
Abstract
Hyperammonemia is the hallmark of the disorders of the urea cycle but occurs episodically also in organic acidurias and disorders of fatty acid oxidation. Routine clinical chemistry is helpful in pointing the direction of the work-up of a patient. Patients with urea cycle defects are alkalotic or normal; those with organic acidurias or disorders of fatty acid oxidation are acidotic. The next step toward a definitive differential diagnosis is the quantitative assay of the concentrations of amino acids in plasma (glutamine, citrulline, arginine) and urine (argininosuccinic acid, orotic acid) or dry blood (argininosuccinic acid).
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
FormalPara Key Facts-
Routine clinical chemistry is helpful in pointing the direction of the work-up of a patient with hyperammonemia. Patients with urea cycle defects are alkalotic or normal; those with organic acidemias or disorders of fatty acid oxidation are acidotic. The presence or absence of ketosis distinguishes the latter two.
-
Urea cycle defects are elucidated by analysis of the amino acids of the plasma (glutamine, citrulline, arginine), urine (argininosuccinic acid, orotic acid), or dried blood (argininosuccinic acid).
-
Urinary orotic aciduria distinguishes ornithine transcarbamylase deficiency from carbamoyl phosphate I synthetase and N-acetylglutamate synthase deficiencies.
Elevated concentrations of ammonia occur episodically in a variety of inherited diseases of metabolism. These include not only the disorders of the urea cycle but also organic acidurias and disorders of fatty acid oxidation. Effective management is predicated on a precise diagnosis and understanding of the nature of the pathophysiology. A systematic progression from routine clinical chemistry to more specific analyses of amino acids, organic acids, and acylcarnitines will lead the clinician to the diagnosis. Liver biopsy has been required for enzymatic diagnosis of carbamoyl phosphate synthetase I deficiency and ornithine transcarbamylase (OTC) deficiency, as well as N-acetylglutamate synthase deficiency. Meanwhile, however, mutational analysis has usually obviated the need for this invasive approach.
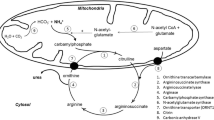
Deficiencies of enzymes of the urea cycle and some other disorders, such as the organic acidurias and disorders of fatty acid oxidation, present with hyperammonemia. Normally, values of NH3 are <110 μmol/L (190 μg/dL) in newborns and below 80 μmol/L (140 μg/dL) in older infants to adults. In the newborn period, a diagnostic work-up for hyperammonemia is warranted at values >150 μmol/L (260 μg/dL) and in older infants to adults at values >100 μmol/L (175 μg/dL). The classic onset of urea cycle defects is with sudden potentially lethal neonatal coma. The male with OTC deficiency exemplifies the classic presentation, but distinct disorders result from deficient activity of each of the enzymes of the urea cycle (Fig. 17.1).

Urea cycle
The work-up of an infant in hyperammonemic coma is shown (Fig. 17.2). The differential diagnosis is very important because different disorders require different treatments. It must proceed with dispatch if the correct diagnosis is to be made and appropriate therapy instituted to prevent death or permanent damage to the brain be done. The initial steps in the algorithm are available in the routine clinical chemistry laboratory.

An approach to the stepwise evaluation of a patient with hyperammonemia
The first step in the evaluation of a patient, especially an infant in coma, is the measurement of the concentration of ammonia in the blood. Since ammonia analysis is very sensitive to various mistakes (e.g., use of test tubes containing ammonium heparin, use of a tourniquet, lack of cooling in ice water, vigorous shaking, delayed start of the analysis), a correct pre-analytical process is indispensable to obtain reliable test results. Very good and reliable readings can be obtained with bedside ammonia checkers. However, some machines have an upper reading fixed at below 300 μmol/L ammonia. Then the sample must be diluted and reanalyzed. We have repeatedly received clinically deteriorating children with supposedly this level where ammonia had in fact already raised >5,000 μmol/L. The next step after verifying hyperammonemia is the quantification of serum concentrations of bicarbonate, sodium, chloride, and the anion gap and testing of the urine for ketones. Metabolic acidosis and/or increased anion gap lowers the likelihood of a urea cycle disorders, which tend to present with respiratory alkalosis, but does not necessarily exclude this diagnosis. The acidotic patient with massive ketosis has an organic aciduria, such as propionic aciduria, methylmalonic aciduria, isovaleric aciduria, glutaric aciduria type II, or multiple carboxylase deficiency (see Chap. 13). A specific diagnosis is made by quantitative analysis of the organic acids of the urine or of the acylcarnitines of the blood. The disorders of fatty acid oxidation, which may present with hyperammonemia, are characteristically hypoketotic (see Chap. 16). However, testing of the urine for ketones may be misleading. We have observed impressively positive urinary KetoStix tests in patients with disorders of fatty acid oxidation. Quantification of concentrations of free fatty acids together with acetoacetic and 3-hydroxybutyric acid in the blood of such patients reliably indicate them to have defective ketogenesis. The acute crises in these patients often display hypoglycemia, and diagnoses of Reye syndrome have been made. A patient with hyperammonemic coma resulting from a urea cycle defect may develop hypoxia, leading to lactic acidosis. Adequate oxygenation and perfusion should be assured before a urea cycle defect is conceptually excluded and a diagnosis of organic aciduria pursued.
Hyperammonemia occurs not only in disorders of the urea cycle but also in organic acidemias and disorders of fatty acid oxidation. Testing for organic acids in urine and acylcarnitine profiles lead to the correct diagnosis.
The definitive diagnosis of a urea cycle abnormality is initiated by the quantitative assay of the concentrations of amino acids in the blood and urine. The plasma concentrations of amino acids provide the diagnosis in patients with argininemia and citrullinemia. Study of the urine is required in argininosuccinic aciduria.
If hyperammonemic patients are found not to have a diagnostic abnormality in the concentration of an amino acid, the urine should be tested for the excretion of orotic acid. This is not reliably performed as a part of organic acid analysis by GCMS, and a specific assay for the compound should be employed. Orotic aciduria is found in patients with OTC deficiency. It is also found in citrullinemia and in argininemia. In a patient without an elevation of a specific amino acid and without orotic aciduria, the usual diagnosis is carbamoyl phosphate synthetase (CPS) I deficiency. N-acetylglutamate synthase deficiency will present with an indistinguishable clinical and biochemical constellation but is much rarer. Very similar is also the mitochondrial carbonic anhydrase VA deficiency which is also associated with low-normal orotic acid excretion and hyperlactatemia. Carbonic anhydrase VA provides bicarbonate to CPS, pyruvate carboxylase, propionyl-CoA carboxylase, and 3-methylcrotonyl-CoA carboxylase. Transient hyperammonemia of the newborn may also present this picture, but for reasons that are not clear, this disorder is nowhere near as commonly encountered as it was 30 years ago. Failure of immediate closure of the ductus venosus after birth is thought to result in (transient) hyperammonemia of the newborn because portal blood bypasses the liver. The definitive diagnoses of CPS, OTC, N-acetylglutamate synthase and mitochondrial carbonic anhydrase VA deficiencies are made by mutation analysis instead of a liver biopsy. If a liver biopsy was planned, it would be well to bring the patient into control of the blood concentration of ammonia and to normalize clotting. The levels of arginine and citrulline in the blood may be helpful in distinguishing CPS I deficiency from transient hyperammonemia of the newborn, in which it is usually normal or elevated. In neonatal CPS, OTC or N-acetylglutamate synthase deficiencies, citrulline is barely detectable. In citrullinemia, concentrations of citrulline in plasma usually exceed 1,000 μmol/L. They are elevated to levels of 150–250 μmol/L in argininosuccinic aciduria, and to 54 ± 22 μmol/L in transient hyperammonemia in the newborn. The normal range is 6–20 μmol/L. Persistent hypocitrullinemia can, in general, be viewed as a marker for disorders of mitochondrial urea cycle enzymes (N-acetylglutamate synthase, CPS I, and ornithine carbamoyltransferase) as well as for deficient pyrroline-5-carboxylate synthetase. Citrulline synthesis is directly coupled to ATP concentration. Consequently, hypocitrullinemia can also be observed in patients with respiratory chain disorders, especially as caused by NARP mutation.
Amino acid analysis also reveals concentrations of glutamine to be regularly elevated in patients with hyperammonemia except for those having classic organic acidurias. Concentrations of alanine are usually elevated, while concentrations of aspartic acid are elevated in some patients. These are nonspecific findings. They are not helpful in the differentiation of the different causes of hyperammonemia. They are potentially helpful in diagnosis, as sometimes an elevated level of glutamine is found in a patient that had not been expected to have hyperammonemia, and while concentrations of ammonia may vary from hour to hour, the elevated concentration of glutamine signifies a state in which there has been more chronic overabundance of ammonia. The transamination of pyruvic acid to alanine and oxaloacetic acid to aspartic acid, as well as 2-oxoglutaric acid to glutamic acid and its subsequent amidation to glutamine are detoxification responses to the presence of excessive quantities of ammonia. Since in patients with classic organic acidurias various enzymes in the tricarboxylic acid cycle are inhibited by toxic metabolic products (e.g., propionyl-CoA, methylcitrate), the availability of 2-oxoglutarate and thus glutamine synthesis is impaired.
Amino acid analysis may also reveal elevations of tyrosine, phenylalanine, and the branched-chain amino acids. If these are substantial, a primary liver disease should be carefully sought.
Some patients with defects of urea cycle and residual enzyme activity, especially many females with OTC deficiency, display completely unremarkable values of ammonia, amino acids, and orotate in between the crises. In female patients with OTC deficiency, ammonia may be normal even during crisis, whereas plasma and cerebral glutamine concentrations are high. It is indispensable that the cause of an unexplained symptomatic episode of hyperammonemia should always be investigated in detail even after the patient recovers, even in adults or aged adults. In those instances, in which the amino acids are normal, an allopurinol loading test may reveal the diagnostic direction.
The differential diagnosis of hyperammonemia also includes the HHH syndrome, which results from deficiency of the ornithine transporter in the mitochondrial membrane, lysinuric protein intolerance, resulting from a (re)absorption defect of the dibasic amino acids or by deficiency of the amino acid transporter citrin which occurs predominantly in east Asians. HHH signifies hyperammonemia, hyperornithinemia, and homocitrullinuria. It is usually suspected first by the identification of large amounts of homocitrulline in the urine. The diagnosis of lysinuric protein intolerance is best made by finding very low levels of lysine in the blood. These patients fail to thrive, and when body stores of lysine are much depleted, the characteristic amino aciduria may not be present; it returns when the diagnosis is made, and blood concentrations of amino acid are brought to normal. The metabolic abnormalities become more obvious by calculating the fractional clearances of lysine and other dibasic amino acids. The concentration of citrulline in the blood may be high. Citrin deficiency presents usually between the second and fourth decade of life as recurrent hyperammonemia with neuropsychiatric symptoms. Onset of symptoms can be rapidly precipitated by medications, surgery, and alcohol consumption.
In older children and adults, a number of acquired disorders can also present with hyperammonemia, especially liver disease, Reye syndrome, drug toxicity, e.g., chemotherapeutics, and hepatotoxins. History, prothrombin time, a urinary toxic screen, and plasma amino acid pattern should help to differentiate these disorders. Symptomatic hyperammonemia may also result from a urinary tract infection or postsurgical superinfection of large hematomas in which the infecting Proteus mirabilis has urease activity, which produces ammonia from urea.
General Suggestions for Reading
Batshaw ML, Brusilow S, Waber L et al (1982) Treatment of inborn errors of urea synthesis: activation of alternative pathways of waste nitrogen synthesis and excretion. N Engl J Med 306:1387
Baumgartner MR, Hörster F, Dionisi-Vici C et al (2014) Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis 9:130
Enns G, Berry SA, Berry GT et al (2007) Survival after treatment with phenylacetate and benzoate for urea-cycle disorders. N Engl J Med 356:2282–2292
Häberle J, Boddaert N, Burlina AB et al (2012) Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis 7:32
Nyhan WL, Barshop BA, Al-Aqeel A (2012) Ornithine transcarbamylase deficiency. In: Atlas of inherited metabolic diseases, 3rd edn. Hodder Arnold, London, pp 191–197
Thoene JG (1999) Treatment of urea cycle disorders. J Pediatr 134:255–256
Van Karnebeek C, Sly WS, Ross CJ et al (2014) Mitochondrial carbonic anhydrase VA deficiency resulting from CA5A alterations present with hyperammonemia in early childhood. Am J Hum Genet 94:453–461
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Nyhan, W.L., Kölker, S., Hoffmann, G.F. (2017). Work-Up of the Patient with Hyperammonemia. In: Hoffmann, G., Zschocke, J., Nyhan, W. (eds) Inherited Metabolic Diseases. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-49410-3_17
Download citation
DOI: https://doi.org/10.1007/978-3-662-49410-3_17
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-49408-0
Online ISBN: 978-3-662-49410-3
eBook Packages: MedicineMedicine (R0)