
Abstract
Age-related macular degeneration remains to be a major cause of irreversible blindness in the world. In this disease, neovascularization plays an important role in disease burden and progression. Current therapies focus on inhibition of neovascularization through suppression of the extracellular VEGF pathway. However, these strategies have several disadvantages including the need for monthly injections and the need for direct injections of drug therapies to the eye. Moreover, none of these strategies currently focus on inhibition of intracellular VEGF signaling. Here, we describe our recent efforts at developing (I) a novel VEGF inhibition strategy that utilizes Flt-1 intracellularly to suppress VEGF before being secreted out of the cell. We also describe our novel strategy of (II) delivering Flt-1 to sites of neovascularization in the eye using a nonviral, intravitreal-injection-free approach, utilizing surface-functionalized nanoparticles. Using this strategy, we are able to suppress choroidal neovascularization and fibrosis and restore visual acuity in several animal models including nonhuman primates. Our results show a promising alternative in our arsenal for anti-VEGF therapies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Intravitreal Injection
- Choroidal Neovascularization
- PLGA Nanoparticles
- Corneal Neovascularization
- Functionalized Nanoparticles
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
8.1 The Road to Treat ARMD
Age-related macular degeneration is a complex disease. In complex problems, several perspectives are needed to understand basic mechanisms underlying disease pathophysiology. Our perspective of ARMD as a neovascular disease stems from our early studies on corneal neovascularization (Ambati et al. 2002). In a chemically induced mouse corneal neovascularization model, we showed the first evidence of using a biological molecule for preventing corneal neovascularization. Since then, we found other molecules important for inhibiting corneal neovascularization in a nonphysiological system (Ambati et al. 2003a, b; Sakurai et al. 2003). At that point, the physiological mechanism of maintaining corneal avascularity was still not known. The dogma then was that multiple redundant pathways controlled avascularity in the cornea.
In 2006, we reported that expression of soluble VEGF receptor-1 (sFLT-1) is necessary and sufficient for maintenance of corneal avascularity (Ambati et al. 2006). Moreover, we showed that the mechanism of sFLT-1 was through sequestration of VEGF-A leading to inhibition of function. In the next few years, we showed that this same pathway was responsible for maintaining the avascular photoreceptor layer of the retina (Luo et al. 2013a). In ARMD, the avascular photoreceptor layer is penetrated by blood vessels, leading to choroidal neovascularization (CNV). This loss of barrier function of the photoreceptor layer may be due in part to loss of sFLT-1 leading to VEGF-A-induced neovascularization.
As sFlt-1 became a clinically attractive platform for inhibiting VEGF-A for several neovascular diseases including ARMD (Lukason et al. 2011; Lai et al. 2012), we continued to search for other strategies for VEGF-A inhibition. Most of the strategies for VEGF-A inhibition including sFlt-1 focused on inhibiting VEGF-A after it is secreted from the cell; however, it has been shown that VEGF can act via an intracellular autocrine loop which is currently not being targeted by current approaches (Gerber et al. 2002). We then began formulating strategies to inhibit VEGF-A intracellularly before the protein can be secreted from the cell. Although the implication of autocrine signaling in the context of neovascularization is unclear, addition of strategies inhibiting this pathway may result in better control of disease, because if vascular endothelial cells express their own growth factors and receptors, extracellular blockade alone may be insufficient.
8.2 Identification of the Gene
Flt-1 (fms-like tyrosine kinase-1) is a transmembrane receptor in the tyrosine kinase family that was first identified in a v-ros oncogene hybridization screen (Shibuya et al. 1990). A couple of years after its identification, it was found that Flt-1 was a high-affinity receptor for VEGF (de Vries et al. 1992).
The 180 kDa Flt-1 protein is known to have seven immunoglobulin (Ig)-like domains in the extracellular region and a tyrosine kinase domain (Shibuya et al. 1990). The extracellular domain is important for ligand binding. Targeted mutation of mouse flt-1 resulted in a disorganized vasculature with embryonic lethality in homozygous animals (Fong et al. 1995). However, deleting only the tyrosine kinase domain was able to produce viable mice that developed normal blood vessels (Hiratsuka et al. 1998). These studies suggested that the ligand-binding domain of Flt-1 was necessary for normal angiogenesis.
The ligand for Flt-1 is VEGF with binding constants in the picomolar range (Davis-Smyth et al. 1996). Crystallographic studies on VEGF-Flt-1 interaction showed that the second and third extracellular domains of Flt-1 are necessary and sufficient for binding VEGF at close to the native binding affinities (Wiesmann et al. 1997).
Since the initial discovery of Flt-1, many studies have looked into the mechanism for VEGF signaling in vivo. Its strong binding affinity with VEGF has enabled us to use Flt-1 as a biological “bait” to sequester VEGF.
8.3 Identification of the Delivery Vector
The current approved treatment for the neovascularization in ARMD is injection of VEGF inhibitors to the vitreous. One limitation for this strategy is the need for recurrent injections (once every 4–6 weeks) into the eye of patients to maintain active VEGF suppression. Current research in this area focuses on (a) developing longer-term strategies for inhibition of VEGF and (b) efficient therapeutic delivery to the eye without the need for direct intravitreal injections (e.g., intravenous-based therapies, oral therapies).
Gene therapy-based strategies are effective for longer-term expression of VEGF inhibitors. Two mechanisms currently exist for gene delivery: using viral vectors (e.g., adeno-associated virus or AAV) and nonviral systems. Several groups are developing AAV-based vectors for delivery of sFlt-1 (soluble Flt-1 receptor) to inhibit VEGF (Lai et al. 2012; Lukason et al. 2011). These studies have shown long-term inhibition of neovascularization after a single subretinal or intravitreal injection of AAV-sFlt in nonhuman primate models of choroidal neovascularization. One disadvantage for using viral vectors is still the need for invasive subretinal or intravitreal injections.
We adopted a nonviral system for Flt-1 delivery to the eye. Specifically, we used poly(lactic-co-glycolic acid) (PLGA) nanoparticles because of its several properties including (a) biodegradability and biocompatibility, (b) possibility to functionalize the nanoparticle to increase target cell specificity, (c) protection of cargo from degradation before reaching the target, and (d) PLGA nanoparticles that are already FDA approved for parenteral administration as drug delivery vehicles (Danhier et al. 2012).
To add target specificity of the PLGA nanoparticles and enhance delivery of its cargo to certain cells, we functionalized the surface of these nanoparticles with a peptide sequence containing the RGD motif (arginine-glycine-aspartic acid) (Singh et al. 2009). It is known that the peptide GRGDSPK binds integrin alpha v beta-3 receptors, which are commonly overexpressed in the blood vessels of patients with ARMD or diabetic retinopathy (Friedlander et al. 1996). We were able to show that intravenous administration of RGD-tagged PLGA nanoparticles containing Flt-1 was able to localize specifically to the neovascular eye of a rat CNV model and inhibit progression of CNV (Singh et al. 2009). We extended our studies to other neovascularization models in murine and nonhuman primates and showed similar results (see below) (Luo et al. 2013b).
8.4 The Construct
Inhibiting VEGF intracellularly necessitates at least two prerequisites: (a) finding a molecule that binds VEGF at high affinity and (b) a molecule that has to be located intracellularly. The VEGF receptor-1 or VEGFR-1/Flt-1 was a good candidate because of its high binding affinity to VEGF. However, Flt-1 is normally secreted from the cell. Therefore, we needed a strategy to keep Flt-1 inside the to bind and sequester VEGF.
Flt-1 is known to have seven domains. Of these, the first domain contained the secretion signal sequence, the second and third domains are known to bind VEGF in nearly wild-type affinity compared to the intact protein, and the fourth domain is also thought to help in VEGF binding. To prevent Flt-1 from being secreted, we made an N-terminal truncation mutation, removing domain 1. Additionally, we engineered the truncated Flt-1 to contain a C-terminal KDEL tag (Singh et al. 2005). The KDEL tag is a peptide sequence (Lys-Asp-Glu-Leu) that binds endoplasmic reticulum receptors, preventing proteins containing this tag from being secreted (Lewis and Pelham 1990).
We initially tested two different constructs, Flt23K (Flt-1 domains 2 and 3 with KDEL tag) and Flt24K (Flt-1 domains 2, 3, and 4 with KDEL tag), for VEGF inhibition in vitro (Singh et al. 2005).
8.5 In Vitro Data
We used a human cornea epithelial cell culture model to determine whether KDEL-tagged Flt23K and Flt24K are able to inhibit VEGF secretion. In this model, we are able to upregulate expression of VEGF by placing the cells in a hypoxic environment. Both constructs were able to remain intracellularly, in close association with the endoplasmic reticulum. After overexpression of Flt23K or Flt24K, we showed that compared to Flt24K and control cell lines, Flt23K is able to significantly reduce VEGF secretion from the cells. These results were promising, and we tested whether KDEL-tagged Flt23K is able to inhibit neovascularization in in vivo animal models. We used the PLGA nanoparticles as vectors for Flt23K.
8.6 The Tests and Results
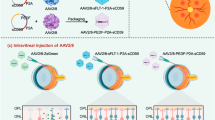
Our overall strategy for VEGF inhibition in vivo used the Flt23K intraceptor loaded in RGD-functionalized PLGA nanoparticles (Fig. 8.1). First, we explored whether RGD-functionalized PLGA nanoparticles can specifically localize to CNV lesions with intravenous loading. Using a laser-induced CNV mouse model with one eye laser treated and the other eye as the control, we intravenously administered nanoparticles labeled with the Nile Red tracer with or without RGD. We showed that nanoparticles can specifically localize to the laser-treated eye but not to the control eye (Fig. 8.2).

Schematic representation of Flt23K plasmid loaded in PLGA nanoparticles. The surface of nanoparticles has been conjugated with the peptide, RGD, which increases specificity of the vector to target areas of neovascularization (Reprinted with permission from Luo et al. (2013a, b). Copyright 2013 American Chemical Society)

Intravenously administered RGD-functionalized nanoparticles are specifically delivered to laser-treated rat eyes. Nanoparticles were injected into the tail veins of Brown Norway (BN) rats 14 days after laser treatment of the right eye. Eyes were harvested 24 h after nanoparticle injection. Representative flatmounts of laser-treated right eyes (bottom row) and control left eyes (top row) were imaged by confocal microscopy. Nanoparticles alone or nanoparticles loaded with the Flt23K plasmid showed minimal targeting to laser-treated eyes. However, nanoparticles functionalized with the RGD peptide loaded with Flt23K plasmid showed increased targeting to laser-treated eyes. Nanoparticles (red, nile red (white arrows)), DAPI (blue) (Reproduced and modified from Singh et al. (2009))
We then proceeded to investigate whether active targeting of our nanoparticles by surface functionalization with RGD could enhance nanoparticle localization and ultimately gene delivery to CNV lesions. Using confocal microscopy, we were able to compare relative concentrations of Nile Red-labeled nanoparticles and green fluorescent protein-labeled Flt23K intraceptor, of both nonfunctionalized and functionalized nanoparticles. Our results show that functionalized nanoparticles increased localization and Flt23K expression to CNV lesions compared to nanoparticles that did not have RGD functionalization (Fig. 8.3).

Flt23K expression in laser-treated rat eyes. Nanoparticles were injected into the tail veins of Brown Norway (BN) rats 14 days after laser treatment of the right eye. Eyes were harvested 24 h after nanoparticle injection. Representative flatmounts of laser-treated right eyes injected with unconjugated Flt23K-plasmid-loaded nanoparticles (Flt23K-NP) and RGD-peptide-conjugated Flt23K-plasmid-loaded nanoparticles (RGD-Flt23K-NP). Only the RGD-conjugated nanoparticles showed increased expression of Flt23K in the laser-treated eyes. Flt23K (green, GFP), nanoparticles (red, nile red (white arrows)), DAPI (blue) (Reproduced and modified from Singh et al. (2009))
Next, we assessed whether functionalized nanoparticles loaded with Flt23K were capable of reducing retinal and choroidal-RPE levels of VEGF in the laser-treated, neovascular eye. At baseline, laser-treated eyes had markedly elevated VEGF levels when compared to control eyes as was expected. Targeted expression of Flt23K inhibited VEGF levels down to baseline levels comparable with the control eye (Fig. 8.4).

Functionalized nanoparticles reduce (a) retinal and (b) choroidal-RPE vascular endothelial growth factor (VEGF) levels. On day 14 after choroidal neovascularization (CNV) induction, the rats were administered one of the following treatments by injection into the tail vein: (1) vehicle, (2) naked Flt23K plasmid, (3) blank nanoparticles, (4) unconjugated Flt23K-plasmid-loaded nanoparticles (Flt23K-NP), and (5) RGD-peptide-conjugated Flt23K-plasmid-loaded nanoparticles (RGD-Flt23K-NP). The rats were euthanized 48 h after nanoparticle injection. Neural retina and choroid-RPE were dissected, and VEGF levels were quantified by sandwich enzyme-linked immunosorbent assay (ELISA). *P < 0.05 as compared to vehicle, naked Flt23K, blank nanoparticles, and nonfunctionalized nanoparticle-treated groups (Reproduced and modified from Singh et al. (2009))
Finally, we wanted to test whether Flt23K-loaded nanoparticles are able to inhibit CNV. Using both histopathologic examination and FITC-dextran analysis of choroidal flatmounts, we were able to quantify the areas of CNV lesions in each eye. We observed a significant reduction in the area of CNV lesions in the laser-treated eyes treated with functionalized nanoparticles delivering Flt23K. These results suggest a possible therapeutic role for Flt23K loaded in RGD-functionalized PLGA nanoparticles in the treatment of neovascular or wet ARMD (Fig. 8.5).

Functionalized nanoparticles reduce laser-induced choroidal neovascular area on histopathologic examination. On day 14 after choroidal neovascularization (CNV) induction, the rats were administered one of the following treatments intravenously: (1) vehicle, (2) naked Flt23K plasmid, (3) blank nanoparticles, (4) unconjugated Flt23K-plasmid-loaded nanoparticles (Flt23K-NP), and (5) RGD-peptide-conjugated Flt23K-plasmid-loaded nanoparticles (RGD-Flt23K-NP). The rats were euthanized 2 weeks after nanoparticle injection. Only the RGD-Flt23K-NP group was able to decrease CNV area significantly. Astricks represents CNV lesions (Reproduced and modified from Singh et al. (2009))
Having refined our method of Flt23K intraceptor delivery and expression utilizing functionalized nanoparticles and showing efficacy at inhibiting laser-induced CNV, we further explored its potential therapeutic benefits in both a mouse and primate ARMD model. Specifically, we were interested in defining the role of our gene therapy strategy on (a) angiogenesis and fibrosis, (b) its safety profile, and (c) exploring its therapeutic potential in restoring visual loss induced by CNV.
Current intravitreal injection protein-based therapies, although successful at reducing CNV-associated angiogenesis, are limited by their inability to address the concomitant fibrosis, which often accompanies CNV in the pathogenesis of ARMD. Thus, in addition to confirming the ability of our particle to inhibit angiogenesis, we wanted to investigate its ability to inhibit fibrosis. Just as we had previously shown in rat, we were able to demonstrate that targeted expression of Flt23K using nanoparticles as a vector was able to reduce angiogenesis in murine and primate laser-induced models as evidenced by decreasing CNV surface volumes (Fig. 8.6). We also showed that this strategy was able to significantly reduce fibrosis in the same model (Fig. 8.6).

RGD-functionalized nanoparticles loaded with Flt23K inhibit CNV and fibrosis. In the laser-induced CNV monkey model, the volumes of CNV lesions (asterisk), including neovessels (stained by perlecan) and fibrosis (stained by collagen I), significantly decreased 4 weeks after RGD.Flt23k.NR.NP treatment. (Reprinted with permission from Luo et al. (2013a, b). Copyright 2013 American Chemical Society)
Although inhibition of CNV is important for decreasing disease burden, functional restoration of visual function is the ultimate goal for patients with ARMD. Unfortunately, with current intravitreal therapy, many patients do not achieve substantial visual improvement, and up to a third of treated eyes progress to legal blindness (Rofagha et al. 2013). While laser-induced CNV models are widely used to study ARMD, they are limited due to the laser’s acute injury and retinal burnout, which results in no potential for recovery of visual function. Consequently, to investigate the role of Flt23K on visual restoration, we created a novel mouse model of ARMD. We induced neovascularization in a mouse by targeting AAV particles containing sFlt-1 shRNA to the retina. By administering a subretinal injection of adeno-associated viral (AAV)-short hairpin RNA (shRNA) targeted against sFlt-1 (a naturally occurring inhibitor of VEGF inhibitor), we were able to create a reversible model of CNV with which we could test nanoparticle-delivered Flt23K. Following intravenous injection of nanoparticles loaded with Flt23K, we were able to observe progressive visual acuity improvement of greater than 10 % in the eyes of the treatment group (Fig. 8.7).

RGD-functionalized nanoparticles loaded with Flt23K improve vision. Optomotor tested vision function was partially restored after 4-week treatment with RGD.Flt23k.NR.NP but not with the vehicle or sham controls. Astricks represents CNV lesions (Reprinted with permission from Luo et al. (2013a, b). Copyright 2013 American Chemical Society)
Lastly, we sought to define the safety profile of RGD.Flt23k.NR.NP. Analyzing the serum 1 day post IV administration, no quantifiable Nile Red was identified, suggesting the amounts of nanoparticles remaining in the bloodstream 24 h after injection was negligible. Additionally, at 30 days postinjection, although present in CNV lesions, no quantifiable amount of Nile Red was found in extraocular tissues including the kidney, lung, liver, and skin. Fundoscopic exam was also performed to assess for the presence of ocular toxicity, and the results showed no evidence of hemorrhage, inflation, retinal detachment, or retinal degeneration. H&E staining also failed to demonstrate any retinal morphologic abnormalities. Finally, using TUNEL staining, we demonstrated the absence of retinal apoptosis outside the area of CNV lesions.
Inspired by the current limitations of intravitreal injections in anti-VEGF therapy, we showed that nanoparticle-mediated delivery of Flt23K is able to inhibit ARMD-associated fibrosis and effectively restore CNV-associated vision loss while simultaneously maintaining a reassuring systemic and ocular safety profile.
8.7 Future Plans
Our recent studies have shown that intravenous injection of Flt23K loaded in surface-functionalized nanoparticles in rat, murine, and primate models of CNV is able to significantly suppress choroidal neovascularization. We have also shown that this strategy is also effective in inhibition of fibrosis. Inhibition of fibrosis is an important clinical problem that is not currently addressed by current therapies. Although the mechanism is unclear, it is likely that suppression of fibrosis is through RGD-mediated inhibition of collagen production as previously reported (Kotoh et al. 2004). Moreover, nanoparticle-based delivery of Flt23K in primates did not show any obvious acute toxicity issues. More studies need to be conducted to assess long-term toxicity issues.
As PLGA nanoparticles are already FDA approved as a nonviral drug delivery vector, it is our vision to conduct a Phase I clinical trial on the safety of RGD-functionalized nanoparticles loaded with Flt23K in humans. Our long-term vision for this strategy is to test whether it is sufficient to inhibit progression or regress neovascularization in CNV and other pathologies including corneal neovascularization either as monotherapy or as an adjunct to current anti-VEGF therapies.
References
Ambati BK, Joussen AM, Ambati J, Moromizato Y, Guha C, Javaherian K, Gillies S, O’Reilly MS, Adamis AP (2002) Angiostatin inhibits and regresses corneal neovascularization. Arch Ophthalmol 120(8):1063–1068
Ambati BK, Anand A, Joussen AM, Kuziel WA, Adamis AP, Ambati J (2003a) Sustained inhibition of corneal neovascularization by genetic ablation of CCR5. Invest Ophthalmol Vis Sci 44(2):590–593
Ambati BK, Joussen AM, Kuziel WA, Adamis AP, Ambati J (2003b) Inhibition of corneal neovascularization by genetic ablation of CCR2. Cornea 22(5):465–467
Ambati BK, Nozaki M, Singh N, Takeda A, Jani PD, Suthar T, Albuquerque RJ, Richter E, Sakurai E, Newcomb MT, Kleinman ME, Caldwell RB, Lin Q, Ogura Y, Orecchia A, Samuelson DA, Agnew DW, St Leger J, Green WR, Mahasreshti PJ, Curiel DT, Kwan D, Marsh H, Ikeda S, Leiper LJ, Collinson JM, Bogdanovich S, Khurana TS, Shibuya M, Baldwin ME, Ferrara N, Gerber HP, De Falco S, Witta J, Baffi JZ, Raisler BJ, Ambati J (2006) Corneal avascularity is due to soluble VEGF receptor-1. Nature 443(7114):993–997
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V (2012) PLGA-based nanoparticles: an overview of biomedical applications. J Control Release 161(2):505–522
Davis-Smyth T, Chen H, Park J, Presta LG, Ferrara N (1996) The second immunoglobulin-like domain of the VEGF tyrosine kinase receptor Flt-1 determines ligand binding and may initiate a signal transduction cascade. EMBO J 15(18):4919–4927
de Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT (1992) The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 255(5047):989–991
Fong GH, Rossant J, Gertsenstein M, Breitman ML (1995) Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376(6535):66–70
Friedlander M, Theesfeld CL, Sugita M, Fruttiger M, Thomas MA, Chang S, Cheresh DA (1996) Involvement of integrins alpha v beta 3 and alpha v beta 5 in ocular neovascular diseases. Proc Natl Acad Sci U S A 93(18):9764–9769
Gerber HP, Malik AK, Solar GP, Sherman D, Liang XH, Meng G, Hong K, Marsters JC, Ferrara N (2002) VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature 417(6892):954–958
Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M (1998) Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A 95(16):9349–9354
Kotoh K, Nakamuta M, Kohjima M, Fukushima M, Morizono S, Kobayashi N, Enjoji M, Nawata H (2004) Arg-Gly-Asp (RGD) peptide ameliorates carbon tetrachloride-induced liver fibrosis via inhibition of collagen production and acceleration of collagenase activity. Int J Mol Med 14(6):1049–1053
Lai CM, Estcourt MJ, Himbeck RP, Lee SY, Yew-San Yeo I, Luu C, Loh BK, Lee MW, Barathi A, Villano J, Ang CL, van der Most RG, Constable IJ, Dismuke D, Samulski RJ, Degli-Esposti MA, Rakoczy EP (2012) Preclinical safety evaluation of subretinal AAV2.sFlt-1 in non-human primates. Gene Ther 19(10):999–1009
Lewis MJ, Pelham HR (1990) A human homologue of the yeast HDEL receptor. Nature 348(6297):162–163
Lukason M, DuFresne E, Rubin H, Pechan P, Li Q, Kim I, Kiss S, Flaxel C, Collins M, Miller J, Hauswirth W, Maclachlan T, Wadsworth S, Scaria A (2011) Inhibition of choroidal neovascularization in a nonhuman primate model by intravitreal administration of an AAV2 vector expressing a novel anti-VEGF molecule. Mol Ther 19(2):260–265
Luo L, Uehara H, Zhang X, Das SK, Olsen T, Holt D, Simonis JM, Jackman K, Singh N, Miya TR, Huang W, Ahmed F, Bastos-Carvalho A, Le YZ, Mamalis C, Chiodo VA, Hauswirth WW, Baffi J, Lacal PM, Orecchia A, Ferrara N, Gao G, Young-Hee K, Fu Y, Owen L, Albuquerque R, Baehr W, Thomas K, Li DY, Chalam KV, Shibuya M, Grisanti S, Wilson DJ, Ambati J, Ambati BK (2013a) Photoreceptor avascular privilege is shielded by soluble VEGF receptor-1. Elife 2:e00324. doi:10.7554/eLife.00324
Luo L, Zhang X, Hirano Y, Tyagi P, Barabás P, Uehara H, Miya TR, Singh N, Archer B, Qazi Y, Jackman K, Das SK, Olsen T, Chennamaneni SR, Stagg BC, Ahmed F, Emerson L, Zygmunt K, Whitaker R, Mamalis C, Huang W, Gao G, Srinivas SP, Krizaj D, Baffi J, Ambati J, Kompella UB, Ambati BK (2013b) Targeted intraceptor nanoparticle therapy reduces angiogenesis and fibrosis in primate and murine macular degeneration. ACS Nano 7(4):3264–3275
Rofagha S, Bhisitkul RB, Boyer DS, Sadda SR, Zhang K, SEVEN-UP Study Group (2013) Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN-UP). Ophthalmology 120(11):2292–2299
Sakurai E, Taguchi H, Anand A, Ambati BK, Gragoudas ES, Miller JW, Adamis AP, Ambati J (2003) Targeted disruption of the CD18 or ICAM-1 gene inhibits choroidal neovascularization. Invest Ophthalmol Vis Sci 44(6):2743–2749
Shibuya M, Yamaguchi S, Yamane A, Ikeda T, Tojo A, Matsushime H, Sato M (1990) Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (flt) closely related to the fms family. Oncogene 5(4):519–524
Singh N, Amin S, Richter E, Rashid S, Scoglietti V, Jani PD, Wang J, Kaur R, Ambati J, Dong Z, Ambati BK (2005) Flt-1 intraceptors inhibit hypoxia-induced VEGF expression in vitro and corneal neovascularization in vivo. Invest Ophthalmol Vis Sci 46(5):1647–1652
Singh SR, Grossniklaus HE, Kang SJ, Edelhauser HF, Ambati BK, Kompella UB (2009) Intravenous transferrin, RGD peptide and dual-targeted nanoparticles enhance anti-VEGF intraceptor gene delivery to laser-induced CNV. Gene Ther 16(5):645–659
Wiesmann C, Fuh G, Christinger HW, Eigenbrot C, Wells JA, De Vos AM (1997) Crystal structure at 1.7 A resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell 91(5):695–704
Compliance with Ethical Requirements
Conflict of Interest
Author Balamurali K. Ambati declares that he has issued a patent on the technologies discussed in the chapter.
Author Samuel F. Passi declares that he has no conflict of interest.
Author Cecinio C. Ronquillo declares that he has no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
Animal Studies
All institutional and national guidelines for the care and use of laboratory animals were followed. All experiments were approved by the IACUCs of Medical College of Georgia, University of Colorado Denver, and University of Utah for the experiments performed at the respective sites.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Ronquillo, C.C., Passi, S.F., Ambati, B.K. (2015). Restoring Physiologic Barriers Against Neovascular Invasion. In: Rakoczy, E. (eds) Gene- and Cell-Based Treatment Strategies for the Eye. Essentials in Ophthalmology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-45188-5_8
Download citation
DOI: https://doi.org/10.1007/978-3-662-45188-5_8
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-45187-8
Online ISBN: 978-3-662-45188-5
eBook Packages: MedicineMedicine (R0)