
Abstract
The metabolic disorders which affect muscle can cause chronic weakness and hypotonia or episodic exercise intolerance cumulating in rhabdomyolysis or both. Rhabdomyolysis disorders can be conveniently separated according to tolerance of short, intense exercise compared to longer, milder efforts. Most metabolic disorders which affect skeletal muscle do so by altering energy metabolism. Muscle at rest uses fatty acids as the main energy source. During intense exercise, there will be anaerobic glycolysis and utilization of muscle glycogen. During sustained exercise, fatty acids become again the source of fuel. Exercise, fasting, cold, infections, and medications may elicit symptoms. Important causes of metabolic myopathy include adenosine monophosphate (myoadenylate) deaminase deficiency, and disorders of glycolysis, glycogenolysis, fatty acid oxidation, and oxidative phosphorylation. In many cases, other organs are involved. Diagnosis requires careful attention to dietary and exercise history and appropriate laboratory investigations. Exercise testing, electromyogram, molecular testing, and muscle biopsy can provide essential information. Treatment depends on avoiding precipitating factors and optimizing muscle energetics.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Malignant Hyperthermia
- Glycogen Storage Disease
- Malignant Hyperthermia
- Pompe Disease
- Glycogen Storage Disease Type
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
FormalPara Key Facts-
The metabolic disorders which affect muscle can cause chronic weakness and hypotonia or episodic exercise intolerance cumulating in rhabdomyolysis or both. Rhabdomyolysis disorders can be conveniently separated according to tolerance of short, intense exercise compared to longer, milder efforts.
-
Most metabolic disorders which affect skeletal muscle do so by altering energy metabolism. Muscle at rest uses fatty acids as the main energy source.
-
During intense exercise, there will be anaerobic glycolysis and utilization of muscle glycogen. During sustained exercise, fatty acids become the source of fuel. Exercise, fasting, cold, infections, and medications may elicit symptoms.
-
Important causes of metabolic myopathy include adenosine monophosphate (myoadenylate) deaminase deficiency and disorders of glycolysis, glycogenolysis, fatty acid oxidation, and oxidative phosphorylation. In many cases other organs are involved.
-
Diagnosis requires careful attention to dietary and exercise history and appropriate laboratory investigations. Exercise testing, electromyogram, molecular testing, and muscle biopsy can provide essential information.
-
Treatment depends on avoiding precipitating factors and optimizing muscle energetics.
1 General Remarks
Many metabolic disorders cause muscle dysfunction or damage (Table 28.1). The pathophysiological basis in most is impairment of energy production when stressed, particularly by exercise, cold, fasting, or infection (especially of viral origin). In some situations, the problem is confined to skeletal muscle, but in many there is cardiac involvement as well. Liver, brain, retina, and kidney, all of which have significant energy requirements, may also be involved. Even more extensive or patchy involvement, e.g., pancreas and bone marrow, is a characteristic of the mitochondrial disorders where heteroplasmy may occur (see also Chaps. 14 and 42).
1.1 Special Aspects of Skeletal Muscle Metabolism
Skeletal muscle relies on different fuel sources at different times and circumstances. Fatty acids are the primary fuel at rest. Glucose from the blood and derived from muscle glycogen is used during short-term intensive exercise. Fatty acids predominate again during prolonged exercise and during fasting. Impairment of muscle energy metabolism will lead to clinical symptoms. The history of events that elicit the symptoms is a guide to the likely area of the biochemical defect.
Triglycerides are stored in the cytoplasm as droplets close to the mitochondria. The mobilization of free fatty acids from the lipid droplets depends on hormone-sensitive lipases and involves the hydrolase ATGL and its activator protein comparative gene identification-58 (CGI-58). Lipid storage myopathies occur with deficiency of these proteins (Wu et al. 2015).
Resting muscle in the fed state uses fatty acids as the primary fuel; glucose is stored as muscle glycogen (in the cytoplasm). Preformed high-energy phosphate compounds and muscle glycogen, in addition to glucose and fatty acids in the blood stream, are a source of energy for short-term intense activity. Lactate is the end product of anaerobic glycolysis. Impaired ability to utilize muscle glycogen (e.g., glycogen storage diseases (GSD) III, debrancher deficiency, and V, muscle phosphorylase deficiency, and muscle phosphorylase kinase deficiency) results in significant limitation when the patient attempts short, intense exercise. There will be diminished production of pyruvate and hence lactate. The situation is magnified if the muscle being tested is deprived of oxygen and continuous fuel by a tourniquet. This is the principle of the ischemic exercise test.
Defects of glycolysis in muscle, e.g., deficiencies of muscle phosphofructokinase (PFK), phosphoglycerate kinase (PGK), phosphoglycerate mutase (PGAM), β-enolase, and lactate dehydrogenase (LDH), can cause symptoms similar to muscle phosphorylase deficiency.
When sustained muscle activity is initiated, there is an initial reliance on glucose and glycogen as a fuel source. Metabolic disorders affecting these pathways typically cause symptoms at this time. After a few minutes, glycogen stores will be depleted, and fatty acids become more important as a fuel. The carnitine cycle and the β-oxidation spiral of fatty acid oxidation are essential at this point, so defects in these pathways can result in easy fatigability and impaired tolerance of sustained exercise.
The pathways of glycogenolysis, glycolysis, and fatty acid oxidation are shown in Fig. 28.1. Not all the steps mentioned in the text are specifically indicated on the pathway figure.

Pathway of carbohydrate and fatty acid metabolism illustrating the relationships of glycogenolysis, glycolysis, and fatty acid oxidation. Major aspects of glucose and fatty acid metabolism. Single arrows represent single steps; sequential arrows (and Roman numerals) represent multiple steps. I Glycogen synthesis—glycogen synthase, brancher enzyme; II Glycogenolysis—phosphorylase kinase, phosphorylase, debrancher enzyme; III Glycolysis—phosphoglycerate kinase, phosphoglycerate mutase, etc. 1 Galactose kinase; 2 galactose-1-P uridyl transferase; 3 epimerase; 4 phosphoglucomutase; 5 glucose transporter; 6 hexokinase; 7 glucose-6-phosphatase; 8 phosphofructokinase; 9 fructose-1,6-bis-phosphatase; 10 fructokinase; 11 fructose aldolase; 12 lactate dehydrogenase; 13 pyruvate carboxylase; 14 pyruvate dehydrogenase complex; 15 citrate synthase; 16 alpha-ketoglutarate dehydrogenase complex; 17 succinyl-CoA synthetase; 18 fumarase; 19 phosphoenolpyruvate carboxykinase; 20 carnitine transporter; 21 carnitine palmitoyltransferase I (CPT I); 22 carnitine-acylcarnitine translocase; 23 carnitine palmitoyltransferase II (CPT II); 24 acyl-CoA dehydrogenases (very-long-chain (VLCAD), long-chain, medium-chain (MCAD), short-chain (SCAD)); 25 2-enoyl-CoA hydratase (trifunctional enzyme, crotonase); 26 3-hydroxyacyl-CoA dehydrogenases (long-chain (trifunctional enzyme/LCHAD), short-chain (SCHAD)); 27 3-ketoacyl-CoA thiolase (long-chain (trifunctional enzyme), short-chain). (Acetoacetyl-CoA is the 4-carbon 3-ketoacyl-CoA.); 28 hydroxymethylglutaryl-CoA (HMG-CoA) synthase; 29 hydroxymethylglutaryl-CoA (HMG-CoA) lyase; 30 3-OH-butyrate (beta-hydroxybutyrate) dehydrogenase
1.2 Basic Patterns of Metabolic Myopathies
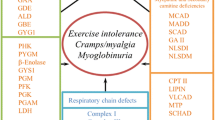
The two major distinct syndromes of muscle metabolic disorders are exercise intolerance and rhabdomyolysis (with or without myoglobinuria) and weakness (with or without hypotonia). Rhabdomyolysis can be further divided into syndromes where it occurs during strenuous exercise and those where it occurs afterward.
Remember
Major symptoms of myopathies are weakness, hypotonia, exercise intolerance, and rhabdomyolysis.
Rhabdomyolysis, the destruction of skeletal muscle cells, often results from failure of energy production, leading to an inability to maintain muscle membranes. The hallmark of rhabdomyolysis is elevation of muscle enzymes in the blood, particularly creatine kinase (CK or CPK). This elevation can persist for several days after an acute event. Chronic elevation of CK indicates continuous damage (Wu et al. 2015; Chan et al. 2015; Sharp and Haller 2014).
Rhabdomyolysis during short-term intensive exercise is a primary feature of the disorders of carbohydrate metabolism, especially muscle phosphorylase deficiency (McArdle disease). After a period of intense pain with or without cramping, however, there may be considerable relief and ability to continue exercise, called the “second wind” phenomenon, as the muscle switches to increased use of fatty acids for fuel. Patients with McArdle disease will also benefit from glucose administration before exercise, because they are able to utilize glucose. In contrast, patients with a metabolic block in glycolysis, e.g., in PGK, cannot utilize glucose or glycogen. Glucose administration even diminishes the concentrations of the alternative fuels triglycerides and ketone bodies (“out of wind” phenomenon).
Postexercise cramps and rhabdomyolysis are the more common pattern in fatty acid disorders, especially deficiencies of carnitine palmitoyltransferase (CPT) II, very-long-chain acyl-CoA dehydrogenase (VLCAD), long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) (see also Chap. 16), and mitochondrial oxidative phosphorylation disorders. Rhabdomyolysis may also be a chronic feature of the various muscular dystrophies, which are usually disorders of the structural proteins of muscle (dystrophin, actin, tropomyosin, the dystroglycan complex, etc.).
Remember
Short, intense exercise stresses glycolysis and glycogen utilization.
Prolonged exercise requires adequate utilization of fatty acids.
Fasting, cold, infection, and medications can worsen many myopathies.
Myoglobinuria is an extreme result of rhabdomyolysis. When muscle cells lyse, myoglobin is released. Visible myoglobin in the urine indicates extensive damage. Typically, there is no myoglobin in the urine if the CK is <10,000 IU, so the absence of myoglobinuria provides no reassurance regarding absence of rhabdomyolysis.
Myoglobinuria is an emergency situation, as the pigment may precipitate in the renal tubules, leading to renal failure, which may become irreversible. Severe rhabdomyolysis can also raise the serum potassium level dangerously high, leading to cardiac rhythm disturbances. Accordingly, dark urine in a patient suffering from muscle symptoms (pain, weakness, cramping, etc.) must be tested for myoglobin using a specific test (to distinguish the pigment from hemoglobin). Hemoglobinuria most often accompanies hematuria, readily detectable by finding erythrocytes on microscopic analysis of the urine. Intravascular hemolysis will occasionally result in hemoglobinuria, without hematuria.
Myoglobinuria is treated with diuresis and careful monitoring of electrolyte, fluid status, and urine output, until the myoglobinuria resolves. Investigation of the underlying cause of myoglobinuria begins at the same time as its treatment.
Chronic weakness and hypotonia are typical features of disorders of endogenous triglyceride catabolism, glycogen breakdown, carnitine availability, fatty acid oxidation, and oxidative phosphorylation. Important causes include lysosomal GSD (acid maltase deficiency – Pompe disease), glycogen debrancher deficiency, carnitine transporter defect and secondary carnitine deficiencies, VLCAD, LCHAD, and mitochondrial myopathies. Chronic weakness may certainly result from rhabdomyolysis and consequent muscle destruction from any cause, especially if recurrent.
2 Approach to Metabolic Myopathies
The most urgent issues in the assessment of myopathy are to determine if there is weakness so severe to impair respiration, if there is sufficient damage to lead to myoglobinuria, and if there is cardiac involvement. Hepatic involvement, often manifest as hypoglycemia and fasting intolerance, occurs in many metabolic disorders, particularly those involving glycogen or fatty acid metabolism. A toxic encephalopathy, including cerebral edema, may also develop.
The history of muscle dysfunction may be easy to elicit from an adult or a child (or the parents), but may be difficult with infants. Hypotonia and weakness may first become evident as developmental delay. Careful assessment may then reveal that social, fine motor, and language skills are appropriate for age, and the only area of delay is in gross motor skills.
As a young child grows older, problems with exercise intolerance and easy fatigability become easier to detect, particularly if there is an unaffected older sibling to serve as a reference point for the parents. Occasionally, a child with a muscle disorder is thought to be “seeking attention” or malingering, but careful history and observation can usually eliminate this possibility quickly. Laboratory tests that convincingly demonstrate ongoing muscle injury (e.g., elevated CK) are most persuasive.
Disorders made worse by fasting may not be evident in infancy, as most infants are fed frequently. An inability to tolerate intense or prolonged exercise will not be evident in infancy and perhaps not until adulthood. Rhabdomyolysis in response to cold also may not become evident until adolescence or adulthood. Rhabdomyolysis triggered by infection (usually viral), or fasting, may present in infancy as sudden weakness, accompanied by dark urine. Rhabdomyolysis is often quite painful, but may be painless.
Some conditions that are not yet completely characterized can cause severe and potentially fatal rhabdomyolysis in children, particularly in the setting of viral infection. Children with such conditions, like children with named disorders of fatty acid oxidation or mitochondrial dysfunction, need to be monitored carefully during infections.
The history of exercise can provide preliminary guidance in determining the most likely causes of a myopathy. An inability to perform sudden intense exercise suggests a problem with glycogenolysis or glycolysis, while inability to perform at a sustained level suggests a problem with fatty acid oxidation.
Many mitochondrial disorders of oxidative phosphorylation first become apparent because of skeletal muscle weakness. Even isolated myopathies can occur. Rhabdomyolysis is uncommon. Mitochondrial disorders may have prominent muscle involvement. Mitochondrial disorders can involve any organ at any age. Other organs commonly affected include the brain, retina, extraocular muscles, cochlea, heart, liver, kidney, pancreas, gut, and bone marrow. Systemic growth may be impaired. Mild hypertrichosis often accompanies systemic lactic acidosis. Despite the diversity of mitochondrial dysfunction, there are several common syndromes in which many patients can conveniently be grouped. They include MERRF, MELAS, and infantile myopathy (Chap. 42).
Two very rare neutral lipid storage diseases have been identified. Neutral lipid storage disease type I or Chanarin–Dorfman syndrome has ichthyosiform nonbullous erythroderma with a slowly progressive proximal myopathy that spares the axial musculature. It is due to mutations in ABHD5. Neutral lipid storage disease type II is due to mutations in the activator gene PNPLA2, again leading to a lipid myopathy associated with cardiac dysfunction and hepatomegaly.
2.1 Genetics
Most metabolic myopathies, like most other metabolic disorders, are inherited in an autosomal recessive manner. All disorders of fatty acid oxidation and most disorders of glycogen and glucose metabolism are inherited this way. However, other mechanisms including X-linked (PGK deficiency and one form of phosphorylase b kinase deficiency), autosomal dominant (heterozygous CPT II deficiency), mitochondrial maternal transmission, and sporadic mitochondrial disorders occur. Specifics of inheritance are mentioned when appropriate in the discussion of the various disorders.
Because of the highly variable nature of most metabolic disorders of muscle, all siblings of patients should be checked for the condition which is in the family. If the disorder may be dominant, X linked, or mitochondrially inherited, other at-risk relatives should also be examined carefully.
2.2 Physical Examination
The general physical examination of a patient suspected of myopathy includes assessment of growth and development and particular attention to other organs. The muscles should be examined for bulk and regional (proximal and distal) or local evidence of wasting, texture and consistency, and tenderness. Deep tendon reflexes, which are generally preserved in myopathies, but lost in peripheral neuropathies, should be tested carefully. Attention should be especially directed to extraocular movements and the retina, hearing, the tongue, the heart, and the size and characteristics of the liver.
2.3 Laboratory Investigations
Laboratory investigation of suspected myopathies should be undertaken during the acute episode if possible and later repeated as indicated. Routine serum electrolytes and measurement of glucose, urea and creatinine, and “muscle enzymes” including CK, LDH including isoforms, aldolase, SGOT (ALT), SGPT (AST), total and free carnitine, plasma or blood spot acylcarnitines profile, plasma lactate and pyruvate, phosphate, calcium, thyroid hormone, plasma and urine amino acids, and urine organic acids may all provide useful information.
Following the assessment of the first-order laboratory tests, further tests may be warranted. Functional testing using ischemic exercise (for suspected glycogen storage and glycolytic disorders and adenosine monophosphate deaminase deficiency) can be most helpful. Graded exercise or bicycle ergometry may help pinpoint the metabolic error, or define the general area of impairment, if history and blood tests have not done so. A “diagnostic fast” to evoke abnormal metabolites or provoke symptoms should only be done if information cannot be obtained by another method – challenges are better put to fibroblasts or tissue samples. However, a fast under controlled circumstances can provide valuable information regarding how long it is safe for a particular child to fast when healthy (see Chap. 41).
Third-order tests include electromyogram (often coupled with nerve conduction studies), chest X-ray, electrocardiogram, echocardiogram, and muscle biopsy (perhaps together with nerve biopsy). Light and electron microscopic examination and special stains for glycogen, lipid, and various enzymes may all be essential. “Classic” lipid storage is found in four conditions: primary carnitine deficiency due to a deficiency of the carnitine transporter, mild multiple acyl-CoA dehydrogenase deficiency (MADD) with secondary coenzyme Q10 deficiency, and the neutral lipid storage diseases types I and II.
Many enzymes can be studied in fibroblasts or lymphocytes, and DNA can be obtained from blood or a buccal brush instead of a tissue biopsy. Mitochondria can be prepared for functional and molecular studies from muscle (the preferred source) but also liver, leukocytes, and other samples. Coenzyme Q is best measured in muscle. Details of these tests are given in (Chap. 43). If an open muscle biopsy is done, a skin biopsy for fibroblast culture and DNA analysis can be taken from the edge of the incision. Some pathologists and mitochondrial laboratories are able to analyze muscle tissue obtained by needle biopsy. Table 28.1 provides a guide to the principal features of the major metabolic myopathies and the usefulness of the various diagnostic materials. Molecular diagnosis using panels of relevant genes is rapidly replacing biochemical and immunohistochemical testing done on biopsy samples and testing of individual genes. The new testing methods are also revealing that some patients have more than one condition contributing to their symptoms.
3 Specific Disorders of Muscle Metabolism
3.1 Exercise Intolerance/Rhabdomyolysis and Myoglobinuria Presentation
3.1.1 Intense Exercise Not Tolerated. Mild, Prolonged Exercise Tolerated. Fasting Tolerated. Dietary Modifications Helpful
3.1.1.1 Muscle Glycogen Phosphorylase (Myophosphorylase) Deficiency: McArdle Disease (GSD Type V)
This dramatic disorder of muscle glycogen metabolism is a relatively common cause of rhabdomyolysis and myoglobinuria. Although it is inherited in an autosomal recessive manner, most symptomatic patients are men. Symptoms usually begin between late childhood and late middle age. Strenuous exercise leads rapidly to cramping and fatigue, but, after a period of rest (adaptation), exercise is tolerated. Some patients with McArdle disease have chronic progressive weakness and wasting, without pain or cramping.
Exceptional cases include a rapidly fatal form in infants or young children with hypotonia and generalized weakness, a late-onset form with chronic weakness, and a late-onset form with severe symptoms (pain, cramping, weakness, and muscle swelling), after decades of normal activity. Diagnosis after inadvertent discovery of elevated CK in a child without symptoms has been reported.
Diagnosis is suspected from the symptoms and response to ischemic exercise. The enzyme is expressed mainly in muscle. Muscle biopsy may show myopathic changes and increased glycogen content. There are two major mutations in the gene PYGM (p.Arg50Ter and p.Gly205Ser) which account for a majority of the mutations in most American and European populations. Other pathogenic variants may be relatively common in specific populations (Martin et al. 1993). Abnormalities in other enzymes may contribute to severity of symptoms (Gonzalez-Freire et al. 2009; Martinuzzi et al. 2003).
Specific treatment is generally not needed, as avoiding strenuous exercise prevents symptoms in most patients. A high-protein, low-carbohydrate diet has been suggested to improve endurance. Others have found increased carbohydrate intake (glucose and fructose) immediately before exercise to be helpful (Orngreen et al. 2009).
3.1.1.2 Muscle Glycogen Phosphorylase Kinase Deficiency (Formerly Phosphorylase b Kinase Deficiency, GSD IX)
There have been a few men with deficiency of muscle phosphorylase kinase. Weakness without cramps and cramps without weakness have both been reported. Increased muscle glycogen content, elevation of CK, and rhabdomyolysis have been reported. Enzyme deficiency was demonstrated in muscle. The gene encoding the alpha subunit of phosphorylase (PHKA1) in muscle is on the X chromosome. It is distinct from the liver isoform (also on X). There are also three autosomal components of the glycogen phosphorylase kinase system. Autosomal recessive defects have been found in the beta and gamma peptides. No defects in the three delta subunit isoforms (which are calmodulins) have been reported to date.
3.1.1.3 Muscle Glycolytic Disorders
Deficiencies of five glycolytic enzymes in muscle are rare causes of myopathy similar to muscle phosphorylase deficiency. They are PFK, PGK, PGAM, β-enolase, and triosephosphate isomerase. Hemolytic anemia can occur in all. PGK deficiency is X linked. Patients may have neurologic problems (mental retardation, behavioral abnormalities, seizures, and strokes). Other disorders are autosomal recessive (Oldfors and DiMauro 2013).
3.1.1.4 Lactate Dehydrogenase (LDH) Deficiency
Lactate dehydrogenase catalyzes the conversion of pyruvate and NADH to lactate and NAD+. The enzyme is a tetramer of H and M peptides, produced from LDHB and LDHA genes. Homozygous deficiency of the M protein results in impaired muscle LDH activity. The result is impaired regeneration of NAD+ for anaerobic glycolysis and impaired production of lactate (and resulting in high levels of pyruvate) with exercise, detectable by the ischemic exercise test. Cramps, weakness, and myoglobinuria can occur with strenuous exercise. Deficiency of LDH can result in a “false-negative” result if LDH is being measured to assess tissue damage in other situations. No syndrome is attributable to LDHB deficiency.
3.1.1.5 Adenosine Monophosphate (Myoadenylate) Deaminase Deficiency
This autosomal recessive disorder of purine metabolism is probably the commonest metabolic myopathy, with impaired exercise tolerance, postexercise cramps, and myalgias. Myoglobinuria is uncommon, but CK is often elevated after exercise. Onset of symptoms (usually pain after exercise) ranges from childhood to later adult life. In the US population perhaps, 2 % are homozygous for deficiency, but most have no symptoms. Because AMP deaminase deficiency is so common, it has sometimes been found coincidentally with a less common muscle disorder that by itself would account for the symptoms, e.g., muscle phosphorylase or PFK deficiency. As the enzyme deficiency will only be discovered by ischemic exercise testing, or specific assay or molecular test, there is a selection bias toward muscle problems.
Many patients discovered to have AMP deaminase deficiency have other symptoms as well, especially neuromuscular disease. Diminished synthesis of AMP deaminase occurs in a variety of situations. This is termed acquired deficiency and does not seem to have a direct genetic basis, and the common mutation is not present at a frequency above the background rate.
AMP deaminase catalyzes the deamination of AMP to IMP (inosine monophosphate) in the purine nucleotide cycle (see Fig. 28.2). During exercise there will be increased production of IMP and ammonia and maintenance of the adenylate energy charge by preventing AMP accumulation. A decrease in ATP and increase in ammonia will also stimulate glycolysis by increasing the activity of PFK. The increase in IMP may also enhance glycogen phosphorylase. Finally, during intense exercise, AMP deaminase moves from the cytosol and becomes bound to myosin, which suggests that it is important in muscle metabolism during such times.

Pathways of synthesis, salvage, and degradation of purines. The enzymatic steps in boxes indicate the sites of the commonly encountered disorders of purine metabolism AMPDA muscle adenosine monophosphate deaminase deficiency, APRT adenine phosphoribosyltransferase deficiency, ASL adenylosuccinate lyase (adenylosuccinase) deficiency, HPRT hypoxanthine–guanine phosphoribosyltransferase deficiency, PNP purine nucleoside phosphorylase deficiency, SO sulfite oxidase deficiency, XO xanthine oxidase (xanthine dehydrogenase) deficiency
The diagnosis of AMP deaminase deficiency is approached by ischemic exercise which ordinarily provokes a rise in blood ammonia level. If AMP is deficient there will be diminished ammonia production. Muscle biopsy may be normal or show some myopathic changes. Specific staining for AMP deaminase is a generally reliable diagnostic test. Enzyme activity in deficient muscle ranges up to 15 % of normal; some authorities regard activity >2 % as adequate to prevent symptoms. A common mutation in AMPD1 accounts for most cases of inherited AMP deaminase deficiency. This common mutation is actually a pair of mutations in linkage disequilibrium (p.Q12X) and p.Pro48Leu. The nonsense mutation in exon 2, p.Q12X, can result in a severely truncated protein. However, an alternative splicing mechanism allows for phenotypic rescue by production of a shortened but functional protein, with p.Pro48Leu in exon 3 retained. This may account for the great variability in symptoms in homozygotes for this mutation. The allele frequency was 0.13 (Caucasians) and 0.19 (African-Americans) in one study, which accounts for the observed homozygote frequency of about 0.02. Symptoms may be due to combined deficiencies of AMP deaminase and another enzyme involved in muscle metabolism (Bruno et al. 1998; Rubio et al. 1997). Polymorphisms of angiotensin I-converting enzyme may contribute to the variability of symptoms seen with AMP deaminase deficiency.
AMP deaminase deficiency has been found in patients with aldolase deficiency, lactate dehydrogenase A (LDHA) deficiency, and β-enolase deficiency.
3.1.2 Short, Intense Exercise Tolerated, Prolonged Exercise Not Tolerated, Fasting Detrimental, Diet Effects Less Pronounced. Symptoms May Be Triggered by Infection. Restriction of Long-Chain Fats, with Supplementation of Medium-Chain Lipids, May Be Helpful, Especially for Cardiomyopathy
3.1.2.1 Carnitine-Acylcarnitine Translocase Deficiency
An inability to import long-chain acylcarnitines into the mitochondrial matrix would be expected to cause serious difficulties, especially in cardiac and skeletal muscle and in the liver. The severe infantile form of translocase deficiency typically does this, starting a day or two after birth. Cardiac (cardiomyopathy, usually hypertrophic) and hepatic dysfunction (hypoglycemia, vomiting, and hyperammonemia) are more prominent than hypotonia and weakness. Urinary organic acids show dicarboxylic aciduria, and the plasma/blood spot acylcarnitine profile is dominated by long-chain species (C16:1, C18:1, and C18:2), which can be formed but not used, and dicarboxylic acylcarnitines.
3.1.2.2 CPT II Deficiency
CPT II deficiency is the commonest disorder of fatty acid oxidation to cause episodic rhabdomyolysis. CPT II is needed to synthesize long-chain acylcarnitines once they have been translocated into the mitochondria, so the clinical features are similar to translocase deficiency. Prolonged exercise, cold, infection, and emotional stress (which will increase catecholamines and fatty acid metabolism) may precipitate episodes, which usually do not occur in children. Cardiac involvement is uncommon in this form of CPT II deficiency.
Plasma or blood spot acylcarnitine analysis shows prominent long-chain species, especially saturated and unsaturated C16 and C18 forms, and there may be dicarboxylic aciduria, similar to translocase deficiency.
A severe form of infantile CPT II deficiency also exists. Hepatic encephalopathy with hypoketotic hypoglycemia, severe cardiac involvement, renal malformations, and low plasma and tissue carnitine levels may be present. This form is usually fatal, from cardiac complications. It is discussed in more detail in Chap. 23.
Treatment includes avoiding fasting and providing adequate fuel for the muscles. Medium-chain fatty acids do not need the carnitine system in order to enter the mitochondria, so they can be used in place of long-chain fats as a source of energy.
Although this is an autosomal recessive disorder, heterozygosity for a mutation may be associated with myopathy (Joshi et al. 2012) and risk of malignant hyperthermia in response to anesthetics or muscle relaxants (Hogan and Vladutiu 2009; Vladutiu et al. 2000).
There is a form of CPT I that is solely expressed in muscle. No clinical deficiency has been recognized so far.
3.1.2.3 VLCAD Deficiency
VLCAD is the first enzyme of the β-oxidation spiral. It is bound to the inner mitochondrial membrane. Deficiency of VLCAD is a common cause of metabolic myopathy and cardiomyopathy. For several years, the enzyme now known as VLCAD was called LCAD (long-chain acyl-CoA dehydrogenase); reports from before about 1993 regarding LCAD deficiency almost always involve what is now called VLCAD. VLCAD utilizes fatty acids of 14–20 carbons. A major source of confusion is that fatty acids called very-long-chain fatty acids, of chain length >20, are metabolized by a different system altogether, in the peroxisomes.
Impairment of VLCAD will lead to variable skeletal, cardiac, liver, and brain symptoms, including recurrent Reye syndrome with coma, and hypoketotic hypoglycemia. Muscle soreness and episodic rhabdomyolysis may be provoked by infection, cold, fasting, or emotional stress (perhaps mediated by catecholamines). There is usually dicarboxylic aciduria, although during severe metabolic derangement it may be overlooked because of excessive lactic aciduria indistinguishable from a primary defect of the respiratory chain. Carnitine depletion, with low plasma and tissue levels, can occur, and acylcarnitine analysis shows prominence of C14:1 (tetradecenoyl) species, derived from oleic acid (C18:1). Hepatic dysfunction may result in hyperammonemia and lipid accumulation. Muscle biopsy may show lipid storage, and the EMG is often myopathic.
VLCAD deficiency, like other disorders of fatty acid oxidation, must be promptly treated during the acute episode with glucose sufficient to maintain the blood glucose level at 6–8 mM or even higher (see also Chap. 19). The use of carnitine supplementation has been controversial on theoretical and experimental grounds, particularly because of fear that long-chain acylcarnitines would accumulate and provoke arrhythmias. However, there are few convincing reports of this actually happening. Long-term management emphasizes adequate calories from carbohydrate, restricting long-chain dietary fats, avoiding fasting and other stressors, and supplementing with medium-chain triglycerides, which will provide a source of fuel that can be metabolized without requiring VLCAD. Triheptanoin, a novel treatment to provide a constant source of ketone bodies and propionyl groups for patients with defects of long-chain fatty acid oxidation, is showing great promise in preliminary studies (Roe and Brunengraber 2015).
The enzyme now known as LCAD is in the mitochondrial matrix. Its major substrates are unsaturated long-chain fatty acids, 12–18 carbons in length. Deficiency has not been convincingly demonstrated in humans.
3.1.2.4 LCHAD (Including Trifunctional Protein) Deficiency
LCHAD deficiency often results in chronic myopathy, with rhabdomyolysis, which may be extensive, particularly during viral infections. Like other disorders of long-chain fatty acids, there is often cardiomyopathy and significant liver dysfunction, both of which may be fulminant. The extent of chronic liver dysfunction can be greater than in other disorders, and fibrosis often occurs. There may be Reye-like episodes of hepatic encephalopathy. In addition there may be peripheral neuropathy and retinopathy. The basis for these complications is not yet completely known.
The enzyme activity called LCHAD is found in an octameric protein (α4β4) called the trifunctional protein, for its ability to catalyze the 2-enoyl-CoA hydration, 3-hydroxyacyl-CoA dehydrogenation, and 3-oxoacyl-CoA thiolysis of long-chain acyl-CoAs. The first two activities reside in the α-subunit and thiolase activity in the β-subunit. The two subunits depend on each other for stability. The trifunctional protein is in the mitochondrial inner membrane.
There is some relationship between mutation and symptoms. The most common mutation (87 % in one study), c.1528 G > C (p.E510Q) in the α-subunit, usually causes liver dysfunction with hypoketotic hypoglycemia in infancy.
LCHAD deficiency is an autosomal recessive disorder, and carriers are generally symptom-free. A particular complication of heterozygous (carrier) status for LCHAD deficiency, especially the p.E510Q mutation, is serious liver disease during pregnancy when carrying an affected infant. The mother may suffer from acute fatty liver of pregnancy (AFLP, with nausea and anorexia, vomiting, and jaundice) or the HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome of hypertension (Ibdah et al. 2000; Jebbink et al. 2012). It may be that the production of abnormal fatty acid metabolites by the fetus overloads the mother’s ability to deal with them, on top of the increased fatty acid mobilization that occurs during pregnancy. Prospective studies of women with AFLP or HELLP syndrome, however, have not always found an increased number of carriers of LCHAD deficiency, indicating there are other causes for these conditions. Women heterozygous for hepatic CPT I deficiency, which may cause a Reye-like syndrome in homozygotes, may also suffer from AFLP when carrying an affected fetus (Innes et al. 2000).
In untreated patients, the urine organic acids reveal increased saturated and unsaturated dicarboxylic and hydroxy species. The plasma acylcarnitine profile typically shows elevation of hydroxy-C18:1 species, which, in combination with an elevation of two of the three long-chain species C14, C14:1, and hydroxy-C16, identifies over 85 % of patients with high specificity (<0.1 % false-positive rate). Blood spot acylcarnitine analysis is not quite as sensitive, because of higher levels of long-chain species in blood samples. Dietary treatment (restriction of long-chain fats and supplementation with medium-chain triglycerides) will lower the long-chain acylcarnitine species, often to normal. Plasma carnitine levels are usually low, especially during acute illness. Carnitine is sometimes given at such times. Its usefulness as a treatment for this myopathy (and whether it might provoke arrhythmias in certain situations – see Chap. 23) is a subject of continuing investigation. A major complication not seen in most organic acidurias is a progressive retinopathy.
3.2 Weakness and Hypotonia Presentation
3.2.1 Carnitine Transporter Deficiency
The carnitine transport defect is the result of deficient activity of the high-affinity carnitine transporter (OCTN2, encoded by the gene SLC22A5), active in kidney, muscle, heart, fibroblasts, and lymphocytes. Renal fractional excretion of carnitine, calculated in relation to creatinine clearance, approaches 100 % (normal <5 %). The severe carnitine depletion that results (plasma levels can be <5 μmol/L, normal ≈ 45 μmol/L) will lead to tissue carnitine depletion as well. Hepatic carnitine depletion results in hypoketotic hypoglycemia. Onset of myopathy and cardiomyopathy can be in the first few months, or not for several years. Urine organic acids are typically normal. Muscle biopsy reveals lipid accumulation, and the muscle carnitine level is extremely low. Response to carnitine supplementation is dramatic, but because of the ongoing renal leak of carnitine, it is extremely difficult to maintain normal plasma carnitine levels or tissue levels, and exercise tolerance may be limited. Oral carnitine supplementation to an amount just short of provoking a fish odor (trimethylamine) by exceeding the oxidizing capacity is the usual approach. Inheritance is autosomal recessive.
3.2.2 Secondary Carnitine Depletion
Adults are able to synthesize all the carnitine they need. The major dietary source of carnitine is meat. The carnitine content of human breast milk is similar to that of plasma. Carnitine deficiency has occurred in several infants on prolonged parenteral nutrition (without added carnitine), resulting in myopathy and cardiomyopathy, impaired ketogenesis, and hepatic steatosis. Carnitine supplementation (intravenous or oral) was rapidly beneficial. This experience suggests that carnitine may be an essential nutrient in the very young and that routine carnitine supplementation of TPN solutions for infants should be considered.
Severe carnitine depletion can result from a generalized Fanconi syndrome, characteristic of cystinosis, Lowe syndrome, mitochondrial disorders (especially cytochrome C oxidase deficiency), etc. (see also Chap. 26). Recognition of the carnitine depletion usually occurs after the discovery of Fanconi syndrome. Plasma carnitine measurement and determination of the fractional excretion should be part of the investigation of all patients with Fanconi syndrome. Restoration of tissue carnitine levels may take a very long time even after correction of the renal leak, as after renal transplantation for cystinosis.
Some medications, such as valproic acid and the pivalic acid component of pivampicillin (and a few other medications), are organic acids and may be excreted as carnitine esters causing significant urinary losses of carnitine as valproylcarnitine or pivaloylcarnitine and consequently secondary systemic carnitine deficiency. Use of pivampicillin has been linked to life-threatening crisis, especially in patients with an underlying metabolic disorder [e.g., medium-chain acyl-CoA dehydrogenase (MCAD) deficiency].
MCAD deficiency is unusual among fatty acid oxidation disorders for having only minimal skeletal muscle symptoms. Hepatic and cerebral symptoms (Reye-like syndrome, hypoketotic hypoglycemia, and sudden unexplained death) are the usual features. However, carnitine depletion can occur. MCAD deficiency accounts for nearly all the patients described with “systemic carnitine deficiency,” before the enzyme deficiency was discovered. This may be the mechanism for chronic weakness and impaired exercise tolerance in some older patients with MCAD deficiency. Long-term carnitine supplementation, whose use in MCAD deficiency is not universally accepted, might be expected to ameliorate this situation.
3.2.3 2,4-Dienoyl-CoA Reductase Deficiency
This extremely rare disorder has been described in at least two infants with hypotonia, normal deep tendon reflexes, poor feeding, and failure to thrive. There was plasma carnitine deficiency, and a unique unsaturated acylcarnitine species (C10:2) shown to be derived from long-chain unsaturated fatty acids, in plasma. Mutations in NADK2 were found in one patient.
3.2.4 MADD–ETF OR ETF-Dehydrogenase Deficiency (Synonymously Glutaric Aciduria Type II)
The severe form of this disorder causes overwhelming acidosis shortly after birth. Impairment of the ETF system blocks many different dehydrogenation systems for fatty acid and amino acid degradation. Milder deficiency of the same system can cause a lipid storage myopathy that may not become apparent for years or decades. Gradual onset of weakness and easy fatigability may be overlooked initially, and the history may first suggest an inflammatory myopathy. There may be liver dysfunction. Urine organic acids can show dicarboxylic aciduria, including ethylmalonic, adipic, and glutaric acids. Plasma acylcarnitine analysis shows elevation of short- and medium-chain species. A secondary deficiency of coenzyme Q10 (CoQ) may occur. Mitochondrial studies may show deficient activity complex (I and II). Response to supplemental riboflavin (50–100 mg/day), the precursor of the cofactor flavin adenine dinucleotide, is often dramatic in the milder forms. A disorder of riboflavin transport is the cause of a similar disorder, Brown–Vialetto–Van Laere syndrome and its allelic variant Fazio Londe syndrome. Both conditions are motor neuron syndromes which respond well to high-dose (10 mg/kg per day) riboflavin supplementation.
3.2.5 Lysosomal GSD Type II (Pompe Disease)
Pompe disease is the severe infantile form of lysosomal α-glucosidase (GAA, or acid maltase) deficiency. It is one of the most common storage diseases presenting in infancy and the first one to be identified. The infant typically presents in the first few months with weakness and profound hypotonia. Feeding and respiratory difficulties are common. There is macroglossia, but minimal hepatomegaly. At least 80 % have significant cardiac involvement. Progressive weakness and cardiomyopathy usually lead to death within a year. Despite the hypotonia, the muscles feel firm or even woody (Howell et al. 2006).
There is a spectrum of deficiency of α-glucosidase, with onset of symptoms reported as late as the eighth decade (Winkel et al. 2005). Various terms, including juvenile and adult onset, are used to describe late-onset patients. The age of onset bears little relation to the rapidity of the course (Lachmann and Schoser 2013). The older the patient, the more likely that symptomatic muscle involvement will be patchy clinically and morphologically. However, enzyme activity will be deficient, regardless of the appearance of the cells. Late-onset Pompe disease may present with weakness of the diaphragm, or as a limb-girdle myopathy. The function of lysosomal glycogen is not known. Mutation analysis of GAA is readily performed on DNA from leukocytes or other sources. α-Glucosidase activity can be measured in leukocytes, as well as in muscle or in liver biopsy, or in cultured skin fibroblasts. Muscle biopsy, which is not necessary for diagnosis if enzyme assay can be performed, shows enlarged lysosomes engorged with glycogen, altering the ultrastructure of the cell.
There was no satisfactory treatment of Pompe disease until the development of enzyme replacement therapy. Recombinant α-glucosidase can arrest and even reverse the disease if begun early enough, so early diagnosis is now an urgent matter (Kishnani et al. 2013). Newborn screening for Pompe disease is now being implemented in the USA.
3.2.6 Glycogen Debrancher Deficiency: GSD Type III (Cori or Forbes Disease)
This relatively common disorder of glycogen metabolism results from varying deficiencies of amylo-1,6-glucosidase, 4-α-glucoanotransferase, the debrancher enzyme, a remarkable peptide which has two separate catalytic activities (transferase and glucosidase). There is always liver involvement, but muscle involvement is variable and does not occur at all in about 15 % (GSD IIIb). In infancy and childhood, the liver symptoms dominate, with hepatomegaly, hyperlipidemia, and fasting hypoglycemia similar to GSD I. Muscle weakness may not be apparent. After puberty the liver symptoms subside, but there is an ongoing risk for cirrhosis. The myopathy may persist as weakness and may worsen with time. CK may be elevated, but may be normal even if there is muscle involvement. There can be distal wasting, myopathic EMG, and peripheral neuropathy. There is usually cardiac involvement (mild) as well.
Diagnosis is suspected from the history. Enzyme assay and gene analysis can be done using fibroblasts, lymphocytes, muscle, or liver. Liver and muscle biopsies are often done to obtain direct information about these organs. Analysis of glycogen structure in muscle or liver can demonstrate abnormalities due to the lack of normal glycogen breakdown. The differences in tissue expression are traceable to alternate splicing of exon 1 of the gene. Differences in phenotype correlate with different mutations. Two mutations in exon 3 account for 90 % of patients with GSD IIIb (i.e., no muscle involvement). Molecular testing, to determine if muscle disease will occur, is replacing muscle biopsy. (If at least one of the mutations involves exon 3, the patient will not have muscle disease.) Support of glucose availability is achieved with cornstarch, similar to GSD I (Kishnani et al. 2010). Treatment of the myopathy using high-protein meals and high-protein enteral feeds overnight has been attempted, but it does not seem to improve the long-term outlook.
3.2.7 Glycogen Brancher Deficiency: GSD Type IV (Anderson Disease)
Type IV GSD, glycogen brancher deficiency, can cause hypotonia and weakness, but the clinical picture is dominated by hepatic fibrosis and dysfunction. Cardiomyopathy may be significant in the severe infantile form (Magoulas and El-Hattab 1993). Partial deficiency of this enzyme is a cause of adult polyglucosan body disease (APBD), a storage myopathy. This condition may be ameliorated by triheptanoin, a compound being studied for disorders of fatty acid oxidation and other conditions with deficient intracellular energy (Roe et al. 2010).
3.2.8 Glycogen Synthase Deficiency: GSD Type 0
This disorder results in insufficient glycogen reserves in the liver, so that fasting hypoglycemia occurs relatively soon after a meal and excessive lactate is produced from dietary glucose. A few families have been reported with a muscle form of the disease (called GSD0B), which caused muscle weakness, reduced exercise capacity, and hypertrophic cardiomyopathy in the first decade.
3.2.9 Mitochondrial Myopathies and Coenzyme Q Deficiency
The mitochondrial myopathies are an extremely heterogeneous group of disorders. Symptoms may be confined to muscle, or may involve other organs, particularly the brain, heart, liver, and kidneys. Symptoms may already be present at birth or not appear for decades. Myopathy is particularly evident in the syndromes of chronic progressive external ophthalmoplegia (CPEO) including the Kearns–Sayre syndrome (KSS) or ophthalmoplegia-plus; mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS); mitochondrial encephalomyopathy with ragged-red fibers (MERFF); fatal infantile mitochondrial myopathy; depletion of the mitochondrial DNA; and autosomal dominant and recessive mitochondrial myopathies. Molecular defects can be in the mitochondrially encoded tRNAs, mitochondrial- and nuclear-encoded subunits of the oxidative phosphorylation complexes, and many other proteins (Delonlay et al. 2013). Some of the most relevant disorders and etiologies are shown in Tables 42.2 and 42.3.
Muscle symptoms are generally those of chronic weakness and impaired exercise tolerance. Cramps are unusual. Rhabdomyolysis can occur, particularly, in the setting of sustained exercise or febrile illness. Malignant hyperthermia may occur with anesthesia or muscle relaxants (Niezgoda and Morgan 2013).
Systemic lactic acidosis may be present at rest or elicited with exercise. Some infants will have acidosis from the work of breathing, but be chemically normal with ventilator support. CK may be elevated. Plasma amino acids may show increased alanine. Urinary organic acids may show increased of lactate, citric acid cycle intermediates, and dicarboxylic fatty acids. The plasma carnitine level may be normal or low. Plasma acylcarnitine analysis may show a generalized increase in short- and medium-chain species, especially acetylcarnitine, or be unrevealing.
EMG may be normal or suggest myopathy; nerve conduction studies may reveal a peripheral neuropathy, usually axonal.
Muscle tissue can be analyzed for carnitine content and acylcarnitine species and coenzyme Q level. Muscle biopsy may show dense clusters of abnormal mitochondria, especially near the surface of the cell membrane (“ragged-red fibers”), as well as increased lipid, but may be normal (see also Chap. 43). Cells may stain strongly for succinate dehydrogenase (complex II) yet not stain for cytochrome oxidase (COX), particularly in CPEO, KSS, and MERFF. Maternally inherited Leigh syndrome patients may have deficient COX staining, but no ragged-red fibers. Electron microscopy may show abnormal mitochondrial morphology, including paracrystalline inclusions.
Studies of oxidative phosphorylation are best carried out in fresh muscle biopsy tissue. Some laboratories will work with frozen muscle tissue or freshly isolated platelets. Mitochondrial DNA studies are optimally performed from muscle biopsy as well. If there is a heteroplasmic disorder in the mtDNA, other tissues (leukocytes and fibroblasts) can sometimes give a misleading normal result. Mitochondrial myopathies are a subset of the overall group of mitochondrial cytopathies, which are discussed in Chap. 42.
Mitochondrial disorders of oxidative phosphorylation are generally treated with a high-fat, low-carbohydrate diet and supplemental vitamins and antioxidants, especially coenzyme Q (ubiquinone) and riboflavin (which may be quite helpful for complex I deficiency myopathy) (Parikh et al. 2013a, b). Vitamin C, thiamin, vitamin E, vitamin K3 (as an artificial electron acceptor-donor), dichloroacetate, carnitine, and succinate have been used in various situations. Responses are generally subtle, but occasionally a patient responds dramatically to CoQ or other therapies.
The synthesis of coenzyme Q involves nine steps; recessive defects have been discovered in the genes SPSS1, SPSS2, CABC1, COQ2, COQ4, COQ6, ADCK3, and COQ9. Besides myopathy, there may be ataxia, deafness, encephalopathy, liver disease, renal tubular dysfunction, and cardiac valvulopathy, depending on the defect (Desbats et al. 2015; Garcia-Cazorla et al. 2015). In many patients suffering from the myopathic form of CoQ deficiency, MAD (multiple acyl-CoA dehydrogenase) deficiency is the primary defect. Secondary CoQ deficiency is also found in the ataxia-oculomotor apraxia disorders with hypoalbuminemia.
3.3 Myopathies with Major Cardiac Involvement
The cardiac manifestations of several disorders discussed in this chapter, including GSD types II and IV; fatty acid oxidation disorders including carnitine transport defect; deficiencies of carnitine-acylcarnitine translocase; CPT II, VLCAD, and LCHAD/trifunctional enzyme; and mitochondrial disorders, are discussed in Chap. 23.
3.4 Malignant Hyperthermia
Malignant hyperthermia (MH) in response to anesthetics occurs in many different situations and myopathies. MH is most commonly due to the failure of regulation of calcium concentration in the sarcoplasmic reticulum. Excess calcium permits continuous muscle contraction, leading to heat generation and a rise in body temperature. Severe myoglobinuria, irreversible kidney damage, and death from hyperthermia or arrhythmia due to hyperkalemia may occur. For these reasons, all patients with a myopathy must be especially carefully monitored during surgery or any other procedure where anesthesia is used, and the most risky anesthetics (e.g., halothane) and muscle relaxants (e.g., suxamethonium) should be avoided. Premedication with dantrolene can lessen the risk of untoward reactions. Several genes are now known for MH syndromes. MHS1 is due to mutations in the ryanodine receptor RYR1 (with or without central core disease) (Robinson et al. 2006). MHS2 is due to mutations in the alpha subunit of the gated sodium channel IV, SCN4A, also altered in hypokalemic periodic paralysis, paramyotonia congenita, and related disorders. MHS3 may be due to mutations in the calcium channel CACNL2A. MHS5 is due to changes in another calcium channel, CACNA1S. All of these conditions can be dominantly transmitted. Patients with muscular dystrophies or the recessive Native American myopathy with cleft palate and congenital contractures are also vulnerable (Stamm et al. 2008; Stewart et al. 1988). Even for these high-risk conditions, MH does not occur with each exposure to a triggering agent. Of the disorders of intermediary metabolism, which are the primary topic of this book, the greatest risk is to patients with CPT II deficiency (perhaps even in the heterozygous state) and with mitochondrial disorders, but all patients with metabolic myopathy should be regarded as at potential risk for MH.
References
Bruno C, Minetti C, Shanske S et al (1998) Combined defects of muscle phosphofructokinase and AMP deaminase in a child with myoglobinuria. Neurology 50:296–298
Chan EK, Kornberg AJ, Ryan MM (2015) A diagnostic approach to recurrent myalgia and rhabdomyolysis in children. Arch Dis Child 100:793–797
Delonlay P, Rotig A, Sarnat HB (2013) Respiratory chain deficiencies. Handb Clin Neurol 113:1651–1666
Desbats MA, Lunardi G, Doimo M et al (2015) Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J Inherit Metab Dis 38:145–156
Garcia-Cazorla A, Mochel F, Lamari F, Saudubray JM (2015) The clinical spectrum of inherited diseases involved in the synthesis and remodeling of complex lipids. A tentative overview. J Inherit Metab Dis 38:19–40
Gonzalez-Freire M, Santiago C, Gomez-Gallego F et al (2009) Does the K153R variant of the myostatin gene influence the clinical presentation of women with McArdle disease? Neuromuscul Dis: NMD 19:220–222
Hogan KJ, Vladutiu GD (2009) Malignant hyperthermia-like syndrome and carnitine palmitoyltransferase II deficiency with heterozygous R503C mutation. Anesth Analg 109:1070–1072
Howell RR, Byrne B, Darras BT et al (2006) Diagnostic challenges for Pompe disease: an under-recognized cause of floppy baby syndrome. Genet Med 8:289–296
Ibdah JA, Yang Z, Bennett MJ (2000) Liver disease in pregnancy and fetal fatty acid oxidation defects. [Review] [44 refs]. Mol Gen Metab 71:182–189
Innes AM, Seargeant LE, Balachandra K et al (2000) Hepatic carnitine palmitoyltransferase I deficiency presenting as maternal illness in pregnancy. Pediatr Res 47:43–45
Jebbink J, Wolters A, Fernando F et al (2012) Molecular genetics of preeclampsia and HELLP syndrome – a review. Biochim Biophys Acta (BBA) – Mol Basis Dis 1822:1960–1969
Joshi PR, Deschauer M, Zierz S (2012) Clinically symptomatic heterozygous carnitine palmitoyltransferase II (CPT II) deficiency. Wien Klin Wochenschr 124:851–854
Kishnani PS, Austin SL, Arn P et al (2010) Glycogen storage disease type III diagnosis and management guidelines. Genet Med: Off J Am Coll Med Genet 12:446–463
Kishnani PS, Amartino HM, Lindberg C et al (2013) Timing of diagnosis of patients with Pompe disease: data from the Pompe registry. Am J Med Genet A 161a:2431–2443
Lachmann R, Schoser B (2013) The clinical relevance of outcomes used in late-onset Pompe disease: can we do better? Orphanet J Rare Dis 8:160
Magoulas PL, El-Hattab AW (1993) Glycogen storage disease type IV. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al (eds) GeneReviews(R). University of Washington, Seattle
Martin MA, Lucia A, Arenas J, et al (1993) Glycogen storage disease type V. In: Pagon RA, Adam MP, Ardinger HH et al (eds) GeneReviews (R). University of Washington, Seattle. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1344/
Martinuzzi A, Sartori E, Fanin M et al (2003) Phenotype modulators in myophosphorylase deficiency. Ann Neurol 53:497–502
Niezgoda J, Morgan PG (2013) Anesthetic considerations in patients with mitochondrial defects. Paediatr Anaesth 23:785–793
Oldfors A, DiMauro S (2013) New insights in the field of muscle glycogenoses. Curr Opin Neurol 26:544–553
Orngreen MC, Jeppesen TD, Andersen ST et al (2009) Fat metabolism during exercise in patients with McArdle disease. Neurology 72:718–724
Parikh S, Goldstein A, Koenig MK et al (2014) Practice patterns of mitochondrial disease physicians in North America. Part 1: diagnostic and clinical challenges. Mitochondrion 14:26–33
Parikh S, Goldstein A, Koenig MK et al (2013) Practice patterns of mitochondrial disease physicians in North America. Part 2: treatment, care and management. Mitochondrion 13:681–687
Robinson R, Carpenter D, Shaw MA et al (2006) Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat 27(10):977–989
Roe CR, Brunengraber H (2015) Anaplerotic treatment of long-chain fat oxidation disorders with triheptanoin: Review of 15 years experience. Mol Genet Metab 116:260–268
Roe CR, Bottiglieri T, Wallace M (2010) Adult polyglucosan body disease (APBD): anaplerotic diet therapy (triheptanoin) and demonstration of defective methylation pathways. Mol Genet Metab 101:246–252
Rubio JC, Martin MA, Bautista J (1997) Association of genetically proven deficiencies of myophosphorylase and AMP deaminase: a second case of ‘double trouble’. Neuromuscul Dis: NMD 7(6–7):387–389
Sharp LJ, Haller RG (2014) Metabolic and mitochondrial myopathies. Neurol Clin 32:777–799, ix
Stamm DS, Aylsworth AS, Stajich JM, Kahler SG et al (2008) Native American myopathy: congenital myopathy with cleft palate, skeletal anomalies, and susceptibility to malignant hyperthermia. Am J Med Genet A 146A:1832–1841
Stewart CR, Kahler SG, Gilchrist JM (1988) Congenital myopathy with cleft palate and increased susceptibility to malignant hyperthermia: King syndrome? Pediatr Neurol 4:371–374
Vladutiu GD, Bennett MJ, Smail D (2000) A variable myopathy associated with heterozygosity for the R503C mutation in the carnitine palmitoyltransferase II gene. Mol Genet Metab 70:134–141
Winkel LP, Hagemans ML, van Doorn PA et al (2005) The natural course of non-classic Pompe’s disease; a review of 225 published cases. J Neurol 252:875–884
Wu JW, Yang H, Wang SP et al (2015) Inborn errors of cytoplasmic triglyceride metabolism. J Inherit Metab Dis 38:85–98
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Kahler, S.G. (2017). Metabolic Myopathies. In: Hoffmann, G., Zschocke, J., Nyhan, W. (eds) Inherited Metabolic Diseases. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-49410-3_28
Download citation
DOI: https://doi.org/10.1007/978-3-662-49410-3_28
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-49408-0
Online ISBN: 978-3-662-49410-3
eBook Packages: MedicineMedicine (R0)