
Abstract
The effect of the extract from Ginkgo biloba tree has been investigated in relation to an extended range of disorders and diseases. Commercial preparations of the extract, over-the-counter dietary supplements, or prescription products worldwide have long been advocated for their ability to improve various cognitive functions. Several extensive reviews on the chemistry, therapeutic effect, and the neuropsychological efficacy of the Ginkgo biloba extract have been published in recent years (Abad et al. 2010; Mahadevan and Park 2008; Crews et al. 2005) and they will not be discussed here. Although the exact molecular mechanisms of Ginkgo biloba extracts’ action remain unclear, the neuromodulatory activity has been attributed to the so-called terpene trilactone fractions. The synthetic and biological aspects of the compounds that comprise the terpene trilactone fraction will be presented here.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
12.1 Introduction
The effect of the extract from Ginkgo biloba tree has been investigated in relation to an extended range of disorders and diseases. Commercial preparations of the extract, over-the-counter dietary supplements, or prescription products worldwide have long been advocated for their ability to improve various cognitive functions. Several extensive reviews on the chemistry, therapeutic effect, and the neuropsychological efficacy of the Ginkgo biloba extract have been published in recent years (Abad et al. 2010; Mahadevan and Park 2008; Crews et al. 2005) and they will not be discussed here. Although the exact molecular mechanisms of Ginkgo biloba extracts’ action remain unclear, the neuromodulatory activity has been attributed to the so-called terpene trilactone fractions. The synthetic and biological aspects of the compounds that comprise the terpene trilactone fraction will be presented here.
12.2 Structure of Ginkgolides and Their Isolation from the Ginkgo biloba Extract
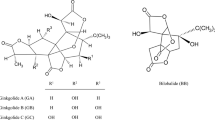
Ginkgo biloba extract contains a variety of phytochemicals, with the main components being flavonoids and terpernoids, primarily terpene trilactones. The terpene trilactone fraction can be further divided into ginkgolides and bilobalide. Ginkgolides are diterpenes with a cage-like skeleton consisting of six 5-membered rings, including a spiro[4.4]nonane carbocyclic ring, three lactones, and a tetrahydrofuran moiety (Fig. 12.1). The number and position of hydroxy groups around the skeleton provide the differentiation among ginkgolides.

Structures of ginkgolides and bilobalide from Ginkgo biloba L.
All ginkgolides have a hydroxy group at C-10 (Fig. 12.1). Ginkgolide A (GA) has only two OH functionalities that are located at C-3 and C-10. In ginkgolide B (GB) an additional OH group at C-1 is present. Ginkgolide C (GC) is the most oxygenated ginkgolide with hydroxy groups at 1, 3, 7, and 10 positions. Ginkgolide J (GJ) is isomeric to GB. Ginkgolide M (GM) lacks the tertiary hydroxy group at the C-3 position. Ginkgolide L (GL) and ginkgolide K (GK) can be viewed as dehydrated versions of GA and GB, respectively. GX, which was first reported in 2002, is the most distinct compound in the set of ginkgolides, and it was isolated from large waste accumulation during the production of the Ginkgo biloba extract (Jensen et al. 2010). Unlike other ginkgolides, GX only contains two lactone groups and features an unsaturated six-membered ring. The lack of hydroxy groups in the 1 and 7 positions might indicate a relationship of GX to GA.
GA, GB, GC, and GM were initially isolated and separated from the root bark of five Ginkgo biloba trees, which were damaged by a typhoon in 1967 (Nakanishi 2005). Their structures were deduced by a combination of chemical and spectroscopic approaches (Maruyama et al. 1967a, b, c, d; Woods et al. 1967). GJ was isolated in 1987 from the Ginkgo biloba leaves (Weinges et al. 1987a).
The sesquiterpenoid bilobalide (BB) is related to ginkgolides by the presence of three lactone rings, one of which contains an α-OH, and the tert-butyl group (Fig. 12.1). Distinctions are made by the absence of the rings A and F, and the tetrahydrofuran ring found on the ginkgolide skeleton is replaced by a lactone. The tert-butyl group found in BB (and in ginkgolides) is very rare for terrestrial natural products, but it is more common in marine natural products (Bisel et al. 2008). Although BB exhibits a spectrum of biological activities (Strømgaard and Nakanishi 2004), from the chemical point of view, BB is much more labile than the ginkgolides, and as a result very few derivatives have been prepared (Nakanishi et al. 1971; Weinges and Bäehr 1972; Weinges et al. 1987b). Therefore, the synthetic and bioorganic aspects of BB will not be discussed here.
Several spectroscopic (van Beek et al. 1993; Li et al. 2004), spectrometric (Lang and Wai 1999), and chromatographic (van Beek 2005) methods for quantification of terpene trilactones in the extract have been developed. However, for the evaluation of the biological activities of ginkgolides and for the preparation of the required derivatives, an access to large quantities of pure compounds is required. Typically, commercially available leaf extracts, e.g., BioGinkgo™ from Pharmanex, are used as a starting point. BB and the main ginkgolides, i.e., GA, GB, and GC, constitute up to 7 wt.% of the whole Ginkgo biloba extract. Several procedures have been reported that allowed for a fast and facile separation of the terpene trilactone fraction from the rest of the extracts’ components. Specifically, boiling the extract in dilute hydrogen peroxide solution was noted to prevent the formation of stable emulsions that usually hamper the subsequent extraction and purification steps (Lichtblau et al. 2002). Additionally, direct solid/liquid extraction of the solid BioGinkgo™ with ethyl acetate quantitatively, and almost selectively, led to isolation of all ginkgolides and BB from the extract (Nakanishi et al. 2005).
Similarities in solubilities and chromatographic behaviors might be considered among the main challenges associated with the separation of ginkgolides. In fact, only BB can be easily separated from the ginkgolides using chromatography. Typically the difficulties arise during separation of GA/GB and GC/CJ pairs, which cannot be separated by routine chromatographic techniques (Weinges and Bäehr 1972; Lobstein-Guth et al. 1983). In GB, for example, separation difficulties are attributed to strong hydrogen bonding between the 1-OH and 10-OH, which makes the polarity of GB very similar to that of GA. However, for the GC/GJ pair, the presence of multiple hydroxy groups in each ginkgolide negates the difference in polarities. The use of NaOAc impregnated silica gel allowed to obtain individual ginkgolides in high purity using medium-pressure liquid chromatography (van Beek and Lelyveld 1997). Derivatization of a GA/GB mixture with an ether functionality to alter polarities, followed by a facile separation and subsequent conversion of the derivatives to pure ginkgolides, was successfully accomplished in the early 1900s. This proved to be an efficient route for separation of larger quantities of ginkgolide mixtures (Corey et al. 1992). Subsequently, a convenient method that involved benzylation, column chromatography separation and debenzylation allowed for a gram-scale access to all individual ginkgolides (Jaracz et al. 2004a).
12.3 Synthetic Studies on Ginkgolides
12.3.1 Modification of the OH Functionalities
Distinct reactivities of the OH groups around the ginkgolide skeleton present an opportunity for a straightforward and selective functionalization of these compounds. It has been demonstrated that selective silylation at 1-OH can be achieved by reaction of ginkgolides with bulky silylating agents, such as t-butyl(chloro)diphenylsilane, in the presence of imidazole as a base (Weinges and Schick 1991). To date, this remains virtually the only route for direct functionalization of the 1-OH group.
Transformations such as alkylation, benzylation, allylation, propargylation, etc., occur almost exclusively at C-10. Furthermore, it was recognized early on that deprotonation of the 10-OH group of GB and GC is much more facile than deprotonation of the 10-OH of GA (Corey et al. 1992). This phenomenon was attributed to the neighboring group participation from 1-OH, and the resulting alkoxides are apparently stabilized by the intramolecular hydrogen bonding. It should be pointed out that alkylations at GA’s 10-OH are still possible, albeit they require an excess of stronger bases, such as NaH or KH.
Acetylation of the hydroxy groups can be tuned by the proper choice of a base (Jaracz et al. 2002). Specifically, acetylation of all 1-, 7-, and 10-OH groups is possible when GC is treated with Ac2O/pyridine, whereas the acetylation at 10-OH or 7-OH takes place when GC is treated with Ac2O/HOBr/pyridine or AcOH/H2SO4, respectively. Significantly, acetylation of GA using Ac2O/NaOAc induces the elimination of the 3-OH group in addition to the acetylation of 10-OH.
Exclusive derivatization of 7-OH was also realized when GC was reacted with triflic anhydride in the presence of pyridine to prepare 7-OTf-GC (Cazaux et al. 1995). The OTf group provides a convenient entry for the preparation of a diverse range of 7-substituted ginkgolides (Vogensen et al. 2003).
12.3.2 Ginkgolide Interconversion
The isolation of ginkgolides from Ginkgo biloba extracts represents the main source of these trilactones for all synthetic, structural, and biological studies. However, some ginkgolides are more abundant in the extracts than others. In addition, the leaf extract contains only GA, GB, GC, and GJ, with GJ being the minor component, whereas GM is only present in the root bark of the Ginkgo trees. In this light, ginkgolide interconversion becomes an attractive way to access minor ginkgolides. Although it would be most appealing to start with the most abundant GA and introduce C–O functionality via selective C–H activation, for example, on a scaffold as complex as the ginkgolides’, it still presents a synthetic challenge. Therefore, a number of accounts have addressed the selective removal of the OH groups from the most oxygenated GC en route to other ginkgolides.
The groups of Weinges (Weinges and Schick 1991), Corey (Corey et al. 1992), and Teng (Cazaux et al. 1995) took advantage of selective silylation at 1-OH, alkylation at 10-OH, and sulfonylation at 7-OH, respectively, to install suitable moieties which aided in the subsequent conversion of GC into GB (Scheme 12.1). In addition, GB was converted to GA via protecting the 10-OH, and subjecting the 1-OH to xanthane formation, following the reduction with tri-n-butyltin hydride and deprotection (Corey and Ghosh 1988).

Interconversions of ginkgolides
Later it was shown that selective functionalization of the 1-OH of GC via formation of phenylthiocarbonate (Scheme 12.1), followed by the deoxygenation under Barton–McCombie conditions could lead to an efficient conversion of GC to GJ in just two steps via the formation of 1.1 (Dzyuba et al. 2005). The selectivity of the thiocarbonation step was solvent controlled, and functionalization of the 10-OH was possible by using CH3CN as the solvent. This derivative was deoxygenated as well to produce a GC analogue that lacked the 10-OH group. In DMF, the thiocarbonation of the 1-OH of GB was achieved via a migration from the position 10 (1.2) to the position 1 (1.3) in the presence of DMAP; subsequent Barton–McCombie deoxygenation yielded GA in a moderate yield.
Access to the rare GM was reported in a few accounts. The first synthesis of GM was accomplished in 1993 via silylation of the 1-OH of GC, followed by a nonselective acetylation, which required a chromatographic isolation of the 7-OAc derivative. Subsequent dehydration, hydrogenation, deacetylation, and DMAP-promoted epimerization afforded GM (Weinges et al. 1993a, b). A more recent example relied on the selective dehydration of the easily accessible 10-Bn-GC, i.e., an intermediate in the isolation of ginkgolides from the extract (Jaracz et al. 2004a) as the starting material (Scheme 12.1). 10-Bn-GC was successfully dehydrated to 10-Bn-GL, and subsequent hydrogenation and DMAP-promoted epimerization of the methyl group in position 14 furnished GM in a moderate overall yield (Bolshakov et al. 2006).
GL is a dehydrated version of GA, and is likely to be produced as an impurity during the isolation of terpene trilacones fraction from the Ginkgo biloba extract. Several protocols demonstrated that GA can be converted to GL directly upon treatment with either pyridine/POCl3 (Weinges et al. 1993a, b) or DAST (Bolshakov et al. 2006) The DAST-mediated dehydration turned out to be generally applicable for the ginkgolides as a clean, high yielded elimination of the 3-OH group was observed upon reaction with GB and GC derivatives.
In 1992, Corey’s group suggested that some of the methodology developed for ginkgolide interconversion could and should be used for introduction of isotopes onto the ginkgolide skeleton (Corey et al. 1992). These isotopically labeled ginkgolides are of interest since they enable metabolic and pharmacological studies. Specifically, the introduction of 3H and 18F isotopes into position 7 was disclosed in the early 2000s via several two-step procedures using GC as a starting material (Strømgaard et al. 2004; Suehiro et al. 2004). Notably, these labeled GB were used for some in vivo studies to map bio-distribution of ginkgolides in various tissues (Suehiro et al. 2005). In addition, the methodology for installing a 14C label on GA was developed (Weinges et al. 2000).
12.3.3 Core-Modified Ginkgolides
Although a great number of accounts on the synthetic transformations of ginkgolides relied on the modification of the OH functionalities, it might be argued that functionalization of the hydroxy groups produces ligands that are drastically larger than the original ginkgolides, and thus would be more likely to alter the mode of interaction with a receptor. In this light, modifications of the ginkgolide skeleton might be appealing. The first set studies on the modification of the ginkgolide core were performed during the structure elucidation in the mid/late 1960s. The majority of efforts focused on using GA as a starting point, in view of its higher abundance in the extract. It was then established that the treatment of GA with LiAlH4, followed by an acidic work-up and subsequent sublimation and/or extensive vacuum drying produced GA-triether (Maruyama et al. 1967b; Woods et al. 1967). This compound was crucial for determining the overall structure of ginkgolides. A more direct and facile synthesis of GA-triether was disclosed in the mid-2000s (Scheme 12.2), when all of GA’s lactone moieties were converted into the corresponding ethers in a stepwise manner (Ishii et al. 2005). The first reduction to the lactol was shown to occur at ring F, followed by the reduction at ring C, with the final reduction taking place at ring E. Except for the reduction of lactol C, mixtures of epimers were obtained, while a single epimer was obtained upon the reduction of the lactone C, which is consistent with the presence of the 10-OH group. The GA-triether was also prepared directly from GA in just two steps using the excess of the reagents. This methodology, however, appeared to be of limited use, as its application to the delactonization of GB and GC proved fairly inefficient. Only GB-monoether was obtained in a moderate yield, and deoxygentation of both lactol of GB-monoether and lactol of GC failed completely.

Reduction of lactones of GA and GB
Notably, the modification of the lactone moieties of 10-Bn-GB derivatives (Scheme 12.2) was also accomplished upon treatment with NaBH4 and subsequent functionalization of the lactol to give 2.1 after chromatographic separation (Tanaka et al. 2005a). The lactol functionality could be liberated upon treatment with K2CO3 in MeOH to give 10Bn-GB-lactol. The lactone C was reduced preferentially due to the neighboring group participation of the 10-O (Scheme 12.2). However, the lactone of ring E was also reduced to the corresponding lactol, albeit at a slower rate. This reduction might be attributed to the close proximity of the 3-OH, since the lactone of ring F, which does not have any neighboring OH groups that were proposed to aid in the coordination of the reducing agent, was not reduced at all. No subsequent deoxygenations or other modifications of the lactol functionalities were carried out.
Additionally, it was demonstrated that ring C of GA is susceptible to various modifications (Scheme 12.3). Oxidation of GA was shown to produce 10-oxo-GA, which upon exposure to UV light produced photodehydro-GA that features a seven five-membered ring structure (Maruyama et al. 1967d). This compound was also obtained as a very minor product upon treating GA with the Jones reagent (Hu et al. 2001). The 10-oxo-GA was also shown to undergo a ring opening of the lactone C upon treatment with various amines to produce α-oxo-amide derivatives (Hu et al. 2001). Furthermore, submitting GA to alkali fusion under elevated temperatures led to the removal of ring C, yielding bisnor GA, while the subsequent oxidation produced an unnatural trilactone, i.e., dehydrobisnor-GA (Maruyama et al. 1967d).

Transformations of GA
It is of interest to note that exposing GA to Ac2O/NaOAc produces an unsaturated, acetylated derivative (Scheme 12.3), whose reactivity was explored in a series of accounts from Weinges’ group (Weinges et al. 1986, 1997a, b). Interesting and unique structures, i.e., 3.1, 3.2, and 3.3, with partially removed and/or modified rings F and D were obtained in several steps (Scheme 12.3).
Ring F of GA could also be easily opened by dehydration of the 3-OH, using POCl3/pyridine which converted GA into GL. The conversion was followed by the oxidation of the alkene functionality into the diol with OsO4/NaClO3, and subsequent oxidation with NaIO4 to produce F-nor-GA (Hu et al. 1999). Similar transformations were also reported for GB.
During the ginkgolide acetylation studies in the early 2000s (Scheme 12.4), it was noted that in the presence of iPr2NEt an efficient translactonization of ring E produced a novel ginkgolide skeleton 4.1 (Jaracz et al. 2002). The availability of the X-ray structure helped in the rationalization of the mechanism for this interesting transformation. It was proposed that the intramolecular ring opening of the E lactone by the deprotonated 7-OH and the capture of the intermediate with acetic anhydride were responsible for the observed formation of the iso-ginkgolide. Further evidence for the observed mechanism was obtained by treating 7-OAc-GC with either iPr2EtN or Ac2O/iPr2EtN, and observing no rearrangement products.

Translactonizations of GC derivatives
In addition, when reacted with 2,6-lutidine/MeOH, 7-OTf-GC was shown to undergo a unique ring opening of lactone C (Scheme 12.4), followed by intramolecular substitution of the triflate group to produce a relactonized product 4.2 (Vogensen et al. 2003). This relatively thermodynamically unstable ginkgolide derivative was cleanly transformed into epi-7-OH-GC under basic conditions.
The special reactivity of the C lactone ring, i.e., α-hydroxy-lactone group (Scheme 12.5), was also explored as a key step en route to some elaborated ginkgolide scaffolds (Tanaka et al. 2005b). Allylation and propargylation of GB produced the corresponding 10-allyl (5.1) or 10-propargyl (5.2) derivatives of GB, which were treated with the allyl Grignard reagent to yeild 5.3 and 5.4, respectively. Subsequently, either alkene or alkene/alkyne metathesis using Grubbs’ second generation catalysts afforded seven-membered, bowl-like structures 5.5 and 5.6 (Scheme 12.5). The newly installed ene/diene moieties may potentially be used as a handle for the subsequent functionalization.

Synthesis of 7-membered ginkgolides via olefin metatheses
12.4 Bioorganic Studies on Ginkgolides and Their Derivatives
The extracts from Ginkgo biloba trees demonstrate a diverse range of activities, albeit with various levels of efficiencies. Arguably, this could be attributed to the heterogeneity of terpene trilactone fractions in various batches. Therefore, studies with ginkgolides might prove to be more viable for establishing and expanding the biological mode of action.
12.4.1 Interactions with Platelet-Activating Factor Receptor
In the mid-1980s, GB was identified as a potent antagonist of the platelet-activating factor receptor (PAFR) in vitro (Braquet 1987; Braquet et al. 1991). PAFR is a member of the G protein-coupled receptor family and its mRNA was detected among several neuronal tissues including hypothalamic, cerebellar, hippocampal, and cortical. The expression of PAFR was noted in both neuronal and glial cells (Maclennan et al. 2002). PAF is a phospholipid (1-O-alkyl-2-acetyl-sm-glycero-3-phosphocholine, with hexadecyl to octadecyl alkyl chain lengths) that is produced in a variety of tissues and is a very potent PAFR agonist that can exert its effects at picomolar concentrations. Binding of PAF to PAFRs is believed to trigger a number of events in the CNS, including Ca2+ mobilization, long-term potentiation, and apoptosis. However, the specific details of PAF and PAFR modulation on neuronal function still remain to be elucidated.
Compared to GB, other ginkgolides either are inactive or exhibit significantly lower levels of activity towards PAFR. From the structure–activity point of view, it is of interest to note that a very fine balance exists between potency and the number of hydroxy groups around the ginkgolide skeleton (Strømgaard and Nakanishi 2004). Specifically, as an antagonist, GA (a ginkgolide that lacks the 1-OH group) is several fold less efficient as compared to GB. GC, which has an OH-group at position 7, is an order of magnitude less potent inhibitor than GB. The importance of the t-Bu group was also established (Corey and Rao 1991).
The majority of studies on ginkgolide–PAFR interactions were done using radioactive ligand displacement assays. However, the nonradioactive microphysiometry assay was developed and proved to be effective in determining the functional activity of native ginkgolides, their derivatives, and the Ginkgo biloba extract (Krane et al. 2003).
Following the initial reports on the antagonistic activity of GB, a variety of synthetic studies that aimed at improving the potency of GB followed. It was established that the introduction of substitutents in positions 1 and 10 led to potent PAFR antagonists. Hence, it was suggested that the incorporation of photoaffinity labels in position 10 might produce potent antagonists, which would allow mapping of the site-specific interactions between ginkgolides and PAFR. Initial studies demonstrated that the presence of benzophenone, trifluoromethyldiazirine, and tetraphenylazide photoactivatable moieties produced potent antagonists (Strømgaard et al. 2002). It was also shown that 10-substituted GB derivatives were several fold more active than the corresponding 10-substituted GC derivatives, which is in accord with the higher potency of GB over GC. In addition, it was demonstrated that the introduction of a fluorescent group, which would aid in the fragment characterization steps such as dansyl, at the 1-OH did not reduce the activity. Photolabeling studies using the synthesized ginkgolide probes have not been reported to date.
A recent account examined the synthesis of photoaffinity biotin containing chimeras of GA and GB (Kato et al. 2007), which could potentially simplify the post-photoaffinity isolation and identification steps significantly. The strategies relied on the incorporation of the benzophenone moiety at C-10, while incorporating biotin via a linker at position 15 of ring F, since modifications of this part of the molecule are known to have no affect on the activity towards PAFR. The activity of these benzophenone–biotin chimeric compounds towards PAFR was not evaluated.
The effect of the nature of the substituent at position 7 and its stereochemistry was investigated in some detail, but proved to be particularly involved (Vogensen et al. 2003). As compared to GB (K i = 0.88 μM), the binding of the 7-NHR derivatives depended strongly on the nature of the R-group. 7-NH2 compound (R = H) showed a drastic decrease in affinity (K i = 8.64 μM) while the alkyl versions, R = Me and Et, appeared to be somewhat equipotent to GB with a K i of 0.61 and 1.62 μM, respectively. Significantly, introduction of chlorine at the 7-position produced a ginkgolide congener with drastically improved binding ability to PARF (K i = 0.11 μM).
Acetylation of some or all hydroxy groups of ginkgolides GA, GB, and GC appeared to produce compounds with similar or reduced antagonistic activities towards PAFR as compared to the native ginkgolides (Jaracz et al. 2002). The acetylated version of the newly synthesized iso-GC (Scheme 12.4) was shown to be an equally inefficient antagonist.
A variety of synthetic derivatives that lacked ring F, for example, were also prepared and proved to be efficient in their ability to inhibit platelet aggregation (Corey and Gavai 1989). Some of the derivatives were used for several theoretical SAR studies on ginkgolide–PAFR interactions. A three-dimensional quantitative SAR investigated the effect of ginkgolides and their analogues (Chen et al. 1998; Zhu et al. 2005). In general, very good correlations between experimental and calculated activity values were observed. Specifically, the calculations demonstrated that bulky, hydrophobic substitutents at 1-OH and 10-OH would lead to more potent analogues. In agreement, the synthesized derivatives of GB with benzyl and BOM moieties at those positions exhibited IC50 values several fold lower than GB itself. Collectively, the aforementioned structure–activity studies are summarized in Fig. 12.2.

Ginkgolide–PAFR structure–activity relationships
12.4.2 Interactions with Glycine Receptors
Glycine gated chloride channels (GlyRs) are one of the major inhibitory receptors in the CNS. These ligand-gated ion channels are found in the hippocampus, cortex, spinal cord, and brainstem. GlyRs are found as either homomeric receptors, which consist of four identical α subunits (α1–α4), or heteromeric ones, which are composed of α and β subunits.
In 2002, GB was demonstrated to be an efficient blocker of GlyRs in pyramidal hippocampal neurons (Kondratskaya et al. 2002). Based on the whole-cell voltage-clamp and concentration-clamp recording techniques, using glycine as an agonist and strychnine as an antagonist of GlyR, it was suggested that GB inhibited glycine-mediated currents by interacting at the pore region of the GlyR. Subsequent studies that introduced a mutation in the pore region of the α1 subunit proved this mode of action (Kondratskaya et al. 2004). In addition, investigations using recombinant GlyRs demonstrated a high affinity of GB toward the β subunit (Kondratskaya et al. 2005). It was also indicated that GB could be used as a probe for discrimination between synaptic and extrasynaptic glycine currents.
The antagonistic effect of GB on GlyRs was further confirmed using embryonic cortical neurons in a series of whole-cell patch-clamp measurements (Ivic et al. 2003). This account also investigated the ability of other ginkgolides to interact with GlyRs. It was determined that GB, GC, and GM were more potent antagonists than GA and GJ, thus indicating the significance of the 1-OH group.
An extensive structure–activity study on the interaction of 49 GC derivatives, including ether, ester, and carbamoyl functionalities with homomeric α1 GlyRs revealed that hydroxy groups were required for the GlyR inhibition as virtually any modification of the hydroxy functionalities around the ginkgolide skeleton produced less potent compounds (Jaracz et al. 2004b). A follow-up study focused on more subtle changes of the ginkgolides’ structure to gage further requirements about the biological activity towards α1, α2, and αβ GlyR using the FLIPR membrane potential assay as well as a patch-clamp technique (Jensen et al. 2007). In addition to native ginkgolides, i.e., GA, GB, GC, GJ, and GM, 29 ginkgolide derivatives were analyzed using in vitro and in silico screenings. Among native ginkgolides, GM was shown to be the most potent antagonist, whereas virtually every synthetic derivative turned out to be less potent than the native compounds. The rigidity of the ginkgolide core was identified as the main determinant of the antagonistic properties towards GlyR. Overall, increasing the activity of ginkgolides toward GlyRs appears to be much more challenging as compared to the PAFR.
A recent account reported on the ability of GX to act as a potent antagonist of GlyR (Jensen et al. 2010). Unlike other ginkgolides, GX exhibited complete selectivity for the homomeric GlyRs over the heteromeric GlyRs. Based on docking studies, different modes of interactions of GX with GlyRs, as compared to other ginkgolides, were proposed. The full potential of this ginkgolide is yet to be elucidated, but its restricted availability might prevent extensive biological studies.
12.4.3 Interactions with Amyloid Peptides and Relations to Alzheimer’s Disease
Alzheimer’s disease is an age-related pathology, which is responsible for the deterioration of cognitive function. The underlying causes of this disease are extremely complex, and are yet to be fully understood. However, it is largely accepted that conformational changes and aggregation of so-called amyloid peptides, i.e., Aβ, are responsible for the occurrence and progression of the disease (Hardy and Selkoe 2002). Typically, full-length amyloid peptides that are composed of 40–42 amino acids, commonly referred to as Aβ1-40 or Aβ1-42, are used for various in vitro and in vivo studies. In view of the difficulties associated with handling these full-length peptides and the reproducibility of the results, a number of truncated versions, such as Aβ25-35, for example, have been used as other suitable, easy-to-handle models.
Studies on developing inhibitors of Aβ aggregation have attracted enormous attention from academic and industrial research laboratories. Ginkgolides have been used in several in vitro and in vivo studies to test their effects on amyloid-induced damage. Initial studies demonstrated that among the main native ginkgolides, GJ was the most efficient compound in suppressing the aggregation of Aβ1-40 peptide in vitro by using the thioflavin T dye binding method (Luo et al. 2002). Subsequently, the neuroprotective effect of GJ was confirmed by preventing Aβ1-42-induced inhibition of long-term potentiation in the CA1 region of mouse hippocampal slices (Vitolo et al. 2009). This ginkgolide was also demonstrated to be efficient in inhibiting cell death of rodent hippocampal neurons caused by the Aβ1-42 oligomers. Other ginkgolides, such as GA and GB as well as the synthetic derivative, GA-triether (Scheme 12.2), were also able to partially block Aβ1-42-induced damage to synaptic plasticity.
Spectroscopic studies indicated that the effect of ginkgolides in modulating the aggregation of Aβ25–35 peptide was relatively small (He et al. 2008). The absence of a specific interaction between ginkgolides, which was established with the aid of some lactone-free derivatives, such as GA-monoether and GA-diether (Scheme 12.2), was attributed to the inability to affect the aggregation of amyloid peptides. Thus, it could be suggested that ginkgolides might not be suitable inhibitors of amyloid aggregation per se and that a direct interaction of ginkgolides with Aβ peptides is an unlikely mode of their neuroprotective actions.
Using neuroblastoma cell lines and primary cortical neurons, it was demonstrated that both GA and GB, at nano- to micromolar concentrations, could inhibit Aβ1-42-induced cell death (Bate et al. 2004). It was also established that GB is somewhat more efficient than GA. The protective effects of ginkgolides were suggested to correlate with reduced caspase-3 responses, a known apoptosis marker, as well as with the ability to suppress PAF-induced effects. In a related study, the effect of GA and GB on Aβ1-42 and PAF-induced reduction of the presynaptic membrane protein, synaptophysin, was investigated (Bate et al. 2008). Although ginkgolides did not alter the binding of Aβ1-42 into neurons, they were able to increase the neuronal survival in the presence of high micromolar concentrations of amyloid peptide.
The effect of GB and GA was also investigated in relation to the Aβ25-35-induced acetylcholine release from hippocampal brain slices (Li et al. 2004). It was demonstrated that while GB could effectively restore the acetylcholine release from rat hippocampal slices to their control levels in a dose-dependent manner, GA was completely inactive. It was proposed that the direct interaction of GB on the cholinergic nerve terminals was responsible for the observed effects.
Recently, Aβ25–35-induced apoptosis was studied in hippocampal neurons (Xiao et al. 2010). The changes in cell viability, morphology, caspase-3 activity, expression of brain-derived neurotrophic factor mRNA, and protein synthesis were detected due to the presence of Aβ25–35 peptides. It was found out that GB could reduce the amyloid-induced insults.
12.5 Conclusions
As the knowledge of ginkgolide–receptor interactions continues to improve and expand, new insights into the molecular mode of action of these compounds and the discovery of novel biological targets should be anticipated. Although ginkgolides were examined in a great wealth of pharmacological assays, it is presently unclear whether any drugs could arise from them. However, it is plausible that the combination of their unique and interesting structure and the great history of these natural products will continue to be the source of inspiration for organic synthesis, bioorganic chemistry, pharmacology, and chemical biology.
References
Abad MJ, Bedoya LM, Bemejo P (2010) An update on drug interactions with the herbal medicine Ginkgo biloba. Curr Drug Metab 11:171–181
Bate C, Salmona M, Williams A (2004) Ginkgolide B inhibits the neurotoxicity of prions or amyloid-β1-42. J Neuoroinflammation. http://www.jneuroinflammation.com/content/1/1/4
Bate C, Tayebi M, Williams A (2008) Ginkgolides protect against amyloid-β1–42-mediated synapse damage in vitro. Mol Neurodegeneration 3:1. http://www.molecularneurodegeneration.com/content/3/1/1
Bisel P, Al-Momani L, Muller M (2008) The tert-butyl group in chemistry and biology. Org Biomol Chem 6:2655–2665
Bolshakov S, Dzyuba SV, Nakanishi K (2006) A concise synthesis of ginkgolide M, a minor component of a terpene trilactone fraction from Ginkgo biloba roots. J Nat Prod 69:429–431
Braquet P (1987) The ginkgolides: potent platelet-activating factor antaginists isolated from Ginkgo biloba L.: chemistry, pharmacology and clinical applications. Drug of the Future 12:643–699
Braquet P, Esanu A, Buisine E, Hosford D, Broquet C, Koltai M (1991) Recent progress in ginkgolide research. Med Res Rev 11:295–355
Cazaux J-B, Dafniet M, Rebollo J, Teng B-p (1995) Conversion of ginkgolide C to ginkgolide B. Brit UK Pat Appl GB 2288599 A 19951025
Chen JZ, Hu LH, Jiang HL, Gu JD, Zhu WL, Chen ZL, Chen KX, Ji RY (1998) A 3D-QSAR study on ginkgolides and their analogues with comparative molecular field analysis. Bioorg Med Chem Lett 8:1291–1296
Corey EJ, Gavai AV (1989) Simple analogues of ginkgolide B which are highly active antagonosts of platelet activating factor. Tetrahedron Lett 30:6959–6962
Corey EJ, Ghosh AK (1988) Total synthesis of ginkgolide A. Tetrahedron Lett 29:3205–3206
Corey EJ, Rao KS (1991) Enantioselective total synthesis of ginkgolide derivatives lacking the tert-butyl group, an essential structural subunit for antagonism of platelet activating factor. Tetrahedron Lett 32:4623–4626
Corey EJ, Rao KS, Ghosh AK (1992) Intramolecular and intermolecular hydroxyl reactivity differences in ginkgolides A, B and C and their chemical applications. Tetrahedron Lett 33:6955–6958
Crews WD, Harrison DW, Griffin ML, Falwell KD, Crist T, Longest L, Hehemann L, Rey ST (2005) The neurophyscological efficacy of Ginkgo preparations in healthy and congnitivily intact adults. HerbalGram 67:43–62
Dzyuba SV, Bolshakov S, Nakanishi K (2005) Regioselective thionocarbonation of ginkgolides: facile preparation of ginkgolide J. Tetrahedron Lett 46:7797–7799
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356
He J, Petrovic AG, Dzyuba SV, Berova N, Nakanishi K, Polavarapy PL (2008) Spectrosopic investigation of Ginkgo biloba trialactones and their interaction with amyloid peptide Aβ(25–35). Spectrochim Acta A 69:1213–1222
Hu L, Chen Z, Cheng Z, Xie Y (1999) Chemistry of ginkgolides: structure–activity relationship as PAF antagonists. Pure Appl Chem 71:1153–1156
Hu LH, Chen ZL, Xie YY (2001) Synthesis and biological activity of amide derivatives of ginkgolide A. J Asian Nat Prod Res 3:219–227
Ishii H, Dzyuba SV, Nakanishi K (2005) Lactone-free ginkgolides via regioselective DIBAL-H reduction. Org Biomol Chem 3:3471–3472
Ivic L, Sands TTJ, Fishkin N, Nakanishi K, Kriegstein AR, Strømgaard K (2003) Terpene trialctones from Ginkgo biloba are antagonists of cortical glycine and GABAA receptors. J Biol Chem 278:49279–49285
Jaracz S, Strømgaard K, Nakanishi K (2002) Ginkgolides: selective acetylations, translactonization, and biological evaluation. J Org Chem 67:4623–4626
Jaracz S, Malik S, Nakanishi K (2004a) Isolation of ginkgolides A, B, C, J and biloblide from G. biloba extracts. Phytochemistry 65:2897–2902
Jaracz S, Nakanishi K, Jensen AA, Strømgaard K (2004b) Ginkgolides and glycine receptors: a structure–activity relationship study. Chemistry 10:1507–1518
Jensen AA, Begum N, Vogensen SB, Knapp KM, Gundertoffe L, Dzyuba SV, Ishii H, Nakanishi K, Kristiansen U, Strømgaard K (2007) Probing the pharmacophore of ginkgolides as glycine receptor antagonists. J Med Chem 50:1610–1617
Jensen AA, Bergmann ML, Sander T, Balle T (2010) Ginkgolide X is a potent antagonist of anionic cys-loop receptors with a unique selectivity profile at glycine receptors. J Biol Chem 285:10141–10153
Kato E, Howitt R, Dzyuba SV, Nakanishi K (2007) Synthesis of novel ginkgolide photoaffinity–biotin probes. Org Biomol Chem 5:3758–3761
Kondratskaya EL, Lishko PV, Krishtal OA, Chatterjee SS (2002) BN52021, A platelet activating factor antagonist, is a selective blocker of glycine-gated chloride channel. Neurochem Int 40:647–653
Kondratskaya EL, Firsunov AI, Chatterjee SS, Kristal OA (2004) Ginkoglide B preferentially blocks chloride channels formed by heterometic glycine receptors in hippocampal pyramidal neurons of rat. Brain Res Bull 63:309–314
Kondratskaya EL, Betz H, Krishtal OA, Laube B (2005) The β subunit increases the ginkgolide B sensitivity of inhibitory glycine receptors. Neuropharmacology 49:945–951
Krane S, Kim SR, Abrell LM, Nakanishi K (2003) Microphysiometric measurement of PAF receptor responses to ginkgolides. Helv Chim Acta 86:3776–3786
Lang Q, Wai CM (1999) An extraction method for determination of ginkgolides and bilobalide in Ginkgo leaf extracts. Anal Chem 71:2929–2933
Li C-Y, Lin C-H, Wu C-C, Lee K-H, Wu T-S (2004) Efficient 1H nuclear magnetic resonance method for improved quality control analyses of ginkgo constituents. J Agric Food Chem 52:3721–3725
Lichtblau D, Berger JM, Nakanishi K (2002) Efficient extraction of ginkgolides and bilobalide from Ginkgo biloba leaves. J Nat Prod 65:1501–1504
Lobstein-Guth A, Briançon-Scheid F, Anton R (1983) Analysis of terpenes from Ginkgo biloba L. by high-performance liquid chromatography. J Chromatogr 267:431–438
Luo Y, Smith JV, Paramasivam V, Burdick A, Curry KJ, Buford JP, Khan I, Netzer WJ, Xu H, Butko P (2002) Inhibition of amyloid-β aggregation and caspase-3 activation by the Ginkgo biloba extract EGb761. Proc Natl Acad Sci USA 99:12197–12202
Maclennan KM, Darlington CL, Smith PF (2002) The CNS effects of Ginkgo biloba extracts and ginkgolide B. Prog Neurobiol 67:235–257
Mahadevan S, Park Y (2008) Multifaceted therapeutic benefits of Ginkgo biloba L.: chemistry, efficacy, safety, and uses. J Food Sci 73:R14–R19
Maruyama M, Terahara A, Itagaki Y, Nakanishi K (1967a) The ginkgolides. I. Isolation and characterization of the various groups. Tetrahedron Lett 4:299–302
Maruyama M, Terahara A, Itagaki Y, Nakanishi K (1967b) The ginkgolides. II. Derivations of partial structures. Tetrahedron Lett 4:303–308
Maruyama M, Terahara A, Nakadaira Y, Woods MC, Nakanishi K (1967c) The ginkgolides. III. The structure of the ginkgolides. Tetrahedron Lett 4:309–313
Maruyama M, Terahara A, Woods MC, Tagaki Y, Nakanishi K (1967d) The ginkgolides. IV. Stereochemistry of the ginkgolides. Tetrahedron Lett 4:315–319
Nakanishi K (2005) Terpene trilactones from Ginkgo biloba: from ancient times to the 21st century. Bioorg Med Chem 13:4987–5000
Nakanishi K, Habaguchi K, Nakadaira Y, Woods MC, Maruyama M, Major RT, Alauddin M, Patel AR, Weinges K, Baehr W (1971) Structure of bilobalide, a rare tert-butyl containing sesquiterpenoid related to the C20-ginkgolides. J Am Chem Soc 93:3544–3546
Nakanishi K, Jaracz S, Malik S, Ishii H, Dzyuba SV (2005) Separation of ginkgolides and bilobalide from G. biloba by column chromatography. PCT Int Appl WO 20005046829
Strømgaard K, Nakanishi K (2004) Chemistry and biology of terpene trilactones from Ginkgo biloba. Angew Chem Int Ed 43:1640–1658
Strømgaard K, Saito DR, Shindou H, Ishii S, Shimizu T, Nakanishi K (2002) Ginkgolide derivatives for photolabeling studies: preparation and pharmacological evaluation. J Med Chem 45:4038–4046
Strømgaard K, Suehiro M, Nakanishi K (2004) Preparation of a tritiated ginkgolide. Bioorg Med Chem Lett 14:5673–5675
Suehiro M, Simpson N, van Heertum R (2004) Radiolabeling of ginkgolide B with 18 F. J Label Compd Radiopharm 47:485–491
Suehiro M, Simpson NR, Underwood CJ, Nakanishi K, van Heertum R (2005) In vivo biodistribution of ginkgolide B, a constituent of Ginkgo biloba, visualized by microPET. Planta Med 71:622–627
Tanaka K, Kester KD, Berova N, Nakanishi K (2005a) Preparation of ginkogolide and F-seco-ginkgolide lactols: the unique reactivity of α-hydroxy lactones towards NaBH4. Tetrahedron Lett 46:531–534
Tanaka K, Pimentel M, Berova NK (2005b) Unique reactivity of α-alkoxy ginkgolide lactones to nucleophilic reagents: preparation of new lactol derivatives. Bull Chem Soc Jpn 78:1843–1850
T-f L, C-f C, Wang LCH (2004) Effect of ginkgolides on β-amyloid-supressed acetylcholine release from rat hippocampal slices. Phytother Res 18:556–560
van Beek TA (2005) Ginkgolides and bilobalide: their physical, chromatographic and spectroscopic properties. Bioorg Med Chem 13:5001–5012
van Beek TA, Lelyveld GP (1997) Preparative isolation and separation procedure for ginkgolide A, B, C, and J and bilobalide. J Nat Prod 60:735–738
van Beek TA, van Veldhuizen A, Lelyveld GP, Pron I, Lankhorst PP (1993) Quantification of bilobalide and ginkgolides A, B, C and J by means of nuclear magnetic resonance spectroscopy. Phytochem Anal 4:261–268
Vitolo O, Gong CZ, Ishii H, Jaracz S, Nakanishi K, Arancio O, Dzyuba SV, Lefort R, Shelanski M (2009) Protection against β-amyloid induced abnormal synaptic function and cell death by ginkgolide J. Neurobiol Aging 30:257–265
Vogensen SB, Strømgaard K, Shindou H, Jaracz S, Suehiro M, Ishii S, Shimizu T, Nakanishi K (2003) Preparation of 7-substituted ginkgolide derivatives: potent platelet activating factor (PAF) receptor antagonists. J Med Chem 46:601–608
Weinges K, Bäehr W (1972) Natural products from medicinal plants. XVI. Comparison of the NMR and mass spectra of bilobalide C15H18O8 and of the ginkgolides C20H24O9-11 (in German). Leibigs Ann Chem 759:158–172
Weinges K, Schick H (1991) Chemistry of ginkgolides. IV. Synthesis of ginkgolide B from ginkgolide C (in German) Leibigs Ann Chem 81–83
Weinges K, Hepp M, Huber-Patz U, Rodewald H, Irngartinger H (1986) Chemistry of the ginkgolides. I. 10-Acetyl-1-(methoxycarbonyl)-3,14,15,16-tetranorginkgolide A, an intermediate for the synthesis of bilobalide (in German). Leibigs Ann Chem 1057–1066
Weinges K, Hepp M, Jaggy H (1987a) Chemistry of ginkgolides. II. Isolation and structural elucidation of a new ginkgolide (in German) Leibigs Ann Chem 521–526
Weinges K, Hepp M, Huber-Patz U, Irngartinger H (1987b) Chemistry of ginkgolides. III. Bilobalide/isobilobalide. Structure determination by X-ray analysis (in German). Leibigs Ann Chem 1079–1085
Weinges K, Rümmler M, Schick H, Schilling G (1993a) Chemistry of the ginkgolides. V. On the preparation of the ginkgolide skeleton. Leibigs Ann Chem 287–291
Weinges K, Rümmler M, Schick H (1993b) Chemistry of the ginkgolides. VI. Preparation of 1,10-dihydroxy- and 1,7,10-trihydroxyginkgolide from 1,3,7,10-tetrahydroxyginkgolide (in German). Leibigs Ann Chem 1023–1027
Weinges K, Schick H, Pritzkow H (1997a) Chemistry of ginkgolides. VII. Preparation and crystal structure analysis of a secondary ozonide from ginkgolide A. Leibigs Ann Chem 1607–1609
Weinges K, Schick H, Irngartinger H, Oeser T (1997b) Chemistry of the ginkgolides. IX. Ozonolysis of (10R)-10-acetoxy-3,14-didehydroginkgolide. Leibigs Ann Chem 1755–1756
Weinges K, Schick H, Irngartinger H, Oeser T (2000) Chemistry of the ginkgolides. Part 10: access to 14C-labelled ginkgolide A. Tetrahedron 56:3173–3176
Woods MC, Miura I, Nakdaira Y, Terahara A, Maruyama M, Nakanishi K (1967) The ginkgolides. V. Some aspects of their NMR spectra. Tetrahedron Lett 4:321–326
Xiao Q, Wang C, Li J, Hou Q, Li J, Ma J, Wang W, Wang Z (2010) Ginkgolide B protects hippocampal neurons from apoptosis induced by beta-amyloid 25–35 partly via up-regulation of brain-derived neurophilic factor. Eur J Pharmacol 647:48–54
Zhu W, Chen C, Hu L, Luo X, Gui C, Luo C, Puah CM, Chen K, Jiang H (2005) QSAR analyses on ginkgolides and their analogues using CoMFA, CoMSIA, and HQSAR. Bioorg Med Chem 13:313–322
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Wien
About this chapter
Cite this chapter
Dzyuba, S.V., Jameson, L.P., Nakanishi, K. (2013). Ginkgolides and Their Derivatives: Synthetic and Bioorganic Studies. In: Wagner, H., Ulrich-Merzenich, G. (eds) Evidence and Rational Based Research on Chinese Drugs. Springer, Vienna. https://doi.org/10.1007/978-3-7091-0442-2_12
Download citation
DOI: https://doi.org/10.1007/978-3-7091-0442-2_12
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-0441-5
Online ISBN: 978-3-7091-0442-2
eBook Packages: MedicineMedicine (R0)