalla PF, Kiriakidis S, Shepard M, Feldmann M, Paleolog EM (2010) Regulation of the Angiopoietin-Tie ligand-receptor system with a novel splice variant of Tie1 reduces the severity of murine arthritis. Rheumatology 49(10):1828–1839
Arsenault AL, Lhotak S, Hunter WL, Banquerigo ML, Brahn E (1998) Taxol involution of collagen-induced arthritis: ultrastructural correlation with the inhibition of synovitis and neovascularization. Clin Immunol Immunopathol 86(3):280–289
Bainbridge J, Madden L, Essex D, Binks M, Malhotra R, Paleolog EM (2007) Methionine aminopeptidase-2 blockade reduces chronic collagen-induced arthritis: potential role for angiogenesis inhibition. Arthritis Res Ther 9(6):R127
Bernier SG, Lazarus DD, Clark E, Doyle B, Labenski MT, Thompson CD, Westlin WF, Hannig G (2004) A methionine aminopeptidase-2 inhibitor, PPI-2458, for the treatment of rheumatoid arthritis. Proc Natl Acad Sci USA 101(29):10768–10773
Hwang SR, Seo DH, Al-Hilal TA, Jeon OC, Kang JH, Kim SH, Kim HS, Chang YT, Kang YM, Yang VC, Byun Y (2012) Orally active desulfated low molecular weight heparin and deoxycholic acid conjugate, 6ODS-LHbD, suppresses neovascularization and bone destruction in arthritis. J Control Release 163(3):374–384
Someno T, Kumagai H, Ohba S, Amemiya M, Naganawa H, Ishizuka M, Ikeda D (2004) ICM0301s, new angiogenesis inhibitors from Aspergillus sp. F-1491. II. Physico-chemical properties and structure elucidation. J Antibiot (Tokyo) 57(2):104–109
Kumagai H, Masuda T, Ohba SI, Ikeda D (2013) Suppression of type II collagen-induced arthritis by ICM0301B, a new angiogenesis inhibitor. J Antibiot (Tokyo) 66(4):243–246
Tanaka K, Morii T, Weissbach L, Horiuchi K, Takeuchi K, Toyama Y, Morioka H (2011) Treatment of collagen-induced arthritis with recombinant plasminogen-related protein B: a novel inhibitor of angiogenesis. J Orthop Sci 16(4):443–450
Chang LH, Huang HS, Wu PT, Jou IM, Pan MH, Chang WC, Wang DD, Wang JM (2012) Role of macrophage CCAAT/enhancer binding protein delta in the pathogenesis of rheumatoid arthritis in collagen-induced arthritic mice. PLoS One 7(9):e45378
Roy A, Kolattukudy PE (2012) Monocyte chemotactic protein-induced protein (MCPIP) promotes inflammatory angiogenesis via sequential induction of oxidative stress, endoplasmic reticulum stress and autophagy. Cell Signal 24(11):2123–2131
Sheikh A, Naqvi SH, Sheikh K (2012) Itraconazole: its possible role in inhibiting angiogenesis in rheumatoid arthritis. Med Hypotheses 79(3):313–314
Mazzone M, Dettori D, Leite de Oliveira R, Loges S, Schmidt T, Jonckx B, Tian YM, Lanahan AA, Pollard P, Ruiz de Almodovar C, De Smet F, Vinckier S, Aragones J, Debackere K, Luttun A, Wyns S, Jordan B, Pisacane A, Gallez B, Lampugnani MG, Dejana E, Simons M, Ratcliffe P, Maxwell P, Carmeliet P (2009) Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 136(5):839–851
Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, Taylor CT (2008) The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 134(1):156–165
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Wien
About this chapter
Cite this chapter
Konisti, S., Kiriakidis, S., Paleolog, E.M. (2013). Angiogenesis in Rheumatoid Arthritis. In: Dulak, J., Józkowicz, A., Łoboda, A. (eds) Angiogenesis and Vascularisation. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1428-5_16
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1428-5_16
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1427-8
Online ISBN: 978-3-7091-1428-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)