
Abstract
Induction of protective immunity in infants has the potential to reduce morbidity and mortality in childhood infections but it has long been known that long-lasting protective immunity is difficult to induce in the neonate. This chapter describe the basis of the neonatal adaptive immune system. It also gives an update on current childhood immunisation programmes on a global level as well as the known side effects to vaccines in children and young adults. Finally, it also provides data on how to improve childhood vaccinations and future much needed vaccines.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The first months of life are a time of high risk for infections in the newborn infant, and it would therefore be useful to induce adaptive immunity by immunizations early in life. However, it has long been known that long-lasting protective immunity is difficult to induce in neonates both upon immunization and also after infection. Some of these problems could in theory be solved by vaccination of the pregnant women to enhance the passage of protective maternal antibodies to the child, but this may later affect the adaptive immune response of the infant. The difficulties to induce proper vaccine responses will be described and put into a context of the neonatal immune system that in many other aspects is capable of protecting the infant against disease [1, 2].
2 Cellular Components of Innate and Adaptive Immunity
A major disadvantage for the neonatal immune system is, independent of species, that most B and T cells are naïve. Although the majority of cells and soluble factors appear early in fetal life, their numbers and relative ratio and activation status differ from adults [3]. There is a lack of knowledge in human infants since the majority of studies in neonatal immunology have been generated in rodent models. It is difficult to translate these findings to humans since newborn mice are underdeveloped compared to human infants [4]. The studies of cells isolated from umbilical cord blood have provided new insight, but these data may not be representative of the circulating cells in the neonatal immune system. Data obtained from cord blood should therefore be combined with data acquired from infants [5].
3 Neonatal and Childhood Antigen-Presenting Cells
Monocytes and dendritic cells (DCs) function as antigen-presenting cells and are key players in innate immunity but also responsible for initiation of adaptive immune responses. Two subtypes of DCs are identified in peripheral blood: the myeloid DC (mDCs) and the plasmacytoid DC (pDCs). The mDC is the main antigen-presenting cell and plays a crucial role in B cell differentiation by the release of cytokines such as IL-12, IL-6, BAFF, and April, which drive the formation of antibody-producing B cells. On the other hand, pDCs produce interferons and thereby play a vital role in antiviral immunity [6]. There are age-associated differences in the proportion of DCs in peripheral blood in children, where pDC numbers are much higher in infants than in older children, probably reflecting their importance for protection against viral illness in early life before protective adaptive immune responses have been initiated [7]. However, that comes at the cost of less efficient B cell activation early in life.
Several studies on cord blood DCs have confirmed an immature phenotype with low, or no basal expression of CD40, co-stimulatory molecules CD80/CD86 or MHC class II molecules [8–10]. Functionally, this translates into suboptimal human neonatal DC responses to most stimuli [11]. In addition, the importance of the toll-like receptors (TLR) pathway on DCs for induction of adaptive immune responses is apparent and well established. Despite comparable expression of TLRs on cord blood DCs and adult cells, the capacity of cord blood DCs to respond to TLR agonists is also significantly reduced and characterized by low production of the pro-inflammatory Th1 cytokine TNFα and IFN [12, 13]. However, recent data suggests that neonatal TLR-mediated impairments are selective since the TLR8 agonist R848 is able to induce a robust immune response in cord blood DCs comparable to adult cells. This finding has also been confirmed in infant cells and may have important implications for the choice of adjuvants in neonatal vaccine research [14, 15].
The innate responses of monocytes and antigen-presenting cells develop within the first year of life [15, 16]. Phenotypic analysis of peripheral blood monocytes and DCs has shown that circulating DCs acquire an adult-like phenotype around 6 months of age. Cytokine production after TLR stimulation at birth is skewed towards a Th2 response with production of IL-6, IL-8, and IL-10 and low levels of the Th1-polarizing IL-12p70 cytokine [17]. However, the IL-6 levels are comparable to adults already at 3 month of age. For IL-10, the production remains significantly higher also at 12 month of age, and the same trend is shown for IL-8 [16].
Thus, small children are impaired in both the maturation of the antigen-presenting cells and the capacity of such cells to respond to bacterial and viral antigens, with the important exception of TLR8. Using TLR8 ligands as vaccine adjuvants may represent a window of opportunity.
4 Neonatal and Childhood T Cells
The peripheral blood T lymphocyte subsets in infants differ from that of adults. At birth, there is a gradual increase in the absolute number of CD3+ T lymphocytes and, from the age of 2 years, a decrease to levels similar to adults [18]. Helper CD4+ T cells and cytotoxic CD8+ T cells also increase in the first months of life and decline after 9–15 months of age [19]. Analysis by flow cytometry has revealed several CD4+ memory cell populations in blood, where only the central memory T cell population is present at birth. As a result of antigenic stimulation, effector memory helper T cells increase during the first year to levels comparable to adults and remain stable during childhood. The recently described population of CXCR5+ memory T cells, also defined as follicular helper T cells, is absent at birth but increase in number during the first year of life in parallel to the increase in serum IgA and IgG [19]. Follicular helper T cells were first described as cells able to efficiently support the differentiation of switched B cells in secondary lymphoid organs with the subsequent production of IgA and IgG [20]. The absolute numbers of regulatory T cells (Treg) increase the first month of life similar to CD4+ memory T cells and remain stable thereafter. Neonatal Treg exert potent immunosuppressive activities and suppress antigen-specific T cell proliferation and IFNγ production [21] which may modulate the development of a memory CD4+ T cell pool later in life [22].
Intrinsic defects in T cell immunity has been described for neonatal T cells. A key feature of signalling via T-cell receptor (TCR)-CD3 on naïve CD4+ T cells is the upregulation of CD40 ligand on the cell surface. Neonatal CD4+ T cells have reduced capacity to express CD40 ligand after TCR-CD3 activation, which in turn negatively affect antibody production, Ig switch, and memory B cell generation [23]. Helper T cell responses after immunizations in newborns have been investigated in many contexts [1], and several factors (antigen dose, adjuvant, routes of immunization) influence whether a predominantly Th1 or Th2 response will be elicited. A majority of current childhood vaccine elicit a predominantly Th2-biased response with the exception of BCG and whole-cell pertussis vaccines [24].
So, the T cell responses early in life are skewed towards a Th2 response, and neonatal helper T cells have reduced capacity to support B cell differentiation and antibody production.
5 Neonatal and Childhood B Cells
Several studies show age-dependant developmental changes in peripheral blood B cell subsets during the first 5 years of life with a significant decrease in total B cells with age. Most striking is the shift from a predominantly naïve and transitional blood B cell pool during infancy to an increase of the memory B cell fraction in the older child and adult. The transitional B cells are increased in infants compared to adults, which may bridge the gap between innate and adaptive immunity early in life. Transitional B cells produce IgM upon TLR 9 stimulation and thus may be an important mechanism for a first line defense against bacteria at birth [25]. The expansion of the memory B cell pool is most evident during the first year of life, where after the absolute number is stable over time. Taken together, these findings suggest that the decrease in total B cells with age is mainly related to a reduction in the output from the bone marrow (BM) of transitional and naïve cells [26, 27].
Using CD27 as a surrogate marker of human memory B cells together with the surface expression of IgD, several memory B cell populations have been characterized. Classical switched memory B cells increase during infancy and reaches a peak between 5 and 10 years of age [27, 28]. Differentiation of classical switched memory B cells occurs in the germinal center (GC) of secondary lymphoid organs; immunohistochemistry studies show that GCs are absent at birth and gradually develop to adult size between 12 and 24 months of life [29]. Interestingly, gut colonization of Escherichia coli promotes the early development (0–4 months) of the CD27+ memory pool in infants [30]. The IgM memory subset appears gradually in circulation from around birth and reaches adult levels at 2 years of age [31]. Several studies have shown that IgM memory B cells confer protection against Streptococcus pneumoniae, both after infection and immunization [32].
Less is known on the terminally differentiated plasma cell pool in infants and children. In preschool children, the plasma cell compartment is similar in size as reported for adults [33]. However, in the mouse model of KLH-NP immunization, it has been shown that survival of plasma cells is impaired in neonatal mice compared to adults [34]. The supportive network of BM stromal cells was less capable of supporting plasma cell survival factors in neonatal mice compared to adults.
In summary, postnatal maturation of the B cell compartment occurs in the presence of antigenic stimulation by microbes and also requires maturation of lymphoid organs and the bone marrow (Fig. 4.1).

Maturation of peripheral blood cell populations involved in adaptive immune responses occurs during the first 2 years in children. At birth, translational (orange) and naive B cells (dark orange) are most abundant, and the T cell compartment is dominated by naive T cells (dark green). After stimulation by environmental antigens, the memory B cell (pale orange), memory CD4 T cell (pale green), and follicular T cell (green) pool increases. Accordingly, the interactions between DCs, B cells, and T cells increase, and the antibody response matures with the production of IgG and IgA. Dendritic cells (DCs) at birth are predominantly plasmacytoid DCs (blue), but the myeloid DCs (pale blue) increase after 1 year
6 Quality of Antibody Responses in Infants
During fetal and early neonatal ontogeny, the peripheral B cell population is much less diverse that that of adults. Early studies showed that the B cell repertoire expressed early in life is skewed towards specific VH genes and that early neonatal cells lack molecular mechanisms utilized by adult cells for diversification [35]. It is also known that in vivo antibody (Ab) responses are of lower affinity and restricted heterogeneity compared to adults.
One important difference in outcome of B cell activation is that neonatal B cells produce less amounts of Abs than adults after antigen-specific activation [36]. The differences in Ab secretion could be due to impaired antigen presentation by DCs or macrophages as well as suboptimal secretion of cytokines by T cells. However, there are also intrinsic B cell differences in that neonatal B cells show little or no proliferation after B cell receptor (BCR) cross-linking [37] even though signal transduction occurs upon Ig ligation [38]. It has been demonstrated that neonatal B cells are more prone to tolerance induction and/or apoptosis after BCR ligation. Neonatal B cells also express less MHC class II and the co-stimulatory molecules CD80/CD86 are not upregulated after BCR triggering [39]. While these impairments may render neonatal B cells hyporesponsive, CD40 ligation and IL-4 stimulation leads to B cell activation and proliferation allowing B cell differentiation. Thus, in the presence of T helper mechanisms, the neonatal B cell response is adequate, although more stimulatory signals may be needed to achieve similar outcomes as for adult B cells [35].
Somatic hypermutation (SHM) occurs predominantly in germinal centers of spleen or lymph node and is essential to diversify and improve the antibody formation as it leads to selection of antibodies with high affinity [40]. This process is dependent on the enzyme activation-induced deaminase (AID), which inserts point mutations in to the Ig heavy and light chain genes and thus, play an essential role in repertoire diversification and affinity maturation [41]. Data on SHM in human infants are rare, but one early study reported SHM in IgG and IgA heavy chain transcripts in cord blood [42]. In peripheral blood of newborns, few or no mutations could be detected when sequencing the VH6 gene, but in older infants (10–60 days), more mutations were found in the same locus [43]. By 8 months of age, the range of mutations reached adults levels, and there were signs of repertoire selection [44].
So, neonatal B cells are less responsive to BCR ligation and more prone to apoptosis or tolerance induction. Antibody maturation is limited. The responses to the majority of antigens will be less efficient due to both T and B cell inabilities (Fig. 4.2). For type 1 T cell-independent antigens, which themselves can activate immature B cells, these are partially hampered by lack of TLR and BCR signalling (Fig. 4.2). For type 2 T cell-independent antigens, which are repetitive structures that can cross-link BCR by multiple binding on mature B cell responses are very limited in young children, both due to few mature B cells and also poor BCR function. Children hence respond poorly to vaccines consisting of such antigens (polysaccharide vaccines).

A schematic figure on adaptive immune responses in young children. At birth, translational B cells respond to certain T-independent antigens (TI-1) with the formation of short-lived plasma cells that mainly produce IgM as a first line defense. IgM+ mature B cells are also able to respond to T cell-independent antigens (TI-2) which cross-link several BCR through binding of repetitive antigenic structures. The response to T-dependent antigens is not present at birth but mature during the first 2 years. The production of switched memory B cells and homing of plasma cells to the bone marrow are therefore impaired in the infant
7 Soluble Factors in Neonatal Blood Affecting Adaptive Immune Responses
Although most components of the immune system appear during fetal development, the concentrations of soluble components can differ markedly from those of adults. In particular, the plasma complement proteins and their activity are low in infants. The complement system is an important part of innate immunity, but it may also impact on adaptive immune responses, it enhances the effects of specific immunoglobulins, it primes antigen-presenting cells and aid their maturation, and finally, it enhances the antigen-driven maturation of antibody responses by B lymphocytes. The level of classical complement components in newborns is decreased compared to adult levels, which probably contributes to the deficit in early adaptive immune responses [45]. During the first 6 months of life, there is an evolution towards adult levels for several of the complement proteins [46].
In recent years, neonatal plasma has been shown to contain other molecules with immune modulatory functions mainly affecting the outcome of TLR activation on antigen-presenting cells. Adenosine, an endogenous purine metabolite, selectively inhibits TNF production from TLR2-activated monocytes while IL-6 production is preserved. Thus, adenosine contributes to the Th2-polarizing properties of neonatal plasma [47]. In addition, yet unidentified factors in neonatal plasma have the capacity to polarize TLR4-mediated cytokine responses with low IL-12p70 production and high IL-10 production, thus mediating immunosuppression during the first month of life [48].
The influence of maternal antibodies on adaptive immune responses is debated. Potential mechanisms by which maternal antibodies could affect infant vaccine responses include specific masking of infant B cell epitopes by maternal antibodies and the uptake of maternal antibodies: antigen complexes by APC [49]. Abundant data in the literature favors these models, and it also fits well with the observation that maternal antibodies lack the capacity to interfere with infant T cell priming in vivo. This issue will be discussed more below.
8 Current Pediatric Vaccines, Worldwide
More than half of the children that die under the age of five worldwide do so because of an infectious disease. Many of these diseases are vaccine preventable, and WHO estimates that around 1.5 million children below the age of 5 died in 2008 in such diseases. S. pneumoniae (pneumococcal) infections and rotavirus infections are the leading causes, followed by infections caused by Haemophilus influenzae B (HIB), Bordetella pertussis (pertussis), measles virus, and Clostridium tetani infection in the neonatal period (neonatal tetanus).
The vaccine schedules used in the world differ due to economic issues and the endemic infection situation of the region. The goals from WHO are focused on reaching a 90 % vaccine coverage rate of each country’s national policy. In almost all parts of the world, these include vaccinations against diphtheria, tetanus, pertussis, and polio, which have lead to a drastic reduction in the incidence the past 30 years. Neonatal tetanus is still quite prevalent, mostly due to low vaccine coverage rates in mothers, hence less antibodies are transferred in utero. The MMR vaccine (measles, mumps, rubella) is used in Europe, America, Australia, and some Eastern Mediterranean countries, whereas the plain measles vaccine is used in most African countries and South East Asia. The measles vaccination coverage is 85 % worldwide; however, the coverage is poor in mid- and southern Africa. Furthermore, the introduction of the pneumococcus vaccine is recently established in Europe, North America, Australia, and many African and South American countries now follow. Vaccines against HIB are also becoming very prevalent worldwide, but only recently, thus less than half of the world population were protected in 2010, though this number is likely to rise. The introduction of the varicella vaccine has been less successful, and it is primarily used in America but also in other countries for risk groups.
In the coming years the rotavirus vaccines will be introduced with a focus on African countries, but also in America, Europe, and Eastern Mediterranean areas. Vaccines against local endemic infections including Neisseria meningitidis (meningococcus), Japanese encephalitis, Mycobacterium tuberculosis, hepatitis, rabies, Salmonella typhi (typhoid fever), and yellow fever may also be included in childhood vaccination schedules. In the older children, utilization of the human papillomavirus (HPV) vaccine, reducing cervical cancer, is increasing in all parts of the world, with the exception of the East Mediterranean countries.
9 Vaccine Side Effects
Side effects of vaccinations are a debated field. Immediate reactions, such as allergy [50] and local reactions, with swelling, sourness, and pain at the site of injection, are easily measured and described. In addition, common early systemic side effects including irritability and fever can be measured on a population basis. Less common side effects, occurring in close proximity to vaccination, are also described. For example, febrile seizures 7–10 days post MMR vaccination have long been recognized occurring in 1 of 600 doses [51]. Similarly, MMR vaccination associates with a 1/50,000 doses risk of immune thrombocytopenic purpura [52].
When it comes to rare and long-term side effects, the burden of proof is more challenging. Furthermore, such reactions/diseases are often multifactorial. There are basically two approaches to long-term side effect investigations: either a purely logical hypothesis based on immunological data from the vaccine or a suspicion from epidemiological data. One example of the first is the effects of childhood vaccination on allergy and atopy. It is known that children who retain the neonatal Th2 profile longer may have an increased likelihood of allergies and that an early Th1 tilted response will decrease incidence of allergy and asthma [17]. Pediatric vaccines that give a predominant Th1 or Th2 response may hence affect development of allergy and atopy. Circumstantial clinical reports suggest such an association for the pertussis vaccine, and controlled clinical trials point away from allergy promoting or preventing effects [50].
One example of the second type, where epidemiological data has prompted further investigation, comes from the increased frequency of childhood narcolepsy in Scandinavia post H1N1 influenza vaccinations in 2009. For the group under 11 years, the frequency rose from around 0 cases/100,000 inhabitants to around 3.4/100,000 in 2010. For 11–16 year olds, it rose from around 1/100,000 to around 8.7/100,000 in 2010 [53]. This occurred almost exclusively in Finland and Sweden and has been attributed to the genetic background and possibly other unknown factors. It has been suggested based on genetic data and the lowered onset age that vaccination brought forward the onset of a disease that normally would have occurred later [53]. In much the same way, the pertussis vaccine has been described as a trigger of severe myoclonic epilepsy (Dravet syndrome) in genetically susceptible individuals. Time for vaccination coincides, however, with characteristic onset time for this disease, and fever (which may be associated with vaccination) is known to trigger first events. Pertussis vaccination does not associate with any altered outcome of disease as compared to unvaccinated genetically susceptible individuals [54]. The proposed link between autism spectrum diseases and vaccination (in particular MMR) is hereto not proven [55, 56].
In parallel, data also suggest that you are more likely to have side effects if you are vaccinated early in a vaccine campaign. One interpretation of this is that people with underlying disease and/or risk factors are vaccinated early, and such individuals appear more prone to side effects. This was shown for the H1N1 vaccine, where the early cohorts had increased frequencies of Bell’s palsy, paresthesia, and inflammatory bowel disease [57].
Finally, it is not only the antigen as such that is important for the side effects but also the formulation. This has been demonstrated by comparing the seizure frequency when giving a combined MMR-varicella (MMRV) vaccination or MMR and varicella (MMR+V) vaccine separately on the same day. Using MMRV the seizure frequency rises to 1/2,300 doses, as compared to MMR+V where the seizure risk is comparable to MMR alone [58].
So when it comes to side effects causing long-term morbidity, these are linked to the onset of a disease that most likely would have occurred in an unvaccinated child as well. Children with underlying disease may be at higher risk of side effects. For the short-term side effects, formulation appears to play a role.
10 Vaccination During Pregnancy
Since vaccinations are less efficient in very young children, one way to protect neonates against severe infection is to vaccinate women during pregnancy, utilizing the transmission of antibodies from the mother to the child in utero. This strategy is already in use in some countries, and US health authorities recommend the seasonal trivalent inactivated influenza vaccine and the tetanus/diphtheria/acellular pertussis vaccine to be used during second and third trimester of pregnancy. Furthermore, a number of other killed vaccines are recommended to pregnant risk groups including vaccines against hepatitis A, hepatitis B, meningococcus, and the 23-valent pneumococcus polysaccharide vaccine. Live viral vaccines are contraindicated in pregnant women.
General concerns include the safety and efficacy of maternal-fetal immunization. Safety has been shown using killed vaccines against seasonal as well as H1N1 influenza [59]. Furthermore, similar data are available for the adult-type tetanus, reduced diphtheria toxoids, and acellular pertussis vaccine [60].
For the discussion of efficacy, several aspects have to be taken into consideration. First, whether fetal immunization is efficient in preventing fetal illness, either from the community or from the mother during fetal life or not. In addition, whether there will be negative effects of the passive antibodies transferred on later immunizations of the child or not. For the efficacy two types of vaccines will be discussed: the vaccines containing T-dependent antigens and the vaccines containing T-independent antigens. One can assume that T-dependent antigens will be more efficient, as they give rise to antibodies that are more efficiently enriched over the placenta. For T-dependent antigens, such as the influenza vaccine, epidemiological data show that children born to vaccinated mothers have milder influenza-like symptoms and a reduced incidence of verified influenza [61].
For T-independent antigens, the efficacy is less clear. It has been shown that pneumococcal antibodies are transferred in utero, at sufficient levels, post maternal immunization with the pneumococcal polysaccharide vaccine [62]. These are results for serotypes 1 and 5, where only 5 can be really seen as T-independent due to the zwitterionic nature of the serotype 1 polysaccharide [63]. Maternal immunization does in this case interfere with early childhood vaccination (7–17 weeks postpartum), in that vaccination does not increase the amount of specific antibodies, if already present at high concentration in the infant. No effect of maternal vaccination was seen on vaccination efficacy at 3 years of age [64]. There is little evidence for a role of these antibodies in preventing disease, and neonatal pneumococcal colonization is not affected [65].
One argument against fetal immunization is the possible interference with later childhood immunizations. This can be mediated both by the ability of neutralizing antibodies to interfere with specific T cell responses and the ability of neutralizing antibodies to interfere with humoral responses upon immunization. As for T cell responses, these are clearly less prominent, as discussed above, in small children as compared to in adults. The presence of maternal neutralizing antibodies does not affect efficacy of T cell responses, measured as IFN-γ production [49, 66].
When it comes to the humoral response, the situation is more complex. It is clear that lower levels of neutralizing antibodies are produced upon vaccination in children that are still retaining maternal antibodies at the age of 9 month [66] and that at 6 months, the ability to produce antibodies upon vaccination is poor in general irrespective of maternal antibodies. The potential interference with childhood immunization at 9 months will then depend of the persistence of maternal antibodies. Antibodies against different antigens show different half-life in vivo, and antibodies against measles virus and rubella virus persist longer than, for example, antibodies against mumps virus. Mumps antibodies also persist even shorter if the mother has been vaccinated as compared to infected naturally [67]. Furthermore, transport across the placenta will not depend only on subclass, as both IgG1 and IgG2 antibodies against pneumococcus are transferred less well than antibodies of similar subclasses against tetanus [68]. Clearly, the potential interference will have to be judged for each vaccine. Most children that were vaccinated while still immune from the mother, however, have a good response upon second dose vaccination [66]. Some studies have found a slightly higher vaccine failure rate upon second vaccination, where the first early vaccination has failed [69]. If this is a result of the child’s intrinsic ability to respond to vaccination or a result of interference by maternal antibodies is not known.
Vaccination with killed vaccines, preferentially in third trimester, appears safe and at least for some vaccines efficient. There are conflicting data on interference with childhood immunizations, but most data point towards little interference. Recommendations will have to be specific for each vaccine, and more studies are required.
11 Immunization Responses in Children with Primary or Acquired Immunodeficiency
A growing number of children survive infancy and early childhood despite severe immunodeficiency including transplantation and chemotherapy, primary inherited immunodeficiency disorders, or congenital HIV infection. These children are vulnerable to infections and thus would benefit from effective immunizations. It is also possible that a better understanding of the molecular deficits behind impaired vaccine responses in these patients could contribute to the development of better vaccines.
Common variable immunodeficiency disorder (CVID) affects antibody production and is characterized by low serum concentrations of IgG and IgA and/or IgM and increased susceptibility to respiratory infections with encapsulated bacteria (H. influenzae, S. pneumoniae). CVID is a heterogeneous disease with several genetic defects involving important molecules for B cell signalling and/or T-B cell interactions. In adult CVID patients, who had switched memory B cells (CD27+) in peripheral blood before immunization, a protective antibody response could be detected against several antigens [70]. Similarly, in a pediatric study, 11/16 children were found to respond to the meningococcal group C polysaccharide vaccine [71]. In addition, vaccine responses against polysaccharide vaccines were associated with the presence of IgM memory B cells in these patients. A similar study of CVID patients indicates that there is a block in the formation of plasmablasts after immunization against both Clostridium tetani and S. pneumoniae [72].
Immunization responses in HIV-1-infected patients are severely impaired, both in adults and children [73] for the majority of antigens. The introduction of highly active antiretroviral therapy (HAART) has improved vaccination outcome for the majority of patients [74]. Immunization guidelines for this vulnerable pediatric group have recently been published [75]. Vaccination is safe with few side effects, and the only vaccine that is contraindicated as of today is BCG for HIV-1-infected children. However, there are still unresolved questions regarding immunizations in pediatric HIV-1 infection and, in particular, how durable the antibody response in HAART-treated children will be compared to healthy individuals.
Re-immunization of children posttransplantation and after chemotherapy is required since the different treatment modalities eradicate protective antibody-mediated memory [76]. As for HIV-1-infected children, most vaccines are safe although revaccination with live vaccines should be postponed 12–24 months after completion of therapy. There is no consensus on the optimal time to start revaccination or how many doses that should be administered to achieve long-lasting protection [77, 78].
12 Development of New Pediatric Vaccines
Traditional vaccines often consist of whole-killed viruses, administered by intra muscular injection (i.m.). For some important childhood pathogens, for example, respiratory syncytial virus (RSV), such attempts have failed. The development of a RSV vaccine will be discussed as an example of a novel vaccine strategy in infants. Initial clinical studies using formalin-inactivated RSV for administration to small children resulted in disease aggravation upon infection, hospitalization, and in some cases death [79]. This has later been attributed to a devastating Th2 response, resulting in lung pathology. RSV causes a localized respiratory disease, without general viremia, resulting in significant hospitalization, morbidity, and mortality rates. From an immunological perspective, RSV is challenging because of the failure of adaptive immunity to prevent reinfection.
This has been ascribed to the poor quality of the T cell response and the short durability of the antibody response. One hypothesis is that the mucosal immunity is to slow for this rapid virus and that the serum antibodies are present at low levels in the tissue. Finding alternative strategies for RSV vaccination is key. One such way is to deliver the vaccine at mucosal linings such as intranasally (i.n.), instead of i.m. Then immune activation will take place within the mucosa-associated lymphoid tissue (MALT) with activation of microfold epithelial cells (M cells) and subsequently underlying antigen-presenting cells such as DCs. This will induce both local IgA and systemic IgG.
Depending on which route that is used, different local immunity will result. For example, using i.n. administration, immunity in the upper respiratory tract and the cervicovaginal tract will prevail, whereas oral administration will induce IgA production mostly in the small intestine and in the mammary glands. Experiments using live-killed RSV nanoemulsions for i.n. administration in mice have shown an IgA response in the lung as well as protective capacity [80]. Current most promising results come from clinical phase II trials where live attenuated RSV strains have been given i.n [79]. Still, RSV vaccine development suffers from the initial failures, and no good vaccine is yet available. Results are to be awaited from clinical studies of naked DNA and vector-expressed DNA vaccines in the pediatric population.
Development of new vaccines also includes the introduction of adjuvants with enhanced immune stimulatory capacity to compensate for low intrinsic immunogenicity of antigens [81]. Novel adjuvants aim at optimizing B cell responses and generating appropriate T cell responses, which could be of particular importance in childhood vaccines. One of new adjuvants is the TLR4 agonist monophosphoryl lipid A (MPL) which of to date is licensed or in phase III clinical trials [82]. MPL, when combined with alum, acts on DCs and promotes IFN-γ production and thus overcomes the Th2-bias response associated with alum. It is now licensed in a human papillomavirus (HPV)-16/18 vaccine and has been shown to induce long-lasting B cell memory and persistent antibodies [83]. In children immunized at 1–4 years of age against Plasmodium falciparum, an MPL-containing adjuvant-induced high anti-parasite antibodies with long-term protection against clinical disease as a result [84]. An additional potential new adjuvant, the non-toxic mutant of heat-labile enterotoxin (LT) of E. coli (LTK63), was shown to overcome delayed maturation of follicular DCs and thus induce germinal centers when given parentally in mice together with a polysaccharide conjugate vaccine [85]. In addition to an improved B cell response, LTK63 upon binding to macrophages, induces a balanced Th1/Th2 cytokine production as well as cytotoxic T cell responses [86].
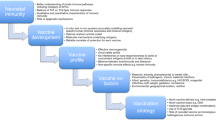
So, the development of new efficient vaccines to be administered early in life may take advantage of novel adjuvants to circumvent the hyporesponsiveness of neonatal adaptive immunity. Finally, a summary of the key points outlined in this chapter is shown in Fig. 4.3.

The key points of this chapter are summarized in a text box
References
Wood, N., Siegrist, C.A.: Neonatal immunization: where do we stand? Curr. Opin. Infect. Dis. 24, 190–195 (2011). doi:10.1097/QCO.0b013e328345d563
Adkins, B., Leclerc, C., Marshall-Clarke, S.: Neonatal adaptive immunity comes of age. Nat. Rev. Immunol. 4, 553–564 (2004). doi:10.1038/nri1394
Ygberg, S., Nilsson, A.: The developing immune system - from foetus to toddler. Acta Paediatr. 101, 120–127 (2012). doi:10.1111/j.1651-2227.2011.02494.x
Siegrist, C.A.: Neonatal and early life vaccinology. Vaccine 19, 3331–3346 (2001)
Hodgins, D.C., Shewen, P.E.: Vaccination of neonates: problem and issues. Vaccine 30, 1541–1559 (2012). doi:10.1016/j.vaccine.2011.12.047
Ueno, H., et al.: Dendritic cell subsets in health and disease. Immunol. Rev. 219, 118–142 (2007). doi:10.1111/j.1600-065X.2007.00551.x
Teig, N., Moses, D., Gieseler, S., Schauer, U.: Age-related changes in human blood dendritic cell subpopulations. Scand. J. Immunol. 55, 453–457 (2002)
Sorg, R.V., Kogler, G., Wernet, P.: Identification of cord blood dendritic cells as an immature CD11c- population. Blood 93, 2302–2307 (1999)
Jones, C.A., Holloway, J.A., Warner, J.O.: Fetal immune responsiveness and routes of allergic sensitization. Pediatr. Allergy Immunol. 13(Suppl 15), 19–22 (2002)
Liu, E., Tu, W., Law, H.K., Lau, Y.L.: Decreased yield, phenotypic expression and function of immature monocyte-derived dendritic cells in cord blood. Br. J. Haematol. 113, 240–246 (2001)
Langrish, C.L., Buddle, J.C., Thrasher, A.J., Goldblatt, D.: Neonatal dendritic cells are intrinsically biased against Th-1 immune responses. Clin. Exp. Immunol. 128, 118–123 (2002)
De Wit, D., et al.: Impaired responses to toll-like receptor 4 and toll-like receptor 3 ligands in human cord blood. J. Autoimmun. 21, 277–281 (2003)
De Wit, D., et al.: Blood plasmacytoid dendritic cell responses to CpG oligodeoxynucleotides are impaired in human newborns. Blood 103, 1030–1032 (2004). doi:10.1182/blood-2003-04-1216
Levy, O., et al.: Selective impairment of TLR-mediated innate immunity in human newborns: neonatal blood plasma reduces monocyte TNF-alpha induction by bacterial lipopeptides, lipopolysaccharide, and imiquimod, but preserves the response to R-848. J. Immunol. 173, 4627–4634 (2004)
Burl, S., et al.: Age-dependent maturation of Toll-like receptor-mediated cytokine responses in Gambian infants. PLoS One 6, e18185 (2011). doi:10.1371/journal.pone.0018185
Nguyen, M., et al.: Acquisition of adult-like TLR4 and TLR9 responses during the first year of life. PLoS One 5, e10407 (2010). doi:10.1371/journal.pone.0010407
Belderbos, M.E., et al.: Skewed pattern of Toll-like receptor 4-mediated cytokine production in human neonatal blood: low LPS-induced IL-12p70 and high IL-10 persist throughout the first month of life. Clin. Immunol. 133, 228–237 (2009). doi:10.1016/j.clim.2009.07.003
de Vries, E., et al.: Longitudinal survey of lymphocyte subpopulations in the first year of life. Pediatr. Res. 47, 528–537 (2000)
Schatorje, E.J., et al.: Pediatric reference values for the peripheral T-cell compartment. Scand. J. Immunol. (2011). doi:10.1111/j.1365-3083.2011.02671.x
Schaerli, P., et al.: CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J. Exp. Med. 192, 1553–1562 (2000)
Wing, K., et al.: CD4+ CD25+ FOXP3+ regulatory T cells from human thymus and cord blood suppress antigen-specific T cell responses. Immunology 115, 516–525 (2005). doi:10.1111/j.1365-2567.2005.02186.x
Rabe, H., et al.: Higher proportions of circulating FOXP3+ and CTLA-4+ regulatory T cells are associated with lower fractions of memory CD4+ T cells in infants. J. Leukoc. Biol. 90, 1133–1140 (2011). doi:10.1189/jlb.0511244
Jullien, P., et al.: Decreased CD154 expression by neonatal CD4+ T cells is due to limitations in both proximal and distal events of T cell activation. Int. Immunol. 15, 1461–1472 (2003)
Fadel, S., Sarzotti, M.: Cellular immune responses in neonates. Int. Rev. Immunol. 19, 173–193 (2000)
Capolunghi, F., et al.: CpG drives human transitional B cells to terminal differentiation and production of natural antibodies. J. Immunol. 180, 800–808 (2008)
Morbach, H., Eichhorn, E.M., Liese, J.G., Girschick, H.J.: Reference values for B cell subpopulations from infancy to adulthood. Clin. Exp. Immunol. 162, 271–279 (2010). doi:10.1111/j.1365-2249.2010.04206.x
Smet, J., Mascart, F., Schandene, L.: Are the reference values of B cell subpopulations used in adults for classification of common variable immunodeficiencies appropriate for children? Clin. Immunol. 138, 266–273 (2011). doi:10.1016/j.clim.2010.12.001
Huck, K., et al.: Memory B-cells in healthy and antibody-deficient children. Clin. Immunol. 131, 50–59 (2009). doi:10.1016/j.clim.2008.11.008
Kruschinski, C., Zidan, M., Debertin, A.S., von Horsten, S., Pabst, R.: Age-dependent development of the splenic marginal zone in human infants is associated with different causes of death. Hum. Pathol. 35, 113–121 (2004)
Lundell, A.C., et al.: Infant B cell memory differentiation and early gut bacterial colonization. J. Immunol. 188, 4315–4322 (2012). doi:10.4049/jimmunol.1103223
Weller, S., et al.: Human blood IgM “memory” B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood 104, 3647–3654 (2004). doi:10.1182/blood-2004-01-0346
Kruetzmann, S., et al.: Human immunoglobulin M memory B cells controlling Streptococcus pneumoniae infections are generated in the spleen. J. Exp. Med. 197, 939–945 (2003). doi:10.1084/jem.20022020
Nilsson, A., et al.: Current chemotherapy protocols for childhood acute lymphoblastic leukemia induce loss of humoral immunity to viral vaccination antigens. Pediatrics 109, e91 (2002)
Pihlgren, M., et al.: Reduced ability of neonatal and early-life bone marrow stromal cells to support plasmablast survival. J. Immunol. 176, 165–172 (2006)
Press, J.L.: Neonatal immunity and somatic mutation. Int. Rev. Immunol. 19, 265–287 (2000)
Siegrist, C.A.: The challenges of vaccine responses in early life: selected examples. J. Comp. Pathol. 137(Suppl 1), S4–S9 (2007). doi:10.1016/j.jcpa.2007.04.004
Tasker, L., Marshall-Clarke, S.: Immature B cells from neonatal mice show a selective inability to up-regulate MHC class II expression in response to antigen receptor ligation. Int. Immunol. 9, 475–484 (1997)
Chang, T.L., Capraro, G., Kleinman, R.E., Abbas, A.K.: Anergy in immature B lymphocytes. Differential responses to receptor-mediated stimulation and T helper cells. J. Immunol. 147, 750–756 (1991)
Marshall-Clarke, S., Reen, D., Tasker, L., Hassan, J.: Neonatal immunity: how well has it grown up? Immunol. Today 21, 35–41 (2000)
McHeyzer-Williams, L.J., McHeyzer-Williams, M.G.: Antigen-specific memory B cell development. Annu. Rev. Immunol. 23, 487–513 (2005). doi:10.1146/annurev.immunol.23.021704.115732
Pan-Hammarstrom, Q., Zhao, Y., Hammarstrom, L.: Class switch recombination: a comparison between mouse and human. Adv. Immunol. 93, 1–61 (2007). doi:10.1016/S0065-2776(06)93001-6
Mortari, F., Wang, J.Y., Schroeder Jr., H.W.: Human cord blood antibody repertoire. Mixed population of VH gene segments and CDR3 distribution in the expressed C alpha and C gamma repertoires. J. Immunol. 150, 1348–1357 (1993)
Ridings, J., et al.: Somatic hypermutation of immunoglobulin genes in human neonates. Clin. Exp. Immunol. 108, 366–374 (1997)
Ridings, J., Dinan, L., Williams, R., Roberton, D., Zola, H.: Somatic mutation of immunoglobulin V(H)6 genes in human infants. Clin. Exp. Immunol. 114, 33–39 (1998)
McGreal, E.P., Hearne, K., Spiller, O.B.: Off to a slow start: under-development of the complement system in term newborns is more substantial following premature birth. Immunobiology 217, 176–186 (2012). doi:10.1016/j.imbio.2011.07.027
Davis, C.A., Vallota, E.H., Forristal, J.: Serum complement levels in infancy: age related changes. Pediatr. Res. 13, 1043–1046 (1979)
Levy, O., et al.: The adenosine system selectively inhibits TLR-mediated TNF-alpha production in the human newborn. J. Immunol. 177, 1956–1966 (2006)
Belderbos, M.E., et al.: Neonatal plasma polarizes TLR4-mediated cytokine responses towards low IL-12p70 and high IL-10 production via distinct factors. PLoS One 7, e33419 (2012). doi:10.1371/journal.pone.0033419
Siegrist, C.A.: Mechanisms by which maternal antibodies influence infant vaccine responses: review of hypotheses and definition of main determinants. Vaccine 21, 3406–3412 (2003)
Gruber, C., Nilsson, L., Bjorksten, B.: Do early childhood immunizations influence the development of atopy and do they cause allergic reactions? Pediatr. Allergy Immunol. 12, 296–311 (2001)
Klein, N.P., et al.: Measles-mumps-rubella-varicella combination vaccine and the risk of febrile seizures. Pediatrics 126, e1–e8 (2010). doi:10.1542/peds.2010-0665
O’Leary, S.T., et al.: The risk of immune thrombocytopenic purpura after vaccination in children and adolescents. Pediatrics 129, 248–255 (2012). doi:10.1542/peds.2011-1111
Partinen, M., et al.: Increased incidence and clinical picture of childhood narcolepsy following the 2009 H1N1 pandemic vaccination campaign in Finland. PLoS One 7, e33723 (2012). doi:10.1371/journal.pone.0033723
McIntosh, A.M., et al.: Effects of vaccination on onset and outcome of Dravet syndrome: a retrospective study. Lancet Neurol. 9, 592–598 (2010). doi:10.1016/S1474-4422(10)70107-1
Uno, Y., Uchiyama, T., Kurosawa, M., Aleksic, B., Ozaki, N.: The combined measles, mumps, and rubella vaccines and the total number of vaccines are not associated with development of autism spectrum disorder: the first case–control study in Asia. Vaccine 30, 4292–4298 (2012). doi:10.1016/j.vaccine.2012.01.093
Demicheli, V., Rivetti, A., Debalini, M.G., Di Pietrantonj, C.: Vaccines for measles, mumps and rubella in children. Cochrane Database Syst. Rev. 2, CD004407 (2012). doi:10.1002/14651858.CD004407.pub3
Bardage, C., et al.: Neurological and autoimmune disorders after vaccination against pandemic influenza A (H1N1) with a monovalent adjuvanted vaccine: population based cohort study in Stockholm, Sweden. BMJ 343, d5956 (2011). doi:10.1136/bmj.d5956
Klein, N.P., et al.: Measles-containing vaccines and febrile seizures in children age 4 to 6 years. Pediatrics 129, 809–814 (2012). doi:10.1542/peds.2011-3198
Oppermann, M., et al.: A(H1N1)v2009: a controlled observational prospective cohort study on vaccine safety in pregnancy. Vaccine 30, 4445–4452 (2012). doi:10.1016/j.vaccine.2012.04.081
Fortner, K.B., Kuller, J.A., Rhee, E.J., Edwards, K.M.: Influenza and tetanus, diphtheria, and acellular pertussis vaccinations during pregnancy. Obstet. Gynecol. Surv. 67, 251–257 (2012). doi:10.1097/OGX.0b013e3182524cee
Eick, A.A., et al.: Maternal influenza vaccination and effect on influenza virus infection in young infants. Arch. Pediatr. Adolesc. Med. 165, 104–111 (2011). doi:10.1001/archpediatrics.2010.192
Quiambao, B.P., et al.: Immunogenicity and reactogenicity of 23-valent pneumococcal polysaccharide vaccine among pregnant Filipino women and placental transfer of antibodies. Vaccine 25, 4470–4477 (2007). doi:10.1016/j.vaccine.2007.03.021
Groneck, L., et al.: Oligoclonal CD4+ T cells promote host memory immune responses to Zwitterionic polysaccharide of Streptococcus pneumoniae. Infect. Immun. 77, 3705–3712 (2009). doi:10.1128/IAI.01492-08
Holmlund, E., Nohynek, H., Quiambao, B., Ollgren, J., Kayhty, H.: Mother-infant vaccination with pneumococcal polysaccharide vaccine: persistence of maternal antibodies and responses of infants to vaccination. Vaccine 29, 4565–4575 (2011). doi:10.1016/j.vaccine.2011.04.068
Lopes, C.R., et al.: Ineffectiveness for infants of immunization of mothers with pneumococcal capsular polysaccharide vaccine during pregnancy. Braz. J. Infect. Dis. 13, 104–106 (2009)
Gans, H., et al.: Measles and mumps vaccination as a model to investigate the developing immune system: passive and active immunity during the first year of life. Vaccine 21, 3398–3405 (2003)
Leuridan, E., Goeyvaerts, N., Hens, N., Hutse, V., Van Damme, P.: Maternal mumps antibodies in a cohort of children up to the age of 1 year. Eur. J. Pediatr. 171, 1167–1173 (2012). doi:10.1007/s00431-012-1691-y
Shahid, N.S., et al.: Serum, breast milk, and infant antibody after maternal immunisation with pneumococcal vaccine. Lancet 346, 1252–1257 (1995)
Redd, S.C., et al.: Comparison of vaccination with measles-mumps-rubella vaccine at 9, 12, and 15 months of age. J. Infect. Dis. 189(Suppl 1), S116–S122 (2004). doi:10.1086/378691
Goldacker, S., et al.: Active vaccination in patients with common variable immunodeficiency (CVID). Clin. Immunol. 124, 294–303 (2007). doi:10.1016/j.clim.2007.04.011
Rezaei, N., et al.: Serum bactericidal antibody response to serogroup C polysaccharide meningococcal vaccination in children with primary antibody deficiencies. Vaccine 25, 5308–5314 (2007). doi:10.1016/j.vaccine.2007.05.021
Chovancova, Z., Vlkova, M., Litzman, J., Lokaj, J., Thon, V.: Antibody forming cells and plasmablasts in peripheral blood in CVID patients after vaccination. Vaccine 29, 4142–4150 (2011). doi:10.1016/j.vaccine.2011.03.087
Cagigi, A., Nilsson, A., Pensieroso, S., Chiodi, F.: Dysfunctional B-cell responses during HIV-1 infection: implication for influenza vaccination and highly active antiretroviral therapy. Lancet Infect. Dis. 10, 499–503 (2010). doi:10.1016/S1473-3099(10)70117-1
Sutcliffe, C.G., Moss, W.J.: Do children infected with HIV receiving HAART need to be revaccinated? Lancet Infect. Dis. 10, 630–642 (2010). doi:10.1016/S1473-3099(10)70116-X
Menson, E. N., et al.: Guidance on vaccination of HIV-infected children in Europe. HIV Med. 13, 333–336; e331–e314 (2012). doi:10.1111/j.1468-1293.2011.00982.x
Patel, S. R., Chisholm, J. C., Heath, P. T.: Vaccinations in children treated with standard-dose cancer therapy or hematopoietic stem cell transplantation. Pediatr. Clin. North. Am. 55, 169–186, xi (2008). doi:10.1016/j.pcl.2007.10.012
Brodtman, D.H., Rosenthal, D.W., Redner, A., Lanzkowsky, P., Bonagura, V.R.: Immunodeficiency in children with acute lymphoblastic leukemia after completion of modern aggressive chemotherapeutic regimens. J. Pediatr. 146, 654–661 (2005). doi:10.1016/j.jpeds.2004.12.043
Lehrnbecher, T., et al.: Revaccination of children after completion of standard chemotherapy for acute lymphoblastic leukaemia: a pilot study comparing different schedules. Br. J. Haematol. 152, 754–757 (2011). doi:10.1111/j.1365-2141.2010.08522.x
Graham, B.S.: Biological challenges and technological opportunities for respiratory syncytial virus vaccine development. Immunol. Rev. 239, 149–166 (2011). doi:10.1111/j.1600-065X.2010.00972.x
Lindell, D.M., et al.: A novel inactivated intranasal respiratory syncytial virus vaccine promotes viral clearance without Th2 associated vaccine-enhanced disease. PLoS One 6, e21823 (2011). doi:10.1371/journal.pone.0021823
Mastelic, B., et al.: Mode of action of adjuvants: implications for vaccine safety and design. Biologicals 38, 594–601 (2010). doi:10.1016/j.biologicals.2010.06.002
Garcon, N., Segal, L., Tavares, F., Van Mechelen, M.: The safety evaluation of adjuvants during vaccine development: the AS04 experience. Vaccine 29, 4453–4459 (2011). doi:10.1016/j.vaccine.2011.04.046
Giannini, S.L., et al.: Enhanced humoral and memory B cellular immunity using HPV16/18 L1 VLP vaccine formulated with the MPL/aluminium salt combination (AS04) compared to aluminium salt only. Vaccine 24, 5937–5949 (2006). doi:10.1016/j.vaccine.2006.06.005
Sacarlal, J., et al.: Long-term safety and efficacy of the RTS, S/AS02A malaria vaccine in Mozambican children. J. Infect. Dis. 200, 329–336 (2009). doi:10.1086/600119
Bjarnarson, S.P., Adarna, B.C., Benonisson, H., Del Giudice, G., Jonsdottir, I.: The adjuvant LT-K63 can restore delayed maturation of follicular dendritic cells and poor persistence of both protein- and polysaccharide-specific antibody-secreting cells in neonatal mice. J. Immunol. 189, 1265–1273 (2012). doi:10.4049/jimmunol.1200761
da Hora, V.P., Conceicao, F.R., Dellagostin, O.A., Doolan, D.L.: Non-toxic derivatives of LT as potent adjuvants. Vaccine 29, 1538–1544 (2011). doi:10.1016/j.vaccine.2010.11.091
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Wien
About this chapter
Cite this chapter
Ygberg, S., Nilsson, A. (2013). Pediatric Immunology and Vaccinology. In: Giese, M. (eds) Molecular Vaccines. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1419-3_4
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1419-3_4
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1418-6
Online ISBN: 978-3-7091-1419-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)