
Abstract
The unrivaled potential of T cells for targeted immune function is central to the eradication of cancer. While their natural anti-tumor response might sometimes be insufficient, several studies and importantly, multiple clinical trials in terminally-ill cancer patients have demonstrated that it is possible to design novel and efficient immunotherapeutic approaches based on the adoptive transfer of autologous tumor-specific T lymphocytes. Herein, we will expand on the development and the use of such strategies using tumor-infiltrating lymphocytes or genetically-engineered T cells. We will also comment on the requirements and potential hurdles encountered when elaborating and implementing such treatments as well as the exciting prospects for this kind of emerging personalized medicine therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Chimeric Antigen Receptor
- Adoptive Cell Therapy
- Functional Avidity
- Chimeric Immune Receptor
- Autologous TILs
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
9.1 Tumor Antigens: Defining the Target
T cells play a central role in the immune response against cancer. Their activation is initiated by the interaction of the T-cell receptor (TCR) with its cognate MHC-peptide complex presented on the surface of the target cell, which activates them specifically [1]. Whether T cells could recognize endogenous tissues was a matter of debate during several decades, especially as T cells are supposed to be tolerant to self-antigens. Nevertheless, molecular and immunological advances in the 1990s led to the discovery of self-originated proteins that could be recognized by T lymphocytes [2]. Accordingly, tumor-specific T cells have been shown to be activated through the binding of their TCR to specific epitopes derived from tumor antigens (TA) presented by a major histocompatibility complex (MHC) molecule [3]. TA are present on some tumor cells but also on normal tissues (in this case, they are termed tumor-associated antigens—TAA), and were shown to represent effective targets for T-cell-based cancer immunotherapy. They can be classified into several categories; this division pertains to the pattern of expression of these antigens (e.g., over-expressed, oncofetal, …) and whether these antigens are “self” or mutated [4]. Several sources indicate different classifications, but five known classes of TA can be broadly described:
-
Cancer/testis antigens (C/T)—they are expressed in various human cancers, but also in normal testis tissues. Some evidences suggest that there may be some level of T-cell tolerance toward these antigens [5].
-
Tissue-specific differentiation antigens—these antigens are typically expressed only by the tumor and its tissue of origin. Known examples of tissue-specific differentiation antigens include the MART-1/Melan-A and gp100, which are expressed in both melanocytes and melanoma cells. These antigens have emerged as very promising target antigens for T-cell-based adoptive immunotherapy, but their presence on normal tissues can be the source of auto-immune manifestations.
-
Mutated self-proteins—usually when mutations occur in the initial cancerous cell (or one of its early daughter cells), this class of tumor antigens can potentially provide targets for T-cell-based immunotherapy of cancer, as they are to be expressed in most of the tumor tissues.
-
Over-expressed antigens—this type of antigens constitutes also an important TA class, which is relevant in both T-cell therapy and antibody-based treatments. Based on clinical data, it seems that their over-expression in several tumor tissues (e.g., Her2/neu) but then again their reduced levels in healthy cells may limit the potential for deleterious autoimmune side-effects [4].
-
Viral antigens—as it is believed that around 20 % of all cancer cases are linked to infectious agents [6], antigens derived from oncogenic viruses would provide a source of “non-self” targets, which would be recognized more efficiently than TAA due to a potential lack of tolerance against the viral epitopes.
9.2 Tumor-Infiltrating Lymphocytes
9.2.1 Presence of Intra-tumoral T Lymphocytes
For several decades, it has been demonstrated that tumor-specific T cells can massively migrate into tumor sites. Some of these tumor-infiltrating lymphocytes (TILs) have thus the ability to specifically recognize tumor antigens expressed on the surface of tumor cells, and may greatly influence directly or indirectly the anti-tumor immune responses and the progression of a variety of solid tumors [7]. The presence of TILs in the tumor vicinity, and the nature of their interactions with target cells, contribute to determine whether a tumor is destroyed or grows unimpeded. It may also correlate with responses to chemotherapy/radiotherapy and disease prognosis. Indeed, high densities of CD3+ T cells, CD8+ cytotoxic T cells, and memory T cells into tumor sites could represent a reliable prognostic factor for the disease-free and overall survival of patients with different tumor types, such as melanoma, and head and neck, breast, bladder, urothelial, ovarian, colorectal, renal, prostatic, and lung cancer [8]. In contrast to the effects of cytotoxic T cells and memory T cells that are associated with a positive clinical outcome, the impact of CD4+ T cell infiltration on survival and prognosis is unclear; for example, there are conflicting data about the role of regulatory T-cells (Tregs) in this context, and their effects on tumor progression have been a matter of debate for the past decade [7, 9]. Moreover, there is a great variability in the density and location of these infiltrating T cells between different patients bearing the same type of cancer [7].
9.2.2 Adoptive TIL Immunotherapy
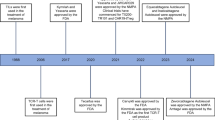
Nonetheless, to harness the potential benefit of tumor-specific T cells in cancer treatment settings, pioneering therapeutic approaches (Fig. 9.1) were developed in the last three decades [10]. Adoptive immunotherapy using autologous TILs has become an appealing strategy for the treatment of mainly melanoma and renal cell carcinoma. This necessitated the development of techniques and systems to grow large numbers of anti-tumor lymphocytes. An important milestone in the development of this kind of immunotherapy occurred in 1987 when tumor-infiltrating lymphocytes from patients with metastatic malignant melanoma were successfully cultured and expanded using the T-cell growth factor interleukin 2 (IL-2) [11]. During this expansion process performed ex vivo, fragments from resected tumors were grown in culture vessels in conditions that favor T-cell growth (using for example high concentrations of IL-2). Tumor-specific T-cell populations can be identified on the basis of their reactivity with MHC-matched tumor cell lines or the autologous tumor. Reactive cultures can be then selected and expanded, and adoptively infused back into cancer patients. Furthermore, to facilitate the engraftment of this autologous T-cell transplant, patients receive high-dose intravenous bolus IL-2 [12, 13]. As exemplified in several studies, the transfer of these cells back into the patient led to dramatic partial or complete clinical responses and durable regression [14, 15].

A summary of different adoptive T-cell therapy approaches. OR objective response, PBLs peripheral blood lymphocytes
The adoptive transfer of TILs is one of the most effective treatments for patients with stage IV melanoma. The first study aimed at directly targeting human tumor using autologous TILs to treat patients with metastatic melanoma was reported in 1988 by Rosenberg et al. at the National Cancer Institute [16], and a significant improvement in the response rate and durability of response was steadily reported in subsequent studies [15]. This improvement occurred when bulk cultures (CD8+ and CD4+) were transferred and more importantly, when a non-myeloablative conditioning regimen (depleting chemotherapy or whole-body irradiation) was administered to the patient prior to T-cell transfer [12]. In that regard, studies reported a significant correlation between the intensity of lymphodepletion and the in-vivo anti-tumor effect of the infused cells [17]. It has been suggested that the positive impact of lymphodepletion prior to TIL transfer is based in part on the elimination of suppressive CD4+CD25+ Tregs as well as of normal endogenous lymphocytes that could compete with the transferred cells for homeostatic cytokines such as IL-7 and IL-15 [18, 19].
Recent results indicate that the objective clinical response observed in patients with metastatic melanoma that were treated with adoptively transferred autologous TILs ranges between 49 % and 72 % [15]. Importantly, objective response was highly associated with the persistence of the transferred cells [20]. Indeed, many patients in the recently reported trials display high levels of persistence, sometimes reaching up to 75 % of all of the circulating CD8+ T cells. Still, it appears that persistence alone was not a sufficient requirement for an effective response [20, 21]. Studies have also shown that the state of differentiation of the transferred cells may be inversely correlated to the effectiveness of these cells in adoptive cell therapy (ACT) settings, and to their capacity to proliferate and persist [12, 22]. In other words, early effector T cells seem to mediate better anti-tumor response than intermediate and late effector T cells.
9.2.3 Tumor Microenvironment and Potential Hurdles
Solid tumors contain many other cell types, including cells derived from the innate and adoptive immune system, stromal cells, and myeloid-derived suppressor cells (MDSCs) [23, 24]. The latter are endowed with potent immunosuppressive properties, and their intratumoral presence at a high frequency correlates with a poor prognosis in patients with different tumor types. Recent findings indicate that targeting these cells, and the supportive environment (for the tumor) they promote, might represent an effective approach to promoting the destruction of cancer cells, leading to tumor elimination [25].
Despite its aforementioned success (especially in melanoma), adoptive cell transfer (ACT) therapy with autologous TILs bears some limitations which include, for example, the requirement to isolate and expand T cells with anti-tumor activity. Even if such cells are generated, adoptive T-cell therapy for some tumors will not necessarily be effective, as these may be poorly antigenic. Other tumors, such as colon and breast tumors, are infiltrated by T cells, but the specificities and functions of the latter are unclear [26, 27]. In this regard, a potential explanation as to why melanoma has been widely studied as a target for therapeutic TILs is that this type of cancer appears to be unique among human cancers because of its ability to promote elevated numbers of lymphocytes with anti-tumor activity. This might be due to the fact that melanoma tumors express a high number of mutated antigens that could help in breaking self-tolerance and were also shown to harbor class II-MHC molecules [10, 28]. Renal cell carcinoma (RCC) is also considered an immunogenic tumor that exhibits rich intra-tumoral lymphocytic infiltration. Still, it seems that T-cell activation is insufficient at the tumor site due to many immunosuppressive mechanisms induced in the microenvironment of RCC [29–32]. This may provide an explanation as to why previous clinical trials with TILs in RCC did not yield substantial benefit compared to melanoma. Nevertheless, current knowledge and experience with TIL generation from—and treatment of—melanoma patients could provide clues to elaborate an improved therapeutic regiment for ACT in RCC and other malignancies [33, 34].
9.2.4 TIL Treatment: Current Status and Future Promises
By utilizing current techniques today, tumor-infiltrating lymphocytes can be detected in approximately 80 % of melanoma patients [35]. However, in most cancer patients, those naturally-occurring TILs fail to destroy the tumor as they are outnumbered, subjected to constant immunosuppression, and due to other factors that are not fully understood. Additionally, the generation of a TIL culture(s) that prove reactive for each patient tumor is not always feasible and requires several weeks. The latter might be overcome, as exemplified in new clinical studies designed to improve the TIL anti-tumor activity, growth, and expansion by generating “young TIL” cultures [36, 37]. In this method, tumor-infiltrating lymphocytes are grown and expanded briefly (around 2–3 weeks compared to 4–6 in the conventional TIL protocol) and are introduced back into patients without testing for selection. Thus, the “young TIL” protocol utilizes bulk unselected TIL which spend minimal time in culture by eliminating the individualized tumor reactivity screening step [38]. As no further selection process is required, all established “young TIL” cultures are technically eligible for treatment [37]. “Young TIL” protocols reduce labor time and can be implemented in most patients, but importantly, recent studies indicate that this approach leads to an objective response rate of 50 %, close to that observed in classical TIL protocols [36].
As immunomodulatory monoclonal antibodies show promise in the clinical trials recently conducted, the combination of T-cell transfer with antibodies blocking CTLA-4 or PD-1 function may help to overcome negative costimulatory signals, which may improve the function of the transferred T cells [39, 40]. In addition, it is possible to manipulate the T-cell differentiation state during culture/expansion to improve TIL-ACT for the treatment of human cancer, using, for example, molecules that may inhibit differentiation processes (e.g., GSK-3b [41]) or by subjecting TIL cultures to different cytokines, such as IL-7, IL-15, or IL-21 alone or in addition to IL-2 [42–47].
While TIL-based clinical trials have demonstrated impressive results in terminally-ill melanoma patients, they require dedicated facilities, and collaboration between surgical and cell therapy teams, which may have limited their implementation to a few clinical centers worldwide. Nonetheless, parallel approaches aimed at exploiting the unrivaled potential of T cells to mediate tumor regression are being developed, and are based on the genetic modification of T cells to express tumor-specific receptors.
9.3 Adoptive Immunotherapy Based on the Genetic Modification of Lymphocytes
9.3.1 TCR Gene Transfer
9.3.1.1 Development and Implementation of TCR Gene Transfer Approaches
As T-cell specificity is solely based on the nature of its TCR, TCR gene transfer therapy represents a promising approach based on the genetic modification of T cells engineered to recognize tumor antigens. A study by Steinmetz and colleagues back in 1986 demonstrated for the first time the feasibility of the TCR gene transfer approach. In this study, T cells were redirected by genetic engineering in order to study the receptor dynamics [48]. Since then, several studies have demonstrated how human T cells can be redirected toward specific antigen by TCR gene transfer using a melanoma-specific TCR in vitro [49], followed by an in-vivo study using a mouse model [50]. In 2006, the first clinical trial involving TCR gene therapy was reported by Morgan et al. involving metastatic melanoma patients, who were treated with autologous peripheral blood lymphocytes (PBLs) retrovirally transduced with a MART-1 specific TCR following a lymphodepletion regimen. An objective clinical response was observed in two out of 17 patients treated in this trial (12 %), demonstrating dramatic tumor regression [51].
Three years later, the results of a second clinical trial were reported by the same group (led by Dr. Steven Rosenberg, NCI); in this trial, metastatic melanoma patients were treated with two high-affinity TCRs against the melanoma antigens MART-1 and gp100 [52]. The expression levels of the TCR and the persistence of modified T cells were markedly increased compared with the first trial, and an objective response rate of 30 % (six out of 20 patients) was reported. Since then, progress has been made towards the clinical testing of additional TCRs, specific to other antigens such as p53 [53], NY-ESO-1 [54], and CEA [55], in order to target cancers other than melanoma.
So far, TCR gene transfer has been proven to be an effective strategy to create specific tumor-reactive T cells, without the restrictions or the need of isolating natural tumor-reactive T cells from the patient. Factors that should be taken into account towards improving the clinical efficacy of this approach, and that will be discussed in part below are, for instance, the persistence of the TCR-modified T cells after infusion, the prolonged expression of the TCR genes, and the need to reach sufficient T-cell functional avidity.
9.3.1.2 How to Select the Appropriate (Suitable) Antigen?
As for other therapeutic treatments, two main factors should be considered to choose the proper target antigen for TCR gene therapy: safety and efficiency. By choosing a target antigen characterized by high levels of tumor-specific expression and lacking any expression levels in the normal tissue, one can limit the possibility of on/off-target effects and the possible dose-limiting toxicity which can result from the destruction of normal tissues that express the aimed target antigen [55].
Currently, over-expressed antigens, cancer-testis (CT) antigens, and differentiation antigens represent the most common target antigens for TCR-based adoptive immunotherapy. NY-ESO-1, a cancer-testis antigen (CT), is one of the most promising targets that have been the subject of a recent clinical trial for TCR gene therapy, which resulted in a 40–60 % objective response in melanoma and synovial cell sarcoma patients [54]. Many CT antigens have been identified in various human cancers is discussed above [5, 56], while they are normally expressed only in the human germ line. The restriction of CTs to cells that partially or do not express human leukocyte antigen (HLA) molecules (in healthy tissues) makes them unsusceptible to recognition by a TCR, thus preventing toxicity to normal tissues when targeting T cells to tumor-associated CT antigens. Two other classes of tumor antigens that may be also taken into account as targets for TCR gene therapy are the mutation antigens and the neo-antigens [57, 58]. Indeed, it seems that the majority of these antigens are to be safe targets owing to their exclusive expression in tumor cells. While the first group is represented by antigens that are common not only to a variety of patients but also shared between several tumor types, the second group is constituted of patient-specific antigens that can be characterized using recent technological advancements such as individual tumor sequencing [57]. Still, as an immune selective pressure builds up, the down-regulation of target antigens could represent a concrete impediment to the therapeutic efficacy of TCR gene therapy [59, 60], especially as it is based on mono-specific T cells. Recently, the study of Kaluza and colleagues demonstrated tumor (B16/Ovalbumin) recurrence after adoptive transfer of specific (OT-1) effector cells, due to the loss of the target tumor antigen [61]. Possible solutions for the down-regulation of target antigen expression may consist in: (1) targeting of proteins that have an essential role in the survival of the tumor [4], (2) combining two (or more) different specificities expressed by the same T cell [61], or (3) using multiple populations of T cells, each expressing a different tumor-specific TCR.
9.3.1.3 Choosing the “Right” TCR for the “Right” pMHC Complex
Several approaches have been described in order to isolate the desirable TCR, which will not only recognize specifically the targeted peptide–MHC complex, but will also endow T cells with superior functional avidity. As mentioned above, the objective response rate observed in the first two clinical TCR-gene therapy trials, in which MART-1-specific TCRs were produced from a melanoma patient [51, 52], was low in comparison to that in TIL therapy trials [17, 38, 62]. This disparity could be due to: (1) low levels of TCR expression of the introduced TCR on the engineered T cells, (2) a diminished persistence of TCR-modified T cells after infusion, and/or (3) the induction of immunological self-tolerance that might hinder a proper response to target antigens with suboptimal affinity to their cognate TCR. Therefore, unmodified TCRs derived from melanoma patients may require further optimization steps to endow T cells with an improved performance.
High-affinity TCRs could be isolated from HLA-mismatched donors, since one does not expect that those TCRs would be subjected to any tolerance mechanism pertaining to the targeted MHC–peptide complex, which thus would be recognized as non-self [63–65]. Similarly, HLA-transgenic mice [66–69] and phage/yeast/T-cell display systems [70–73] also provide platforms that could be exploited to isolate “non-tolerized TCR.” The TCR phage display technique, for example, yielded TCRs with high affinity specific for human telomerase reverse transcriptase (hTERT), human T-cell lymphotropic virus type 1 (HTLV-1), TAX antigen, and additional antigens [73, 74].
Additionally, a human-TCR repertoire transgenic mice system was recently established. In this system, the entire human TCR loci was cloned into HLA-A2-transgenic mice [75], and this resulted in the reconstitution of a potentially broad human TCR repertoire in the mouse recipient which can provide a platform to isolate human high-affinity TCRs, provided the targeted epitope is not expressed by the mouse recipient.
9.3.1.4 TCR Expression Systems
In most of the clinical trials reported, TCR gene therapy made use of γ-retroviral vectors which are common viral expression systems that facilitate transgene integration into the genome of the host cells [76–78]. MFG/SFG-, MP71/SF91-, and MSGV1- are examples for such γ-retroviral vectors that in pre-clinical studies and clinical trials exhibit high transduction efficiency together with minimal vector-associated toxicity. Lentiviral vectors are another viral expression platform that, unlike γ-retroviral vectors, is largely independent from cells’ dividing status and thus could successfully infect minimally activated T cells [79, 80]. Moreover, lentiviral vectors display a greater gene insertion capacity, allowing the transfer of larger and highly complex gene constructs into T cells.
There are also several non-viral alternatives for TCR-gene transfer into T-cells. One main advantage of the latter is that, unlike viral platforms, they require a minimal production and testing time from a regulatory standpoint. The Sleeping Beauty and the piggyBac are example of transposon-based systems that have been used to alternatively redirect T cells to express antigen-specific receptors [81, 82]. This approach relies on the expression of the transposase in the target cell, together with the transfer of the transposon that encodes the genes of interest [83, 84]. Transfer of mRNA molecules encoding TCR chains by electroporation may also be used as a non-viral expression system to modify T cells; it eliminates the risk of insertional mutagenesis. Still, the main downside of this approach is the short-term expression of the transgene (a few days), which necessitates repetitive injection of electroporated cells to achieve in-vivo effects [85].
9.3.1.5 Off-Target and Safety Risks Involved in TCR Gene Transfer Strategy
Off-target events following TCR gene therapy may be due to self/cross-reactivity of the transduced TCR and/or the formation of mixed dimmers between the two α and two β chains that are co-expressed in the transduced cell, which may potentially lead to new auto-immune specificity [86]. Four different TCR combinations can form when mixing the chains that originated from the exogenous α/β TCR with the two chains that originate from natural/endogenous α/β TCR. The two mispaired heterodimeric TCRs may result either in a non-functioning TCR or a receptor with a new specificity that can prove self-reactive. In this regard, a recent study demonstrated how the formation of mixed TCRs can result in self-reactive T cells that engendered autoimmune manifestations in a mouse model [87].
Several strategies have been devised to increase the expression of the introduced TCRs, which are often based on molecular approaches aiming for better pairing/association of the α/β chains of the introduced-exogenous TCR [86, 88]. For example, hybrid human TCRs that are composed of parts of/entire murine constant regions [89–93] mediated an improved expression of the transferred TCR. The inclusion of an additional disulfide bond within the constant region of the TCR [94, 95], molecular “knob into holes” inversions in the constant regions of the TCR chains [96], single-chain TCRs [97], and the use of TCR/CD3ζ fusion products [98] were also recently demonstrated as potential pairing-optimization strategies. Since α/β and γ/δ TCR chains cannot mutually pair [99], the use of γδ T cells that are transduced with an αβ TCR is also an alternative approach [100]. Silencing the endogenous TCRs is another strategy, which can be achieved by co-transferring siRNAs/shRNAs targeting the endogenous TCR [101] or by making use of zinc-finger nucleases (ZFNs) that are specific for the endogenous TCR chains [102].
9.3.1.6 How to Further Improve the Anti-tumor Efficacy of TCR Gene Transfer?
In addition to the aforementioned strategies to improve adoptive T-cell therapy (such as lymphodepletion and cytokine polarization), several approaches are being developed in order to enhance functional and durable responses by TCR gene therapy. TCR affinity enhancement, which is believed to lead to an improved functional avidity, could be achieved by introducing selective modifications in the CDR3 region of the TCR α or β chain, which has been shown to be crucial for the recognition and binding of the antigen [70, 73]. The use of pairing (see above) and codon optimization (to improve protein expression) may also contribute to enhancing antigen-specific reactivity in T cells [68, 103]. Additionally, it has been demonstrated that reduced TCR glycosylation can elevate functional avidity and prevent the internalization of the transduced TCRs [104]. Recently, we demonstrated that it is possible to greatly enhance T-cell functional avidity against tumor cells by mutating three transmembrane residues in the TCRα chain into hydrophobic amino acid, which led to increased TCR stability and expression and augmented TCR expression in the transduced T cells [105]. In addition, the design of the gene expression cassette may also influence TCR expression: the use of P2A or IRES elements, which link the α and β chains, has been shown to improve TCR expression and to reduce the risk of induced autoimmune pathology [87, 106].
Beyond the engineering of T-cell specificity using TCR transgenes, several genetic approaches to further amplify/generate important T-cell functions (such as co-stimulation, cytokine secretion, expression of chemokine receptors and homing factors) have been described (reviewed in [107]). For example, though the administration of IL-12 in tumor mouse models can improve host survival and tumor regression rate [108, 109], the associated toxicities are a major drawback. Engineering gene-modified T cells to produce IL-12 in vivo using an inducible retroviral vector demonstrated intensified anti-tumor activity against B16 murine melanoma tumors [110]. Alternatively, the use of T cells that are conjugated to adjuvant cytokine-loaded nanoparticles is another potential way to lead to a local production/delivery of cytokines, while reducing toxicity [111]. The (sub-) type of T-cell to be transduced is also of importance; recent studies have demonstrated the superior properties of other kinds of lymphocytes, such as memory T cells, naïve T cells, memory stem cells and central-memory T cells [41, 112–114].
In addition to TCR signaling, T-cell function is controlled by both positive and negative regulation. The tumor microenvironment has been shown to greatly induce immune suppression. For example, the immunosuppressive role of transforming growth factor-β (TGF-β) involves the inhibition of proliferation and function of T cells [115, 116]. By expressing a non-functional TGF-β receptor, tumor cells may also escape the apoptotic effects of TGF-β [117, 118]. In order to diminish the inhibition induced by TGF-β, it is possible to express in the genetically engineered T cells a truncated (dominant negative) form of TGF-β receptor [119], or to use a decoy-soluble TGF-β receptor II [120]. Bollard et al. recently reported that human T cells transduced with a dominant negative form of TGF-β receptor were resistant to the anti-proliferative and anti-cytotoxic effects of exogenous TGF-β [121, 122]. More recently, several groups [120, 123] have shown that this strategy is also effective in vivo, though the sustained effects of this might not last as expected [123].
9.3.2 Chimeric Antigen Receptor Gene Transfer
In parallel to the TCR gene transfer approach, it is possible to redirect the specificity of T-cells using chimeric antigen receptors (CARs). These CARs, also known as “T-bodies” or “chimeric immune receptors” are fusion proteins that generally contain an extracellular targeting domains based on an antibody single-chain variable fragment (scFv) that is fused to intracellular signaling elements. As mentioned above for TCRs, transduction of peripheral blood T cells with CARs allows the redirection of T-cell specificity against tumor cell surface antigen.
9.3.2.1 CAR Development
The development of antibody-based chimer receptor, was first reported in 1989 in the pioneering studies by Gross and Eshhar [124]. They generated a chimeric T-cell receptor assembled from the TCR constant domains fused to the variable domains of an antibody specific for anti-2,4,6-trinitrophenyl (TNP). T cells that expressed this chimeric receptor successfully recognized TNP, which led to the production of IL-2 and cell-mediated cytotoxicity of TNP-expressing targets. Thus, the use of CARs enables the targeting of tumor in an HLA-independent manner, which suggests the possibility, in theory, of treating a larger part of the population, compared to TCR-based therapies. Moreover, CARs allow the targeting of not only protein-based antigens but also carbohydrates and glycolipids, provided targeting moieties/monoclonal antibodies can be generated against these. Another advantage of the CAR approach, as these function in an MHC-independent way, is their ability to stimulate both CD8+ and CD4+ T cells, which have been shown to act synergistically in enhancing the T-cell anti-tumor effect [125]. Still, it is important to remember that technically CARs can target only surface expressed antigens (though intracellular antigens could be also detected by CARs based on antibodies that target a specific pMHC (peptide–MHC) complex, and thus can mimic the mode of action of the TCR [126, 127]).
9.3.2.2 CAR Structure
As mentioned earlier, the common design of CARs is based on a binding domain, an extracellular spacer/hinge element, a trans-membrane region, and an intracellular singling domain (Fig. 9.2). Most of the CAR targeting domains are scFv (i.e., the variable regions of heavy and light chains joined together by a short linker peptide). If the scFv is derived from a murine antibody, it is possible to “humanize” it by replacing the mouse framework regions by their human counterparts. Another possible design for the targeting moiety of CARs (instead of scFv) are protein receptor/ligands; such alternatives include, for instance, a vascular endothelial growth factor polypeptide [128], an integrin binding peptide heregulin [129], interleukin—13 mutein [130], NKp30 (NCR3/CD337) [131], and the NKG2D receptor [132].

Schematic representation of the different CAR generations
The second component in this design is the hinge region that serves as spacer, which increases the distance of the binding domain from the transmembrane region, providing more flexibility for the binding domain. The nature of the hinge region can influence cytokine secretion and cell-mediated killing of target cells by CAR-modified T cells [133]. Some common examples for hinge region are immunoglobulin domains such as the fragment crystallizable (Fc) regions of antibodies, or immunoglobulin-like domains derived from CD8α and CD28 molecules. It has been found that the function of the hinge region in the CAR is dependent on the binding site on the antigen itself; if the binding site is a membrane-proximal epitope, the use of a hinge region will be beneficial. In contrast, when the binding site is a membrane-distal epitope, improved cytokine release and cytotoxicity will be higher in the absence of a hinge region [134].
The third component in the CARs is the transmembrane region: in most cases, it is based on transmembrane domains derived from co-receptor/costimulatory molecules such as CD8 and CD28.
The fourth module in the structure of the CARs is the intracellular signaling domain. Importantly, a lot of effort is being invested in order to develop optimal conformation of the intracellular signaling portions to achieve the best activation. The first generation of CARs included only one signaling domain (Fig. 9.2) derived either from the CD3ζ or FcRγ chains, which are the common signal-transducing subunits of the TCR or the immunoglobulin receptor respectively [135]. One main difference between these two subunits is the number of the immunoreceptor tyrosine-based activation motifs (ITAMs); while the CD3ζ chain contains three ITAMs, the FcRγ chain contains only one and this feature has been shown to impact on T-cell function and survival [136].
9.3.2.3 CAR Development and Generations
When first compared, the ζ and γ subunits were fused to single-chain variable domain chimeric receptors recognizing the carcinoembryonic antigen (CEA). Although similar levels of expression were detected after transduction, some significant functional difference was found after co-culture with target cells [137]. These assays demonstrated the superiority of the chimeric receptors that contained the CD3ζ, mainly in improved cytokine production and enhanced ability to mediate lysis of target cells. Additionally, it was revealed that CD3ζ-based chimeric receptors displayed a better ability to eradicate human tumors in vivo. While it has been postulated that the anti-tumor activity mediated by the CD3ζ moiety might result in activation-induced T-cell death (AICD) because of the numerous ITAMs (3), these claims have been refuted [138], and so far most of the CAR designs include a CD3ζ moiety as their main signaling domain.
Despite the encouraging results that were obtained in the studies with the first-generation CARs (that contained only the CD3ζ chain in the intracellular singling domain) and which demonstrated anti-tumor activity against a range of target cells [139], the lack of co-stimulatory signals (“signal 2”) led to inefficient cytokine production, reduced proliferation, and even a state of T-cell anergy [140, 141]. A second generation of CAR was designed to include a co-stimulatory portion in addition to the CD3ζ signaling domain. The most common co-stimulatory molecule that fills this role is CD28, the first isolated co-stimulatory molecule, which is essential to prevent anergy and to drive increased cytokine secretion [142]. Still, the possibility of generating two chimeras that express the ζ chain and the CD28 separately was explored, and this approach did mediate increased secretion of IL-2 in vitro [143]. More recently, a similar concept to reduce CAR side-effects made use of a first-generation CAR transduced in conjunction with a CCR (chimeric co-stimulatory receptor) specific for a second antigen, which enabled safer in-vivo targeting of tumors which expressed both cognate antigens [144]. So far, a more widespread concept is to combine both signaling moieties in the same receptor [145]. From a structural standpoint, a better surface expression of the CAR can be achieved by positioning the CD28 domain in proximity to the CD3ζ domain and immediately after the transmembrane region [146]. Several studies have demonstrated the improved function of second-generation CAR-modified T cells in mediating increased proliferation [147] and cytokine secretion (IL-2, interferon-γ, granulocyte–macrophage colony-stimulating factor) [148, 149]. Furthermore, this kind of design promoted the up-regulation of anti-apoptotic proteins such as Bcl-2 (which would contribute to reduce AICD) and better resistance to immunosuppressive conditions prevalent in the tumor microenvironment; studies have shown that second-generation CAR-modified T cells are less sensitive to TGF-β-mediated suppression [150], and could increase the expression of NFκB counteracting Tregs-induced inhibition [151].
There does not seem to be an optimal signaling moiety for CARs, and thus there is often a need to evaluate empirically several combinations for each given targeting moiety. Although most of the CARs use the CD28 signaling domain, alternative co-stimulatory molecules that were tested include the inducible T-cell costimulator (ICOS) B7 family member, and CD27, CD137 (4-1BB), and CD134 (OX-40) from the TNFR family members, which can enhance effector functions also in resting human T cells [152–154]. However, to further improve second-generation CARs, several studies have shown that it was possible to include another co-stimulatory moiety in addition to CD3ζ chain and CD28 in the signaling domain, leading to the design of third-generation CARs [155]. For example, a CAR for prostate-specific membrane antigen (PSMA), which contains CD28+ 4-1BB+ CD3ζ signaling domain, showed an increased cytokine production and mediated an improved prostate tumor regression in vivo [154]. Furthermore, third-generation CARs can induce PI3Kinase/Akt activation and BclXL expression and can help to reduce T-cell apoptosis. Another study showed that a CAR that contained the antigen-binding domain of the anti-GD2a fused to a CD28/OX40/ζ signaling domain endowed T-cells with improved proliferative capacity and anti-tumor function [156]. Still, the presence of the three activation/stimulation motifs in a single signaling domain may theoretically cause a lower sensitivity threshold, which should be taken into account when designing future clinical applications.
9.3.2.4 Driving the CARs into the Clinic
Results from in-vitro and in-vivo (in animal models) studies that show the potential of CARs in mediating tumor regression in several types of cancer—such as medulloblastoma, prostate [157] and colon carcinoma [158]—,facilitated their translation into the clinic. In the first clinical trial that made use of first-generation CAR-modified T cells, Lamers et al. treated three patients with metastatic renal cell carcinoma (RCC) using a CAR that recognizes carboxy-anhydrase-IX (CAIX), which is over-expressed by RCC tumors. All three patients were reported to suffer from liver toxicity, which was apparently caused by on-target effects of CAR-modified T cells against the CAIX+ bile duct epithelial cells and no clinical responses were observed [159]. In another trial, 14 patients with metastatic ovarian cancer were treated with CAR-modified T cells against the ovarian cancer-associated antigen α-folate receptor (FR) [160]. Analysis of the CAR-modified T-cell presence in the circulation showed it quickly declined in the majority of the patients after 1 month, and also in this case no clinical response was observed in any of the patients treated.
Pule et al. engineered Epstein–Barr virus (EBV)-specific CTLs to express a first-generation CAR directed to the diasialoganglioside GD2 antigen, which is expressed on neuroblastoma cells. Infusion of these CAR-modified T cells seemed safe, and resulted in encouraging tumor regressions in half of the subjects tested [161]. Whereas these three clinical trials used retroviral transduction, in a clinical trial reported by Till et al., CAR-modified T cells were generated by electroporation with a vector plasmid encoding a CAR specific to CD20, to target indolent B-cell lymphoma (or mantle cell lymphoma). Out of seven patients treated, two achieved complete responses, one had a partial response, and four had stable disease [162]. Another notable clinical study was carried out recently by Kalos et al., in which three patients with advanced chronic lymphocytic leukemia (CLL) were treated with an anti-CD19 second-generation CAR that contained a CD3ζ chain coupled with CD137 domain. CAR-modified T-cells expanded over 1,000-fold in vivo, trafficked to the bone marrow and remained detectable 6 months post-infusion; a fraction of these cells even differentiated into memory T cells. Ten months after treatment, all the patients demonstrated an objective clinical response, with two of the three patients treated showing complete remission and one partial response [163]. A recent clinical trial using a third-generation CAR was conducted by Till et al. using a CAR targeting CD20 (which is expressed on indolent B-cell and mantle cell lymphomas) [164]. This third-generation CAR contained two co-stimulatory domains, CD28 and CD137, in addition to CD3ζ. CAR-modified T cells were detected for up to 1 year in patients’ blood. Moreover, one out of four patients treated had an objective partial response (later relapsed a year after infusion), one patient developed transient infusional symptoms, and two patients remained progression-free for 12 and 24 months. Thus, some 20 years after they were initially developed, chimeric antigen receptors have entered the clinic and are showing promising results.
Nevertheless, one has to bear in mind that side-effects may arise, and unfortunately these may on rare occasions be lethal. In a trial that made use of a trastuzumab (Herceptin)-based third-generation CAR to target breast tumors, infusion of CAR-modified T cells led to the death of one patient. This was attributed to a “cytokine storm,” possibly linked to the widespread expression of the targeted antigen, Her2/neu (ERBB2), by normal lung cells [165]. Another fatality was noted after using a second-generation CAR targeting CD19, in combination with cyclophosphamide lymphodepleting chemotherapy [166]. This treatment led to hypotension, dyspnea, and renal failure in the treated patient, and 4 days after the initial infusion the patient died. This suggests the need to include suicide genes in the CAR-bearing viral construct, or to use a dual-CAR/CCR design [144] to potentially provide another layer of safety. In addition, knocking down the expression of the endogenous TCR might prove valuable in order to prevent undesired/non-specific responses of CAR-activated T-cells [167].
9.4 Conclusions
In the past 25 years, adoptive T-cell transfer has established itself as a promising immunotherapeutic strategy for the treatment of advanced cancer. The basic idea, that the (autologous) immune system can be manipulated in order to promote tumor regression and remission, is appealing as it may provide long-lasting protection. Still, from the “bench-side” of things, additional targets/antigens have to be defined/characterized to provide safer treatments targeting a broad spectrum of tumors. From a clinical standpoint, there is a need to speed up processing times [168] and to ease regulatory requirements [169]. Improving the success rate of adoptive T-cell transfer will also require its combination with multi-modal therapies targeting, for instance, the tumor micro-environment as well as immunosuppressive agents. Much has to be done also to encourage partnership with the industry in order to commercialize this kind of immunotherapy that requires cell manipulation and conditioning [170]. Several studies also suggest that these concepts can be applied to treat other conditions than cancer [88]. Adoptive T-cell immunotherapy is certainly earning a respected place in the “Hall of Fame” of personalized medicine treatments.
Abbreviations
- ACT:
-
Adoptive cell transfer
- AICD:
-
Activation-induced T-cell death
- CAIX:
-
Carboxy-anhydrase-IX
- CAR:
-
Chimeric antigen receptor
- CEA:
-
Carcino embryonic antigen
- CT:
-
Cancer/testis
- EBV:
-
Epstein–Barr virus
- HLA:
-
Human leukocyte antigen
- hTERT:
-
Human telomerase reverse transcriptase
- HTLV-1:
-
Human T-cell lymphotrophic virus type I
- IL:
-
Interleukin
- ITAM:
-
Immunoreceptor tyrosine-based activation motif
- MDSC:
-
Myeloid-derived suppressor cell
- MHC:
-
Major histocompatibility complex
- PBL:
-
Peripheral blood lymphocyte
- RCC:
-
Renal cell carcinoma
- scFv:
-
Single-chain variable fragment
- TA:
-
Tumor antigen
- TCR:
-
T-cell receptor
- TGF-β:
-
Transforming growth factor-β
- TIL:
-
Tumor-infiltrating lymphocyte
- Tregs:
-
Regulatory T-cells
References
Zhang N, Bevan MJ (2011) CD8(+) T cells: foot soldiers of the immune system. Immunity 35:161–168
Seremet T, Brasseur F, Coulie PG (2011) Tumor-specific antigens and immunologic adjuvants in cancer immunotherapy. Cancer J 17:325–330
Renkvist N, Castelli C, Robbins PF et al (2001) A listing of human tumor antigens recognized by T cells. Cancer Immunol Immunother 50:3–15
Linnemann C, Schumacher TN, Bendle GM (2011) T-cell receptor gene therapy: critical parameters for clinical success. J Invest Dermatol 131:1806–1816
Simpson AJ, Caballero OL, Jungbluth A et al (2005) Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer 5:615–625
Zur HH (2009) The search for infectious causes of human cancers: where and why. Virology 392:1–10
Fridman WH, Pages F, Sautes-Fridman C et al (2012) The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 12:298–306
Pages F, Galon J, Dieu-Nosjean MC et al (2010) Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene 29:1093–1102
Curiel TJ, Coukos G, Zou L et al (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10:942–949
Restifo NP, Dudley ME, Rosenberg SA (2012) Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12:269–281
Muul LM, Spiess PJ, Director EP et al (1987) Identification of specific cytolytic immune responses against autologous tumor in humans bearing malignant melanoma. J Immunol 138:989–995
Rosenberg SA, Dudley ME (2009) Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol 21:233–240
Rosenberg SA, Spiess P, Lafreniere R (1986) A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 233:1318–1321
Atkins MB, Lotze MT, Dutcher JP et al (1999) High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 17:2105–2116
Rosenberg SA, Yang JC, Sherry RM et al (2011) Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17:4550–4557
Rosenberg SA, Packard BS, Aebersold PM et al (1988) Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med 319:1676–1680
Dudley ME, Yang JC, Sherry R et al (2008) Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol 26:5233–5239
Gattinoni L, Finkelstein SE, Klebanoff CA et al (2005) Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med 202:907–912
Muranski P, Boni A, Wrzesinski C et al (2006) Increased intensity lymphodepletion and adoptive immunotherapy—how far can we go? Nat Clin Pract Oncol 3:668–681
Robbins PF, Dudley ME, Wunderlich J et al (2004) Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol 173:7125–7130
Zhou J, Dudley ME, Rosenberg SA et al (2005) Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J Immunother 28:53–62
Klebanoff CA, Gattinoni L, Torabi-Parizi P et al (2005) Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci USA 102:9571–9576
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12:253–268
Kerkar SP, Restifo NP (2012) Cellular constituents of immune escape within the tumor microenvironment. Cancer Res 72:3125–3130
Gattinoni L, Klebanoff CA, Restifo NP (2012) Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer 12:671–684
Ogino S, Galon J, Fuchs CS et al (2011) Cancer immunology—analysis of host and tumor factors for personalized medicine. Nat Rev Clin Oncol 8:711–719
Ruffell B, Au A, Rugo HS et al (2012) Leukocyte composition of human breast cancer. Proc Natl Acad Sci USA 109:2796–2801
Walia V, Mu EW, Lin JC et al (2012) Delving into somatic variation in sporadic melanoma. Pigment Cell Melanoma Res 25:155–170
Biswas K, Richmond A, Rayman P et al (2006) GM2 expression in renal cell carcinoma: potential role in tumor-induced T-cell dysfunction. Cancer Res 66:6816–6825
Storkel S, Keymer R, Steinbach F et al (1992) Reaction patterns of tumor infiltrating lymphocytes in different renal cell carcinomas and oncocytomas. Prog Clin Biol Res 378:217–223
Uzzo RG, Rayman P, Kolenko V et al (1999) Renal cell carcinoma-derived gangliosides suppress nuclear factor-kappaB activation in T cells. J Clin Invest 104:769–776
Zhang J, Chen Y, Li J et al (2006) Renal tubular epithelial expression of the coinhibitory molecule B7-DC (programmed death-1 ligand). J Nephrol 19:429–438
Goedegebuure PS, Douville LM, Li H et al (1995) Adoptive immunotherapy with tumor-infiltrating lymphocytes and interleukin-2 in patients with metastatic malignant melanoma and renal cell carcinoma: a pilot study. J Clin Oncol 13:1939–1949
Markel G, Cohen-Sinai T, Besser MJ et al (2009) Preclinical evaluation of adoptive cell therapy for patients with metastatic renal cell carcinoma. Anticancer Res 29:145–154
Dudley ME, Wunderlich JR, Shelton TE et al (2003) Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother 26:332–342
Besser MJ, Shapira-Frommer R, Treves AJ et al (2010) Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res 16:2646–2655
Tran KQ, Zhou J, Durflinger KH et al (2008) Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J Immunother 31:742–751
Dudley ME, Gross CA, Langhan MM et al (2010) CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res 16(24):6122–6131
Hershkovitz L, Schachter J, Treves AJ et al (2010) Focus on adoptive T cell transfer trials in melanoma. Clin Dev Immunol 2010:260267
Topalian SL, Hodi FS, Brahmer JR et al (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443–2454
Gattinoni L, Zhong XS, Palmer DC et al (2009) Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med 15:808–813
Hinrichs CS, Spolski R, Paulos CM et al (2008) IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood 111:5326–5333
Kerkar SP, Muranski P, Kaiser A et al (2010) Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res 70:6725–6734
Klebanoff CA, Finkelstein SE, Surman DR et al (2004) IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci USA 101:1969–1974
Pellegrini M, Calzascia T, Elford AR et al (2009) Adjuvant IL-7 antagonizes multiple cellular and molecular inhibitory networks to enhance immunotherapies. Nat Med 15:528–536
Pouw N, Treffers-Westerlaken E, Kraan J et al (2010) Combination of IL-21 and IL-15 enhances tumour-specific cytotoxicity and cytokine production of TCR-transduced primary T cells. Cancer Immunol Immunother 59:921–931
Refaeli Y, Van Parijs L, London CA et al (1998) Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity 8:615–623
Dembic Z, Haas W, Weiss S et al (1986) Transfer of specificity by murine alpha and beta T-cell receptor genes. Nature 320:232–238
Clay TM, Custer MC, Sachs J et al (1999) Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol 163:507–513
Kessels HW, Wolkers MC, van dB et al (2001) Immunotherapy through TCR gene transfer. Nat Immunol 2:957–961
Morgan RA, Dudley ME, Wunderlich JR et al (2006) Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314:126–129
Johnson LA, Morgan RA, Dudley ME et al (2009) Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114:535–546
Davis JL, Theoret MR, Zheng Z et al (2010) Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials. Clin Cancer Res 16:5852–5861
Robbins PF, Morgan RA, Feldman SA et al (2011) Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29:917–924
Parkhurst MR, Yang JC, Langan RC et al (2011) T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 19:620–626
Jager E, Chen YT, Drijfhout JW et al (1998) Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1: definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J Exp Med 187:265–270
Metzker ML (2010) Sequencing technologies — the next generation. Nat Rev Genet 11:31–46
Warren RL, Holt RA (2010) A census of predicted mutational epitopes suitable for immunologic cancer control. Hum Immunol 71:245–254
Dudley ME, Wunderlich JR, Yang JC et al (2005) Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 23:2346–2357
Yee C, Thompson JA, Byrd D et al (2002) Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA 99:16168–16173
Kaluza KM, Thompson JM, Kottke TJ et al (2012) Adoptive T cell therapy promotes the emergence of genomically altered tumor escape variants. Int J Cancer 131:844–854
Dudley ME, Wunderlich JR, Robbins PF et al (2002) Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298:850–854
Amir AL, van der Steen DM, van Loenen MM et al (2011) PRAME-specific Allo-HLA-restricted T cells with potent antitumor reactivity useful for therapeutic T-cell receptor gene transfer. Clin Cancer Res 17:5615–5625
Sadovnikova E, Stauss HJ (1996) Peptide-specific cytotoxic T lymphocytes restricted by nonself major histocompatibility complex class I molecules: reagents for tumor immunotherapy. Proc Natl Acad Sci USA 93:13114–13118
Savage P, Gao L, Vento K et al (2004) Use of B cell-bound HLA-A2 class I monomers to generate high-avidity, allo-restricted CTLs against the leukemia-associated protein Wilms tumor antigen. Blood 103:4613–4615
Chinnasamy N, Wargo JA, Yu Z et al (2011) A TCR targeting the HLA-A*0201-restricted epitope of MAGE-A3 recognizes multiple epitopes of the MAGE-A antigen superfamily in several types of cancer. J Immunol 186:685–696
Cohen CJ, Zheng Z, Bray R et al (2005) Recognition of fresh human tumor by human peripheral blood lymphocytes transduced with a bicistronic retroviral vector encoding a murine anti-p53 TCR. J Immunol 175:5799–5808
Parkhurst MR, Joo J, Riley JP et al (2009) Characterization of genetically modified T-cell receptors that recognize the CEA:691–699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin Cancer Res 15:169–180
Theobald M, Biggs J, Dittmer D et al (1995) Targeting p53 as a general tumor antigen. Proc Natl Acad Sci USA 92:11993–11997
Chlewicki LK, Holler PD, Monti BC et al (2005) High-affinity, peptide-specific T cell receptors can be generated by mutations in CDR1, CDR2 or CDR3. J Mol Biol 346:223–239
Holler PD, Holman PO, Shusta EV et al (2000) In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc Natl Acad Sci USA 97:5387–5392
Kessels HW, van dB, Spits H et al (2000) Changing T cell specificity by retroviral T cell receptor display. Proc Natl Acad Sci USA 97:14578–14583
Li Y, Moysey R, Molloy PE et al (2005) Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol 23:349–354
Zhao Y, Bennett AD, Zheng Z et al (2007) High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J Immunol 179:5845–5854
Li LP, Lampert JC, Chen X et al (2010) Transgenic mice with a diverse human T cell antigen receptor repertoire. Nat Med 16:1029–1034
Hughes MS, Yu YY, Dudley ME et al (2005) Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther 16:457–472
Riviere I, Brose K, Mulligan RC (1995) Effects of retroviral vector design on expression of human adenosine deaminase in murine bone marrow transplant recipients engrafted with genetically modified cells. Proc Natl Acad Sci USA 92:6733–6737
Schambach A, Swaney WP, van der Loo JC (2009) Design and production of retro- and lentiviral vectors for gene expression in hematopoietic cells. Methods Mol Biol 506:191–205
Yang S, Cohen CJ, Peng PD et al (2008) Development of optimal bicistronic lentiviral vectors facilitates high-level TCR gene expression and robust tumor cell recognition. Gene Ther 15:1411–1423
Yang S, Rosenberg SA, Morgan RA (2008) Clinical-scale lentiviral vector transduction of PBL for TCR gene therapy and potential for expression in less-differentiated cells. J Immunother 31:830–839
Peng PD, Cohen CJ, Yang S et al (2009) Efficient nonviral Sleeping Beauty transposon-based TCR gene transfer to peripheral blood lymphocytes confers antigen-specific antitumor reactivity. Gene Ther 16:1042–1049
Williams DA (2008) Sleeping beauty vector system moves toward human trials in the United States. Mol Ther 16:1515–1516
Huang X, Wilber AC, Bao L et al (2006) Stable gene transfer and expression in human primary T cells by the Sleeping Beauty transposon system. Blood 107:483–491
Ivics Z, Hackett PB, Plasterk RH et al (1997) Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 91:501–510
Zhao Y, Moon E, Carpenito C et al (2010) Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res 70:9053–9061
Govers C, Sebestyen Z, Coccoris M et al (2010) T cell receptor gene therapy: strategies for optimizing transgenic TCR pairing. Trends Mol Med 16:77–87
Bendle GM, Linnemann C, Hooijkaas AI et al (2010) Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med 16:565–570
Daniel-Meshulam I, Ya’acobi S, Ankri C et al (2012) How (specific) would like your T-cells today? Generating T-cell therapeutic function through TCR-gene transfer. Front Immunol 3:186
Bialer G, Horovitz-Fried M, Ya’acobi S et al (2010) Selected murine residues endow human TCR with enhanced tumor recognition. J Immunol 184:6232–6241
Cohen CJ, Zhao Y, Zheng Z et al (2006) Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res 66:8878–8886
Sommermeyer D, Uckert W (2010) Minimal amino acid exchange in human TCR constant regions fosters improved function of TCR gene-modified T cells. J Immunol 184:6223–6231
Stanislawski T, Voss RH, Lotz C et al (2001) Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat Immunol 2:962–970
Thomas S, Xue SA, Cesco-Gaspere M et al (2007) Targeting the Wilms tumor antigen 1 by TCR gene transfer: TCR variants improve tetramer binding but not the function of gene modified human T cells. J Immunol 179:5803–5810
Cohen CJ, Li YF, El Gamil M et al (2007) Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res 67:3898–3903
Kuball J, Dossett ML, Wolfl M et al (2007) Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood 109:2331–2338
Voss RH, Willemsen RA, Kuball J et al (2008) Molecular design of the Calphabeta interface favors specific pairing of introduced TCRalphabeta in human T cells. J Immunol 180:391–401
Aggen DH, Chervin AS, Schmitt TM et al (2012) Single-chain ValphaVbeta T-cell receptors function without mispairing with endogenous TCR chains. Gene Ther 19:365–374
Sebestyen Z, Schooten E, Sals T et al (2008) Human TCR that incorporate CD3zeta induce highly preferred pairing between TCRalpha and beta chains following gene transfer. J Immunol 180:7736–7746
Saito T, Hochstenbach F, Marusic-Galesic S et al (1988) Surface expression of only gamma delta and/or alpha beta T cell receptor heterodimers by cells with four (alpha, beta, gamma, delta) functional receptor chains. J Exp Med 168:1003–1020
van der Veken LT, Coccoris M, Swart E et al (2009) Alpha beta T cell receptor transfer to gamma delta T cells generates functional effector cells without mixed TCR dimers in vivo. J Immunol 182:164–170
Okamoto S, Mineno J, Ikeda H et al (2009) Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res 69:9003–9011
Provasi E, Genovese P, Lombardo A et al (2012) Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med 18(5):807–815
Jorritsma A, Gomez-Eerland R, Dokter M et al (2007) Selecting highly affine and well-expressed TCRs for gene therapy of melanoma. Blood 110:3564–3572
Kuball J, Hauptrock B, Malina V et al (2009) Increasing functional avidity of TCR-redirected T cells by removing defined N-glycosylation sites in the TCR constant domain. J Exp Med 206:463–475
Haga-Friedman A, Horovitz-Fried M, Cohen CJ (2012) Incorporation of transmembrane hydrophobic mutations in the TCR enhance its surface expression and T cell functional avidity. J Immunol 188:5538–5546
Uckert W, Schumacher TN (2009) TCR transgenes and transgene cassettes for TCR gene therapy: status in 2008. Cancer Immunol Immunother 58:809–822
Merhavi-Shoham E, Haga-Friedman A, Cohen CJ (2012) Genetically modulating T-cell function to target cancer. Semin Cancer Biol 22:14–22
Brunda MJ, Luistro L, Warrier RR et al (1993) Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med 178:1223–1230
Cavallo F, Di Carlo E, Butera M et al (1999) Immune events associated with the cure of established tumors and spontaneous metastases by local and systemic interleukin 12. Cancer Res 59:414–421
Zhang L, Kerkar SP, Yu Z et al (2011) Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther 19:751–759
Krieg C, Letourneau S, Pantaleo G et al (2010) Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci USA 107:11906–11911
Berger C, Jensen MC, Lansdorp PM et al (2008) Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest 118:294–305
Hinrichs CS, Borman ZA, Cassard L et al (2009) Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci USA 106:17469–17474
Turtle CJ, Riddell SR (2011) Genetically retargeting CD8+ lymphocyte subsets for cancer immunotherapy. Curr Opin Immunol 23:299–305
Gorelik L, Constant S, Flavell RA (2002) Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J Exp Med 195:1499–1505
Gorelik L, Flavell RA (2002) Transforming growth factor-beta in T-cell biology. Nat Rev Immunol 2:46–53
Knaus PI, Lindemann D, DeCoteau JF et al (1996) A dominant inhibitory mutant of the type II transforming growth factor beta receptor in the malignant progression of a cutaneous T-cell lymphoma. Mol Cell Biol 16:3480–3489
Park K, Kim SJ, Bang YJ et al (1994) Genetic changes in the transforming growth factor beta (TGF-beta) type II receptor gene in human gastric cancer cells: correlation with sensitivity to growth inhibition by TGF-beta. Proc Natl Acad Sci USA 91:8772–8776
Gorelik L, Flavell RA (2001) Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med 7:1118–1122
Zhang L, Yu Z, Muranski P et al (2013) Inhibition of TGF-beta signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther 20(5):575–580
Bollard CM, Rossig C, Calonge MJ et al (2002) Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood 99:3179–3187
Foster AE, Dotti G, Lu A et al (2008) Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J Immunother 31:500–505
Chou CK, Schietinger A, Liggitt HD et al (2012) Cell-intrinsic abrogation of TGF-beta signaling delays but does not prevent dysfunction of self/tumor-specific CD8 T cells in a murine model of autochthonous prostate cancer. J Immunol 189:3936–3946
Gross G, Waks T, Eshhar Z (1989) Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA 86:10024–10028
Moeller M, Haynes NM, Kershaw MH et al (2005) Adoptive transfer of gene-engineered CD4+ helper T cells induces potent primary and secondary tumor rejection. Blood 106:2995–3003
Cohen CJ, Denkberg G, Lev A et al (2003) Recombinant antibodies with MHC-restricted, peptide-specific, T-cell receptor-like specificity: new tools to study antigen presentation and TCR-peptide-MHC interactions. J Mol Recognit 16:324–332
Willemsen RA, Ronteltap C, Chames P et al (2005) T cell retargeting with MHC class I-restricted antibodies: the CD28 costimulatory domain enhances antigen-specific cytotoxicity and cytokine production. J Immunol 174:7853–7858
Niederman TM, Ghogawala Z, Carter BS et al (2002) Antitumor activity of cytotoxic T lymphocytes engineered to target vascular endothelial growth factor receptors. Proc Natl Acad Sci USA 99:7009–7014
Muniappan A, Banapour B, Lebkowski J et al (2000) Ligand-mediated cytolysis of tumor cells: use of heregulin-zeta chimeras to redirect cytotoxic T lymphocytes. Cancer Gene Ther 7:128–134
Kahlon KS, Brown C, Cooper LJ et al (2004) Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res 64:9160–9166
Zhang T, Wu MR, Sentman CL (2012) An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J Immunol 189:2290–2299
Sentman CL, Barber MA, Barber A et al (2006) NK cell receptors as tools in cancer immunotherapy. Adv Cancer Res 95:249–292
Sadelain M, Brentjens R, Riviere I (2009) The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol 21:215–223
Guest RD, Hawkins RE, Kirillova N et al (2005) The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother 28:203–211
Eshhar Z, Waks T, Gross G et al (1993) Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA 90:720–724
Holst J, Wang H, Eder KD et al (2008) Scalable signaling mediated by T cell antigen receptor-CD3 ITAMs ensures effective negative selection and prevents autoimmunity. Nat Immunol 9:658–666
Haynes NM, Snook MB, Trapani JA et al (2001) Redirecting mouse CTL against colon carcinoma: superior signaling efficacy of single-chain variable domain chimeras containing TCR-zeta vs Fc epsilon RI-gamma. J Immunol 166:182–187
Ren-Heidenreich L, Mordini R, Hayman GT et al (2002) Comparison of the TCR zeta-chain with the FcR gamma-chain in chimeric TCR constructs for T cell activation and apoptosis. Cancer Immunol Immunother 51:417–423
Gilham DE, O’Neil A, Hughes C et al (2002) Primary polyclonal human T lymphocytes targeted to carcino-embryonic antigens and neural cell adhesion molecule tumor antigens by CD3zeta-based chimeric immune receptors. J Immunother 25:139–151
Brocker T (2000) Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood 96:1999–2001
Felix NJ, Suri A, Salter-Cid L et al (2010) Targeting lymphocyte co-stimulation: from bench to bedside. Autoimmunity 43:514–525
Zang X, Allison JP (2007) The B7 family and cancer therapy: costimulation and coinhibition. Clin Cancer Res 13:5271–5279
Alvarez-Vallina L, Hawkins RE (1996) Antigen-specific targeting of CD28-mediated T cell co-stimulation using chimeric single-chain antibody variable fragment-CD28 receptors. Eur J Immunol 26:2304–2309
Kloss CC, Condomines M, Cartellieri M et al (2012) Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 31:71–75
Maher J, Brentjens RJ, Gunset G et al (2002) Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol 20:70–75
Geiger TL, Nguyen P, Leitenberg D et al (2001) Integrated src kinase and costimulatory activity enhances signal transduction through single-chain chimeric receptors in T lymphocytes. Blood 98:2364–2371
Beecham EJ, Ma Q, Ripley R et al (2000) Coupling CD28 co-stimulation to immunoglobulin T-cell receptor molecules: the dynamics of T-cell proliferation and death. J Immunother 23:631–642
Hombach A, Wieczarkowiecz A, Marquardt T et al (2001) Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J Immunol 167:6123–6131
Spear P, Barber A, Rynda-Apple A et al (2012) Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-gamma and GM-CSF. J Immunol 188:6389–6398
Loskog A, Giandomenico V, Rossig C et al (2006) Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia 20:1819–1828
Koehler H, Kofler D, Hombach A et al (2007) CD28 costimulation overcomes transforming growth factor-beta-mediated repression of proliferation of redirected human CD4+ and CD8+ T cells in an antitumor cell attack. Cancer Res 67:2265–2273
Finney HM, Akbar AN, Lawson AD (2004) Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol 172:104–113
Song DG, Ye Q, Poussin M et al (2012) CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 119:696–706
Zhong XS, Matsushita M, Plotkin J et al (2010) Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther 18:413–420
Wilkie S, Picco G, Foster J et al (2008) Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol 180:4901–4909
Pule MA, Straathof KC, Dotti G et al (2005) A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther 12:933–941
Gade TP, Hassen W, Santos E et al (2005) Targeted elimination of prostate cancer by genetically directed human T lymphocytes. Cancer Res 65:9080–9088
Haynes NM, Trapani JA, Teng MW et al (2002) Rejection of syngeneic colon carcinoma by CTLs expressing single-chain antibody receptors codelivering CD28 costimulation. J Immunol 169:5780–5786
Lamers CH, Sleijfer S, Vulto AG et al (2006) Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 24:e20–e22
Kershaw MH, Westwood JA, Parker LL et al (2006) A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 12:6106–6115
Pule MA, Savoldo B, Myers GD et al (2008) Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14:1264–1270
Till BG, Jensen MC, Wang J et al (2008) Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 112:2261–2271
Kalos M, Levine BL, Porter DL et al (2011) T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3:95ra73
Till BG, Jensen MC, Wang J et al (2012) CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood 119:3940–3950
Morgan RA, Yang JC, Kitano M et al (2010) Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 18:843–851
Brentjens R, Yeh R, Bernal Y et al (2010) Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther 18:666–668
Torikai H, Reik A, Liu PQ et al (2012) A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 119:5697–5705
Feldman SA, Goff SL, Xu H et al (2011) Rapid production of clinical-grade gammaretroviral vectors in expanded surface roller bottles using a “modified” step-filtration process for clearance of packaging cells. Hum Gene Ther 22:107–115
Bear AS, Morgan RA, Cornetta K et al (2012) Replication-competent retroviruses in gene-modified T cells used in clinical trials: is it time to revise the testing requirements? Mol Ther 20:246–249
Costandi M (2013) Kite and NCI partner on T cells. Nat Biotechnol 31:10
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Wien
About this chapter
Cite this chapter
Shamalov, K., Tal, Y., Ankri, C., Cohen, C.J. (2014). Adoptive T-Cell Immunotherapy: Perfecting Self-Defenses. In: Klink, M. (eds) Interaction of Immune and Cancer Cells. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1300-4_9
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1300-4_9
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1299-1
Online ISBN: 978-3-7091-1300-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)