
Abstract
Acute lymphoblastic leukaemia (ALL) rises from lymphoid progenitors. ALL cells show vast antigens linked to normal B-cell development and may be used as targets for immunotherapy. Since CD20 is expressed in B-ALL cells, rituximab, an anti-CD20 antibody used in B-cell non-Hodgkin’s lymphomas (B-NHLs), has also been used in the ALL. Reports have shown that the addition of CD20 antibodies to conventional chemotherapy leads to a higher rate of complete response as well as a better overall survival. CD22 molecule expression is found in more than 90 % of B-lineage ALL and is highly attractive for toxin-linked antibodies. Inotuzumab ozogamicin, an anti-CD22 antibody linked to calicheamicin, has shown high activity in phase II trials. CD19 is expressed in nearly all B-ALLs. Blinatumomab is a structured monoclonal antibody combining two single-chain antibodies to CD19 B cells and to CD3 T cells. It has shown encouraging results in the treatment of ALL. More recently, cell-based, especially T-cell-based, strategies have gained clinical interest, due to the curative effect of allogeneic stem cell transplantation, which is still the most effective immunotherapy for ALL and is mediated by donor T cells as “graft-versus-leukaemia (GVL)” effect; nonetheless, it also causes graft-versus-host disease (GVHD). The most promising approach is to target chimeric antigen receptors (CARs). CARs are composed of a single-chain variable-fragment (scFv) antibody specific to tumour antigen, fused to a transmembrane domain and a T-cell signalling moiety, most commonly either the CD3-ζ or Fc receptor γ cytoplasmic signalling domains. By choosing CD19 as the immunological target, primary clinical studies have shown high activity in ALL patients.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
6.1 Introduction
Acute lymphatic leukaemia (ALL) is a disorder occurring from a lymphoid progenitor cell. Mostly ALL is known as leukaemia occurring in children, but there are a number of adult patients suffering from ALL. While both age groups can be affected by the “same” disease, the outcome is often different. There are plenty of molecular changes that can be found in ALL – however their prognostic impact may vary between both patient groups.
Mostly ALL is classified as leukaemia of the B-cell lineage, which is the case in 85 %; we therefore focused on the B-cell ALL and their biological background and immune therapeutical options.
This chapter will discuss the different pathobiological changes that occur in the development of ALL as well as their implication on the prognosis of the diseases. The second part will focus on the progress that has been made on different immune therapeutical approaches to treat and cure ALL. The therapies range from tyrosine kinase inhibitors, antibodies against different lymphatic antigens to cellular approaches like haematopoietic stem cell transplantation and chimeric antigen receptors (CARs)-transduced T cells. By incorporating the different therapeutic options, the treatment and opportunities have dramatically changed.
6.2 Immunopathology of Lymphoblastic Leukaemia
6.2.1 General Considerations
The incidence of acute lymphoblastic leukaemia (ALL) is about 1–4/100.000 persons per year. Most of the cases occur in children below 6 years or in adults aged 80 years and more. Approximately 85 % of all ALL cases are of a B-cell phenotype.
6.2.2 Lymphocyte Development as Biological Basis of Disease
Acute lymphatic leukaemia rises from lymphoid progenitors. In humans LIN−/CD34+/CD38− cells are recognised as a stem and progenitor population in which three different sub-compartments can be found: CD90+/CD45RA−, CD90−/CD45RA− and CD90−/CD45RA+. The LIN−/CD34+/CD38−/CD90+/CD45RA− fraction is highly enriched for haematopoietic stem cells HST [1]. The common lymphoid progenitor (CLP) can be defined as LIN−/CD10+/CD34+ [2]. Originating from this CLP, B cells continue to differentiate into pre-pro-B cells, which then turned into pro-B cells, large pre-B cells and small pre-B cells and finally differentiate into immature B cells. This differentiation is highly regulated by various transcription factors which are specifically expressed over a time period to ensure the correct development of B cells (reviewed in [3]).
B-ALLs are heterogenic diseases, with an accumulation of abnormal cells. Traditionally B-ALL cells have been compared to their normal counter-partners in B-cell development. This was mostly done because of similarities in morphology and immune phenotype. However, this head-to-head comparison misses some ALL features, for example, up to 30 % of ALL cases express myeloid markers [4]. Research on chromosome changes in ALL has shown that some of the initiating changes occur very early (e.g. being of parental origin), while others occur at a later stage of development [3].
Genetical changes leading to the development of B-ALL are discussed below. The increasing role of the signalling of the pre-B-cell receptor and signal transduction by this receptor should be mentioned. During B-cell development, the pre-B-cell receptor has a dual function. It promotes survival and proliferation, and subsequently it induces differentiation in the B-cell compartment [5]. Two downstream targets which are mainly important for the tumor suppressive function of the pre-B-cell receptor are IKAROS and AIOLOS [6]. Interestingly, in 80 % of the BCR-ABL+ ALL, the gene responsible for IKAROS (IKZF1) has been deleted [7], underlining the importance of those events in the signal transduction for the development of ALL.
Similar to AML, there is a two-hit model for ALL. The first “hit” is posed to be a chromosomal abnormality (the major are listed below); nonetheless, this first “hit” is not sufficient for the induction of an ALL. Therefore, a second hit such as the deletion of tumor suppressor genes is needed to fully generate an ALL.
6.2.3 Genetics in Acute Lymphatic Leukaemia
6.2.3.1 Numerical Chromosome Changes
6.2.3.1.1 Hyperdiploid
Hyperploidy (>50 chromosomes) can be found in up to 30 % of all cases in children [8]. In contrast, the number of hyperploid cases in adults is significantly lower (about 10 %). Hyperploidy is associated with good prognosis in children. This might be explained by the higher sensitivity to chemotherapy [9]. The impact of hyperploidy in adults is less clear; while some reports found a benefit, others deny this finding [10, 11].
6.2.3.1.2 Hypodiploid
Hypoploidy is defined as the presence of less than 46 chromosomes in a cell. Approximately 10 % of children and nearly 10 % of adult cases show a hypoploid chromosome content [12]. Patients with hypoploidy have a worse prognosis compared to those with a normal or hyperploid leukaemia [13]. This is even more of importance since the event-free survival depends on the number of chromosomes and patients having less than 44 chromosomes showed 8-year EFS of 30 % [14].
6.2.3.2 Structural Changes
6.2.3.2.1 MLL Rearrangements
Several rearrangements involving the MLL gene at chromosome 11q23 are present in ALL cells. The most common are t(4;11)(q21;q23), t(11;19)(q23;p13.3) and t(9;11)(p22;q23) which lead to the fusion of the 5′portion of the MLL with the 3′portion of AFF1, MLLT1 and MLLT3 [15, 16]. Beside these frequent translocation patterns, over 50 other translocations are known which fuse to MLL. Interestingly, there are two major breaking clustering regions in the MLL gene between exon 5 and exon 11 [17]. Regardless of the other fusion partner (e.g. AFF1), fusion proteins will keep the transcription repressing domain of MLL and gain the 3′portion of the partner, which is mostly a transcription factor. MLL rearrangement is usually associated with poor outcome [18–20].
6.2.3.2.2 BCR-ABL
The translocation of 9q to chromosome 22q leads to the formation of the Philadelphia (Ph) chromosome. This fusion protein is the hallmark of the chronic myeloid leukaemia (CML) but is also found in ALL. Around 5 % of the children and up to nearly 30 % of adults will show the t(9;22)(q34;q11.2) translocation which can be detected by conventional cytogenetics, FISH or PCR [12, 21, 22]. The latest is often used for quantification which makes this method extremely interesting for detection of minimal residual disease (MRD) [23].
Genetically the Ph chromosome is a fusion of the 5′portion of the BCR gene to the 3′portion of the Abelson leukaemia virus proto-oncogene (ABL1). The breaking points of BCR cluster occur in two regions. The major clustering region (M-bcr) is mostly found in CML, and the minor cluster (m-bcr) is predominantly seen in ALL. Therefore, two different translocation products can result depending on the involved breaking point, the p210 kDa and the smaller p180-190 kDa. Patients will show only one of the two possible fusion samples [24, 23]. Detection of the t(9;22) in patients with ALL leads to an adverse disease prognosis [25, 26].
Tyrosine kinase inhibitors are active in Ph+ ALL; however, the majority of patients will relapse after initial response to treatment and even during treatment [27, 28].
6.2.3.2.3 ETV6-RUNX1
Translocation t(12;21)(p13:q22) leads to the production of the fusion protein ETV6-RUNX1. This is the most frequent structural chromosome change in paediatric ALL, which occurs in nearly 30 % of the cases [29, 30]. While being quite frequent in childhood lymphatic leukaemia, ETV6-RUNX1 transcripts are rare in adults with a frequency below 5 % [31, 32]. RUNX1 is a transcription factor that regulates various genes important for human haematopoiesis [33].
Occurrence of the ETV6-RUNX1 translocation is associated with a good prognosis in childhood ALL. This is especially seen in younger children (1–9 years of age) rather than in those older than 10 [14, 32, 34, 35]. A hypothesised mechanism is that the occurrence of the translocation sensitises malignant cells to classical chemotherapeutic drugs used in ALL protocols [36, 37].
6.3 Immune Phenotype and Targets in Lymphatic Leukaemia
6.3.1 Cell Surface Marker
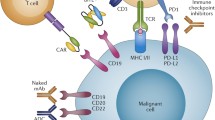
ALL cells express a variety of antigens that are linked to normal B-cell development. In a more simplified way, three major subgroups can be defined which can be classified by their immune phenotype. The early precursor or pro-ALL is characterised by the expression of CD19, cytoplasmic CD79a, cytoplasmic CD22 and nuclear TdT. The intermediate stage or common ALL is recognised by CD10, and the late precursor or pre-B-ALL stage is marked by the cytoplasmic expression of the μ chain. Typical phenotypes are listed in Table 6.1.
Lymphoblasts are positive for CD10, surface CD22, CD24, Pax5 and TdT in most cases. The expression of CD34 and CD20 varies. CD45 may be absent. Myeloid markers such as CD13, CD15, CD33 and CD68 can also be expressed on lymphoblasts [38].
6.3.2 Tumor Antigens
6.3.2.1 WT1
Wilms tumor gene 1 (WT1) is a zinc finger transcription factor that was originally found as a mutated tumor suppressor in Wilms tumor. The expression of the transcription factor was also found in haematopoiesis, and the expression and relevance of its expression has been extensively studied in AML and to a lesser extent in ALL. Reports showed that a great number of ALL have an WT1 expression, but in contrast to childhood ALL where WT1 was inversely correlated to an inferior prognosis [39], a study on nearly 300 adult ALL cases missed to show any impact as an individual prognostic marker [40]. The expression varies in different ALL subtypes, and matured B-ALL were negative or showed low WT1 expression, while aberrant expression of myeloid marker led to the highest WT1 levels. However, in adult T-ALL an inferior outcome for patients harbouring a WT1 mutation in exon 7 was reported [41]. While data for WT1-specific T-cell therapy in ALL are missing, a report in CML patients used WT1-specific T cells to prevent relapse of leukaemia [42].
6.3.2.2 BCR-ABL
Most work with BCR-ABL as a tumor antigen was done in CML. Numbers of reports have shown that T cells specific for BCR-ABL contribute to the immune vs. CML effect [42–44]. However, in BCR-ABL-positive ALL, the potential use of BCR-ABL as an immune target is less promising. Data of allografted BCR-ABL-positive ALL patients showed a better overall survival (OS) and less relapse compared to patients treated with conventional chemotherapy [45, 46], suggesting that a graft-versus-leukaemia (GvL) effect also exists for BCR-ABL ALL; however the relapse rate is still 30 % [47], and ALL has shown to be less sensitive for donor lymphocyte infusion [48]. BCR-ABL-positive ALL has also been included into trail with bi-specific antibodies (discussed below), resulting in a considerable rate of relapse which however was short lasting [49]. These results suggest that either BCR-ABL itself is less immunogenic or that priming and expansion of BCR-ABL-specific T cells take too long in acute leukaemia to be effective as therapeutical approach.
6.3.3 Cancer/Testis Antigens
Cancer/testis antigens (CTAs) are a group of tumor antigens being limitedly expressed in somatic tissues and represent an attractive target for immunotherapy in cancer since the gonads are immune privileged organs and anti-CTA immune response can be tumor specific (reviewed in [50]). While CTA represents an attractive target in AML [51], the expression and practicability as an immunological target in ALL is less clear. In a small study, the expression of CTA in ALL patients could be detected [52].
6.4 Immunotherapy for Lymphatic Leukaemia
6.4.1 Cellular Approaches
6.4.1.1 T Cells and Modified T Cells
T cells as part of the adoptive immune system have the ability to recognise and kill tumor cells. This quality is part of the concept of donor lymphocyte infusions (DLI, discussed below) as cellular therapy against various types of haematological cancers [53–55]. However, the response rates of ALL to DLI are inferior compared to other haematological cancer types and mostly lower than 15 % [56, 57]. Possible explanations may be that ALL is an aggressive disease where time for priming the naïve T cells is lacking and ALL cells are missing co-stimulatory molecules [58]. Even ex vivo pre-stimulation of T cells with CD3/CD28 antibodies did not enhance the benefit of DLI for ALL patients [59]. Another approach to enhance the T-cell toxicity towards ALL is an ex vivo priming against known tumor antigens. WT1 and BCR-ABL are the best studied antigens so far, and first reports are promising that the priming may lead to a better control of tumor cells [60].
An alternative to conventional T cells for adoptive immunotherapy is the application of genetically modified T cells. Here the α and β subunits of the T-cell receptor (TCR) of a tumor-specific T-cell clone are used. First results in solid tumors such as melanoma were promising [61]; however, its use in haematological malignancies was limited due to the antigen restriction of the T-cell clone [62, 63]. In addition, many tumor cells downregulate HLA molecules and thereby lower the ability of recognition by T cells [64].
A possibility to avoid the limitations of TCR gene transfer may be the use of chimeric antigen receptors (CARs). CARs are composed of a single-chain variable-fragment (scFv) antibody specific to tumor antigen, fused to a transmembrane domain and a T-cell signalling moiety, most commonly either the CD3-ζ or Fc receptor γ cytoplasmic signalling domains [65]. The resulting receptor, when expressed on the surface of the T cell, mediates binding to the target tumor antigen through the scFv domain, which subsequently mediates an activating signal to the T cell inducing target cell lysis. Major advantages are the ability to produce large amounts of modified T cells in the lab and the ability that those cells kill HLA-independent T cells and that CAR-modified T cells can be further manipulated by co-expressing cytokines or co-stimulatory molecules [66–68].
By choosing the CD19 as an immunological target, some preclinical work reported beneficial effects of viral-transduced CD19-targeted CAR T cells [69, 70]. Further modifications of CAR T cells with co-stimulatory receptors have enhanced their potential in mice models [71–73]. Ongoing phase I trials are investigating the benefit of CAR-modified T cells in the context of ALL (NCT01044069 and NCT01029366), and it will be very interesting to see which impact this modified T cells will have on the management and cure of ALL. In a first proof-of-principle report, Porter et al. treated a patient with refractory CLL with modified autologous T cells. T cells were transduced with CD19, CD137 and CD3-ζ and infused at a dosage of 1.5 × 105/kg BW. A remarkable remission for 10 months was noted [74]. Interestingly in patients treated with CAR-modified T cells, a portion of memory CAR T cells could be found after 6 months [75].
In a more recent study, Grupp and co-workers used CD19 CAR-modified T cells with dosages of 1.4 × 106 to 1.2 × 107 T cells per kg/BW to treat two children with relapsed and refractory pre-B-ALL. Both children reached complete remission (CR) after treatment, and one remained in CR for 11 months, while the other child relapsed with a clone of non-CD19-expressing blasts [76]. Therefore alternative targets are investigated, and first reports show that CD22 can also be used as an immunological target for CAR in ALL [77]. However, this point remains critical as the chosen antigen determines the success of the cellular therapy.
6.4.1.2 NK Cell Approaches
NK cells are part of the innate immune system. In contrast to B or T cells, NK cells do not have receptors rearranged during their maturation, making them less specific for antigens. Indeed, receptors expressed on the NK cell surface have more function of carefully controlling NK cell activation. One of those receptors is the killer-cell immunoglobulin-like recepor (KIR, CD158) family, which consists of different members that have activating as well as inhibitory functions on NK cells. NK cell cytotoxicity is triggered by tumor cells, which lack the of expression of some self-MHC class I molecules referred to as “missing self” hypothesis [78]. Inhibitory KIRs recognise groups of HLA-A, HLA-B and HLA-C alleles. If KIR inhibitory NK cells target cells lacking the corresponding HLA-class I ligand, the target cell will be lysed (KIR-ligand model) [79].
Up to now, NK cell alloreactivity does not seem to be beneficial in the treatment of ALL [80], but some reports with genetic modified NK cells provide some encouraging data. Retroviral or electroporation of NK cells to induce a CD19 targeting CAR led to increased NK cell-mediated killing of ALL cell lines, as well as primary ALL blasts [81–83].
6.4.2 Antibodies (See Table 6.2)
6.4.2.1 CD20 Antibodies
The CD20 molecule is an integral membrane protein that is specific for B cells and seems to be important for calcium transport across the cell membrane [84]. The expression of CD20 is linked to poor prognosis [85]. CD20 is expressed on leukemic blast cells of about 50 % of the patients with B-lineage ALL.
Rituximab is a chimeric mouse/humane antibody that has dramatically changed the therapy of NHL. Since CD20 is also expressed in B-ALL cells, the antibodies have also been used in the ALL setting. Reports have shown that the addition of CD20 antibodies to conventional chemotherapy leads to a higher rate of complete response as well as a better overall survival. Of note the advantage seems only to be true in younger ALL patients [86, 87].
6.4.2.2 CD22 Antibody
CD22 molecule expression is found in more than 90 % of B-lineage ALL. Functionally, CD22 leads to downregulation of CD19 after its phosphorylation. CD22 is rapidly internalised after activation and therefore is highly attractive for toxin-linked antibodies [88]. Inotuzumab ozogamicin is an anti-CD22 antibody linked to calicheamicin. Calicheamicin is a toxic antibiotic which causes double-strand breaks in the DNA. In a first phase II trial of nearly 60 patients, 57 % responded to the immunotoxin and showed an OS of 5.1 months [89].
Epratuzumab is an unconjugated CD22 antibody. In a very small study with 15 paediatric patients, nine achieved CR with only moderate toxicity [90]. Furthermore, the addition of epratuzumab to standard chemotherapy improved the CR rate in a Children’s Oncology Group (COG) study [91]. Moxetumomab pasudotox is a new-generation toxin-linked anti-CD22 antibody that is currently being investigated in a phase I trial [92].
6.4.2.3 CD52 Antibody
CD52 is a glycoprotein on the surface of lymphoid cells. CD52 can be found on T and B cells, making the antigen interesting for application in T- and B-ALL. Campath-1H is a humanised IgG1k antibody that showed major efficiency in NHL and CLL.
A small series of six patients with advanced ALL who had been treated with alemtuzumab (three times 30 mg IV) was reported, and nearly all patients showed infectious complications [93]. CALGB presented data from a phase 1 study including 24 patients in CR1 with alemtuzumab as treatment. The OS was 55 months, and DFS was 53 months. Interestingly minimal residual disease (MRD) levels were 1 log lower in alemtuzumab-treated patients [94]. Again infection complications were a major side effect in the treatment.
6.4.2.4 CD19 Antibody
CD19 is expressed in nearly all B-cell malignancies due to the early expression of the CD19 molecule in B-cell development. Blinatumomab is a structured monoclonal antibody combining two single-chain antibodies to CD19 B cells and to CD3 T cells [95]. This antibody increases the contact of cytotoxic CD3 to malignant B cells which thereby gets lysed. The GMALL showed data on 21 patients who achieved MRD negativity after blinatumomab therapy. The response rate was 80 % with a probability of relapse-free survival of 78 % and only mild side effects [96].
6.5 Stem Cell Transplantation
6.5.1 Allogeneic Stem Cell Transplantation (Allo SCT)
Allogeneic stem cell transplantation is still the most effective immunotherapy for ALL. Donor T cells contribute to controlling malignant cells. This effect of GvL was first described in acute leukaemias including ALL [97]. However, there are also significant limiting factors in the treatment of ALL by allo SCT; even with the improved supportive care therapy, there is up to 30 % treatment-related mortality (TRM) [98], and GVHD accounts for up to 70 % of the cases [99].
The majority of ALL patients will achieve CR after the induction therapy [99], but if patients profit more from allo SCT or conventional chemotherapy as consolidation is still not fully answered. There are some patient subgroups defined by genetical features, delayed response (> day 28) and high leukocyte count at diagnosis that have been summarised by the term high-risk patients. In these patients allo SCT seems to be favourable as consolidation therapy [100–102]. Nevertheless, it has to be underlined that these data are mainly based on myeloablative treatment protocols and related donor transplantation. Of note some studies failed to support the beneficial use of allo SCT in high-risk ALL patients [103, 104].
Besides the group of high-risk ALL patients, debate exists on whether standard-risk ALL patients should be transplanted. The British MRC analysed 1,646 patients who were negative for the t(9;22). If a CR was achieved and the patient was eligible for an allo SCT, patients were biologically randomised on the donor/no donor base. Interestingly in this cohort of standard-risk ALL patients, the 5-year OS was significantly better in the transplant group compared to non-transplanted patients (62 % vs. 52 %, p = 0.02) [103].
High-resolution typing of the HLA locus resulted in improved survival after matched unrelated donor transplantation, and results become similar to those of HLA-identical sibling transplantation [105–107].
As an alternative stem cell source, umbilical cord blood (UCB) or haploidentical donors can be used for allogeneic stem cell transplantation. Retrospective studies resulted in similar outcome after UCB and matched related or unrelated stem cell transplantation [108, 109], and haploidentical donor transplantation has become a reasonable transplant option for those patients lacking a suitable HLA-matched donor [110].
One way to lower TRM is to Reduced-intensity conditioning (RIC). Patients in advanced stage of ALL, with older age or heavily pretreated can be transplanted after RIC [111, 112]. Registry data from the EBMT showed that using RIC protocols reduced TRM in the context of ALL, but also in this data set, there was an increase in relapse rate (RIC, 47 % vs. MAC 31 %, p < 0.001) [113].
6.6 Concluding Remarks
ALL are acute leukaemias arising from a common lymphatic progenitor. In a number of cases, chromosomal changes occur; some of them like BCR-ABL have direct impact on the biology of the disease. As the neoplastic cells are closely related to normal lymphoid development, ALL express a vast of antigens, which are a target of antibody-based therapies. Incorporation, especially of the chimeric blinatumomab, has improved therapeutical outcome in ALL patients. Stem cell transplantation is mostly the therapy of choice in the case of an ALL relapse after conventional therapy or in case of high-risk features of the disease in upfront treatment. By replacing the haematopoietic system, the donor immune system is thought to control leukaemia growth by the GvL effect. This is mostly mediated by T cells as one of the effector arms of the adoptive immune system. Therefore the development of “more specialised” T cells by genetical modification of the T-cell receptor (CAR-Ts) is a logical progress. CAR-modified T cells have already shown high efficiency in the use against lymphatic leukaemias. In summary immune therapeutic approaches held great promise to optimise ALL therapy and lead to a longer and better survival of ALL patients.
References
Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1(6):635–45.
Haddad R, Guardiola P, Izac B, Thibault C, Radich J, Delezoide AL, et al. Molecular characterization of early human T/NK and B-lymphoid progenitor cells in umbilical cord blood. Blood. 2004;104(13):3918–26.
Campos-Sanchez E, Toboso-Navasa A, Romero-Camarero I, Barajas-Diego M, Sanchez-Garcia I, Cobaleda C. Acute lymphoblastic leukemia and developmental biology: a crucial interrelationship. Cell Cycle. 2011;10(20):3473–86.
Suggs JL, Cruse JM, Lewis RE. Aberrant myeloid marker expression in precursor B-cell and T-cell leukemias. Exp Mol Pathol. 2007;83(3):471–3.
Nahar R, Muschen M. Pre-B cell receptor signaling in acute lymphoblastic leukemia. Cell Cycle. 2009;8(23):3874–7.
Thompson EC, Cobb BS, Sabbattini P, Meixlsperger S, Parelho V, Liberg D, et al. Ikaros DNA-binding proteins as integral components of B cell developmental-stage-specific regulatory circuits. Immunity. 2007;26(3):335–44.
Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453(7191):110–4.
Moorman AV, Harrison CJ, Buck GA, Richards SM, Secker-Walker LM, Martineau M, et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. 2007;109(8):3189–97.
Whitehead VM, Vuchich MJ, Lauer SJ, Mahoney D, Carroll AJ, Shuster JJ, et al. Accumulation of high levels of methotrexate polyglutamates in lymphoblasts from children with hyperdiploid (greater than 50 chromosomes) B-lineage acute lymphoblastic leukemia: a Pediatric Oncology Group study. Blood. 1992;80(5):1316–23.
Cytogenetic abnormalities in adult acute lymphoblastic leukemia: correlations with hematologic findings outcome. A Collaborative Study of the Group Francais de Cytogenetique Hematologique. Blood. 1996;87(8):3135–42.
Wetzler M, Dodge RK, Mrozek K, Carroll AJ, Tantravahi R, Block AW, et al. Prospective karyotype analysis in adult acute lymphoblastic leukemia: the cancer and leukemia Group B experience. Blood. 1999;93(11):3983–93.
Chessells JM, Hall E, Prentice HG, Durrant J, Bailey CC, Richards SM. The impact of age on outcome in lymphoblastic leukaemia; MRC UKALL X and XA compared: a report from the MRC Paediatric and Adult Working Parties. Leukemia. 1998;12(4):463–73.
Heerema NA, Nachman JB, Sather HN, Sensel MG, Lee MK, Hutchinson R, et al. Hypodiploidy with less than 45 chromosomes confers adverse risk in childhood acute lymphoblastic leukemia: a report from the children’s cancer group. Blood. 1999;94(12):4036–45.
Schultz KR, Pullen DJ, Sather HN, Shuster JJ, Devidas M, Borowitz MJ, et al. Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children’s Cancer Group (CCG). Blood. 2007;109(3):926–35.
Domer PH, Fakharzadeh SS, Chen CS, Jockel J, Johansen L, Silverman GA, et al. Acute mixed-lineage leukemia t(4;11)(q21;q23) generates an MLL-AF4 fusion product. Proc Natl Acad Sci U S A. 1993;90(16):7884–8.
Gu Y, Nakamura T, Alder H, Prasad R, Canaani O, Cimino G, et al. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell. 1992;71(4):701–8.
Felix CA, Hosler MR, Slater DJ, Parker RI, Masterson M, Whitlock JA, et al. MLL genomic breakpoint distribution within the breakpoint cluster region in de novo leukemia in children. J Pediatr Hematol Oncol. 1998;20(4):299–308.
Pui CH, Gaynon PS, Boyett JM, Chessells JM, Baruchel A, Kamps W, et al. Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet. 2002;359(9321):1909–15.
Pui CH, Chessells JM, Camitta B, Baruchel A, Biondi A, Boyett JM, et al. Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia. 2003;17(4):700–6.
Marchesi F, Girardi K, Avvisati G. Pathogenetic, clinical, and prognostic features of adult t(4;11)(q21;q23)/MLL-AF4 positive B-cell acute lymphoblastic leukemia. Adv Hematol. 2011;2011:621627.
Bloomfield CD, Goldman AI, Alimena G, Berger R, Borgstrom GH, Brandt L, et al. Chromosomal abnormalities identify high-risk and low-risk patients with acute lymphoblastic leukemia. Blood. 1986;67(2):415–20.
Mullighan CG. The molecular genetic makeup of acute lymphoblastic leukemia. Hematol Am Soc Hematol Educ Program. 2012;2012:389–96.
Bruggemann M, Schrauder A, Raff T, Pfeifer H, Dworzak M, Ottmann OG, et al. Standardized MRD quantification in European ALL trials: proceedings of the second international symposium on MRD assessment in Kiel, Germany, 18–20 September 2008. Leukemia. 2010;24(3):521–35.
De Klein A, Hagemeijer A, Bartram CR, Houwen R, Hoefsloot L, Carbonell F, et al. bcr rearrangement and translocation of the c-abl oncogene in Philadelphia positive acute lymphoblastic leukemia. Blood. 1986;68(6):1369–75.
Fielding AK. How i, treat Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 2010;116(18):3409–17.
Stock W. Current treatment options for adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Leuk Lymphoma. 2010;51(2):188–98.
Ottmann OG, Pfeifer H. Management of Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL). Hematology Am Soc Hematol Educ Program. 2009;371–81.
Hunger SP. Tyrosine kinase inhibitor use in pediatric Philadelphia chromosome-positive acute lymphoblastic anemia. Hematol Am Soc Hematol Educ Program. 2011;2011:361–5.
Pui CH, Evans WE. Acute lymphoblastic leukemia. N Engl J Med. 1998;339(9):605–15.
Barry E, DeAngelo DJ, Neuberg D, Stevenson K, Loh ML, Asselin BL, et al. Favorable outcome for adolescents with acute lymphoblastic leukemia treated on Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium Protocols. J Clin Oncol. 2007;25(7):813–9.
Jabber Al-Obaidi MS, Martineau M, Bennett CF, Franklin IM, Goldstone AH, Harewood L, et al. ETV6/AML1 fusion by FISH in adult acute lymphoblastic leukemia. Leukemia. 2002;16(4):669–74.
Lee DS, Kim YR, Cho HK, Lee CK, Lee JH, Cho HI. The presence of TEL/AML1 rearrangement and cryptic deletion of the TEL gene in adult acute lymphoblastic leukemia (ALL). Cancer Genet Cytogenet. 2005;162(2):176–8.
Hart SM, Foroni L. Core binding factor genes and human leukemia. Haematologica. 2002;87(12):1307–23.
Loh ML, Goldwasser MA, Silverman LB, Poon WM, Vattikuti S, Cardoso A, et al. Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood. 2006;107(11):4508–13.
Borkhardt A, Cazzaniga G, Viehmann S, Valsecchi MG, Ludwig WD, Burci L, et al. Incidence and clinical relevance of TEL/AML1 fusion genes in children with acute lymphoblastic leukemia enrolled in the German and Italian multicenter therapy trials. Associazione Italiana Ematologia Oncologia Pediatrica and the Berlin-Frankfurt-Munster Study Group. Blood. 1997;90(2):571–7.
Krishna Narla R, Navara C, Sarquis M, Uckun FM. Chemosensitivity of TEL-AML1 fusion transcript positive acute lymphoblastic leukemia cells. Leuk Lymphoma. 2001;41(5–6):615–23.
Ramakers-van Woerden NL, Pieters R, Loonen AH, Hubeek I, van Drunen E, Beverloo HB, et al. TEL/AML1 gene fusion is related to in vitro drug sensitivity for L-asparaginase in childhood acute lymphoblastic leukemia. Blood. 2000;96(3):1094–9.
Sabattini E, Bacci F, Saqramoso C, Pileri SA. WHO classification of tumors of haematopoietic and lymphoid tissues in 2008: an overview. Pathologica. 2010;102(3):83–7.
Boublikova L, Kalinova M, Ryan J, Quinn F, O’Marcaigh A, Smith O, et al. Wilms’ tumor gene 1 (WT1) expression in childhood acute lymphoblastic leukemia: a wide range of WT1 expression levels, its impact on prognosis and minimal residual disease monitoring. Leukemia. 2006;20(2):254–63.
Busse A, Gokbuget N, Siehl JM, Hoelzer D, Schwartz S, Rietz A, et al. Wilms’ tumor gene 1 (WT1) expression in subtypes of acute lymphoblastic leukemia (ALL) of adults and impact on clinical outcome. Ann Hematol. 2009;88(12):1199–205.
Heesch S, Goekbuget N, Stroux A, Tanchez JO, Schlee C, Burmeister T, et al. Prognostic implications of mutations and expression of the Wilms tumor 1 (WT1) gene in adult acute T-lymphoblastic leukemia. Haematologica. 2010;95(6):942–9.
Bornhauser M, Thiede C, Platzbecker U, Kiani A, Oelschlaegel U, Babatz J, et al. Prophylactic transfer of BCR-ABL-, PR1-, and WT1-reactive donor T cells after T cell-depleted allogeneic hematopoietic cell transplantation in patients with chronic myeloid leukemia. Blood. 2011;117(26):7174–84.
Cai A, Keskin DB, DeLuca DS, Alonso A, Zhang W, Zhang GL, et al. Mutated BCR-ABL generates immunogenic T-cell epitopes in CML patients. Clin Cancer Res. 2012;18(20):5761–72.
Gannage M, Abel M, Michallet AS, Delluc S, Lambert M, Giraudier S, et al. Ex vivo characterization of multiepitopic tumor-specific CD8 T cells in patients with chronic myeloid leukemia: implications for vaccine development and adoptive cellular immunotherapy. J Immunol. 2005;174(12):8210–8.
Fielding AK, Richards SM, Chopra R, Lazarus HM, Litzow MR, Buck G, et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109(3):944–50.
Laport GG, Alvarnas JC, Palmer JM, Snyder DS, Slovak ML, Cherry AM, et al. Long-term remission of Philadelphia chromosome-positive acute lymphoblastic leukemia after allogeneic hematopoietic cell transplantation from matched sibling donors: a 20-year experience with the fractionated total body irradiation-etoposide regimen. Blood. 2008;112(3):903–9.
Lee HJ, Thompson JE, Wang ES, Wetzler M. Philadelphia chromosome-positive acute lymphoblastic leukemia: current treatment and future perspectives. Cancer. 2011;117(8):1583–94.
Kolb HJ, Schattenberg A, Goldman JM, Hertenstein B, Jacobsen N, Arcese W, et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood. 1995;86(5):2041–50.
Hoelzer D. Novel antibody-based therapies for acute lymphoblastic leukemia. Hematol Am Soc Hematol. 2011;2011:243–9.
Lim SH, Zhang Y, Zhang J. Cancer-testis antigens: the current status on antigen regulation and potential clinical use. Am J Blood Res. 2012;2(1):29–35.
Atanackovic D, Luetkens T, Kloth B, Fuchs G, Cao Y, Hildebrandt Y, et al. Cancer-testis antigen expression and its epigenetic modulation in acute myeloid leukemia. Am J Hematol. 2011;86(11):918–22.
Niemeyer P, Tureci O, Eberle T, Graf N, Pfreundschuh M, Sahin U. Expression of serologically identified tumor antigens in acute leukemias. Leuk Res. 2003;27(7):655–60.
Sprangers B, Van Wijmeersch B, Fevery S, Waer M, Billiau AD. Experimental and clinical approaches for optimization of the graft-versus-leukemia effect. Nat Clin Pract Oncol. 2007;4(7):404–14.
Chang YJ, Huang XJ. Donor lymphocyte infusions for relapse after allogeneic transplantation: when, if and for whom? Blood Rev. 2013;27(1):55–62.
Roddie C, Peggs KS. Donor lymphocyte infusion following allogeneic hematopoietic stem cell transplantation. Expert Opin Biol Ther. 2011;11(4):473–87.
Choi SJ, Lee JH, Lee JH, Kim S, Lee YS, Seol M, et al. Treatment of relapsed acute lymphoblastic leukemia after allogeneic bone marrow transplantation with chemotherapy followed by G-CSF-primed donor leukocyte infusion: a prospective study. Bone Marrow Transplant. 2005;36(2):163–9.
Collins Jr RH, Goldstein S, Giralt S, Levine J, Porter D, Drobyski W, et al. Donor leukocyte infusions in acute lymphocytic leukemia. Bone Marrow Transplant. 2000;26(5):511–6.
Loren AW, Porter DL. Donor leukocyte infusions for the treatment of relapsed acute leukemia after allogeneic stem cell transplantation. Bone Marrow Transplant. 2008;41(5):483–93.
Porter DL, Levine BL, Bunin N, Stadtmauer EA, Luger SM, Goldstein S, et al. A phase 1 trial of donor lymphocyte infusions expanded and activated ex vivo via CD3/CD28 costimulation. Blood. 2006;107(4):1325–31.
Rezvani K, Yong AS, Savani BN, Mielke S, Keyvanfar K, Gostick E, et al. Graft-versus-leukemia effects associated with detectable Wilms tumor-1 specific T lymphocytes after allogeneic stem-cell transplantation for acute lymphoblastic leukemia. Blood. 2007;110(6):1924–32.
Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–9.
Dossett ML, Teague RM, Schmitt TM, Tan X, Cooper LJ, Pinzon C, et al. Adoptive immunotherapy of disseminated leukemia with TCR-transduced, CD8+ T cells expressing a known endogenous TCR. Mol Ther. 2009;17(4):742–9.
Xue SA, Gao L, Hart D, Gillmore R, Qasim W, Thrasher A, et al. Elimination of human leukemia cells in NOD/SCID mice by WT1-TCR gene-transduced human T cells. Blood. 2005;106(9):3062–7.
Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3(11):999–1005.
Park JH, Brentjens RJ. Adoptive immunotherapy for B-cell malignancies with autologous chimeric antigen receptor modified tumor targeted T cells. Discov Med. 2010;9(47):277–88.
Singh H, Figliola MJ, Dawson MJ, Huls H, Olivares S, Switzer K, et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res. 2011;71(10):3516–27.
Tammana S, Huang X, Wong M, Milone MC, Ma L, Levine BL, et al. 4-1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell malignancies. Hum Gene Ther. 2010;21(1):75–86.
Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24(6):1160–70.
Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9(3):279–86.
Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13(18 Pt 1):5426–35.
Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–84.
Loskog A, Giandomenico V, Rossig C, Pule M, Dotti G, Brenner MK. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia. 2006;20(10):1819–28.
Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–64.
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–33.
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–18.
Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121(7):1165–74.
Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319(6055):675–8.
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097–100.
Park JH, Sauter C, Brentjens R. Cellular therapies in acute lymphoblastic leukemia. Hematol Oncol Clin North Am. 2011;25(6):1281–301.
Li L, Liu LN, Feller S, Allen C, Shivakumar R, Fratantoni J, et al. Expression of chimeric antigen receptors in natural killer cells with a regulatory-compliant non-viral method. Cancer Gene Ther. 2010;17(3):147–54.
Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106(1):376–83.
Shimasaki N, Campana D. Natural killer cell reprogramming with chimeric immune receptors. Methods Mol Biol. 2013;969:203–20.
Walshe CA, Beers SA, French RR, Chan CH, Johnson PW, Packham GK, et al. Induction of cytosolic calcium flux by CD20 is dependent upon B Cell antigen receptor signaling. J Biol Chem. 2008;283(25):16971–84.
Borowitz MJ, Shuster J, Carroll AJ, Nash M, Look AT, Camitta B, et al. Prognostic significance of fluorescence intensity of surface marker expression in childhood B-precursor acute lymphoblastic leukemia. A Pediatric Oncology Group Study. Blood. 1997;89(11):3960–6.
Thomas DA, O’Brien S, Faderl S, Garcia-Manero G, Ferrajoli A, Wierda W, et al. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J Clin Oncol. 2010;28(24):3880–9.
Thomas DA, Faderl S, O’Brien S, Bueso-Ramos C, Cortes J, Garcia-Manero G, et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer. 2006;106(7):1569–80.
de Vries JF, Zwaan CM, De Bie M, Voerman JS, den Boer ML, van Dongen JJ, et al. The novel calicheamicin-conjugated CD22 antibody inotuzumab ozogamicin (CMC-544) effectively kills primary pediatric acute lymphoblastic leukemia cells. Leukemia. 2012;26(2):255–64.
Kantarjian H, Thomas D, Jorgensen J, Jabbour E, Kebriaei P, Rytting M, et al. Inotuzumab ozogamicin, an anti-CD22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: a phase 2 study. Lancet Oncol. 2012;13(4):403–11.
Raetz EA, Cairo MS, Borowitz MJ, Blaney SM, Krailo MD, Leil TA, et al. Chemoimmunotherapy reinduction with epratuzumab in children with acute lymphoblastic leukemia in marrow relapse: a Children’s Oncology Group Pilot Study. J Clin Oncol. 2008;26(22):3756–62.
Raetz EA, Cairo MS, Borowitz MJ. Reinduction chemoimmunotherapy with epratuzumab in relapsed acute lymphoblastic leukemia (ALL) in children, adolescents and young adults: results from Children’s Oncology Group (COG) study ADVL04P2 [abstract] Blood (ASH Annual Meeting Abstracts) 2011;118(21):573.
Kreitman RJ, Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res. 2011;17(20):6398–405.
Tibes R, Keating MJ, Ferrajoli A, Wierda W, Ravandi F, Garcia-Manero G, et al. Activity of alemtuzumab in patients with CD52-positive acute leukemia. Cancer. 2006;106(12):2645–51.
Stock W, Sanford B, Lozanski G, Vij R, Byrd JC, Powell BL et al., editors. Alemtuzumab can be incorporated into front-line therapy of adult Acute Lymphoblastic Leukemia (ALL): Final Phase I results of a Cancer and Leukemia Group B Study (CALGB 10102). ASH annual meeting abstracts. 2009;114:abstract 838.
Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321(5891):974–7.
Topp MS, Kufer P, Gokbuget N, Goebeler M, Klinger M, Neumann S, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29(18):2493–8.
Weiden PL, Flournoy N, Thomas ED, Prentice R, Fefer A, Buckner CD, et al. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N Engl J Med. 1979;300(19):1068–73.
Gokbuget N, Hoelzer D. Treatment of adult acute lymphoblastic leukemia. Semin Hematol. 2009;46(1):64–75.
Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354(2):166–78.
Hahn T, Wall D, Camitta B, Davies S, Dillon H, Gaynon P, et al. The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute lymphoblastic leukemia in adults: an evidence-based review. Biol Blood Marrow Transplant. 2006;12(1):1–30.
Yanada M, Matsuo K, Suzuki T, Naoe T. Allogeneic hematopoietic stem cell transplantation as part of postremission therapy improves survival for adult patients with high-risk acute lymphoblastic leukemia: a metaanalysis. Cancer. 2006;106(12):2657–63.
Thomas X, Boiron JM, Huguet F, Dombret H, Bradstock K, Vey N, et al. Outcome of treatment in adults with acute lymphoblastic leukemia: analysis of the LALA-94 trial. J Clin Oncol. 2004;22(20):4075–86.
Goldstone AH, Richards SM, Lazarus HM, Tallman MS, Buck G, Fielding AK, et al. In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the International ALL Trial (MRC UKALL XII/ECOG E2993). Blood. 2008;111(4):1827–33.
Ribera JM, Oriol A, Bethencourt C, Parody R, Hernandez-Rivas JM, Moreno MJ, et al. Comparison of intensive chemotherapy, allogeneic or autologous stem cell transplantation as post-remission treatment for adult patients with high-risk acute lymphoblastic leukemia. Results of the PETHEMA ALL-93 trial. Haematologica. 2005;90(10):1346–56.
Chim CS, Lie AK, Liang R, Au WY, Kwong YL. Long-term results of allogeneic bone marrow transplantation for 108 adult patients with acute lymphoblastic leukemia: favorable outcome with BMT at first remission and HLA-matched unrelated donor. Bone Marrow Transplant. 2007;40(4):339–47.
Dahlke J, Kroger N, Zabelina T, Ayuk F, Fehse N, Wolschke C, et al. Comparable results in patients with acute lymphoblastic leukemia after related and unrelated stem cell transplantation. Bone Marrow Transplant. 2006;37(2):155–63.
Nishiwaki S, Terakura S, Yasuda T, Imahashi N, Sao H, Iida H, et al. Outcome of allogeneic bone marrow transplantation from unrelated donors for adult Philadelphia chromosome-negative acute lymphocytic leukemia in first complete-remission. Int J Hematol. 2010;91(3):419–25.
Atsuta Y, Suzuki R, Nagamura-Inoue T, Taniguchi S, Takahashi S, Kai S, et al. Disease-specific analyses of unrelated cord blood transplantation compared with unrelated bone marrow transplantation in adult patients with acute leukemia. Blood. 2009;113(8):1631–8.
Kumar P, Defor TE, Brunstein C, Barker JN, Wagner JE, Weisdorf DJ, et al. Allogeneic hematopoietic stem cell transplantation in adult acute lymphocytic leukemia: impact of donor source on survival. Biol Bone Marrow Transplant. 2008;14(12):1394–400.
Aversa F. Haploidentical haemotopoietic stem cell transplantation for acute leukaemia in adults: experience in Euvope and the united states. Bone Marrow Transplant. 2008;41(5):473–81.
Ram R, Storb R, Sandmaier BM, Maloney DG, Woolfrey A, Flowers ME, et al. Non-myeloablative conditioning with allogeneic hematopoietic cell transplantation for the treatment of high-risk acute lymphoblastic leukemia. Haematologica. 2011;96(8):1113–20.
Stein AS, Palmer JM, O’Donnell MR, Kogut NM, Spielberger RT, Slovak ML, et al. Reduced-intensity conditioning followed by peripheral blood stem cell transplantation for adult patients with high-risk acute lymphoblastic leukemia. Biol Blood Marrow Transplant. 2009;15(11):1407–14.
Mohty M, Labopin M, Volin L, Gratwohl A, Socie G, Esteve J, et al. Reduced-intensity versus conventional myeloablative conditioning allogeneic stem cell transplantation for patients with acute lymphoblastic leukemia: a retrospective study from the European Group for Blood and Marrow Transplantation. Blood. 2010;116(22):4439–43.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Stübig, T., Kröger, N. (2015). Immunopathology and Immunotherapy of Lymphoblastic Leukaemia. In: Rezaei, N. (eds) Cancer Immunology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-46410-6_6
Download citation
DOI: https://doi.org/10.1007/978-3-662-46410-6_6
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-46409-0
Online ISBN: 978-3-662-46410-6
eBook Packages: MedicineMedicine (R0)