
Abstract
Opioids are considered the gold standard for the treatment of moderate to severe pain. However, heterogeneity in analgesic efficacy, poor potency and side effects are associated with opioid use, resulting in dose limitations and suboptimal pain management. Traditionally thought to exhibit their analgesic actions via the activation of the neuronal G-protein-coupled opioid receptors, it is now widely accepted that neuronal activity of opioids cannot fully explain the initiation and maintenance of opioid tolerance, hyperalgesia and allodynia. In this review we will highlight the evidence supporting the role of non-neuronal mechanisms in opioid signalling, paying particular attention to the relationship of opioids and immune signalling.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
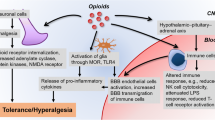
Opioids not only suppress pain, they also activate endogenous counter-regulatory mechanisms that, for example, actively oppose opioid-induced pain suppression, enhance analgesic tolerance wherein repeated opioid doses lose their ability to suppress pain, and enhance dependence as continued opioid exposure is required to stave off drug withdrawal (Watkins et al. 2009). Despite the continual clinical use of opioids over several millennia, and intense scientific research in the past century, a complete understanding of opioid action remains elusive. Of particular importance to this review are those opioid-induced systems that counter-regulate the beneficial and wanted opioid actions. For example, hypertrophy of the cyclic AMP system, enhancement of N-methyl-d-aspartate (NMDA) receptor activity, upregulation of P-glycoprotein and hetero-dimerisation and trafficking of μ-opioid/δ-opioid receptors have been shown to contribute to opioid tolerance and addiction. However, in recent years immune signalling within the central nervous system (CNS), such as that derived from non-neuronal cells, has become the focus of many groups (Johnston et al. 2004; Shavit et al. 2005; Hutchinson et al. 2008a). This ‘central immune signalling’ profoundly affects all types of cells within the CNS, contributing to the development of the negative side effects of opioids, such as tolerance and enhanced pain states.
2 Opioid-Induced Initiation of Non-neuronal Cell Intracellular Signalling in the Central Nervous System
It is now thought that exclusively considering neuronal activity provides an incomplete understanding of the initiation and maintenance of opioid tolerance, hyperalgesia and allodynia. In recent times, one of the most prominently reported cascades in the CNS influenced by opioid exposure is the mitogen-activated protein kinase (MAPK) signalling pathway. The MAPK pathway comprises a collection of secondary messengers that are recruited by cell surface receptors in response to extracellular stimuli to elicit various cellular responses, such as gene expression. Phosphorylation of the three key kinases of this pathway, p38, JNK, and ERK, results in an active functional signalling complex. Morphine has been shown to activate both p38 and ERK within microglia, which can be prevented by administration of (−)-naloxone (an opioid antagonist) and minocycline (a glial attenuator) (Cui et al. 2006; Xie et al. 2010). The role of JNK is less clear; Guo and co-workers have reported morphine-mediated phosphorylation of JNK in astrocytes in an NMDA receptor-dependent fashion (Guo et al. 2009), whereas others have reported it to be unaffected (Wang et al. 2009). In addition to the MAPK pathway, the IP3/Akt pathway is also activated by opioid exposure and appears to be involved in activation of microglial ERK (Takayama and Ueda 2005; Horvath and DeLeo 2009). The common downstream consequence of MAPK and related pathway signalling is activation of NF-κB, which is responsible for the transcriptional activation of a large number of immune factors, such as IL-1β, IL-6 and TNF-α (Baeuerle and Henkel 1994). Classical G-protein-coupled opioid receptors play a fundamental role in opioid pharmacology; however, as discussed below, a key role for nonclassical opioid sites has been for the most part overlooked.
3 Non-neuronal Central Immune Cells
It has recently been recognised that non-neuronal immunocompetent cells (glia–astrocytes and microglia [‘glia’] and endothelial cells) of the CNS and brain play a powerful modulatory role in pain and opioid pharmacodynamics (Hutchinson et al. 2011). Activation of these immunocompetent cells is thought to enhance spinal nociceptive transmission and behavioural responsiveness via the release of central immune signals such as cytokines, chemokines, ATP, nitric oxide and excitatory amino acids (Watkins et al. 2005, 2009; Hutchinson et al. 2008a). It has been suggested that μ-opioid receptor (MOR) agonists are responsible for the glial activation. However, findings by Kao and colleagues indicate that MOR expression is absent from spinal cord astrocytes and microglia, suggesting that these cell types are indirectly activated by MOR agonists under chronic opioid tolerance conditions (Kao et al. 2012).
3.1 Microglia
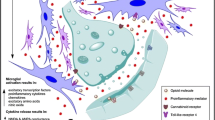
Microglia are a subset of glial cells within the brain and CNS that make up 5–12 % of all cells and 5–10 % of all glia. Microglia are the resident immune cells of the CNS and under basal conditions scan their microenvironment, performing immune surveillance (Raivich 2005). Microglia are believed to be the most reactive and mobile cells of the CNS and a shift to an activated state can occur within minutes (Morioka et al. 1991). Given their immunological roles, it is no surprise microglia share many of the same immune signalling and response systems as peripheral immune cells. Critical to sensing their microenvironment, microglia express key innate-immune receptors and accompanying response pathways such as the innate-immune pattern recognition toll-like receptors (TLRs). The sensing capabilities of the innate-immune receptors are key in activating early response pathways to molecules such as endogenous danger signals (e.g. heat shock proteins) and xenobiotics (e.g. opioids) (Buchanan et al. 2010). Activation of microglia results in changes in morphology, rapid proliferation, upregulated receptor expression (e.g. complement receptors, TLRs) and changes in function (e.g. migration to sites of damage, phagocytosis and release of proinflammatory mediators). After the activation stimulus has resolved, microglia can either return to their basal state or enter a ‘primed’ state. Primed microglia do not constitutively produce proinflammatory mediators but may overrespond to new challenges, both in speed and magnitude of release of proinflammatory mediators (Perry et al. 1985; Watkins et al. 2007).
3.2 Astrocytes
Astrocytes are the most abundant glial cell, significantly outnumber neurons, populate all regions of the CNS and, for a long time, were considered exclusively as the metabolic supporting cells for neurons. Astrocytes have highly dynamic processes and are well suited to share synaptic functions with neurons due to their location, organisation and the morphology of their processes that, in combination with microglia, are capable of completely encapsulating neuronal synapses to form the tetrapartite synapse (De Leo et al. 2006; Watkins et al. 2009). The intimate contact astrocytes and microglia have with neurons allows these cells to directly modulate neuron-to-neuron synaptic communication. Astrocytes are known to play an important structural and metabolic role in the homeostasis of the extracellular environment, providing the required conditions for the function of neurons and synapses. Some key roles include forming the blood–brain barrier; metabolic support to neurons, supplying nutrients and neurotransmitters; maintenance of the extracellular environment such as uptake and release of neurotransmitters; regulation of ion concentrations; and detection of neuronal damage (Johnston et al. 2004; Shavit et al. 2005; Hutchinson et al. 2008a, 2011; Araque and Navarrete 2010; Smith 2010). Like microglia, upon stimulation, astrocytes are capable of changing from their basal but active state to elicit a proinflammatory response profile characterised by changes in morphology, proliferation and expression of inflammatory factors such as cytokines and chemokines (Cui et al. 2006; Ben Achour and Pascual 2010; Xie et al. 2010). The immune signalling can further activate other nearby glia and ultimately leads to altered homeostatic balance resulting in the release of various soluble factors involved in neuronal hyperexcitability and the development of abnormal pain.
3.3 Other Cell Types
Microglia and astrocytes are not likely to be the only non-neuronal cell types able to elicit a proinflammatory response profile. Endothelial cells, fibroblasts, oligodendrocytes and other cell types in both the spinal cord and overlying meninges can also produce many of the same neuroexcitatory substances as astrocytes and microglia. For example, it has recently been hypothesised that following the release of central immune signals, the tight junctions of endothelial cells of the blood–brain barrier become leaky, exposing the CNS to peripheral immune signals (Guo et al. 2009; Grace et al. 2011). However, blood–brain barrier endothelial cells remain a significant, yet largely uncharacterised, source of central immune signalling and contributor to altered neuronal function (Quan et al. 2003; Wang et al. 2009).
3.4 Central Immune Synergy
Glia and their released products can work in synergy resulting in an enhanced state of activation and further release of central immune products. With regard to activation, microglia release substances that induce astrocyte activation, expression of adhesion molecules and release of glutamate, TNF, IL-1β and nitric oxide. Astrocytes in return can release substances that stimulate microglial activation, proliferation and production of IL-1β, TNF-α, IL-6 and nitric oxide. The release of these central immune products can synergise and induce the release of others. For example, proinflammatory cytokines can synergise with each other as well as with neurotransmitters and neuromodulators, such as norepinephrine, prostaglandin E2 (PGE2) and nitric oxide. The synergy of TNF and IL-1β with ATP can enhance PGE2 release (Loredo and Benton 1998; Takayama and Ueda 2005; Horvath and DeLeo 2009). Nitric oxide potentiates IL-1β, which can induce PGE2 production and substance P release from sensory afferent terminals in the spinal cord. Substance P can potentiate IL-1β-induced release of IL-6 and PGE2 from human spinal cord astrocytes (Baeuerle and Henkel 1994; Morioka et al. 2002; Watkins et al. 2007). The synergy of these products can therefore lead to the production and release of substances that further activate central immune cells within the CNS leading to enhanced modulation of excitatory neurotransmission.
4 Involvement of Immunocompetent Cells in Opioid Pharmacodynamics
It is now widely accepted that neuronal activity of opioids cannot fully explain the initiation and maintenance of opioid tolerance, hyperalgesia and allodynia. A greater understanding of the role of non-neuronal immunocompetent cells of the CNS and brain is required to fully understand the intricate mechanisms behind pain and opioid pharmacodynamics (Hutchinson et al. 2011). Increased astrocyte activation in the spinal cord following chronic systemic morphine administration was the first report linking glia to morphine tolerance (Song and Zhao 2001; Watkins et al. 2005, 2009; Hutchinson et al. 2008a). Importantly, co-administration of fluorocitrate (a glial metabolic inhibitor) with morphine significantly attenuated not only glial activation but also morphine tolerance. Further work has demonstrated that morphine activates both microglia and astrocytes (Song and Zhao 2001; Cui et al. 2008; Bland et al. 2009; Kao et al. 2012) which is associated with the upregulation and release of proinflammatory cytokines (Raghavendra et al. 2002, 2004; Johnston et al. 2004; Raivich 2005; Hutchinson et al. 2008a, 2009). This opioid-induced non-neuronal cell-mediated anti-analgesia is significantly reduced by co-administration with either the glial attenuators minocycline, ibudilast or fluorocitrate (Morioka et al. 1991; Song and Zhao 2001; Cui et al. 2008; Hutchinson et al. 2008a, b, 2009), or by directly blocking proinflammatory cytokine actions (Fairbanks and Wilcox 2000; Shavit et al. 2005; Hutchinson et al. 2008a, c; Buchanan et al. 2010).
5 Stereoselective and Non-stereoselective Receptor Binding
In the infancy of opioid research, attention was focused directly towards the stereoselective receptors that were shown to be critical for opioid analgesic responses. Classical opioid receptors are stereoselective, as they bind (−)-opioid isomers but not (+)-opioid isomers. Intriguingly, nonclassical opioid actions were observed in one of the first studies that used synthesised inactive opioid stereoisomers (Takagi et al. 1960; Perry et al. 1985; Watkins et al. 2007). Takagi et al. (1960) demonstrated that co-administration of (+)-morphine with (−)-morphine gave rise to naïve tolerance with significantly reduced (−)-morphine-induced analgesia. Further evidence for nonclassical opioid receptors was established in early opioid binding studies conducted by Goldstein et al. (1971). Goldstein demonstrated that the classical stereoselective opioid receptors only made up a small portion of total opioid binding (2 %). Non-stereoselective nonclassical opioid binding sites were responsible for more than half (53 %), while 46 % was attributed to non-specific trapped and dissolved binding. Since then, several studies have reported that (+)-opioid agonists suppress (−)-opioid analgesia (Wu et al. 2007), an effect attributed to glial activation (Wu et al. 2005) which is independent of classical MORs (Wu et al. 2006). To further highlight the involvement of nonclassical opioid receptors, continuous infusion of morphine, oxymorphone or fentanyl administered to opioid receptor knockout mice initiated immediate and steady declines in nociceptive thresholds culminating in several days of unremitting hyperalgesia (Juni et al. 2007; Waxman et al. 2009). Not only did this suggest an involvement of nonclassical opioid receptors but also indicated such receptors are likely responsible for activating endogenous counter-regulatory mechanisms that actively oppose opioid-induced pain suppression and enhance analgesic tolerance and opioid dependence.
6 Non-stereoselective Activation of Central Immune Cells
Recent work by Hutchinson and colleagues has suggested that the innate-immune toll-like receptor-4 (TLR4) is involved in the non-stereoselective binding of opioids (Watkins et al. 2009; Hutchinson et al. 2010; Wang et al. 2012). In vivo, in vitro and in silico approaches provided converging lines of evidence that members of each structural class of opioids activate TLR4 and that opioid antagonists such as naloxone and naltrexone non-stereoselectively block TLR4 signalling (Hutchinson et al. 2010). It was demonstrated that acute pharmacological blockade of TLR4, genetic knockout of TLR4 or blockade of TLR4 downstream signalling leads to a marked potentiation of the magnitude and duration of opioid analgesia, with TLR4 modulation of opioid actions in wild-type animals occurring within minutes (Hutchinson et al. 2010). Furthermore, Wang et al. (2012) demonstrated that morphine binds the human TLR4 accessory protein, MD-2, inducing TLR4 oligomerisation to activate TLR4 signalling. Within the CNS, TLR4 is predominantly expressed by microglia and astrocytes, but expression has also been demonstrated on other non-neuronal cells such as endothelial cells (Tanga et al. 2005; Wang et al. 2010).
7 Soluble Contributors to Opioid Analgesia Opposition
As outlined below, there have been numerous studies looking at soluble factors that either reduce or contribute to opioid-induced pain enhancement. This suggests that, for many situations of abnormal pain, it is not generally just one mediator contributing to the initiation and maintenance but rather a combination thereof. Here, we will highlight the evidence that suggests acute opioid analgesia is substantially modified by the rapid opioid-induced initiation of central immune signalling and that, upon repeated opioid exposure, continued central immune signalling leads to analgesic tolerance and enhanced pain states.
7.1 Cytokines
Cytokines are proteins involved in paracrine and autocrine communication. Cytokines bind to specific receptors on the surface of target cells, which generally activate intracellular signalling and second messenger cascades. One of the key features of immune-derived cytokines is their ability to trigger the feedforward release of more proinflammatory cytokines, which is an important feature of inflammation. In the CNS, cytokines are an effective means for inducing physiological responses to stress, immunological challenges and pathological conditions, but cytokine signalling can also have detrimental effects on neuronal signalling. Within the CNS, astrocytes, microglia, oligodendrocytes and endothelial cells are robust producers of cytokines, in particular, IL-1β, IL-6 and TNF-α. These proinflammatory cytokines are known to be involved in initiating and maintaining states of enhanced pain in pathologies such as neuropathic pain (Milligan et al. 2003; Milligan and Watkins 2009). Importantly however, the levels of cytokine signalling required to elicit a behavioural response in the CNS is far below the quantitative threshold considered as a classical inflammatory response. Hence, the concepts of central immune signalling and homeostatic immune signalling have been proposed (Hutchinson et al. 2011). A recent major development in opioid pharmacology was the demonstration that these key molecules are also significantly involved in opioid analgesia (Johnston et al. 2004; Shavit et al. 2005; Hutchinson et al. 2008a).
Proinflammatory cytokines are gaining traction in the involvement in opioid-induced tolerance and enhanced pain states. Of particular interest is the evidence that proinflammatory cytokines can substantially modify opioid analgesia following a single administration. Blockade of the key inflammatory cytokine IL-1β using an exogenous IL-1 receptor antagonist (IL-1ra) potentiated both acute and chronic, systemic and intrathecal morphine analgesia (Fairbanks and Wilcox 2000; Johnston et al. 2004; Shavit et al. 2005; Hutchinson et al. 2008a). These results have been confirmed using three separate genetically modified strains of mice lacking IL-β function: a transgenic knock-in of IL-1ra such that IL-1ra is over-expressed, IL-1 receptor knockout leaving IL-1β without its cognate receptor and IL-1 receptor accessory protein knockout preventing IL-1 receptor signalling due to the lack of an intracellular link to the associated toll/IL-1 receptor signalling cascade (Shavit et al. 2005). In each case, morphine analgesia was significantly potentiated and prolonged (Shavit et al. 2005). The acute morphine-induced IL-1β signalling caused nearly an eightfold decrease in morphine analgesic potency as demonstrated by a profound leftward shift in the morphine dose–response curve in the presence of IL-1ra (Hutchinson et al. 2008a). IL-1β not only reduces the potency of morphine, it also reduces the duration of effect. Administration of IL-1ra after the normal analgesic response returned to pre-drug baseline unmasked significant analgesia (Shavit et al. 2005; Hutchinson et al. 2008a). This proinflammatory mediated anti-analgesic effect is not limited to IL-1β, as unmasking and/or potentiation of morphine analgesia is also observed by blocking the action of IL-6 and TNF-α (Johnston et al. 2004; Hutchinson et al. 2008a).
Potentiating/unmasking opioid analgesia can also be achieved using less-specific pharmacological interventions such as minocycline or ibudilast that globally disrupt glial cell activation and subsequently the release of inflammatory mediators (Hutchinson et al. 2008a, 2009). Attenuation of glial activation with ibudilast resulted in a fivefold increase in analgesic potency (Hutchinson et al. 2009). This induction of anti-analgesic central immune signalling is not a phenomenon limited to morphine, as oxycodone analgesia was also potentiated threefold by ibudilast (Hutchinson et al. 2009). In addition, the activation of endogenous anti-inflammatory systems that result in elevations of IL-10 or administration of exogenous IL-10 was also capable of potentiating acute morphine analgesia (Fairbanks and Wilcox 2000; Johnston et al. 2004; Hutchinson et al. 2008a). Thus, acute opioid-induced proinflammatory central immune signalling can be pharmacologically modified to enhance acute opioid analgesia.
While numerous studies have demonstrated clear opioid-induced activation of central immune signalling responses, repeated attempts to quantify short-term transcriptional and/or translational events of these proinflammatory central immune signals, after acute in vivo opioid administration, have failed (Johnston et al. 2004; Hutchinson et al. 2008a). Cytokine receptors and their ligands exhibit very high affinity and potency; thus, very low quantities of opioid-induced cytokine release (at levels undetectable by current cytokine quantification techniques) can potentially cause a significant biological effect. Moreover, it is possible that these short-term effects result from the activation of stored immature protein and therefore do not require transcription and translation.
7.2 Proinflammatory Cytokine-Mediated Neuronal Excitation
The release of proinflammatory products can result in enhanced neuronal excitation in the dorsal horn of the spinal cord (Kawasaki et al. 2008) and actively oppose opioid analgesia (Johnston et al. 2004; Shavit et al. 2005; Hutchinson et al. 2008a). Neurons of the spinal cord express receptors for proinflammatory cytokines and chemokines and exhibit increased neuronal excitability in response to these immune signals (Oka et al. 1994; Dame and Juul 2000; Holmes et al. 2004; Kawasaki et al. 2008). These immune-derived cytokines contribute to a phenomenon called central sensitisation, which has been well studied in the area of neuropathic pain (Kawasaki et al. 2008). IL-1β release has been shown to induce the phosphorylation of NMDA receptors on neurons which leads to an increase in calcium conductivity (Viviani et al. 2003; Broom et al. 2004). TNF-α increases α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor conductivity while also increasing spontaneous neurotransmitter release from neuronal presynaptic terminals (De et al. 2003). IL-1β and TNF-α synergistically upregulate neuronal cell surface expression of both NMDA and AMPA receptors while downregulating gamma-aminobutyric acid (GABA) receptors (Stellwagen et al. 2005). TNF-α also enhances neuroexcitability in response to glutamate (Emch et al. 2001), and IL-1β induces the release of the neuroexcitant ATP via an NMDA-mediated mechanism (Sperlágh et al. 2004). Beyond these actions, proinflammatory cytokines also lead to the release of a host of neuroexcitatory substances, including more proinflammatory cytokines, nitric oxide, prostaglandins, nerve growth factors, reactive oxygen species, proinflammatory chemokines (e.g. CCL2/MCP-1, CXCL8/IL-8, CXCL10/IP-10) and BDNF (Watkins et al. 1999; Samad et al. 2001; Inoue 2006). Proinflammatory cytokines can also indirectly lead to elevations in extracellular glutamate levels via downregulation of glial and neuronal glutamate transporters (Tawfik et al. 2006). Thus, taken together, opioid-induced release of proinflammatory cytokines exert multiple effects resulting in increased neuronal excitatory tone, which is, in part, the basis behind enhancing the development of hyperalgesia and tolerance.
7.3 Chemokines
Chemokines are a family of small proteins characterised as chemotactic cytokines, involved in cellular migration and intercellular communication. Chemokine receptors are members of the G-protein-coupled receptor (GPCR) superfamily. Chemokines and their receptors have four subclasses of families: C, CC, CXC and CX3C (Murphy et al. 2000). Some chemokines are considered to be proinflammatory and released during an immune response to recruit cells of the immune system to specific sites, while other chemokines are considered homeostatic and control the migration of cells during normal processes of tissue maintenance and development. Some key CNS and brain-derived chemokines include, but are not limited to, CCL2/MCP-1, CCL3/MIP-1α, CCL5/RANTES, CXCL12/SDF-1, CX3CL1/fractalkine and CXCL10. The distributions of chemokines within the CNS are heterogeneous where many have an established involvement in the modulation of pain and opioid pharmacodynamics. There is now a large amount of data indicating that chemokines and their receptors can influence both the acute and chronic stages of pain and opioid analgesia (Szabo et al. 2002; Johnston et al. 2004; Chen et al. 2007a; Triantafilou et al. 2008).
Fractalkine is unique among the typically promiscuous chemokines in that it only binds one known receptor, CX3C receptor-1 (CX3CR1), and this receptor binds only fractalkine (Murphy et al. 2000). Spinal fractalkine is expressed and tethered to the extracellular surface of sensory afferents and intrinsic neurons (Asensio and Campbell 1999), while the fractalkine receptor is predominantly expressed by microglia (Verge et al. 2004). Fractalkine can be cleaved and released, forming a diffusible neuron-to-microglial signal, where binding of fractalkine to its receptor results in activation of microglia (Chapman et al. 2000). Fractalkine binding to its receptor leads to NFκB and p38 MAPK activation (Stievano et al. 2004) followed by production of proinflammatory cytokines and chemokines (Stievano et al. 2004; Johnston et al. 2004). An intrathecal injection of exogenous fractalkine produces both thermal hyperalgesia and mechanical allodynia (Milligan et al. 2004, 2005) and does so via the actions of activated glia. This suggests that fractalkine can facilitate nociception independent of opioid receptor desensitisation and that microglia play a large role in chemokine-mediated pain facilitation.
The involvement of fractalkine in opioid pharmacology was highlighted with experiments demonstrating that co-administration of morphine with an intrathecal fractalkine receptor neutralising antibody potentiated acute morphine analgesia and attenuated the development of tolerance, hyperalgesia and allodynia (Johnston et al. 2004). Similarly, Hutchinson et al. (2008a) also demonstrated fractalkine’s ability to oppose acute morphine analgesia. Hutchinson et al. (2008a) further examined opioid-induced release of fractalkine. Analysis of both lumbar dorsal spinal cord sections and cerebrospinal fluid demonstrated that chronic treatment with morphine or methadone caused significant elevations of fractalkine (Hutchinson et al. 2008a). However, fractalkine is not the only chemokine that can oppose opioid analgesia. A study by Szabo et al. (2002) found that pretreatment with CCL5/RANTES (the ligand for CCR1 and CCR5) or SDF-1/CXCL12 (the ligand for CXCR4) followed by opioid administration into the periaqueductal grey (PAG) matter of the brain resulted in a significantly reduced antinociceptive effect.
Another chemokine that is likely to be important in the pharmacodynamics of opioid analgesia is CCL2 (formerly monocyte chemoattractant protein-1, MCP-1). A growing body of evidence ranging from in vitro molecular profiling studies in dorsal root ganglia and spinal cord to data from in vivo assessment, including studies in knockout mice, indicated that CCL2 and its receptor CCR2 contribute to enhanced pain. Studies using chronic morphine or the selective μ-opioid agonist [d-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO) have shown an increase in expression of CCL2 (Rock et al. 2006). Intrathecal administration of CCL2 induces microglial activation, which is abolished in CCR2 (CCL2 receptor) knockout mice (Zhang et al. 2007). Given that opioids induce CCL2 upregulation in the spinal cord, which closely precedes microglial reactivity, it is likely that CCL2 may play a role in initiating a neuron–glial central immune signalling process causing microglial reactivity leading to counter-regulation of opioid-induced analgesia.
Further studies have been conducted implicating chemokines in opposing acute opioid analgesia. For example, the antinociceptive actions of μ-, δ- and κ-opioid receptor agonists are blocked or significantly reduced when the chemokines RANTES/CCL5; the ligand for CCR1, CCR5 or SDF-1α/CXCL12; and the ligand for CXCR4 or CX3CL1/fractalkine are either administered into the PAG of rat 30 min before or co-administered with the opioid agonists (Szabo et al. 2002; Chen et al. 2007a, b). While these behavioural studies do not directly implicate heterologous desensitisation (see Sect. 8), a study by Zhang et al. (2004) demonstrated that proinflammatory chemokines are capable of desensitising MORs on peripheral sensory neurons.
7.4 Cholecystokinin
Cholecystokinin (CCK) was first recognised as a major gastrointestinal hormone responsible for gallbladder contraction and pancreatic enzyme secretion. It has since been discovered in the brain and spinal cord where it is thought to function as a neurotransmitter. CCK exerts its physiological effects via two different GPCRs, CCKB receptor, the predominant receptor found in the brain and in the terminals of neurons, and CCKA receptors, which are abundant in the peripheral tissues (Raiteri and Paudice 1993).
Several forms of CCK have been detected; however, sulphated octapeptide C terminal, CCK8, is the most predominant form of CCK in the CNS (Vanderhaeghen et al. 1975). Within the CNS, the distribution of CCK is heterogeneous. Under normal conditions, CCK is not found in either the DRG or terminals of primary afferents but has been found in the superficial laminae of the spinal cord (Wiesenfeld-Hallin and Xu 1996; Ossipov et al. 2003). However, the peptide is particularly concentrated in regions involved in nociceptive transmission such as the PAG, thalamus, raphe nuclei and the medullary reticular formation (Raiteri and Paudice 1993; Wiesenfeld-Hallin and Xu 1996). In addition, CCK-containing projections from the RVM to the spinal cord have been identified (Mantyh and Hunt 1984). Interestingly, the areas where CCK neurons are located are involved in the mediation of the supraspinal and spinal analgesic effect of morphine. In these areas the actions of CCK have been shown to counteract opioid antinociception (Xie et al. 2005).
The spinal and supraspinal administration of CCK produces behavioural signs of hyperalgesia and enhanced activity of dorsal horn neurons consistent with a pronociceptive role. Systemic or perispinal CCK potently antagonised opioid analgesia produced by foot shock and morphine (Itoh et al. 1982; Faris et al. 1983; Li and Han 1989). CCK antagonists produce significant enhancements of exogenous and endogenous opioid analgesia and, interestingly, could slow or prevent the development of opioid tolerance in some paradigms (Watkins et al. 1985a, b; Dourish et al. 1990; Rezayat et al. 1994; Chapman et al. 1995). In addition, antisense oligodeoxynucleotide ‘knock-down’ of the CCKB receptor also enhances morphine antinociception (Vanderah et al. 1994).
During the extensive studies on the behavioural effects of CCK-opioid interactions, it was found that regions with well-documented roles in analgesia, such as the PAG and the RVM, were involved in CCK-mediated opposition of analgesia. For example, opioid tolerance induced by repeated microinjections of morphine into the PAG or via systemic morphine injections was reversed by PAG microinjection of proglumide, a CCK receptor antagonist (Vanderah et al. 1994). Within the RVM, only one group of neurons, OFF cells, is activated by μ-opioid agonists (Fields et al. 1983; Heinricher et al. 1994), while ON-cell firing is directly inhibited by opioid agonists (Bederson et al. 1990; Pan et al. 1990; Fields 1992; Heinricher et al. 1992). This is sufficient to produce behaviourally measurable antinociception (Heinricher et al. 1994; Heinricher and Tortorici 1994). However, low dose of CCK microinjected into the RVM blocked the analgesic effect of systemically administered morphine by preventing activation of OFF cells (Heinricher et al. 2001). At this dose, CCK had no effect on the spontaneous activity of these neurons or on the activity of ON cells. The same research group later demonstrated that microinjection of a higher dose of CCK into the RVM selectively activated ON cells and produced behavioural hyperalgesia (Heinricher 2004). This indicates that the pronociceptive actions of the peptide are mediated by neural elements distinct from those mediating the anti-opioid effects (Heinricher 2004).
There is considerable evidence that while CCK modulates the antinociceptive activity of opioids, the opioids in turn promote CCK release. In vivo microdialysis demonstrated that systemic and spinal administration of morphine increased cerebrospinal levels of CCK (de Araujo Lucas et al. 1998). Following a single systemic injection of morphine, CCK mRNA was significantly increased in the hypothalamus and spinal cord (Ding and Bayer 1993). Prolonged exposure to morphine resulted in an accelerated increase in CCK mRNA and CCK peptide (Zhou et al. 1992; Ding and Bayer 1993). For example, after 1, 3 and 6 days of exposure to morphine, whole brain levels of pro-CCK mRNA increased by 52 %, 62 % and 97 %, respectively (Zhou et al. 1992). Since opioids are known to induce the release of endogenous CCK, it is thought that this could be sufficient to activate both the anti-opioid (OFF-cell inhibition) and pronociceptive (ON-cell activation) circuits in parallel (Ossipov et al. 2003).
7.5 ATP
Of all the known neurotransmitters involved in enhanced pain modulation, ATP and a subset of spinal cord ATP receptors, termed purinergic P2X receptors, have gained focus in the facilitation of pain. The activation of the purinergic P2X4 receptor in microglia by ATP results in phosphorylation of p38 MAPK (Trang et al. 2009), which has been shown to be critical for microglial signalling and neuropathic pain sensitisation (Ji et al. 2009). Activation of p38 by phosphorylation leads to the synthesis and release of several glial products, such as the proinflammatory cytokines IL-1β and TNF-α (Ji et al. 2009; McMahon and Malcangio 2009) and the neurotrophin BDNF (Trang et al. 2009). As reviewed earlier, these mediators have been shown to modulate both excitatory and inhibitory synaptic transmission in the spinal cord nociceptive circuitry, leading to an increase in pain sensitivity.
The Horvath group has presented several lines of compelling evidence that demonstrate a critical role of P2X4 in morphine tolerance. Within minutes, morphine administration increased microglial migration via a novel interaction between μ-opioid and P2X4 receptors, which is dependent upon PI3K/Akt pathway activation (Horvath and DeLeo 2009). Persistent morphine infusion in rats induced a marked increase in the expression of spinal P2X4 receptors, the microglial surface marker CD11b and astrocytic GFAP levels (Horvath and DeLeo 2009; Horvath et al. 2010). Intrathecal injections of P2X4 antisense oligodeoxynucleotides inhibit morphine-induced P2X4 receptor expression. Importantly, the antisense oligodeoxynucleotide treatment almost completely prevents the development of antinociceptive tolerance to systemically administered morphine (Horvath et al. 2010). This suggests spinal cord microglial P2X4 signalling modulates the spinal cord neuronal plasticity underlying morphine tolerance. The activation of P2X4 receptor may elicit morphine tolerance by producing glial mediators TNF-α, IL-1β and BDNF via p38 activation; intrathecal infusion of a p38 inhibitor has also been shown to prevent morphine tolerance (Cui et al. 2006; Chen and Sommer 2009).
7.6 Nitric Oxide
Nitric oxide (NO) is a free radical that, among other functions, behaves as an intracellular and intercellular messenger in the nervous system (Snyder 1992). It is synthesised by nitric oxide synthase (NOS), of which three isoforms have been characterised. The neuronal and endothelial isoforms are constitutively expressed in the CNS, whereas the third form is inducible and found in macrophages and inflammatory cells (González-Hernández and Rustioni 1999). Previous studies have suggested a possible role for nitric oxide in acute nociception as well as the development of chronic pain (Tao et al. 2003). For instance, persistent thermal hyperalgesia induced by sciatic nerve injury can be reversed by the administration of the NOS inhibitor NG-nitro-l-arginine methyl ester (l-NAME) (Meller et al. 1992). Moreover, the NOS/nitric oxide system also participates in the development of opioid tolerance and withdrawal. NOS activity is increased in chronic morphine-treated mouse brains and the NOS mRNA level is greater in morphine-tolerant rat spinal cords (Machelska et al. 1997) where the inducible nitric oxide synthase (iNOS) isoform is likely to be the key enzyme responsible for increased NO production (Célérier et al. 2006).
In a study by Kolesnikov et al. (1993), the co-administration of the NOS inhibitor NG-nitro-l-arginine (l-NOARG) with morphine slowly reverses established morphine tolerance over 5 days despite the continued administration of morphine. In addition, a single dose of l-NOARG was shown to retard the development of morphine tolerance for several days. Similarly, intrathecal co-administration of the NOS inhibitor l-NAME with morphine significantly potentiated acute tail-flick and hind paw analgesia compared to morphine alone (Hutchinson et al. 2008a). It has been shown that glial activation can occur via reactive oxygen species, including NO (Meller and Gebhart 1993; Freeman et al. 2008), which suggests that opioid-induced NO release could indirectly modulate opioid-induced analgesia via glial activation and further release of proinflammatory mediators (Holguin et al. 2004).
7.7 Sphingomyelins
Sphingomyelins are a class of membrane sphingolipids found largely in the brain and nervous tissue (Bryan et al. 2008). The sphingomyelin degradation pathway produces ceramide, which is broken down into sphingosine and ceramide-1-phosphate. Sphingosine is further phosphorylated into sphingosine-1-phosphate (S1P) by the action of isoenzymes, sphingosine kinases (SphK) 1 and 2 (Pyne et al. 2009). S1P can act as a second messenger intracellularly and as a ligand for GPCRs (S1P1, S1P2, S1P3, S1P4, S1P5). In the CNS, SphK1/S1P signalling plays a key role in neuron-specific functions such as the regulation of neurotransmitter release from neurons and in the proliferation and survival of neurons and glia (Okada et al. 2009). However, it has recently been shown that under chronic morphine conditions, ceramide and its metabolic pathway contribute to morphine tolerance and hyperalgesia. In a study by Muscoli et al. (2010), chronic morphine was shown to upregulate both the sphingolipid ceramide in spinal astrocytes and microglia, but not neurons, or spinal S1P, the end product of ceramide metabolism. Co-administering morphine with intrathecal administration of pharmacological inhibitors of ceramide and S1P attenuated the development of hyperalgesia and tolerance and blocked increased formation of glial-related proinflammatory cytokines such as TNF-α, IL-1β and IL-6 which, as discussed before, are known modulators of neuronal excitability (Muscoli et al. 2010). SphK1, a key enzyme of the sphingolipid metabolic pathway, can alter the expression and production of proinflammatory cytokines and nitric oxide in microglia. LPS treatment was shown to increase SphK1 mRNA and protein expression, while suppression of SphK1 by its inhibitor, N,N-dimethylsphingosine (DMS), resulted in decreased mRNA expression of TNF-α, IL-1β and iNOS and release of TNF-α and NO in LPS-activated microglia. The addition of S1P increased the expression levels of TNF-α, IL-1β and iNOS and production of TNF-α and NO in activated microglia suggesting that suppression of SphK1 in activated microglia inhibits the production of proinflammatory cytokines and NO (Nayak et al. 2010).
In addition to playing a role in the production of proinflammatory cytokines, ceramide is involved in the production of reactive nitroxidative species, including superoxide, nitric oxide and peroxynitrite (Muscoli et al. 2007). These species can increase steady-state concentrations of ceramide by activating sphingomyelinases and by increasing the degradation of ceramidases, the enzymes responsible for the degradation of ceramide (Pautz et al. 2002). Peroxynitrite can nitrate mitochondrial manganese superoxide dismutase (MnSOD) (an enzyme responsible for regulating concentrations of superoxide dismutase (SOD)) to inactivate the enzyme. This results in an increase in superoxide levels, thereby favouring peroxynitrite formation (Muscoli et al. 2007). In addition to MnSOD, it is thought that peroxynitrite inactivates proteins of central importance in glutamate homeostasis, including glutamate transporters and glutamine synthase. Loss of the transport function leads to increased glutamate levels in the synaptic cleft, overstimulation of NMDA receptor and neurotoxicity. The involvement of peroxynitrite in the development of morphine tolerance and hyperalgesia appears to be of importance as co-administration of morphine with the peroxynitrite decomposition catalyst, Fe(III) 5,10,15,20-tetrakis (N-methylpyridinium-4-yl) porphyrin, blocked protein nitration and prevented the development of tolerance in a dose-dependent manner (Muscoli et al. 2007, 2010). In addition, it is now thought that the neuroprotective actions of minocycline are mediated by direct and specific scavenging of peroxynitrite. A study by Schildknecht et al. (2011) demonstrated that minocycline acts as a highly selective scavenger of peroxynitrite at submicromolar concentrations in various cellular models, including human neurons. This could be of particular importance as in addition to potentiating opioid analgesia, minocycline has been shown to have neuroprotective properties in a variety of chronic neurodegenerative diseases such as Alzheimer disease, Parkinson disease and amyotrophic lateral sclerosis (Schildknecht et al. 2011).
The connection between chronic administration of morphine and the activation of the ceramide metabolic pathway is hypothesised to be via TLR4. LPS activation of TLR4 receptors expressed on monocytes and macrophages activates enzymes in the de novo and sphingomyelinase pathways, leading to increased production of ceramide that, in turn, activates NF-κB and MAPKs to increase the production of proinflammatory products discussed previously (Muscoli et al. 2010).
8 Understanding the Molecular Mechanisms of Receptor Crosstalk
As mentioned earlier, opioids, cytokines and chemokines mediate their biological effects via their cognate GPCRs. GPCRs are the largest family of cell surface receptors implicated in signal transduction. Historically it was understood that these seven-transmembrane (7TM) receptors existed and functioned as monomeric units, acting like ‘on and off’ switches to transduce extracellular signals in a linear G-protein-dependent manner. However, it is now widely accepted that GPCRs can influence the signalling outcomes, and hence the biological response, of other unrelated receptors at multiple levels and this is often referred to as receptor ‘crosstalk’. Receptor crosstalk can be achieved through diverse mechanisms which, although not fully understood, offer the tantalising opportunity for developing highly selective pharmaceutical drugs.
One such mechanism is ‘heterologous desensitisation’. Desensitisation is a regulatory mechanism which can completely or partially abolish signal transduction. It has evolved to prevent overstimulation of GPCRs in the presence of continuous agonist stimulation and is important in both physiological and pharmacological settings. Desensitisation can be classified as either homologous or heterologous in nature. Homologous desensitisation occurs when a given GPCR is activated by its cognate ligand and is then desensitised to prevent further signal transduction. Heterologous desensitisation however describes the situation where the activation of one GPCR can lead to the desensitisation of other unrelated and often inactivated GPCRs. An in-depth review of desensitisation mechanisms is beyond the scope of this chapter; please see reviews (Freedman and Lefkowitz 1996; Gainetdinov et al. 2004). However, in general, it is believed that second messenger-dependent protein kinases such as cAMP-dependent protein kinase A (PKA) and protein kinase C (PKC) are primarily responsible for heterologous desensitisation.
The observation that morphine and heroin administration in patients often results in decreased resistance to infections, taken together with the overlapping expression patterns of opioid and chemokine/cytokine receptors and their ligands, has led to much interest in the identification of crosstalk between these receptors. Indeed it has been well documented that the μ- and δ-opioid receptor agonists exert an inhibitory effect on both antibody and cellular immune response (Pellis et al. 1986; Taub et al. 1991) and cytokine expression (Peterson et al. 1987; Chao et al. 1993; Belkowski et al. 1995). Studies conducted by Liu and co-workers in the early 1990s concluded that opioid pretreatment results in the inhibition of the complement-derived chemotactic factor-dependent chemotactic response of leukocytes (Liu et al. 1992). These findings were further supported by the work of Grimm and researchers, extending this inhibitory effect of opioid pretreatment to the responses mediated by the chemokines CCL3, CCL5, CCL2 and CXCL8 (Grimm et al. 1998). Interestingly, with the use of selective μ- or δ-opioid receptor agonists, these inhibitory effects were attributed to the activation of only the μ- and δ-opioid receptors and not the κ-opioid receptor. Furthermore, Grimm and colleagues demonstrated that the chemokine receptors CXCR1 and CXCR2 could be phosphorylated by opioid treatment (Grimm et al. 1998). These findings provided strong evidence of heterologous desensitisation of the chemokine CXCR1 and CXCR2 receptors by opioid receptor activation and a potential mechanism by which opioids may exhibit their immunosuppressive effects.
Not surprisingly, there is evidence in literature supporting the hypothesis that desensitisation is bidirectional and chemokines can influence the perception of pain and inhibit opioid-induced analgesia. Szabo and co-workers investigated the effects of chemokines CCL5 (CCR1 and CCR5 ligand) and CXCL12 (CXCR4 ligand) pretreatment on opioid-induced analgesia (Szabo et al. 2002). In the tail-flick test, rats that were pretreated with CXCL12 followed by DAMGO treatment (MOR agonist) exhibited a dose-dependent reduction in analgesic responses compared to control rats pretreated with saline. When receptor phosphorylation was investigated, CCL5 treatment resulted in the phosphorylation of the MOR at a similar level to that induced by DAMGO treatment. It is important to note the desensitisation of the μ- or δ-opioid receptor was due to the activation of CCR5, CCR2, CCR7 and CXCR4 receptors but not CXCR1 or CXCR2 (Szabo et al. 2002). This suggests that heterologous desensitisation is not indiscriminate but provides another level of highly precise regulation.
It has been previously reported that in vitro activation of the MOR increases the expression of the neuroprotective chemokine CCL5 (Wetzel et al. 2000; Avdoshina et al. 2010). In a recent study conducted by Campbell and researchers, it was determined that naltrexone, an opioid receptor antagonist, could block this morphine-mediated increase of CCL5 (Campbell et al. 2013). The exact mechanism behind this is yet unknown but it is clear that it is mediated by opioid receptors. The inability of morphine to activate glia in the absence of CCR5, the receptor for CCL5 (El-Hage et al. 2008), strongly suggests that complex signalling mechanisms beyond heterologous desensitisation are in place.
Receptors can also crosstalk via a phenomenon known as heteromerisation. Defined as a ‘macromolecular complex composed of at least two (functional) receptor units with biochemical properties that are demonstrably different from those of its individual components’ (Ferré et al. 2009), it is an elaborate mechanism by which GPCRs can influence the signalling outcomes of one or more receptors (please see reference for an example, Mustafa et al. 2012). In keeping with the theme of this chapter, heteromerisation may also offer an explanation for early observations that opioids can directly act as chemoattractants (Simpkins et al. 1984; van Epps and Saland 1984). Sophisticated techniques have been developed to identify and investigate heteromerisation and these have been summarised elsewhere (Mustafa and Pfleger 2011).
Based on overlapping expression patterns and the findings described above, Heinisch and colleagues conducted studies to identify co-localisation, and hence interactions, between the chemokine receptors, CXCR4 and CX3CR1, and the MOR. The findings from these studies demonstrated that the MOR co-localised with both the CXCR4 and CX3CR1 receptors on individual neurons in several regions of the brain including cingulate cortex (Heinisch et al. 2011). Interestingly, in whole-cell patch-clamp recordings of periaqueductal grey neurons in a rat brain slice preparation, morphine-induced membrane hyperpolarisation was either blocked or reduced in the presence of CXCL12 (CXCR4 ligand) or CX3CL1 (CX3CR1 ligand), respectively (Heinisch et al. 2011). Heteromerisation of the MOR with CXCR4 or CX3CR1, hence close proximity of these receptors, may explain the CXCR4 or CX3CR1-induced heterologous desensitisation of the MOR and therefore potentially also shed light on the limited benefits of opioid analgesics for the treatment of inflammatory pain.
As different cell types or tissues will express a unique combination of receptors, heteromerisation may indeed explain why heterologous desensitisation is not indiscriminate but occurs only between specific receptor types. It also provides an opportunity to target heteromers in a tissue-specific or even temporal manner with the tantalising prospect of understanding and reducing ‘off-target’ effects experienced with current pharmaceuticals. The identification and development of heteromer-selective ligands, which only activate a specific heteromer combination, is fast becoming an important research objective (Mustafa et al. 2010). Another approach to selectively activate heteromers is through the synthesis of bivalent ligands, which combine the pharmacophores of ligands for the respective constituent receptor units.
Multiple publications have supported the interactions between the MOR and chemokine receptor CCR5 (Suzuki et al. 2002; Szabo et al. 2002, 2003; Chen et al. 2004). In order to further understand the implications of these interactions, Yuan and colleagues synthesised a bivalent ligand incorporating the pharmacophores of naltrexone (MOR antagonist) and maraviroc (a CCR5 antagonist) (Yuan et al. 2013). In a study designed to investigate HIV-1 entry into human astrocytes, the bivalent ligand was effective in significantly inhibiting viral entry when compared to maraviroc treatment alone (Yuan et al. 2013). Naltrexone treatment did not have any effect. This example highlights the importance of identifying physiological relevant heteromers and investigating their function, as these receptor complexes may be the real pharmacological target for many pathophysiological states.
Although GPCR–GPCR interactions are likely to play an important role in the actions of opioids and chemokines, recent evidence suggesting that morphine may also activate TLR4 to mediate proinflammatory response (Wang et al. 2012), analgesia (Hutchinson et al. 2010) and opioid drug reward (Thomas and Hutchinson 2012) should not be overlooked but investigated to further understand the complex mechanisms in place. Avdoshina and co-workers have demonstrated that the TLR4 activator endotoxin LPS increases CCL5 release in primary cultures of microglia (Avdoshina et al. 2010). This knowledge taken together with the findings from a study conducted by Roscic-Mrkic, which suggest that morphine’s ability to induce proliferation is via the MAPK pathway, which has also been activated by CCL5 (Roscic-Mrkic et al. 2003), highlights the complex signalling pathways involved. In order to fully understand these elaborate mechanisms, hence the relationship between the opioid, chemokine and TLRs, future studies should be designed with the aim of testing the hypothesis that that heteromerisation these receptors may play a crucial role in modulating analgesia and addiction.
9 Immediate Clinical Implications of Opioid-Induced Cytokine Signalling
The rank-order analgesic potency for opioids commonly utilised in medical settings has been determined both experimentally and through decades of clinical experience (Analgesic Expert Group 2007). To avoid complications associated with high-potency opioids, lower-potency opioids, such as codeine, are employed preferentially in the community, resulting in widespread use. In fact, guidelines, including the World Health Organization’s ‘pain ladder’, routinely recommend the use of ‘mild opioids’ before stepping up to high-potency opioids like morphine (World Health Organization 1996). The general perception of greater safety and reduced abuse potential with lower-potency opioids has lead not only to the frequent prescribing of codeine but also to prevalent codeine self-medication in countries where the drug is available over the counter (Abbott and Fraser 1998; Harrison et al. 2012). Despite the well-understood differences in acute analgesic efficacy, the variability in central neuroimmune signalling between opioids of differing analgesic potency remains to be elucidated.
In the clinical setting, a condition in which differences in the ability of opioids to initiate neuroimmune signalling may be of particular importance is opioid-overuse headache. Opioid-overuse headache is a particularly onerous subtype of medication-overuse headache, wherein frequent analgesic intake results in exacerbation of a headache disorder (Headache Classification Committee of the International Headache Society 2006). As practice guidelines recommend against the use of potent opioids in the management of headache (Kennis et al. 2012) and many patients with headache disorders elect to self-medicate, a significant proportion of opioid-overuse headache patients develop the condition following the use of over-the-counter codeine products (Ravishankar 2008); thus, the propensity for codeine to induce opioid-overuse headache relative to other opioids is of importance.
Notably, opioid-overuse headache is confined to patients who already suffer from a pre-existing primary headache disorder such as migraine (Lance et al. 1988). Thus, it cannot be considered simply an adverse effect of opioid therapy and should instead be understood as an interaction between the headache disorder and opioid exposure. Although the pathophysiology behind opioid-overuse headache has not yet been confirmed, it has been hypothesised that the selective tendency of headache patients to develop opioid-overuse headache may arise due to alterations in neuroimmune signalling (Johnson et al. 2012), and a number of clinical observations and experimental findings support involvement of the neuroimmune system in this disorder (Meng and Cao 2007).
In headache patients, glial activation is thought to contribute to neuronal hypersensitivity, even in the absence of opioid use (Thalakoti et al. 2007). It is well established that calcitonin-gene-related peptide (CGRP) is released during migraine attacks, and when exposed to CGRP, glial cells release a variety of proinflammatory cytokines including IL-1β and IL-6 which could facilitate headache pain (Thalakoti et al. 2007; Capuano et al. 2009). The cumulative glial activation resulting from CGRP release and opioid exposure is likely to be greater than that caused by CGRP alone, potentially explaining the increase in headache observed following regular opioid treatment of migraine.
Preclinically the role of glial activation in headache following opioid exposure has been clearly demonstrated (Wieseler et al. 2011). Using a rodent model in which headache pain was assessed via the surrogate marker of facial allodynia, it was found that pre-exposure to morphine leads to allodynia when inflammatory ‘soup’ is applied to the dura in doses that do not cause allodynia in opioid-naïve rats (Wieseler et al. 2011). The involvement of glial activation in the facilitation of head pain was ascertained through administration of the glial-attenuating drug ibudilast concurrently with morphine, which prevented the presentation of facial allodynia (Wieseler et al. 2010).
Evidence also exists suggesting a role specifically for the TLR signalling pathway in medication-overuse headache. In a clinical study the TLR signalling pathway was identified using gene ontology in an analysis of the genomic expression patterns in medication-overuse headache patients that respond to medication withdrawal, alluding again to altered immunity in this condition (Hershey et al. 2011). Moreover, in silico docking simulations indicate codeine may dock to MD2 (Johnson et al. 2012) as morphine does (Eidson and Murphy 2013), suggesting codeine has potential to induce TLR4-dependent pain enhancement, independent of metabolic conversion to morphine. If codeine is able to directly activate the TLR4–MD2 complex, it may lead to far greater increases in pain sensitivity as compared to equianalgesic doses of morphine, as much larger doses of codeine must be administered to provide the same degree of pain relief.
The vast majority of studies investigating the neuroimmune consequences of opioids have focused upon glial activation within the spinal cord; however, to fully appreciate the potential risks, it must be determined if these actions can be generalised to other regions, for example, the trigeminal ganglion, a region of importance in headache pathology. Further research evaluating the neuroimmune actions of different opioids must be conducted to allow the risks to be weighed against the benefits of each treatment option, allowing appropriate drug selection and safe and effective clinical use.
10 Sex Differences in Analgesics
Sex differences in pain and analgesia are now well documented within both the experimental and clinical pain literature. Considerable evidence indicates diverse effectiveness of opioid analgesics in females versus males. Animal studies demonstrate greater analgesia in males; however, human studies reveal the opposite, with robust analgesic responses to opioids in females compared to males (Fillingim and Ness 2000). The existence of developmental and cycling hormone pain and analgesia profiles strongly suggest gonadal steroid hormone manipulation of nociception (Stoffel et al. 2003).
The hormones produced by the ovaries and testes are collectively referred to as the gonadal steroid hormones. The testes are responsible for the production of androgens: testosterone and dihydrotesterone. The ovaries produce both oestrogens (e.g. oestradiol, oestrone and oestriol) and progestins (e.g. progesterone). Although the majority of gonadal steroid hormones are produced in each sex from their respective sex organs, both oestrogens and androgens are present in both sexes; the adrenal cortex is responsible for the production of androgens, the testes known to produce oestrogens and the ovaries in turn producing testosterone (Craft et al. 2004).
The precise mechanisms underlying the role of gonadal steroid hormone manipulation of opioid analgesia are not completely understood. However, their ability to influence nociceptive sensitivity has been recognised both during development (organisational effects) and throughout adulthood (activational effects) (Craft et al. 2004). In addition to altering reproductive physiology and behaviour, the addition of testosterone or oestrogen, in addition to the surgical removal of the ovaries and testes, has been demonstrated to alter opioid analgesia. Androgenisation of neonatal females has been shown to produce more robust morphine analgesia comparable to intact adult males. Moreover, gonadectomy has desensitised morphine analgesia in males, reporting analogous results to adult intact females (Krzanowska and Bodnar 1999; Cicero et al. 2002). Evidently, the manipulation of neonatal gonadal hormones alters and removes the physiological sex differences in opioid analgesia. Furthermore, as (Fillingim and Ness 2000), these results suggest the pathways involved in opioid analgesia are sensitive to gonadal steroid hormones during development.
Studies that have investigated the activational role of gonadal steroid hormones have further demonstrated their contribution to opioid analgesia. Preclinical investigations have revealed this steroid hormone influence, investigating opioid analgesia in (a) gonadally intact and gonadectomised rodents, (b) gonadectomised rodents with and without steroid hormone replacement and (c) across the female menstrual and rodent oestrous cycles (Craft et al. 2004). In a significant number of studies, opioid analgesia was found significantly more potent in intact male rodents compared to gonadectomised subjects. The addition of testosterone in gonadectomised males has also confirmed an association between gonadal steroid hormones and nociceptive pathways, with greater opioid analgesia following testosterone replacement in several preclinical investigations (Ratka 1984; Rao and Saifi 1985; Stoffel et al. 2003). Despite these findings, it must also be noted that some have in fact demonstrated the opposite effect or the failure to influence opioid analgesia utilising testosterone (Kepler et al. 1989, 1991; Candido et al. 1992).
A gonadal hormone contribution to opioid analgesia has further been established in cycling female rodents. Largely, these studies demonstrate reduced sensitivity to opioids throughout oestrus, the oestrus cycle phase characterised by low levels of 17β oestradiols (Banerjee et al. 1983; Stoffel et al. 2003). Preclinical investigations which have investigated gonadal steroid hormone replacement in gonadectomised females have further implicated an association between 17beta oestradiol and opioid analgesia, with reduced analgesia in 17β oestradiol-supplemented females compared to gonadectomised female subjects (Ryan and Maier 1988; Berglund and Simpkins 1988; Ratka and Simpkins 1990, 1991; Ali et al. 1995). Consequently, despite variability across studies, the majority of findings imply that oestrogen may be responsible for the changes in opioid analgesia across the rodent oestrus cycle. Notably, these findings further suggest that the reduced effectiveness of opioid analgesia in females may result from an association between the 17beta oestradiol and the nociceptive pathways.
As previously discussed, opioids, including morphine, activate not only classical opioid receptors but also TLRs, specifically TLR4, resulting in the production of pain-enhancing proinflammatory cytokines (Hutchinson et al. 2010; Wang et al. 2012). This exacerbated release of proinflammatory cytokines has been demonstrated to counteract the analgesic efficacy of opioids (Hutchinson et al. 2008a, 2010, 2012). Considering that TLR4-mediated responses are more robust in the female sex (Berglund and Simpkins 1988; Kahlke et al. 2000; Marriott et al. 2006; Rettew et al. 2009) and the predominant female sex hormone 17beta oestradiol has been found to stimulate the activation of TLR4 signalling components such as NFκB, resulting in the release of proinflammatory mediators known to play a role in nociception (Soucy et al. 2005; Rettew et al. 2009; Calippe et al. 2010), researchers have hypothesised whether the reduced potency of opioids in females lies in part to an association between oestrogens and opioids on TLR4. Current unpublished data from the University of Colorado investigating the links between the analgesic efficiency of opioids and TLR4 have in fact revealed a correlation between elevated oestrogen and opioid-induced hyperalgesia, resulting in the reduced efficacy of opioids in females. Although unpublished, these results highlight gonadal steroid hormone manipulation of nociception and the need to further investigate the mechanistic link between gonadal steroid hormones and TLRs in the aim to better treat female pain.
11 Conclusion
It is now widely accepted that exclusively considering neuronal opioid activity provides an incomplete understanding of the initiation and maintenance of opioid tolerance, hyperalgesia and dependence. In this chapter, we have highlighted evidence supporting the hypothesis that the release of proinflammatory mediators is initiated by activation of a non-stereoselective receptor such as the innate-immune toll-like receptor 4. The activation of non-neuronal cells within the CNS can profoundly affect the neuronal homeostatic environment leading to significant alterations in neuronal firing. There is now accumulating evidence to suggest that the initiation and maintenance of tolerance and enhanced pain states are not likely to be attributed to one mediator but rather a combination of many. Of note, the consequence of such central immune signalling substantially modifies not only the development of opioid tolerance, hyperalgesia and dependence but also acute opioid analgesia.
The current work on central immune signalling complements the existing body of published findings on neuronal-mediated opioid side effects, such as receptor internalisation and recycling. Of note, the potential of separating the negative side effects from the beneficial actions by targeting opioid-induced glial activation using blood–brain barrier permeable pharmacotherapies such as minocycline, ibudilast or (+)-opioid antagonists has immense clinical utility. For example, development of non-opioid treatments for chronic pain and opioid dependence would enable co-administration with opioids with the possibility of greater efficacy and decreased side effects. The development of pharmaceuticals which selectively regulate signalling pathways implicated in the desired or undesired effects in a tissue- or cell-specific manner promises greater success for opioid use in the future.
References
Abbott FV, Fraser MI (1998) Use and abuse of over-the-counter analgesic agents. J Psychiatry Neurosci 23:13–34
Ali BH, Sharif SI, Elkadi A (1995) Sex differences and the effect of gonadectomy on morphine-induced antinociception and dependence in rats and mice. Clin Exp Pharmacol Physiol 22:342–344
Analgesic Expert Group (2007) Getting to know your analgesics and adjuvants. In: Therapeutic guidelines analgesic. Version 5. Therapeutic guidelines limited, Melbourne, p 44
Araque A, Navarrete M (2010) Glial cells in neuronal network function. Philos Trans R Soc Lond B Biol Sci 365:2375–2381. doi:10.1098/rstb.2009.0313
Asensio VC, Campbell IL (1999) Chemokines in the CNS: plurifunctional mediators in diverse states. Trends Neurosci 22:504–512
Avdoshina V, Biggio F, Palchik G et al (2010) Morphine induces the release of CCL5 from astrocytes: potential neuroprotective mechanism against the HIV protein gp120. Glia 58:1630–1639. doi:10.1002/glia.21035
Baeuerle PA, Henkel T (1994) Function and activation of NF-kappa B in the immune system. Annu Rev Immunol 12:141–179
Banerjee P, Chatterjee TK, Ghosh JJ (1983) Ovarian steroids and modulation of morphine-induced analgesia and catalepsy in female rats. Eur J Pharmacol 96:291–294
Bederson JB, Fields HL, Barbaro NM (1990) Hyperalgesia during naloxone-precipitated withdrawal from morphine is associated with increased on-cell activity in the rostral ventromedial medulla. Somatosens Mot Res 7:185–203
Belkowski SM, Alicea C, Eisenstein TK et al (1995) Inhibition of interleukin-1 and tumor necrosis factor-alpha synthesis following treatment of macrophages with the kappa opioid agonist U50, 488H. J Pharmacol Exp Ther 273:1491–1496
Ben Achour S, Pascual O (2010) Glia: the many ways to modulate synaptic plasticity. Neurochem Int 57:440–445. doi:10.1016/j.neuint.2010.02.013
Berglund LA, Simpkins JW (1988) Alterations in brain opiate receptor mechanisms on proestrous afternoon. Neuroendocrinology 48:394–400
Bland ST, Hutchinson MR, Maier SF et al (2009) The glial activation inhibitor AV411 reduces morphine-induced nucleus accumbens dopamine release. Brain Behav Immun 23:492–497. doi:10.1016/j.bbi.2009.01.014
Broom DC, Samad TA, Kohno T et al (2004) Cyclooxygenase 2 expression in the spared nerve injury model of neuropathic pain. Neuroscience 124:891–900. doi:10.1016/j.neuroscience.2004.01.003
Bryan L, Kordula T, Spiegel S, Milstien S (2008) Regulation and functions of sphingosine kinases in the brain. Biochim Biophys Acta 1781:459–466. doi:10.1016/j.bbalip.2008.04.008
Buchanan MM, Hutchinson M, Watkins LR, Yin H (2010) Toll-like receptor 4 in CNS pathologies. J Neurochem 114:13–27. doi:10.1111/j.1471-4159.2010.06736.x
Calippe B, Douin-Echinard V, Delpy L et al (2010) 17Beta-estradiol promotes TLR4-triggered proinflammatory mediator production through direct estrogen receptor alpha signaling in macrophages in vivo. J Immunol 185:1169–1176. doi:10.4049/jimmunol.0902383
Campbell LA, Avdoshina V, Rozzi S, Mocchetti I (2013) CCL5 and cytokine expression in the rat brain: differential modulation by chronic morphine and morphine withdrawal. Brain Behav Immun 34:130–140. doi:10.1016/j.bbi.2013.08.006
Candido J, Lutfy K, Billings B et al (1992) Effect of adrenal and sex hormones on opioid analgesia and opioid receptor regulation. Pharmacol Biochem Behav 42:685–692
Capuano A, De Corato A, Lisi L et al (2009) Proinflammatory-activated trigeminal satellite cells promote neuronal sensitization: relevance for migraine pathology. Mol Pain 5:43. doi:10.1186/1744-8069-5-43
Célérier E, González JR, Maldonado R et al (2006) Opioid-induced hyperalgesia in a murine model of postoperative pain: role of nitric oxide generated from the inducible nitric oxide synthase. Anesthesiology 104:546
Chao CC, Molitor TW, Close K et al (1993) Morphine inhibits the release of tumor necrosis factor in human peripheral blood mononuclear cell cultures. Int J Immunopharmacol 15:447–453
Chapman GA, Moores K, Harrison D et al (2000) Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci 20:RC87
Chapman V, Honoré P, Buritova J, Besson JM (1995) Cholecystokinin B receptor antagonism enhances the ability of a low dose of morphine to reduce c-Fos expression in the spinal cord of the rat. Neuroscience 67:731–739
Chen C, Li J, Bot G et al (2004) Heterodimerization and cross-desensitization between the μ-opioid receptor and the chemokine CCR5 receptor. Eur J Pharmacol 483:175–186. doi:10.1016/j.ejphar.2003.10.033
Chen X, Geller EB, Rogers TJ, Adler MW (2007a) Rapid heterologous desensitization of antinociceptive activity between mu or delta opioid receptors and chemokine receptors in rats. Drug Alcohol Depend 88:36–41. doi:10.1016/j.drugalcdep.2006.09.010
Chen X, Geller EB, Rogers TJ, Adler MW (2007b) The chemokine CX3CL1/fractalkine interferes with the antinociceptive effect induced by opioid agonists in the periaqueductal grey of rats. Brain Res 1153:52–57. doi:10.1016/j.brainres.2007.03.066
Chen Y, Sommer C (2009) The role of mitogen-activated protein kinase (MAPK) in morphine tolerance and dependence. Mol Neurobiol 40:101–107. doi:10.1007/s12035-009-8074-z
Cicero TJ, Nock B, O’Connor L, Meyer ER (2002) Role of steroids in sex differences in morphine-induced analgesia: activational and organizational effects. J Pharmacol Exp Ther 300:695–701
Craft RM, Mogil JS, Aloisi AM (2004) Sex differences in pain and analgesia: the role of gonadal hormones. Eur J Pain 8:397–411. doi:10.1016/j.ejpain.2004.01.003
Cui Y, Chen Y, Zhi J-L et al (2006) Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res 1069:235–243. doi:10.1016/j.brainres.2005.11.066
Cui Y, Liao X-X, Liu W et al (2008) A novel role of minocycline: attenuating morphine antinociceptive tolerance by inhibition of p38 MAPK in the activated spinal microglia. Brain Behav Immun 22:114–123. doi:10.1016/j.bbi.2007.07.014
Dame JB, Juul SE (2000) The distribution of receptors for the pro-inflammatory cytokines interleukin (IL)-6 and IL-8 in the developing human fetus. Early Hum Dev 58:25–39
De A, Krueger JM, Simasko SM (2003) Tumor necrosis factor alpha increases cytosolic calcium responses to AMPA and KCl in primary cultures of rat hippocampal neurons. Brain Res 981:133–142
de Araujo Lucas G, Alster P, Brodin E, Wiesenfeld-Hallin Z (1998) Differential release of cholecystokinin by morphine in rat spinal cord. Neurosci Lett 245:13–16
De Leo JA, Tawfik VL, LaCroix-Fralish ML (2006) The tetrapartite synapse: path to CNS sensitization and chronic pain. Pain 122:17–21. doi:10.1016/j.pain.2006.02.034
Ding XZ, Bayer BM (1993) Increases of CCK mRNA and peptide in different brain areas following acute and chronic administration of morphine. Brain Res 625:139–144
Dourish CT, O’Neill MF, Coughlan J et al (1990) The selective CCK-B receptor antagonist L-365,260 enhances morphine analgesia and prevents morphine tolerance in the rat. Eur J Pharmacol 176:35–44
Eidson LN, Murphy AZ (2013) Blockade of toll-like receptor 4 attenuates morphine tolerance and facilitates the pain relieving properties of morphine. J Neurosci 33:15952–15963. doi:10.1523/JNEUROSCI. 1609-13.2013
El-Hage N, Bruce-Keller AJ, Knapp PE, Hauser KF (2008) CCL5/RANTES gene deletion attenuates opioid-induced increases in glial CCL2/MCP-1 immunoreactivity and activation in HIV-1 Tat-exposed mice. J Neuroimmune Pharmacol 3:275–285. doi:10.1007/s11481-008-9127-1
Emch GS, Hermann GE, Rogers RC (2001) TNF-alpha-induced c-Fos generation in the nucleus of the solitary tract is blocked by NBQX and MK-801. Am J Physiol Regul Integr Comp Physiol 281:R1394–R1400
Fairbanks CA, Wilcox GL (2000) Spinal plasticity of acute opioid tolerance. J Biomed Sci 7:200–212
Faris PL, Komisaruk BR, Watkins LR, Mayer DJ (1983) Evidence for the neuropeptide cholecystokinin as an antagonist of opiate analgesia. Science 219:310–312
Ferré S, Baler R, Bouvier M et al (2009) Building a new conceptual framework for receptor heteromers. Nat Chem Biol 5:131–134. doi:10.1038/nchembio0309-131
Fields H (1992) Is there a facilitating component to central pain modulation? APS J 1:139–141
Fields HL, Vanegas H, Hentall ID, Zorman G (1983) Evidence that disinhibition of brain stem neurones contributes to morphine analgesia. Nature 306:684–686
Fillingim RB, Ness TJ (2000) Sex-related hormonal influences on pain and analgesic responses. Neurosci Biobehav Rev 24:485–501
Freedman NJ, Lefkowitz RJ (1996) Desensitization of G protein-coupled receptors. Recent Prog Horm Res 51:319–351; discussion 352–3
Freeman SE, Patil VV, Durham PL (2008) Nitric oxide-proton stimulation of trigeminal ganglion neurons increases mitogen-activated protein kinase and phosphatase expression in neurons and satellite glial cells. Neuroscience 157:542–555. doi:10.1016/j.neuroscience.2008.09.035
Gainetdinov RR, Premont RT, Bohn LM et al (2004) Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci 27:107–144. doi:10.1146/annurev.neuro.27.070203.144206
Goldstein A, Lowney LI, Pal BK (1971) Stereospecific and nonspecific interactions of the morphine congener levorphanol in subcellular fractions of mouse brain. Proc Natl Acad Sci U S A 68:1742–1747
González-Hernández T, Rustioni A (1999) Expression of three forms of nitric oxide synthase in peripheral nerve regeneration. J Neurosci Res 55:198–207
Grace PM, Hutchinson MR, Bishop A et al (2011) Adoptive transfer of peripheral immune cells potentiates allodynia in a graded chronic constriction injury model of neuropathic pain. Brain Behav Immun 25:503–513. doi:10.1016/j.bbi.2010.11.018
Grimm MC, Ben-Baruch A, Taub DD et al (1998) Opiates transdeactivate chemokine receptors: delta and mu opiate receptor-mediated heterologous desensitization. J Exp Med 188:317–325
Guo R-X, Zhang M, Liu W et al (2009) NMDA receptors are involved in upstream of the spinal JNK activation in morphine antinociceptive tolerance. Neurosci Lett 467:95–99. doi:10.1016/j.neulet.2009.10.013
Harrison CM, Charles J, Henderson J, Britt H (2012) Opioid prescribing in Australian general practice. Med J Aust 196:380–381. doi:10.5694/mja12.10168
Headache Classification Committee of the International Headache Society (2006) New appendix criteria open for a broader concept of chronic migraine. Cephalalgia 26(6):742–746
Heinisch S, Palma J, Kirby LG (2011) Interactions between chemokine and mu-opioid receptors: anatomical findings and electrophysiological studies in the rat periaqueductal grey. Brain Behav Immun 25:360–372. doi:10.1016/j.bbi.2010.10.020.Interactions
Heinricher M (2004) Neural basis for the hyperalgesic action of cholecystokinin in the rostral ventromedial medulla. J Neurophysiol 92:1982–1989
Heinricher M, McGaraughty S, Tortorici V (2001) Circuitry underlying antiopioid actions of cholecystokinin within the rostral ventromedial medulla. J Neurophysiol 85:280–286
Heinricher M, Morgan M, Fields HL (1992) Direct and indirect actions of morphine on medullary neurons that modulate nociception. Neuroscience 48(3):533–543
Heinricher MM, Morgan MM, Tortorici V, Fields HL (1994) Disinhibition of off-cells and antinociception produced by an opioid action within the rostral ventromedial medulla. Neuroscience 63:279–288
Heinricher MM, Tortorici V (1994) Interference with GABA transmission in the rostral ventromedial medulla: disinhibition of off-cells as a central mechanism in nociceptive modulation. Neuroscience 63:533–546
Hershey AD, Burdine D, Kabbouche MA, Powers SW (2011) Genomic expression patterns in medication overuse headaches. Cephalalgia 31:161–171. doi:10.1177/0333102410373155
Holguin A, O'Connor KA, Biedenkapp J et al (2004) HIV-1 gp120 stimulates proinflammatory cytokine-mediated pain facilitation via activation of nitric oxide synthase-I (nNOS). Pain 110:517–530. doi:10.1016/j.pain.2004.02.018
Holmes GM, Hebert SL, Rogers RC, Hermann GE (2004) Immunocytochemical localization of TNF type 1 and type 2 receptors in the rat spinal cord. Brain Res 1025:210–219. doi:10.1016/j.brainres.2004.08.020
Horvath RJ, DeLeo JA (2009) Morphine enhances microglial migration through modulation of P2X4 receptor signaling. J Neurosci 29:998–1005. doi:10.1523/JNEUROSCI. 4595-08.2009
Horvath RJ, Romero-Sandoval EA, De Leo JA (2010) Inhibition of microglial P2X4 receptors attenuates morphine tolerance, Iba1, GFAP and mu opioid receptor protein expression while enhancing perivascular microglial ED2. Pain 150:401–413. doi:10.1016/j.pain.2010.02.042
Hutchinson MR, Coats BD, Lewis SS et al (2008a) Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav Immun 22:1178–1189. doi:10.1016/j.bbi.2008.05.004
Hutchinson MR, Lewis SS, Coats BD et al (2009) Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic AV411 (ibudilast). Brain Behav Immun 23:240–250. doi:10.1016/j.bbi.2008.09.012
Hutchinson MR, Northcutt AL, Chao LW et al (2008b) Minocycline suppresses morphine-induced respiratory depression, suppresses morphine-induced reward, and enhances systemic morphine-induced analgesia. Brain Behav Immun 22:1248–1256. doi:10.1016/j.bbi.2008.07.008
Hutchinson MR, Northcutt AL, Hiranita T et al (2012) Opioid activation of toll-like receptor 4 contributes to drug reinforcement. J Neurosci 32:11187–11200. doi:10.1523/JNEUROSCI. 0684-12.2012
Hutchinson MR, Shavit Y, Grace PM et al (2011) Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev 63:772–810. doi:10.1124/pr.110.004135
Hutchinson MR, Zhang Y, Brown K et al (2008c) Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4). Eur J Neurosci 28:20–29. doi:10.1111/j.1460-9568.2008.06321.x
Hutchinson MR, Zhang Y, Shridhar M et al (2010) Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun 24:83–95. doi:10.1016/j.bbi.2009.08.004
Inoue K (2006) The function of microglia through purinergic receptors: neuropathic pain and cytokine release. Pharmacol Ther 109:210–226. doi:10.1016/j.pharmthera.2005.07.001
Itoh S, Katsuura G, Maeda Y (1982) Caerulein and cholecystokinin suppress beta-endorphin-induced analgesia in the rat. Eur J Pharmacol 80:421–425
Ji R-R, Gereau RW, Malcangio M, Strichartz GR (2009) MAP kinase and pain. Brain Res Rev 60:135–148. doi:10.1016/j.brainresrev.2008.12.011
Johnson JL, Hutchinson MR, Williams DB, Rolan P (2012) Medication-overuse headache and opioid-induced hyperalgesia: a review of mechanisms, a neuroimmune hypothesis and a novel approach to treatment. Cephalalgia 33:52–64. doi:10.1177/0333102412467512
Johnston IN, Milligan ED, Wieseler-Frank J et al (2004) A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci 24:7353–7365. doi:10.1523/JNEUROSCI. 1850-04.2004
Juni A, Klein G, Pintar JE, Kest B (2007) Nociception increases during opioid infusion in opioid receptor triple knock-out mice. Neuroscience 147:439–444. doi:10.1016/j.neuroscience.2007.04.030
Kahlke V, Angele MK, Ayala A et al (2000) Immune dysfunction following trauma-haemorrhage: influence of gender and age. Cytokine 12:69–77. doi:10.1006/cyto.1999.0511
Kao S-C, Zhao X, Lee C-Y et al (2012) Absence of μ opioid receptor mRNA expression in astrocytes and microglia of rat spinal cord. Neuroreport 23:378–384. doi:10.1097/WNR.0b013e3283522e1b
Kawasaki Y, Zhang L, Cheng J-K, Ji R-R (2008) Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci 28:5189–5194. doi:10.1523/JNEUROSCI. 3338-07.2008
Kennis K, Kernick D, O’Flynn N (2012) Diagnosis and management of headaches in young people and adults: NICE guideline. Br J Gen Pract 63(613):443–445. doi:10.3399/bjgp13X670895
Kepler KL, Kest B, Kiefel JM et al (1989) Roles of gender, gonadectomy and estrous phase in the analgesic effects of intracerebroventricular morphine in rats. Pharmacol Biochem Behav 34:119–127
Kepler KL, Standifer KM, Paul D et al (1991) Gender effects and central opioid analgesia. Pain 45:87–94
Kolesnikov YA, Pick CG, Ciszewska G, Pasternak GW (1993) Blockade of tolerance to morphine but not to kappa opioids by a nitric oxide synthase inhibitor. Proc Natl Acad Sci U S A 90:5162–5166
Krzanowska EK, Bodnar RJ (1999) Morphine antinociception elicited from the ventrolateral periaqueductal gray is sensitive to sex and gonadectomy differences in rats. Brain Res 821:224–230
Lance F, Parkes C, Wilkinson M (1988) Does analgesic abuse cause headaches de novo? Headache 28:61–62
Li Y, Han JS (1989) Cholecystokinin-octapeptide antagonizes morphine analgesia in periaqueductal gray of the rat. Brain Res 480:105–110
Liu Y, Blackbourn DJ, Chuang LF et al (1992) Effects of in vivo and in vitro administration of morphine sulfate upon rhesus macaque polymorphonuclear cell phagocytosis and chemotaxis. J Pharmacol Exp Ther 263:533–539
Loredo GA, Benton HP (1998) ATP and UTP activate calcium-mobilizing P2U-like receptors and act synergistically with interleukin-1 to stimulate prostaglandin E2 release from human rheumatoid synovial cells. Arthritis Rheum 41:246–255. doi:10.1002/1529-0131(199802)41:2<246::AID-ART8>3.0.CO;2-I
Machelska H, Ziólkowska B, Mika J et al (1997) Chronic morphine increases biosynthesis of nitric oxide synthase in the rat spinal cord. Neuroreport 8:2743–2747
Mantyh PW, Hunt SP (1984) Evidence for cholecystokinin-like immunoreactive neurons in the rat medulla oblongata which project to the spinal cord. Brain Res 291:49–54
Marriott I, Bost KL, Huet-Hudson YM (2006) Sexual dimorphism in expression of receptors for bacterial lipopolysaccharides in murine macrophages: a possible mechanism for gender-based differences in endotoxic shock susceptibility. J Reprod Immunol 71:12–27. doi:10.1016/j.jri.2006.01.004
McMahon SB, Malcangio M (2009) Current challenges in glia-pain biology. Neuron 64:46–54. doi:10.1016/j.neuron.2009.09.033
Meller S, Pechman PS, Gebhart GF, Maves TJ (1992) Nitric oxide mediates the thermal hyperalgesia produced in a model of neuropathic pain in the rat. Neuroscience 50(1):7–10
Meller ST, Gebhart GF (1993) Nitric oxide (NO) and nociceptive processing in the spinal cord. Pain 52:127–136
Meng ID, Cao L (2007) From migraine to chronic daily headache: the biological basis of headache transformation. Headache 47(8):1251–1258
Milligan E, Zapata V, Schoeniger D et al (2005) An initial investigation of spinal mechanisms underlying pain enhancement induced by fractalkine, a neuronally released chemokine. Eur J Neurosci 22:2775–2782. doi:10.1111/j.1460-9568.2005.04470.x
Milligan ED, Twining C, Chacur M et al (2003) Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci 23:1026–1040
Milligan ED, Watkins LR (2009) Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 10:23–36. doi:10.1038/nrn2533
Milligan ED, Zapata V, Chacur M et al (2004) Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci 20:2294–2302. doi:10.1111/j.1460-9568.2004.03709.x
Morioka N, Inoue A, Hanada T et al (2002) Nitric oxide synergistically potentiates interleukin-1 beta-induced increase of cyclooxygenase-2 mRNA levels, resulting in the facilitation of substance P release from primary afferent neurons: involvement of cGMP-independent mechanisms. Neuropharmacology 43:868–876
Morioka T, Kalehua AN, Streit WJ (1991) The microglial reaction in the rat dorsal hippocampus following transient forebrain ischemia. J Cereb Blood Flow Metab 11:966–973. doi:10.1038/jcbfm.1991.162
Murphy PM, Baggiolini M, Charo IF et al (2000) International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev 52:145–176
Muscoli C, Cuzzocrea S, Ndengele MM et al (2007) Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J Clin Invest 117:3530–3539. doi:10.1172/JCI32420
Muscoli C, Doyle T, Dagostino C et al (2010) Counter-regulation of opioid analgesia by glial-derived bioactive sphingolipids. J Neurosci 30:15400–15408. doi:10.1523/JNEUROSCI. 2391-10.2010
Mustafa S, Ayoub MA, Pfleger KDG (2010) Uncovering GPCR heteromer-biased ligands. Drug Discov Today Technol 7:e77–e85. doi:10.1016/j.ddtec.2010.06.003
Mustafa S, Pfleger KDG (2011) G protein-coupled receptor heteromer identification technology: identification and profiling of GPCR heteromers. J Lab Autom 16:285–291. doi:10.1016/j.jala.2011.03.002
Mustafa S, See HB, Seeber RM et al (2012) Identification and profiling of novel α1A-adrenoceptor-CXC chemokine receptor 2 heteromer. J Biol Chem 287:12952–12965. doi:10.1074/jbc.M111.322834
Nayak D, Huo Y, Kwang WXT et al (2010) Sphingosine kinase 1 regulates the expression of proinflammatory cytokines and nitric oxide in activated microglia. Neuroscience 166:132–144. doi:10.1016/j.neuroscience.2009.12.020
Oka T, Aou S, Hori T (1994) Intracerebroventricular injection of interleukin-1 beta enhances nociceptive neuronal responses of the trigeminal nucleus caudalis in rats. Brain Res 656:236–244
Okada T, Kajimoto T, Jahangeer S, Nakamura S-I (2009) Sphingosine kinase/sphingosine 1-phosphate signalling in central nervous system. Cell Signal 21:7–13. doi:10.1016/j.cellsig.2008.07.011
Ossipov MH, Lai J, Vanderah TW, Porreca F (2003) Induction of pain facilitation by sustained opioid exposure: relationship to opioid antinociceptive tolerance. Life Sci 73:783–800
Pan ZZ, Williams JT, Osborne PB (1990) Opioid actions on single nucleus raphe magnus neurons from rat and guinea-pig in vitro. J Physiol Lond 427:519–532
Pautz A, Franzen R, Dorsch S et al (2002) Cross-talk between nitric oxide and superoxide determines ceramide formation and apoptosis in glomerular cells. Kidney Int 61:790–796. doi:10.1046/j.1523-1755.2002.00222.x
Pellis NR, Harper C, Dafny N (1986) Suppression of the induction of delayed hypersensitivity in rats by repetitive morphine treatments. Exp Neurol 93:92–97
Perry VH, Hume DA, Gordon S (1985) Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience 15:313–326. doi:10.1016/0306-4522(85)90215-5
Peterson PK, Sharp B, Gekker G et al (1987) Opioid-mediated suppression of interferon-gamma production by cultured peripheral blood mononuclear cells. J Clin Invest 80:824–831. doi:10.1172/JCI113140
Pyne S, Lee SC, Long J, Pyne NJ (2009) Role of sphingosine kinases and lipid phosphate phosphatases in regulating spatial sphingosine 1-phosphate signalling in health and disease. Cell Signal 21:14–21. doi:10.1016/j.cellsig.2008.08.008
Quan N, He L, Lai W (2003) Endothelial activation is an intermediate step for peripheral lipopolysaccharide induced activation of paraventricular nucleus. Brain Res Bull 59:447–452
Raghavendra V, Rutkowski MD, DeLeo JA (2002) The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci 22:9980–9989
Raghavendra V, Tanga FY, DeLeo JA (2004) Attenuation of morphine tolerance, withdrawal-induced hyperalgesia, and associated spinal inflammatory immune responses by propentofylline in rats. Neuropsychopharmacology 29:327–334. doi:10.1038/sj.npp.1300315
Raiteri M, Paudice P (1993) Release of cholecystokinin in the central nervous system. Neurochem Int 26:519–527
Raivich G (2005) Like cops on the beat: the active role of resting microglia. Trends Neurosci 28:571–573. doi:10.1016/j.tins.2005.09.001
Rao SS, Saifi AQ (1985) Influence of testosterone on morphine analgesia in albino rats. Indian J Physiol Pharmacol 29:103–106
Ratka A (1984) Interaction of morphine and steroid hormones in the postirradiation disease in rats. Pol J Pharmacol Pharm 36(1):41–49
Ratka A, Simpkins JW (1990) A modulatory role for luteinizing hormone-releasing hormone in nociceptive responses of female rats. Endocrinology 127:667–673. doi:10.1210/endo-127-2-667
Ratka A, Simpkins JW (1991) Effects of estradiol and progesterone on the sensitivity to pain and on morphine-induced antinociception in female rats. Horm Behav 25:217–228
Ravishankar K (2008) Medication overuse headache in India. Cephalalgia 28:1223–1226. doi:10.1111/j.1468-2982.2008.01731.x
Rettew JA, Huet YM, Marriott I (2009) Estrogens augment cell surface TLR4 expression on murine macrophages and regulate sepsis susceptibility in vivo. Endocrinology 150:3877–3884. doi:10.1210/en.2009-0098
Rezayat M, Nikfar S, Zarrindast MR (1994) CCK receptor activation may prevent tolerance to morphine in mice. Eur J Pharmacol 254:21–26
Rock RB, Hu S, Sheng WS, Peterson PK (2006) Morphine stimulates CCL2 production by human neurons. J Neuroinflammation 3:32. doi:10.1186/1742-2094-3-32
Roscic-Mrkic B, Fischer M, Leemann C et al (2003) RANTES (CCL5) uses the proteoglycan CD44 as an auxiliary receptor to mediate cellular activation signals and HIV-1 enhancement. Blood 102:1169–1177. doi:10.1182/blood-2003-02-0488
Ryan SM, Maier SF (1988) The estrous cycle and estrogen modulate stress-induced analgesia. Behav Neurosci 102:371–380
Samad TA, Moore KA, Sapirstein A et al (2001) Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 410:471–475. doi:10.1038/35068566
Schildknecht S, Pape R, Müller N et al (2011) Neuroprotection by minocycline caused by direct and specific scavenging of peroxynitrite. J Biol Chem 286:4991–5002. doi:10.1074/jbc.M110.169565
Shavit Y, Wolf G, Goshen I et al (2005) Interleukin-1 antagonizes morphine analgesia and underlies morphine tolerance. Pain 115:50–59. doi:10.1016/j.pain.2005.02.003
Simpkins CO, Dickey CA, Fink MP (1984) Human neutrophil migration is enhanced by beta-endorphin. Life Sci 34:2251–2255
Smith K (2010) Neuroscience: settling the great glia debate. Nature 468:160–162. doi:10.1038/468160a
Snyder SH (1992) Nitric oxide: first in a new class of neurotransmitters? Science 257:494–496. doi:10.1126/science.1353273
Song P, Zhao ZQ (2001) The involvement of glial cells in the development of morphine tolerance. Neurosci Res 39:281–286
Soucy G, Boivin G, Labrie F, Rivest S (2005) Estradiol is required for a proper immune response to bacterial and viral pathogens in the female brain. J Immunol 174:6391–6398
Sperlágh B, Baranyi M, Haskó G, Vizi ES (2004) Potent effect of interleukin-1 beta to evoke ATP and adenosine release from rat hippocampal slices. J Neuroimmunol 151:33–39. doi:10.1016/j.jneuroim.2004.02.004
Stellwagen D, Beattie EC, Seo JY, Malenka RC (2005) Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 25:3219–3228. doi:10.1523/JNEUROSCI. 4486-04.2005
Stievano L, Piovan E, Amadori A (2004) C and CX3C chemokines: cell sources and physiopathological implications. Crit Rev Immunol 24:205–228
Stoffel EC, Ulibarri CM, Craft RM (2003) Gonadal steroid hormone modulation of nociception, morphine antinociception and reproductive indices in male and female rats. Pain 103:285–302. doi:10.1016/s0304-3959(02)00457-8
Suzuki S, Chuang LF, Yau P et al (2002) Interactions of opioid and chemokine receptors: oligomerization of mu, kappa, and delta with CCR5 on immune cells. Exp Cell Res 280(2):192–200. doi:10.1006/excr.2002.5638
Szabo I, Chen X-H, Xin L et al (2002) Heterologous desensitization of opioid receptors by chemokines inhibits chemotaxis and enhances the perception of pain. Proc Natl Acad Sci U S A 99:10276–10281. doi:10.1073/pnas.102327699
Szabo I, Wetzel MA, Zhang N et al (2003) Selective inactivation of CCR5 and decreased infectivity of R5 HIV-1 strains mediated by opioid-induced heterologous desensitization. J Leukoc Biol 74(6):1074–1082. doi:10.1189/jlb.0203067.1
Takagi K, Fukuda H, Watanabe M (1960) Studies on antitussives. III. (+)-morphine. Yakugaku Zasshi 80:1506–1509
Takayama N, Ueda H (2005) Morphine-induced chemotaxis and brain-derived neurotrophic factor expression in microglia. J Neurosci 25:430–435. doi:10.1523/JNEUROSCI. 3170-04.2005
Tanga FY, Nutile-McMenemy N, DeLeo JA (2005) The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A 102:5856–5861. doi:10.1073/pnas.0501634102
Tao F, Tao Y-X, Mao P et al (2003) Intact carrageenan-induced thermal hyperalgesia in mice lacking inducible nitric oxide synthase. Neuroscience 120:847–854
Taub DD, Eisenstein TK, Geller EB et al (1991) Immunomodulatory activity of mu- and kappa-selective opioid agonists. Proc Natl Acad Sci U S A 88:360–364
Tawfik VL, LaCroix-Fralish ML, Bercury KK et al (2006) Induction of astrocyte differentiation by propentofylline increases glutamate transporter expression in vitro: heterogeneity of the quiescent phenotype. Glia 54:193–203. doi:10.1002/glia.20365
Thalakoti S, Patil VV, Damodaram S et al (2007) Neuron-glia signaling in trigeminal ganglion: implications for migraine pathology. Headache 47:1008–1023. doi:10.1111/j.1526-4610.2007.00854.x; discussion 24–5
Thomas J, Hutchinson MR (2012) Exploring neuroinflammation as a potential avenue to improve the clinical efficacy of opioids. Expert Rev Neurother 12:1311–1324. doi:10.1586/ern.12.125
Trang T, Beggs S, Wan X, Salter MW (2009) P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci 29:3518–3528. doi:10.1523/JNEUROSCI. 5714-08.2009
Triantafilou M, Lepper PM, Briault CD et al (2008) Chemokine receptor 4 (CXCR4) is part of the lipopolysaccharide “sensing apparatus”. Eur J Immunol 38:192–203. doi:10.1002/eji.200636821
van Epps DE, Saland L (1984) Beta-endorphin and met-enkephalin stimulate human peripheral blood mononuclear cell chemotaxis. J Immunol 132:3046–3053
Vanderah TW, Lai J, Yamamura HI, Porreca F (1994) Antisense oligodeoxynucleotide to the CCKB receptor produces naltrindole- and [Leu5]enkephalin antiserum-sensitive enhancement of morphine antinociception. Neuroreport 5:2601
Vanderhaeghen JJ, Signeau JC, Gepts W (1975) New peptide in the vertebrate CNS reacting with antigastrin antibodies. Nature 257:604–605
Verge GM, Milligan ED, Maier SF et al (2004) Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci 20:1150–1160. doi:10.1111/j.1460-9568.2004.03593.x
Viviani B, Bartesaghi S, Gardoni F et al (2003) Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci 23:8692–8700
Wang X, Loram LC, Ramos K et al (2012) Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc Natl Acad Sci U S A 109:6325–6330. doi:10.1073/pnas.1200130109
Wang Z, Ma W, Chabot J-G, Quirion R (2009) Cell-type specific activation of p38 and ERK mediates calcitonin gene-related peptide involvement in tolerance to morphine-induced analgesia. FASEB J 23:2576–2586. doi:10.1096/fj.08-128348
Wang Z, Ma W, Chabot J-G, Quirion R (2010) Morphological evidence for the involvement of microglial p38 activation in CGRP-associated development of morphine antinociceptive tolerance. Peptides 31:2179–2184. doi:10.1016/j.peptides.2010.08.020
Watkins L, Kinscheck I, Mayer D (1985a) Potentiation of morphine analgesia by the cholecystokinin antagonist proglumide. Brain Res 327:169–180
Watkins LR, Hansen MK, Nguyen KT et al (1999) Dynamic regulation of the proinflammatory cytokine, interleukin-1beta: molecular biology for non-molecular biologists. Life Sci 65:449–481
Watkins LR, Hutchinson MR, Johnston IN, Maier SF (2005) Glia: novel counter-regulators of opioid analgesia. Trends Neurosci 28:661–669. doi:10.1016/j.tins.2005.10.001
Watkins LR, Hutchinson MR, Ledeboer A et al (2007) Glia as the “bad guys”: implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun 21:131–146. doi:10.1016/j.bbi.2006.10.011
Watkins LR, Hutchinson MR, Rice KC, Maier SF (2009) The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci 30:581–591. doi:10.1016/j.tips.2009.08.002
Watkins LR, Kinscheck I, Kaufman E, Miller J (1985b) Cholecystokinin antagonists selectively potentiate analgesia induced by endogenous opiates. Brain Res 327:181–190
Waxman AR, Arout C, Caldwell M et al (2009) Acute and chronic fentanyl administration causes hyperalgesia independently of opioid receptor activity in mice. Neurosci Lett 462:68–72. doi:10.1016/j.neulet.2009.06.061
Wetzel MA, Steele AD, Eisenstein TK et al (2000) μ-opioid induction of monocyte chemoattractant protein-1, RANTES, and IFN-γ-inducible protein-10 expression in human peripheral blood mononuclear cells. J Immunol 165(11):6519–6524
Wieseler J et al. (2010) Facial allodynia: involvement of glia and potentiation by prior morphine. Program No. 780.4/MM4. Neuroscience Meeting Planner. Society for Neuroscience, San Diego. Online
Wieseler J et al. (2011) Facial allodynia potentiation by supradural inflammatory mediators and morphine: a model of medication overuse headache. Program no. 178.09/NN19. Neuroscience Meeting Planner. Society for Neuroscience, Washington, DC. Online
Wiesenfeld-Hallin Z, Xu XJ (1996) The role of cholecystokinin in nociception, neuropathic pain and opiate tolerance. Regul Pept 65:23–28
World Health Organization (1996) Cancer pain relief. World Health Organization, Geneva
Wu H-E, Hong J-S, Tseng LF (2007) Stereoselective action of (+)-morphine over (-)-morphine in attenuating the (-)-morphine-produced antinociception via the naloxone-sensitive sigma receptor in the mouse. Eur J Pharmacol 571:145–151. doi:10.1016/j.ejphar.2007.06.012
Wu H-E, Sun H-S, Terashivili M et al (2006) dextro- and levo-morphine attenuate opioid delta and kappa receptor agonist produced analgesia in mu-opioid receptor knockout mice. Eur J Pharmacol 531:103–107. doi:10.1016/j.ejphar.2005.12.012
Wu H-E, Thompson J, Sun H-S et al (2005) Antianalgesia: stereoselective action of dextro-morphine over levo-morphine on glia in the mouse spinal cord. J Pharmacol Exp Ther 314:1101–1108. doi:10.1124/jpet.105.087130
Xie JY, Herman DS, Stiller C-O et al (2005) Cholecystokinin in the rostral ventromedial medulla mediates opioid-induced hyperalgesia and antinociceptive tolerance. J Neurosci 25:409–416. doi:10.1523/JNEUROSCI. 4054-04.2005
Xie N, Li H, Wei D et al (2010) Glycogen synthase kinase-3 and p38 MAPK are required for opioid-induced microglia apoptosis. Neuropharmacology 59:444–451. doi:10.1016/j.neuropharm.2010.06.006
Yuan Y, Arnatt CK, El-Hage N, Dever SM, Jacob JC, Selley DE, Hauser KF, Zhang Y (2013) A bivalent ligand targeting the putative mu opioid receptor and chemokine receptor CCR5 heterodimers: binding affinity versus functional activities. Medchemcomm 4(5):847–851. doi:10.1039/c3md00080j
Zhang J, Shi XQ, Echeverry S et al (2007) Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci 27:12396–12406. doi:10.1523/JNEUROSCI. 3016-07.2007
Zhang N, Rogers TJ, Caterina M, Oppenheim JJ (2004) Proinflammatory chemokines, such as C-C chemokine ligand 3, desensitize mu-opioid receptors on dorsal root ganglia neurons. J Immunol 173:594–599
Zhou Y, Sun YH, Zhang ZW, Han JS (1992) Accelerated expression of cholecystokinin gene in the brain of rats rendered tolerant to morphine. Neuroreport 3:1121–1123
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Thomas, J., Mustafa, S., Johnson, J., Nicotra, L., Hutchinson, M. (2015). The Relationship Between Opioids and Immune Signalling in the Spinal Cord. In: Schaible, HG. (eds) Pain Control. Handbook of Experimental Pharmacology, vol 227. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-46450-2_11
Download citation
DOI: https://doi.org/10.1007/978-3-662-46450-2_11
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-46449-6
Online ISBN: 978-3-662-46450-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)