
Abstract
Members of the genus Colletotrichum cause anthracnose diseases on nearly every crop grown for food, fiber, and forage worldwide. Colletotrichum fungi display a broad range of lifestyles, including plant associations occupying a continuum from necrotrophy to intracellular hemibiotrophy (IH) to endophytism. There are at least three major variants of IH, differing in the duration of biotrophy and synchronization of the switch to necrotrophy. Comparative genomic analyses may uncover how these lifestyles evolved and their functional relationships, identify commonalities as potential conserved targets for control and management, and transform our current understanding of Colletotrichum taxonomy. The genome sequences of four species were recently published: C. graminicola; C. higginsianum; C. obiculare; and C. fructicola (reported as C. gloeosporioides). These species occupy distinct monophyletic lineages in the genus and represent three different lifestyles (two variants of IH, and necrotrophy). The Colletotrichum genomes are relatively large (58-88 Mb), and encode between 11,000 and 16,000 genes. They share little synteny, suggesting that large-scale genome rearrangements were common during the evolutionary history of the genus. Several gene families are expanded in Colletotrichum relative to other sequenced ascomycetes, including those encoding carbohydrate-active enzymes, secondary metabolism enzymes, secreted proteases, and putative secreted effectors. Analysis of the in planta transcriptomes of C. higginsianum, C. graminicola, and C. orbiculare suggested that appressoria and biotrophic intracellular hyphae function as platforms for the secretion of effectors and secondary metabolites to establish host compatibility, while hyphae developing after the switch to necrotrophy are primarily involved in secreting cell wall degrading enzymes and nutrient uptake.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Long Terminal Repeat
- Colletotrichum Species
- Host Cell Wall
- Host Cell Death
- Secondary Metabolism Gene Cluster
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
3.1 Introduction
The fungal genus Colletotrichum includes more than 100 species responsible for anthracnose foliar blight and rot diseases of nearly every crop grown for food, fiber, and forage worldwide (Cannon et al. 2012b; Hyde et al. 2009). Because of their ubiquity, substantial capacity for destruction, and scientific importance as model pathosystems, fungi in the genus Colletotrichum are collectively ranked by the international plant pathology community among the top ten most important fungal phytopathogens (Dean et al. 2012).
Economically important diseases caused by Colletotrichum are widespread, occurring on maize, beans, strawberries, coffee, chili peppers, cucurbits, potatoes, and countless other cultivated plants (e.g., Bergstrom and Nicholson 1999; Hyde et al. 2009; Lees and Hilton 2003; Legard 2000; Melotto et al. 2000; Prihastuti et al. 2009; Singh and Schwartz 2010; Than et al. 2008; Ureña-Padilla et al. 2002; Varzea et al. 2002; Waller 1992; Wasilwa et al. 1993; Xie et al. 2010). Colletotrichum postharvest fruit rots are responsible for major economic losses, with severe infections resulting in up to 100 % loss during storage (Prusky 1996). Colletotrichum diseases also produce substantial damage on important subsistence crops including lentil, cowpea, yam, banana, sorghum, and cassava (Adegbite and Amusa 2008; Chona 1980; Chongo et al. 2002; Finlay and Brown 1993; Green and Simons 1994; Moses et al. 1996; Moura-Costa et al. 1993).
Colletotrichum diseases can negatively impact many of the most important monocots targeted as candidate bioenergy crops , including switchgrass, miscanthus, maize, sorghum, indiangrass, and sugarcane (Crouch 2013; Crouch and Beirn 2009; Crouch et al. 2009a, b; Cortese and Bonos 2012; Dahlberg et al. 2011; Hartman et al. 2011; King et al. 2011; Waxman and Bergstrom 2011a, b; Zeiders 1987). Plants in a wide variety of uncultivated terrestrial and aquatic biomes may also be impacted by Colletotrichum infections, including forests, grasslands, prairie, shrub land, savannahs, and deserts (Abang et al. 2006; Ammar and El-Naggar 2011; Crouch 2013; Crouch et al. 2009b; Damm et al. 2012a; Dingley and Gilmour 1972; Lubbe et al. 2004; Soares et al. 2009).
Colletotrichum occupies a noteworthy place in the history of plant pathology and mycology. The first description of physiological races and cultivar specificity involved the causal agent of bean anthracnose, C. lindemuthianum (Barrus 1911), with that work leading to some of the first resistance breeding efforts using race differentials (reviewed in Geffroy et al. 1999). Subsequent work with the bean anthracnose pathosystem has greatly advanced our understanding of the gene-for-gene system (López et al. 2003; Melotto and Kelly 2001). Work with the teleomorph of C. gloeosporioides pioneered early investigations of fungal sexual determination and development (Lucas et al. 1944, Chilton et al. 1945, Chilton and Wheeler 1949a, b; Driver and Wheeler 1955; Edgerton et al. 1945; Lucas 1946; Wheeler 1950, 1954; Wheeler et al. 1948; Wheeler and McGahan 1952). In the 1960s and 1970s, Colletotrichum studies were at the cutting edge of our understanding of the nature of systemic induced resistance , the chemistry of host defense, and the importance of phytoalexins in the defense response, and they enabled purification of elicitor molecules from fungal cell walls for the first time (Kuc 1972; Sticher et al. 1997). The development and function of melanized appressoria has been substantially elucidated using Colletotrichum (Kubo and Takano 2013). Key components of the cyclic-AMP , MAP kinase , and calcium-mediated signaling pathways have been cloned and characterized from Colletotrichum species (e.g., Chen and Dickman 2002, 2004; Dickman and Yarden 1999; Ha et al. 2003; Kim et al. 2000; Takano et al. 2000; Warwar and Dickman 1996; Yang and Dickman 1997, 1999a, b). Today, Colletotrichum species continue to serve as important models for studies of the molecular and cellular basis of pathogenicity (Kubo and Takano 2013; O’Connell et al. 2012; O’Connell and Panstruga 2006; Perfect et al. 1999).
3.2 Systematics of Colletotrichum
Colletotrichum is an asexual fungus, with the sexual state traditionally classified in the Ascomycete genus Glomerella (Sordariomycetes; Hypocreomycetidae; Glomerellaceae; Glomerellales) (Réblová et al. 2011). With the adoption of single name nomenclature for pleomorphic fungi established by the 2013 Melbourne Code of the International Code of Nomenclature for algae, fungi, and plants (www.iapt-taxon.org/nomen/main.php), it is unlikely that the Glomerella name will continue to be used in the future. Although several species in the genus are known that produce the teleomorph readily (e.g., G. cingulata, G. acutata), Colletotrichum species are predominantly observed in the vegetative or asexual state, with the sexual morph rarely identified for most species (Vaillancourt et al. 2000b). Since plant pathologists and mycologists working with the fungus typically encounter the anamorph, the Colletotrichum name more accurately communicates biological information about the organism. Furthermore, Colletotrichum is the older of the two genera and has priority (1831 vs. 1903; www.mycobank.org). Final resolution of the sole adopted genus name will go through the formal channels established by the International Subcommission of Colletotrichum Taxonomy (www.fungaltaxonomy.org/ subcommissions) to ensure community consensus. In this chapter we will use Colletotrichum to refer both to the anamorphic and the teleomorphic phases.
Colletotrichum is the sole member of the Glomerellaceae , one of three families that collectively make up the order Glomerellales in the Sordariomycete subclass Hypocreomycetidae (Réblová et al. 2011). Earlier reports suggested Colletotrichum as a sister group to Verticillium (Zhang et al. 2006), but more comprehensive research has shown that this inferred relationship reflected insufficient sampling rather than an actual close phylogenetic association, as Verticillium is a member of the Plectosphaerellaceae (Cannon et al. 2012a; Réblová et al. 2011; Zare et al. 2000).
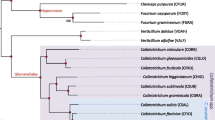
During the past several years, Colletotrichum taxonomy has been the subject of several substantive revisions. Species concepts are still in a state of flux, but it is now well-established that the genus consists of several major monophyletic clades that are referred to as species aggregates, described by the name and attributes of the most prominent representative species in the group (O’Connell et al. 2012; Cannon et al. 2012b; Fig. 3.1). To date, nine aggregates have been described based on multilocus molecular phylogenetics, namely acutatum, graminicola, spaethianum, destructivum, dematium, gloeosporioides, boninense, truncatum, and orbiculare (Cannon et al. 2012b). Although the Colletotrichum aggregates carry no formal taxonomic rank, they provide a convenient way to connect widely used, but outdated and overly broad species concepts with the revised taxonomy. For example, the gloeosporioides aggregate consists of at least 22 species traditionally referred to as C. gloeosporioides, including C. gloeosporioides sensu stricto (Weir et al. 2012). Under the new, more accurate molecular-based taxonomy , C. gloeosporioides sensu stricto is now known to be much less common in the environment than previously thought (e.g., Phoulivong et al. 2010; Weir et al. 2012). The aggregate terminology is especially useful for disease diagnostics that still rely on ITS sequence similarity and/or morphology to identify causal agents. Several of the aggregate groups have been broadly characterized through multilocus phylogenies (Crouch et al. 2009a; Damm et al. 2009; 2012a, b; Weir et al. 2012), yielding a framework for understanding evolutionary relationships across the genus as a whole (Cannon et al. 2012b).

Bayesian phylogenetic tree of the Colletotrichum genus, showing the evolutionary relationship between the four Colletotrichum species with genome sequences, as described in this chapter. The phylogeny was constructed from DNA sequences of four markers (chitin synthase, actin, β-tubulin 2, rDNA ITS; 1514-bp total). The analysis was performed using BEAST for 20 million generations, under the following model: GTR + i, empirical base frequencies, and yule process mode of speciation. The analysis is summarized as a maximum clade credibility tree from 20,001 trees, with the first 2,000 trees ignored (10 %) as burn-in. Posterior probability support values are listed on branches. Branches with thick lines were highly supported by posterior probability values, relative to branches with thin lines. Datasets for included species were generated from alignments of sequences from type specimens, following Cannon et al. (2012a, b). Number of hosts is given following the species name where known. Hosts are dicots except where otherwise noted
3.3 Colletotrichum Lifestyles and Modes of Infection
Fungi in the genus Colletotrichum display a range of nutritional strategies and lifestyles, including plant associations that occupy a continuum from necrotrophy to hemibiotrophy and endophytism. Some species employ a saprotrophic lifestyle to obtain nutrients from soil and organic matter. Colletotrichum are also known to colonize organisms outside the plant kingdom, including insects and humans.
Plant-associated Colletotrichum species typically use a melanized appressorium to penetrate host tissues (Kubo and Takano 2013) (Fig. 3.2). The melanin is required for appressorial function, permitting the accumulation of significant turgor pressure that facilitates mechanical penetration of the host cell wall (Bechinger et al. 1999; Kubo and Furusawa 1991). The appressorium also secretes pectinases and cell wall-degrading enzymes that are likely to play diverse roles in preparing the infection court, adhesion, signaling , and softening the host cell wall (Kleemann et al. 2008; Mendgen et al. 1996). The appressorium of Colletotrichum is morphologically and functionally similar to that formed by Magnaporthe, in spite of the evolutionary distance between these two fungal genera (Mendgen et al. 1996). Appressorial ultrastructure is a taxonomically informative trait in Colletotrichum. Members of the destructivum and graminicola aggregates have a “pore wall overlay” structure surrounding the penetration pore that is similar to that found in Magnaporthe (Howard and Valent 1996), whereas the orbiculare and gloeosporioides aggregates have a distinctive cone-shaped structure associated with the appressorial pore (Fig. 3.2). This cone is surrounded by the appressorial plasma membrane, and consists of modified cell wall material lacking chitin that is similar in structure to the pore wall overlay (O’Connell and Ride 1990). The cone and the pore wall overlay are both continuous with the cell wall of the penetration peg (Fig. 3.2). The functions of the cone and pore wall overlay structures are unknown, but they may serve to reinforce the penetration pore, to focus turgor pressure, or to direct secretion of enzymes and other proteins to the host-pathogen interface.

Appressoria. a Melanized C. orbiculare appressoria (A) formed on glass. Bar = 10 µm, C = conidia. b Penetration peg (PP) emerging from the base of a C. orbiculare appressorium (A) and entering cucumber epidermal cell wall (CW). Bar = 1 µm. c In C. orbiculare, a cone-shaped elaboration of the appressorial cell wall (arrows) surrounds the penetration pore (asterisk). Bar = 0.5 µm. d In C. higginsianum, a pore wall overlay (arrows) surrounds the penetration pore (asterisk). Bar = 0.5 µm. Photos by Richard O’Connell
At one extreme of the spectrum of plant-associated lifestyles, some Colletotrichum fungi rely on a primarily necrotrophic lifestyle to obtain nutrients. These species include most of the causal agents of fruit rots. Necrotrophic Colletorichum do not appear to colonize living host tissue; instead, they first become established either as latent infections confined to unpenetrated appressoria, or as a subcuticular mycelium, before eventually switching to a pathogenic lifestyle, killing host tissue in advance of colonization and inciting significant damage. Colletotrichum species from the acutatum and gloeosporioides aggregates are the most common causes of necrotrophic Colletotrichum diseases (Liao et al. 2012; Prusky 1996; Walker 1921).
At the opposite end of the plant-associated lifestyle continuum are Colletotrichum that colonize host tissue using intracellular hemibiotrophy (IH), a stealthy, highly orchestrated and remarkably effective infection strategy. Intracellular hemibiotrophic Colletotrichum produce specialized infection structures called primary hyphae that are used to invade living host cells, with or without the initial formation of an infection vesicle (O’Connell et al. 2000; Perfect et al. 1999; Perfect and Green 2001). Primary hyphae are thickened or bulbous and are surrounded by a host-derived membrane separating the fungal cell wall from the living host cytoplasm (Mims and Vaillancourt 2002; O’Connell et al. 2000; Perfect et al. 1999; Perfect and Green 2001; Wharton and Julian 1996). The host cell remains alive for a variable period of time that may last for just a few hours up to several days, depending on the interaction (Mims and Vaillancourt 2002; O’Connell et al. 2000; Perfect et al. 1999; Perfect and Green 2001). During biotrophy , the fungus appears to evade plant defenses (Vargas et al. 2012). The transient symptomless phase is followed by a shift to a destructive form of necrotrophic development, accompanied by production of a distinct hyphal morphology that is thinner, not surrounded by a membrane, and exhibits a different wall composition (Mims and Vaillancourt 2002; O’Connell et al. 1996; Pain et al. 1994; Perfect and Green 2001; Perfect et al. 2001; Wharton et al. 2001). These necrotrophic stage hyphae secrete substantial amounts of lytic enzymes, resulting in host tissue collapse and symptom development (O’Connell et al. 2000).
There are at least three major variants of IH utilized by pathogenic Colletotrichum (Fig. 3.3). The first IH strategy is typified by C. higginsianum and other members of the destructivum species aggregate. In this model, there is a limited biotrophic phase confined to the first infected epidermal cell, followed by a complete switch to necrotrophy, marked by development of narrower secondary hyphae, killing of host tissues in advance of fungal colonization , and development of symptoms (Latunde-Dada and Lucas 2007; O’Connell et al. 2004, 2012). The second IH model is employed by members of the orbiculare species aggregate (O’Connell et al. 2000; Perfect et al. 1999; Perfect and Green 2001). Here, the biotrophic phase persists during sequential colonization of several cells. Cells behind the advancing colonization front die gradually, but there is no widespread destruction of cells, and the infection remains symptomless. At some point, the colonizing fungus switches to production of narrow necrotrophic hyphae that kill cells in advance of colonization, and symptoms appear. The third IH strategy is typified by the graminicola species aggregate (Mims and Vaillancourt 2002; Wharton and Julian 1996; Wharton et al. 2001). The graminicola IH model is initially similar to that of orbiculare IH, but differs at the switch to necrotrophy. In the graminicola IH model, narrow secondary necrotrophic hyphae are produced as branches from the thicker primary intercalary hyphae in the cells behind the advancing colony front, which continues to invade new cells biotrophically. Thus, in the graminicola model, biotrophy and necrotrophy exist simultaneously in different parts of the colony (Fig. 3.4). Disease symptoms appear soon after the emergence of necrotrophic hyphae, as the centers of the colonies collapse and die. Relatively few Colletotrichum fungi have been subjected to detailed cytological analysis, and so it is not clear if these models are found in other species aggregates, or even if they are typical of all members of a single aggregate, or of all tissues in a single host. Much more work is needed in this area.

Three variants of intracellular hemibiotrophy. Row 1 C. destructivum model (C. higginsianum). Biotrophic primary hyphae (PH) colonize only one epidermal cell, followed by a complete switch to necrotrophy, with thinner secondary hyphae (SH) killing host cells ahead of infection. Row 2 C. orbiculare model. Biotrophic primary hyphae colonize multiple host cells (first infected cells dead, later infected cells alive), followed by a complete switch to necrotrophy, with secondary hyphae killing host cells ahead of infection. Row 3 C. graminicola model. Biotrophic primary hyphae colonize multiple host cells, as with C. orbiculare, but biotrophy persists at the advancing colony edge while necrotrophy is confined to the colony center. AP = appressorium; IV = infection vesicle. Diagrams by Guillaume Robin

Trypan-blue stained hyphae of C. graminicola colonizing maize leaf sheath epidermal cells during the necrotrophic phase of development. a In the center of the colony, the thicker primary hyphae (white arrow) give rise to thinner necrotrophic hyphae (black arrows) that define this stage of development. b The edge of the same colony is still being colonized biotrophically, evidenced by the ability of the newly invaded and surrounding cells to plasmolyze (black arrows). Scale bars = 50 microns. Photos by Maria Torres
Beyond their notoriety as destructive agricultural pathogens, members of the genus Colletotrichum are among the most common endophytic fungi associated with plants (e.g., Crouch et al. 2009c; Gazis et al. 2011; Hyde et al. 2009; Rodriguez and Redman 2008; Rojas et al. 2010; Vega et al. 2010). For example, the most common endophytic fungi recovered from asymptomatic leaves of forest trees are Colletotrichum species, especially members of the gloeosporioides aggregate (Cannon and Simmons 2002; Arnold et al. 2003). Similarly, a recent survey of wild Arabidopsis thaliana populations identified five different Colletotrichum species as foliar endophytes (García et al. 2013). Asymptomatic colonization of host tissue by Colletotrichum endophytes may lead to any of several outcomes for the plant, including enhanced growth, drought and heat tolerance, and/or disease resistance (e.g., Arnold et al. 2003; Prusky et al. 1994; Redman et al. 2001, 2002). Based on environmental cues such as host senescence, wounding, or other factors associated with changes in plant physiology (Rodriguez et al. 2009), endophytic Colletotrichum may also adopt a saprotrophic lifestyle (Promputtha et al. 2007, 2010), or induce disease symptoms or rots that are only manifested after an extended period of asymptomatic colonization (Freeman et al. 2001; Photita et al. 2004; Rodriguez et al. 2009). Latency/endophytism in association with plants may be an important component of the life cycle of many or most Colletotrichum fungi, but this aspect of development is poorly understood. In particular, there are relatively few cytological studies and so the degree of host colonization by Colletotrichum endophytes is usually unknown. It could range from unpenetrated appressoria (latency) to extended systemic colonization. Much more work is needed in this area.
Surveys of foliar epiphytes show that Colletotrichum fungi are also widespread in the phyllosphere (Alvinidia and Natsuaki 2008; Freeman et al. 2001; Osono 2007, 2008; Santamaría and Bayman 2005). Several Colletotrichum species function as saprophytes, surviving in organic matter or soil; however, this free-living lifestyle may be strictly facultative. In general, Colletotrichum appears to be ill-equipped for long term survival in soil (e.g., Bergstrom and Nicholson 1999; Ripoche et al. 2008), although there are notable exceptions (e.g., Dillard and Cobb 1998; Freeman et al. 2002), and melanized microsclerotia have been observed in several species including C. truncatum, C. sublineola, and C. coccodes (e.g., Boyette et al. 2007; Dillard and Cobb 1998; Sukno et al. 2008). In addition, members of the genus are occasionally reported as opportunistic pathogens of organisms outside the plant kingdom, including insects, turtles, cats, and humans (Cano et al. 2004; Manire et al. 2002; Marcellino et al. 2008, 2009; O’Quinn et al. 2001; Shivaprakash et al. 2011; Winter et al. 2010).
The functional relationships between Colletotrichum lifestyles along the plant-associated continuum are unclear. The mechanisms regulating the developmental switches, and the role of host signals, are also mysterious. It is possible that all of the plant-associated lifestyles are manifestations of a similar underlying interaction, with details of timing dependent on the degree of host resistance. Thus, high levels of resistance may result in latency/endophytism, while reduced resistance could lead to IH, and a further reduction in resistance could trigger a switch to necrotrophy. The duration of the initial biotrophic phase could also partially explain the spectrum of lifestyles . The length of the biotrophic phase may be dependent on the relative ability of the pathogen to mask infection structures from detection. True endophytes that colonize extensively without causing symptoms are likely to have highly developed stealth strategies, for example low expression of lytic enzymes, masking of chitin and other pathogen-associated molecular pattern (PAMP) elicitors , and production of protein and secondary metabolite (SM) effectors to suppress defense.
3.3.1 Colletotrichum Genomics
Genome-scale analyses of Colletotrichum strains with contrasting lifestyles could help us to identify commonalities and provide conserved targets for the management and control of these fungi. Unfortunately, very few Colletotrichum species have been studied in depth at the molecular level, and relatively little is known about those species of the greatest economic importance, or those species that cause the most significant damage to subsistence crops. Individual findings in one pathosystem have only rarely been validated or tested in other systems, so it is not clear to what extent mechanisms of pathogenicity are similar across all lineages. As the genomes of other economically important plant pathogenic fungi were sequenced, beginning with M. oryzae in 2005 (Dean et al. 2005), Colletotrichum remained in the background of the genomics revolution, primarily because it was difficult to make a case for sequencing any single species to represent all the others, without understanding how the pathogenic models related to one another. Problematic taxonomy , and a woefully inadequate understanding of species boundaries and evolutionary relationships, also initially limited our ability to identify the most suitable subjects for genome analysis.
As genome sequencing technologies became faster and cheaper, whole genome sequences were finally generated and published in quick succession for four species of Colletotrichum: C. higginsianum, C. graminicola, C. orbiculare, and C. fructicola (formerly C. gloeosporioides; Fig. 3.1; Gan et al. 2013; O’Connell et al. 2012). According to our current understanding of Colletotrichum taxonomy, these four species are each positioned within distinct monophyletic lineages in the Colletotrichum phylogenetic tree (Fig. 3.1; Cannon et al. 2012a, b). C. graminicola is a member of the graminicola aggregate. C. higginsianum is part of the destructivum aggregate, the sister clade to the graminicola aggregate. C. orbiculare is a close relative of the bean anthracnose pathogen C. lindemuthianum, and together they occupy the orbiculare species aggregate. C. fructicola is a member of the gloeosporioides aggregate, one of the most diverse groups of Colletotrichum, encompassing numerous species associated with a huge number of hosts worldwide (Weir et al. 2012). Although the Nara-gc5 strain was considered a member of C. gloeosporioides sensu lato at the time the genome sequence was published, recent revisions to the taxonomy of the gloeosporioides aggregate now enable a precise species identification (Weir et al. 2012). As shown in the genus-wide multilocus phylogenetic tree in Fig. 3.1, Nara-gc5 is a member of C. fructicola, a globally distributed species within the gloeosporioides aggregate that has been isolated from eight different plant families to date (Weir et al. 2012; Prihastuti et al. 2009). Accordingly, in this review we will refer to Nara-gc5 as C. fructicola to reflect the revised, more accurate taxonomy.
C. graminicola strain M1.001 (aka FGSC 10212, CBS 130836, M2; formerly known as Glomerella graminicola) was the first species of Colletotrichum to have a complete genome sequence available, and is also one of the last of the fungal genomes to be substantially sequenced using Sanger dideoxy technology. C. graminicola is among the best characterized and most tractable of the Colletotrichum fungi, and is one of very few species in the genus in which sexual crosses can be made (Vaillancourt and Hanau 1991). C. graminicola causes one of the most destructive diseases of maize, anthracnose stalk rot , resulting in annual losses of more than $1 billion in the United States (Bergstrom and Nicholson 1999; Frey et al. 2011). Sequences for C. graminicola M1.001 (Forgey et al. 1978) and for M5.001 (aka CBS 130839), a strain that is sexually compatible with M1.001 (Vaillancourt and Hanau 1991), are available.
C. higginsianum strain IMI349063 causes anthracnose on Brassica and Raphanus crops, as well as wild cruciferous species. Although the fungus may occasionally cause significant crop losses, C. higginsianum is generally of only minor importance in commercial agricultural production (Horie et al. 1988; Lin and Huang 2002). However, this species is of considerable scientific interest because of its ability to cause disease on certain ecotypes of the model plant Arabidopsis (O’Connell et al. 2004; Narusaka et al. 2004). The sequenced strain of C. higginsianum was originally isolated from Brassica chinensis, but it also readily infects and causes disease in Arabidopsis (O’Connell et al. 2004, 2012). As such, C. higginsianum provides an experimental system in which both host and fungal partners can be genetically manipulated. In particular, the availability of powerful genetic tools and resources on the plant side facilitate the analysis of host resistance and susceptibility (e.g., Birker et al. 2009; Narusaka et al. 2009).
C. orbiculare 104-T (aka CBS 514.97, LARS414; formerly known as C. lagenarium) is a common and significant problem on cucurbits, causing anthracnose lesions on vegetative tissue and fruit (Westcott 2001; Kubo and Takano 2013). At one point, anthracnose was among the most common and destructive diseases of cucumbers and melons in the United States (Gardner 1918). Today, losses due to C. orbiculare are kept in check through improved crop management strategies, although anthracnose remains a prevalent disease of commercial watermelons grown in regions of high humidity (Maynard and Hopkins 1999). The sequenced strain of C. orbiculare also infects the model plants Nicotiana benthamiana and N. tabacum (Shen et al. 2001), which are amenable to transient gene expression and silencing assays. Techniques for genetic manipulation of C. orbiculare have been established, including gene targeting or random gene insertion . In addition, C. orbiculare pathogenesis is stable and is well characterized cytologically, making this pathosystem an attractive platform for experimental studies (Kubo 2012).
C. fructicola strain Nara-gc5 (formerly known as C. gloeosporioides) causes crown rot of strawberry (Okayama and Tsujimoto 1994, 2007), a disease responsible for substantial losses for strawberry producers worldwide. Strawberry anthracnose can result in up to 80 % losses for nursery plants, and up to 50 % losses in the field (Howard and Albregts 1983; Xie et al. 2010). Other members of C. fructicola are important pathogens of a broad range of commercially grown crops, including coffee, apples, yams, pears, and avocados (Prihastuti et al. 2009; Weir et al. 2012). Comparisons between members of this species, and within the larger gloeosporioides aggregate, promise to be informative in determining factors contributing to adaptation to particular hosts and lifestyles.
3.3.2 Colletotrichum Comparative Genomics
Genome assembly statistics for the four sequenced Colletotrichum strains are summarized in Table 3.1. Differences in the read lengths generated by the different sequencing methods are reflected in the quality of the resulting genome assemblies, with short read technologies producing more fragmented assemblies than those that incorporated Sanger and Roche-generated data. Despite the differences in sequencing approaches, overall gene coverage was high for all four assemblies (Table 3.1; Gan et al. 2013; O’Connell et al. 2012) when assessed using the CEGMA pipeline (Core Eukaryotic Genes Mapping Approach; Parra et al. 2007). Nonetheless, direct comparisons between these four genomes should be made with caution, given that different sequencing strategies and methodologies were used, and different computational tools were employed to assemble and annotate the genomes.
Three of the four sequenced Colletotrichum strains, (C. graminicola M1.001, C. higginsianum IMI349063, and C. fructicola Nara-gc5), yielded genome assemblies with estimated sizes ranging from 53 to 58 Mb, somewhat larger than the average 38 Mb sequenced Pezizomycota genome (Table 3.1; Cuomo and Birren 2010; Gan et al. 2013; Kelkar and Ochman 2012; O’Connell et al. 2012). At 88 Mb, the C. orbiculare 104-T assembly is considerably larger than the other three Colletotrichum species (Gan et al. 2013). It also dwarfs the genomes of most ascomycetes sequenced to date, surpassed in size only by the biotrophic powdery mildew fungi and Tuber melanosporum (Cuomo and Birren 2010; Gan et al. 2013; Spanu et al. 2010). The large genome of C. orbiculare strain 104-T is the result of blocks of low-complexity AT-rich sequences dispersed among the coding sequences, accounting for nearly half of the genome assembly (Gan et al. 2013). These AT-rich sequence blocks may have arisen from modification of transposable elements by repeat-induced point mutation (RIP ; Galagan and Selker 2004).
Despite the differences observed in overall genome size , predicted gene numbers were similar for the four Colletotrichum species, ranging from 12,006 (C. graminicola) to 16,172 (C. higginsianum); all were larger than the average set of 11,281 genes observed in other sequenced Pezizomycota (Table 3.1; Cuomo and Birren 2010; Gan et al. 2013; O’Connell et al. 2012). The reduced number of genes observed in C. graminicola, relative to the other three sequenced Colletotrichum genomes, was largely due to the presence of fewer gene paralogs, with the C. graminicola genome appearing to have undergone less gene duplication . In particular, more than twice as many multicopy genes were identified in the C. fructicola genome as in that of C. graminicola (Table 3.1).
A remarkably low level of synteny was observed among the four sequenced Colletotrichum genomes, much less than that displayed between members of two different genera (Botrytis and Sclerotinia; Amselem et al. 2011). Synteny between C. graminicola and C. higginsianum was only 35 %, while the more distantly related C. orbiculare and C. fructicola shared only 40 % synteny (Gan et al. 2013; O’Connell et al. 2012). These low levels of shared gene order, which appear to be independent of the degree of taxonomic relatedness, suggest that major genome rearrangements have been a common feature during the history of the Colletotrichum genus. Unfortunately, this also means that the value of the high quality assembly of the C. graminicola M1.001 genome as a reference for assembling other Colletotrichum species may be limited.
In marked contrast with the low degree of synteny documented between Colletotrichum species, intraspecific chromosomal rearrangements may be rare. Thus, two strains of C. graminicola, one isolated in North America in 1972 (M1.001), and a second strain isolated in South America in 1989 (M5.001), appeared to be highly syntenic with relatively few sequence polymorphisms (O’Connell et al. 2012). This indicates that major chromosomal rearrangements within species may be uncommon. This may also suggest that genome rearrangements play a role in speciation, perhaps by promoting reproductive isolation (Aguileta et al. 2009).
3.4 Repetitive DNA
Prior to the availability of genomic resources, relatively little was known about Colletotrichum transposable elements (TEs) and their impact on the host genome. Even with the availability of genome assemblies, Colletotrichum TEs have been subject to little detailed analysis, but some general characterizations are possible. All four sequenced Colletotrichum genomes contained signatures of common long terminal repeat (LTR) and DNA transposon classes. Summary data showed that the percentages of these repetitive sequences were higher for C. graminicola and C. orbiculare (12.2 and 8.3 %, respectively) than for C. higginsianum (1.2 %) or C. fructicola (0.75 %), but this may be an artifact derived from the more complete genome assemblies and sequencing strategies used for C. graminicola and C. orbiculare (Gan et al. 2013; O’Connell et al. 2012). Several TE sequences in the Colletotrichum genomes were similar to the Ccret1, Ccret2, Ccret3, and Cgret Metaviridae family LTR retrotransposons , the non-LTR LINE-like retroelement CgT1, and the Collect1 DNA TE sequences described previously from C. cereale and C. gloeosporioides (Crouch et al. 2008; He et al. 1996; Zhu and Oudemans 2000).
Clustering of TEs in the context of rapidly evolving genome regions undergoing high rates of duplication is a trait that Colletotrichum holds in common with other phytopathogenic ascomycetes, including M. oryzae, Verticillium oxysporum, and V. dahlia (Amyotte et al. 2012; Gan et al. 201; Hua-Van et al. 2000; O’Connell et al. 2012; Thon et al. 2006). In C. graminicola, TEs were organized in clusters distributed throughout the genome. C. graminicola supernumerary minichromosomes (see below) and unanchored scaffolds were particularly enriched in TEs, relative to the ten primary chromosomes. Almost 23 % of the three minichromosomes and 50 % of the unanchored scaffolds were composed of predicted TE sequences, while the primary chromosome assemblies contained only 5.5 % repetitive DNA. In the C. graminicola genome, there was a statistically significant correlation between the location of TEs and paralogous gene families, genes encoding secreted proteins, and genes without orthologues in C. higginsianum (O’Connell et al. 2012). Similarly, in the C. orbiculare genome, AT-rich blocks, which may represent relics of TEs mutated through repeat-induced point (RIP) mutation , were associated with small unique secreted protein genes (Gan et al. 2013). Unfortunately, the C. fructicola and C. higginsianum assemblies were too fragmented to perform a similar analysis. The observed transposon clustering in Colletotrichum may reflect selection against harmful integration into gene-rich regions, as described for Saccharomyces cerevisiae Ty3 LTR elements (Voytas and Boeke 1993). There is no obvious evidence that TE integration has played a role in the duplication of adjacent genes. Regardless of the mechanism(s) responsible for TE clustering , the observed patterns suggest that TEs may play a role in the generation of effector diversity and novel genes in Colletotrichum. Further research is needed to investigate these possibilities.
TEs populating the genomes of C. graminicola, C. fructicola, and C. orbiculare showed the signature of widespread RIP mutation, consistent with the mutation patterns documented from elements described previously from C. cereale (Crouch et al. 2008). TpA and ApT dinucleotides were both amplified in the TEs of all three genomes, with the corresponding depletion of CpA, CpG, and CpC dinucleotides resulting in the canonical footprint of the RIP process as first described in Neurospora crassa (Cambareri et al. 1989). Comparative genome profiles between C. graminicola and 48 additional filamentous ascomycetes showed that RIP distortion of dinucleotides exhibited by C. graminicola TEs was exceptionally pronounced (Clutterbuck 2011).
3.5 Supernumerary Chromosomes
Supernumerary minichromosomes (aka B-chromosomes) are a common feature in the genus Colletotrichum. These small chromosomes are typically conditionally dispensable for growth in fungi, and highly variable from one strain to the next (Covert 1998; Stukenbrock et al. 2010). In some fungi, including Alternaria alternata, Cochliobolus heterostrophus, Fusarium oxysporum, M. oryzae, Mycosphaerella graminicola, and Nectria haematococca, minichromosomes are enriched in secreted genes that encode proteins involved in niche or host adaptation (Chuma et al. 2003; Coleman et al. 2009; Hatta et al. 2002; Ma et al. 2010; Stukenbrock et al. 2010). For several fungi, horizontal transfer of minichromosomes has been demonstrated, often resulting in expanded pathogenicity on new hosts (Mehrabi et al. 2011). In Colletotrichum for example, minichromosomes of C. gloeosporioides sensu lato infecting Stylosanthes guianensis were transferred between different strains of the fungus, conferring novel pathogenicity (Masel et al. 1996). The sequenced strains of C. higginsianum and C. graminicola are known to possess minichromosomes (Table 3.1; O’Connell et al. 2012). However, the minichromosomes in each case were very poorly assembled due to high levels of repetitive sequences, extensive tracts of AT-rich sequences, and reduced gene density relative to the rest of genome. Although it is not known whether C. fructicola has minichromomes, C. fructicola sequences shared some similarity with those documented from the minichromosomes of another member of the gloeosporioides aggregate pathogenic to S. guianensis (Gan et al. 2013; Masel et al. 1996). In contrast, C. graminicola M1.001 minichromosome sequences did not match the genome sequence from a second strain of the same species, M5.001 (O’Connell et al. 2012), confirming earlier hybridization analyses that showed that the minichromosomes were not conserved between the M1.001 and M5.001 strains of C. graminicola (Rollins 1996). In both Colletotrichum genomes, the minichromosomes possessed such poor quality sequence assemblies that it was not possible to determine if they were enriched for pathogenicity genes.
3.6 Mating Type Genes
Sexual reproduction is rarely documented from the genus Colletotrichum. Some species for which the Glomerella morph has never been observed in nature, including C. graminicola, have been induced to mate in the laboratory (Politis 1975; Vaillancourt and Hanau 1991). The genetics underlying Colletotrichum mating are perplexing, in that fungi in this genus do not employ the canonical bipolar mating system characteristic of other ascomycete fungi (Vaillancourt et al. 2000a, b). In the standard bipolar model used by most ascomycetes to regulate sexual compatibility, mating can occur when both idiomorphs of the mating type gene, Mat1, are present (Mat1-1 and Mat1-2). This requirement may be met in a single homothallic individual carrying both idiomorphs , or in a combination of two heterothallic individuals, each carrying one of the two different idiomorphs (Ni et al. 2011). Colletotrichum does not conform to this system. To date, only the Mat1-2 idiomorph, with the characteristic conserved high mobility group (HMG) binding domain, is known from any Colletotrichum species surveyed, regardless of whether the strains are heterothallic or homothallic (Crouch et al. 2006; Du et al. 2005, Rodríguez-Guerra et al. (2005); García-Serrano et al. (2008); Vaillancourt et al. 2000b). However, genetic evidence does point to at least two unlinked loci acting as mating determinants in crosses involving C. graminicola strains M1.001 and M5.001 (Vaillancourt et al. 2000a).
Early attempts to identify the Mat1-1 gene were made by using Southern hybridizations, degenerate primer pairs, and primer walking in cosmid libraries. These experiments, performed by multiple laboratories, focused on detection of the highly conserved alpha DNA binding domain that characterizes the Mat1-1 idiomorph in other ascomycete fungi. None of these approaches provided any evidence for the presence of a Mat1-1 gene, even from homothallic strains of Colletotrichum (Crouch et al. 2006; Du et al. 2005, Rodríguez-Guerra et al. 2005; García-Serrano et al. 2008). BLAST searches of the four Colletotrichum genome sequences confirmed the absence of any sequence with significant identity to the Mat1-1 gene. The Mat1-1 gene was not found in the genomes of C. graminicola strains M1.001 and M5.001, even though these two strains can be mated in vitro to produce fertile progeny (Vaillancourt and Hanau 1991; Vaillancourt et al. 2000a).
Evaluation of 90 genes proximal to the Mat1-2 gene (~134 Kb) showed that C. graminicola strains M1.001 and M5.001 share 99.8 % nucleotide sequence similarity in genic regions (99.6 % overall for the region), and the ordering and orientation of genes in this region were identical between the two strains. The Mat1-2 gene is highly conserved between M1.001 and M5.001, sharing 99.5 % nucleotide identity. Only four of the 840 nucleotides vary between these two strains of C. graminicola, and none of the Mat1-2 base changes are located within the HMG box DNA binding domain. Three of the four variable Mat1-2 nucleotides are located in the first intron, while the fourth base change introduces an amino acid change from asparagine in M1.001 to aspartic acid in M5.001 in exon 2. The M5.001 aspartic acid residue at this site is also found in C. higginsianum IMI 349063, while the asparagine residue of M1.001 is also found at this site in the Mat1-2 genes of C. lindemuthianum and C.gloeosporioides (Du et al. 2005; García-Serrano et al. 2008).
Pairwise comparisons of the Mat1-2 coding sequence from the four sequenced Colletotrichum genomes show that outside of the conserved HMG-box , the coding sequences are quite different among the four species. The Mat1-2 exons are considerably longer in the C. higginsianum genome: 987-bp, relative to the smaller genes encoded by C. fructicola, C. graminicola, and C. orbiculare (726-, 840-, and 750-bp, respectively). C. fructicola and C. orbiculare share 66 % nucleotide identity at the Mat1-2 locus, consistent with their closer evolutionary relationship (Fig. 3.1). The more distantly related C. graminicola shared 51 % identity with C. fructicola and C. orbiculare at this locus. However, the C. higginsianum Mat1-2 coding sequence displays extensive sequence divergence relative to the other three species, sharing only 30–35.5 % identity. Differences between Mat1-2 encoded by C. higginsianum and the other three Colletotrichum species is attributable to the presence of numerous insertions throughout the predicted coding sequence—between 193 and 319 nucleotide gaps.
Despite the high level of interspecific differences in the coding sequences of the Mat1-2 genes, the region surrounding the Mat1 locus shows a high level of conserved synteny between C. fructicola, C. graminicola, and C. orbiculare. Comparison of the genes proximal to the Mat1-2 locus shows that this region is 100 % conserved in gene content and gene order in these three Colletotrichum species (Msa1/Cia30/Apc5/Cox13/Apn2/Mat1/Sla2/L21e 60 s ribosomal protein/S4-9 40 s ribosomal protein /Slu7/Rev3/Tex2/Ami1). A similar comparison with the C. higginsianum genome could not be completed, as the genome assembly is fragmented, with no more than three genes on a single contig, several genes incomplete/truncated, and some genes predicted that seem unlikely to actually exist.
3.7 Expanded Gene Families
Several gene families are expanded in the genomes of the four sequenced Colletotrichum strains, relative to other sequenced ascomycetes. Expansions included genes predicted to encode carbohydrate-active enzymes (CAZymes) , secondary metabolism (SM) enzymes , secreted proteases, and putative secreted effectors (Gan et al. 2013; O’Connell et al. 2012).
CAZYmes Expanded enzyme arsenals capable of degrading cellulose and other polysaccharides contained within plant cell walls are a common theme for hemibiotrophic and necrotrophic plant pathogens, including Colletotrichum species, M. oryzae and Fusarium species (Cuomo et al. 2007; Dean et al. 2005; Ma et al. 2010). The expansion of cell wall degrading enzymes is a defining feature of the four Colletotrichum genomes (Fig. 3.5). The overall abundance of these proteins in Colletotrichum is unmatched in any ascomycete sequenced to date, even the destructive necrotrophic gray and white rot fungi Botrytis cinerea and Sclerotinia sclerotiorum (Fig. 3.5) (Amselem et al. 2011; Gan et al. 2013; O’Connell et al. 2012).

Relative numbers of genes encoding CAZymes targeting different plant cell wall components in Colletotrichum, and in other related fungi with various lifestyles. Black bars pectin. Gray bars pectin and hemicellulose. Speckled bars hemicellulose. White bars cellulose. Blumeria graminis (biotroph); Ustilago maydis (biotroph); Botrytis cinerea (necrotroph); Sclerotinia sclerotiorium (necrotroph); Fusarium oxysporum (hemibiotroph); Fusarium graminearum (hemibiotroph); Colletotrichum fructicola (hemibiotroph); Colletotrichum orbiculare (hemibiotroph); Colletotrichum higginsianum (hemibiotroph); Colletotrichum graminicola (hemibiotroph); Neurospora crassa (saprophyte); Magnaporthe oryzae (hemibiotroph). Data from Gan et al. (2013), and Pamela Gan
In dicots, pectin comprises approximately 35 % of the cell walls, while the cell walls of monocots such as maize generally contain only 10 % pectin (Vogel 2008). There was an extremely large number of unique pectinases encoded by the genomes of the dicot-infecting C. higginsianum, C. fructicola, and C. orbiculare, likely reflecting an important adaptive trait for these pathogens. In particular, C. orbiculare and C. fructicola each encode more than 100 different pectinases, greatly surpassing other sequenced fungal genomes (Fig. 3.5). The genome of the maize pathogen, C. graminicola, possesses a reduced cohort of pectinase genes compared with the other Colletotrichum species: on average, 46 % fewer than the dicot-infecting species. Also consistent with overall pectinase abundance, gene expression profiles during the necrotrophic phase revealed that 51 pectinases were deployed by C. higginsianum during necrotrophy, versus only sixteen utilized by C. graminicola (O’Connell et al. 2012).
Secondary metabolism genes . Another highly expanded class of genes in the Colletotrichum genome encodes putative SM enzymes (Fig. 3.6). SM enzymes are low molecular weight molecules that are not essential for growth and survival of the organism, but may become important for niche adaptation. Production of these metabolites is often associated with successful competition for host resources through toxic and/or inhibitory effects on other organisms (Bölker et al. 2008; Shwab and Keller 2008). There are numerous examples of SM genes that function in this manner, including the well-known trichothecene gene cluster of Fusarium graminearum and the AAL toxin cluster of tomato-infecting Alternaria alternata (Akagi et al. 2009; Procter et al. 2009). Large numbers of SM-associated genes are usually found in the genomes of necrotrophic plant pathogens (Amselem et al. 2011), and SM enzymes are often implicated as phytotoxins with direct roles in pathogenicity (e.g., Daub 1982; Gengenbach et al. 1973; Matthews et al. 1979; Scott-Craig et al. 1992). In contrast, a reduced cohort of SM genes is commonly observed in biotrophic fungi such as Blumeria graminis (Spanu et al. 2010) and Ustilago maydis (Kämper et al. 2006; Bölker et al. 2008).

Relative numbers of genes encoding putative SM-related enzymes in Colletotrichum, and in other fungi with various lifestyles. Black bars PKS and PKS-like, Gray bars NRPS and NRPS-like, Speckled bars PKS-NRPS hybrids, White bars DMATs. Aspergillus nidulans, saprophyte; Stagonospora nodorum, hemibiotroph; Sclerotinia sclerotiorum, necrotroph; Magnaporthe oryzae, hemibiotroph; Neurospora crassa, saprophyte; Fusarium graminearum, hemibiotroph; Ustilago maydis, biotroph; Colletotrichum graminicola, hemibiotroph; Colletotrichum higginsianum, hemibiotroph; Colletotrichum fructicola, hemibiotroph; Colletotrichum orbiculare, hemibiotroph. Data from Gan et al. (2013); O’Connell et al. (2012)
Relatively little is known about the role of SM in hemibiotrophic plant pathogens (Collemare et al. 2008; Böhnert et al. 2004). Colletotrichum species have been reported to produce a variety of SM genes, including flavones, peptides, and terpenes, as well as the polyketide-derived DHN (1,8-dihydroxynaphthalene) melanin , an essential requirement for appressorium-mediated host penetration (Kubo et al. 1991; Singh et al. 2010). Additional examples include the siderophore ferricrocin, isolated from C. gloeosporioides, which has phytotoxic activity in grass cotyledons (Ohra et al. 1995), colletotrichins A, B, and C from C. nicotianae, which produce symptoms resembling tobacco anthracnose when infiltrated into tobacco leaves (Goddard et al. 1976; Kimura et al. 1977, 1978; García-Pajón and Collado 2003), and a tetrahydroxylated compound with antioxidant properties from C. gloeosporioides (Femenía-Ríos et al. 2006). Several secondary metabolites have been characterized from C. graminicola, including the antifungal compounds monorden and monicillins I, II, and III (Wicklow et al. 2009), and mycosporine-alanine, a spore germination inhibitor (Leite and Nicholson 1992). It was recently reported that deletion of PPT1, a gene encoding a cofactor essential for the enzymatic function of all polyketide synthases (PKS) and non-ribosomal peptide synthetases (NRPS) , resulted in decreased pathogenicity in C. graminicola, providing support for the idea that SM genes play an important role in the regulation of pathogenicity to maize (Horbach et al. 2009). Studies have also shown that some Colletotrichum fungi, including C. gloeosporioides, are able to synthesize the plant growth hormone auxin , which is likely to be important in host manipulation (Chung et al. 2003; Robinson et al. 1998). All four sequenced Colletotrichum species contain genes with the potential to encode for production and efflux of auxin (Gan et al. 2013; O’Connell et al. 2012). Genes for synthesis of auxin via an IAM intermediate are present in C. fructicola and C. graminicola. Auxin synthesis by C. higginsianum and C. obiculare may occur via a different intermediate.
Genes encoding putative polyketide synthases (PKS) and PKS-like genes and dimethylallyl transferases (DMATs) are especially abundant in the four sequenced Colletotrichum genomes (Gan et al. 2013; O’Connell et al. 2012). C. higginsianum and C. fructicola possess many more PKS genes, in particular, than any other sequenced fungi (although it is important to point out that these two assemblies had the lowest qualities, and therefore PKS numbers may be somewhat inflated by gene fragmentation). Apart from genes involved in the production of melanin, few of the predicted SM genes have orthologs outside of the Colletotrichum genus, and many appear to be specific to individual Colletotrichum strains.
In all fungal species, SM genes tend to be organized into clusters; often these clusters include additional enzymes, cytochrome P450 genes, transcription factor genes, and transporter genes (Shwab and Keller 2008). The number of SM clusters predicted from the four sequenced Colletotrichum strains is exceptionally high, ranging from 42 clusters in C. orbiculare to 56 clusters in C. fructicola. This abundance of SM clusters is substantially greater than other plant pathogenic fungi sequenced to date (Gan et al. 2013; O’Connell et al. 2012). Many of the Colletotrichum SM clusters are not well conserved among the four species (Table 3.2).
3.7.1 The Colletotrichum Secretome
Fungal secreted proteins are known to have many important roles in plant-fungal interactions, including making plant nutrients accessible to the fungus, or inducing host cell susceptibility or host cell death (Choi et al. 2010; Lowe and Howlett 2012). The predicted secretomes of the four sequenced Colletotrichum fungi are large and diverse, ranging in size from 1650 proteins for C. graminicola (14 % of the proteome) to 2356 proteins for C. fructicola (15 % of the proteome) (Table 3.3). This is comparable to the predicted average of 1798 secreted proteins for Pezizomycotina (Choi et al. 2010). Hemibiotrophic pathogens typically have a larger percentage of their genomes (>10 %) devoted to secreted proteins than other fungi, and Colletotrichum fits this pattern (Lowe and Howlett 2012). Approximately 1,500 secreted protein genes were shared by all four Colletotrichum species. Many other secreted proteins appeared to be species-specific, including 248 found only in C. orbiculare, 225 specific to C. fructicola, 227 found only in C. higginsianum, and 123 specific to C. graminicola.
Compared with other sequenced ascomycetes, the three dicot-infecting species of Colletotrichum were highly enriched in genes encoding secreted proteases , particularly the serine proteases known as subtilisins (MEROPS family S8A) that are known in some other pathosystems to function as effectors (Prusky et al. 2001; Olivieri et al. 2002). The relative expansion of these in C. fructicola, C. higginsianum, and C. orbiculare, but not in C. graminicola, may reflect the narrower host range of the latter species. Some of the subtilisins appear to cluster more closely with those of plant origin, suggesting that they could have been acquired by horizontal transfer from their plant hosts (Gan et al. 2013; Jaramillo et al. 2013).
The Colletotrichum secretomes contain homologs of genes that encode known fungal pathogenicity effectors , including several necrosis-inducing proteins (Fellbrich et al. 2002; Gijzen and Nuernberger 2006; Kanneganti et al. 2006), and the biotrophy-associated BAS2 and BAS3 proteins from M. oryzae (Mosquera et al. 2009) (Table 3.4). Genes encoding members of the necrosis- and ethylene- inducing peptide (NEP) 1-like protein family (Gijzen and Nuernberger 2006) were identified in C. higginsianum (Kleemann et al. 2012). Only three of these were able to cause cell death in N. benthamiana: the others lacked crucial amino acids and did not function to induce necrosis (Kleemann et al. 2012). Two NEP protein genes in C. orbiculare also did not contain necrosis-inducing motifs (Gan et al. 2013). These two had peak expression levels in early biotrophy.
A screen for C. orbiculare proteins that induced cell death in N. benthamiana led to the identification of NIS1, a protein secreted by primary hyphae (Yoshino et al. 2012). Cell death induced by NIS1 is mediated by interaction with the plant heat shock protein 90 (Hsp90), known to be important in R-gene mediated HR-response (Zhang et al. 2010). Homologs of the NIS1 effector gene are found in all four sequenced Colletotrichum species.
Screening of an EST library derived from nitrogen-starved mycelium of C. gloeosporioides resulted in the identification of CgDN3, a gene predicted to encode a small secreted protein that is required for the successful establishment of this pathogen on Stylosanthes guianensis leaves (Stephenson et al. 2000). During infection, the CgDN3 transcript accumulated in biotrophic infection vesicles. CgDN3 knockout mutants failed to penetrate or form primary infection hyphae, and they rapidly induced localized cell death. Homologs of CgDN3 were found in the genomes of C. fructicola, and also in C. orbiculare, and C. higginsianum, but not in C. graminicola. The C. orbiculare and C. higginsianum homologs suppressed cell death induced by the necrosis-inducing effector NIS1 when they were transiently expressed in N. benthamiana leaves (Kleemann et al. 2012; Yoshino et al. 2012).
Another conserved secreted effector is the Nudix hydrolase previously identified as an induced gene during the transition to necrotrophy in the cowpea anthracnose pathogen C. truncatum (Bhadauria et al. 2013). Overexpression of CtNudix in C. truncatum induced localized host cell death and loss of pathogenicity. Localization studies in N. benthamiana indicated that the protein is located in the plant plasma membrane, suggesting that it might alter integrity of host cells by affecting stability of the host plasma membrane. Homologs of the Nudix effector are present in other hemibiotrophic pathogens including M. oryzae, and P. infestans, but absent in biotrophic and necrotrophic pathogens, suggesting that it might be important specifically for this lifestyle.
Compared with other sequenced ascomycetes, all four Colletotrichum genomes contain an expanded family of genes encoding proteins containing CBM50 carbohydrate binding modules, also known as LysM motifs. These genes appear to be highly divergent among the species and thus to be evolving rapidly. They may act as chitin-binding lectins and serve to “mask” the biotrophic hyphae from host recognition by binding to the fungal wall chitin (de Jonge and Thomma 2009). In C. lindemuthianum, a LysM protein called CIH1 was localized to the surface of biotrophic hyphae using a monoclonal antibody (Pain et al. 1994; Perfect et al. 1998). All four sequenced species have homologs of CIH1.
Biotrophic plant pathogens are known to produce large numbers of effector candidates, in the form of small secreted proteins (SSP) that act to establish a compatible interaction with the host by suppressing host defenses and reprogramming host cells to accommodate the pathogen (Göhre and Robatzek 2008). These SSP effectors are typically less than 300 amino acids, cysteine-rich, and lineage-specific (Stergiopoulos and de Wit 2009). All four Colletotrichum genome annotations include many SSP effector candidates, including a large number that are cysteine-rich and/or unique to each species (Table 3.3). Interestingly, numerous additional candidate effectors were identified after deep 454 pyrosequencing of the in planta transcriptome of C. higginsianum (Kleeman et al. 2012). It was observed that only about a quarter of these transcripts had been annotated in the initial genome-based analysis, suggesting that the annotated effectors in the four Colletotrichum genomes may represent only the tip of the iceberg.
Overall, the genome sequences of the hemibiotrophic Colletotrichum fungi are more similar to the genomes of necrotrophic fungi rather than biotrophs, having expanded families of secondary metabolites and CAZymes. Indeed, Colletotrichum fungi may have some of the largest and most diverse repertoires of lytic enzymes and secondary metabolites yet found among the pathogenic fungi. In common with necrotrophs, Colletotrichum also encode secreted toxin effectors associated with the induction of cell death. At the same time, Colletotrichum also encode large and diverse repertoires of putative small, lineage-specific secreted effectors, a hallmark of biotrophic fungal genomes, that may have a similar function, to manipulate host defenses and induce compatibility. We can speculate that this combination of gene arsenals reflects the “schizophrenic” hemibiotrophic existence of Colletotrichum, in which they must function almost as two distinct organisms at different stages of their lifecycles.
3.7.2 Colletotrichum Transcriptomics During Biotrophy and Necrotrophy
The availability of whole genome sequences enabled genome-wide analysis of the Colletotrichum transcriptome at different stages of hemibiotrophic infection. Deep Illumina RNA sequencing was performed for C. higginsianum and C. graminicola at three stages of development in planta: prepenetration (appressoria); biotrophic hyphae; and necrotrophic hyphae (O’Connell et al. 2012). For C. orbiculare, whole genome microarrays were produced based on the annotated genome assembly, and were used to investigate gene expression during prepenetration, biotrophic, and necrotrophic growth phases (Gan et al. 2013). In earlier studies, C. graminicola biotrophic hyphae were isolated by laser-capture microscopy (LCM) and analyzed using microarrays designed from a limited gene set based on the preliminary 2X shotgun sequence of strain M5.001 produced by DuPont (Tang et al. 2006). C. higginsianum appressoria formed on artificial surfaces, and primary hyphae isolated from the host tissues by fluorescence-activated cell sorting (FACS), were also analyzed by sequencing expressed sequence tags (ESTs) , and by Roche 454 sequencing (Kleemann et al. 2008; Kleemann et al. 2012; O’Connell et al. 2012; Takahara et al. 2009).
In interpreting and comparing these various Colletotrichum transcriptome datasets, it is important to recall that in C. higginsianum, only the first invaded cell contains biotrophic hyphae, followed by a complete switch to necrotrophy. In C. orbiculare and C. graminicola, the necrotrophic switch is delayed until several cells have been colonized. Thus, the biotrophic phase in these two species consists of a heterogeneous cell population that includes hyphal tip cells advancing into living host cells and intercalary fungal cells occupying dead or dying host cells. Moreover, in C. graminicola, the necrotrophic phase is also heterogeneous, composed of necrotrophic colony centers and biotrophic colony margins. This variation in the timing and extent of host cell death caused by Colletotrichum species is likely to be reflected in the representation of biotrophy- and necrotrophy-related genes in their transcriptomes.
A common theme that has emerged from studies comparing transcription in vitro to transcription in vivo is that a large number of genes in Colletotrichum are plant-induced (Gan et al. 2013; Kleemann et al. 2012; O’Connell et al. 2012; Tang et al. 2006). For example, comparison of gene expression in morphologically identical C. higginsianum appressoria produced in vitro versus in planta revealed that more than 1,500 genes were significantly induced in planta compared with their expression in vitro (O’Connell et al. 2012). Many of these induced genes encoded secreted proteins , including SSP and putative effectors , and many others encoded SM enzymes. Gene Ontology (GO) categories that were over-represented in planta included those involved in carbohydrate binding and degradation, protein degradation, and transmembrane transport (Gan et al. 2013). In C. orbiculare, most of the genes that were upregulated in planta were located in GC-rich, rather than AT-rich, regions (Gan et al. 2013). It is clear that Colletotrichum is able to detect and respond to plant signals, although the nature of these signals remains mysterious.
The dome-shaped melanized appressoria of Colletotrichum function in penetration of the host cuticle and epidermal cell wall (Fig. 3.2). Cutinases were over-represented in prepenetration appressoria in comparison with later phases of development, as might be expected (O’Connell et al. 2012). Genes involved in cAMP signaling were upregulated in the preinvasion stage of C. orbiculare, in agreement with previous reports showing that this is an important signaling pathway in the regulation of germination and appressorium formation (Yang and Dickman 1997, 1999a, b; Yamauchi et al. 2004; Gan et al. 2013). Melanin deposited in the appressorial wall allows accumulation of glycerol and high turgor pressures that facilitate mechanical rupture of the host cell wall (Bechinger et al. 1999; Bastmeyer et al. 2002). Transcriptome analysis of appressorial stages of C. higginsianum, C. graminicola, and C. orbiculare confirmed increased expression of genes of the PKS SM cluster involved in melanin production in all three species (Gan et al. 2013; O’Connell et al. 2012).
A large number of genes encoding putative secreted effector proteins are also expressed in unpenetrated Colletotrichum appressoria (Kleemann et al. 2008; 2012; O’Connell et al. 2012; Gan et al. 2013). In C. graminicola, homologs of the M. oryzae BAS2 and BAS3 effectors were among the most highly expressed genes in appressoria (O’Connell et al. 2012). Lineage-specific SSP effectors were particularly enriched during the early stages of development (appressorial and biotrophic) versus necrotrophy, suggesting they might play a role in establishment of a compatible interaction (Gan et al. 2013; Kleeman et al. 2012; O’Connell et al. 2012). In C. orbiculare, 28 of the 100 most highly upregulated genes at the appressorial stage were SSPs (Gan et al. 2013). C. higginsianum appressoria were shown to deliver candidate effectors by targeted secretion through pores at the host-appressorial interface (Kleemann et al. 2012). Numerous SM gene clusters were also expressed during prepenetration and early penetration stages in the appressoria of C. graminicola, C. higginsianum, and C. orbiculare (Gan et al. 2013; O’Connell et al. 2012). These findings suggest that in addition to mechanical breaching of the host cell wall, appressoria play an important role in the secretion of protein and small molecule effectors, which may prepare the infection court for subsequent invasion.
Analysis of genes expressed during biotrophy in the three Colletotrichum species identified hundreds of differentially regulated genes, including more than 300 upregulated genes in C. graminicola, and more than 700 upregulated genes in C. higginsianum. Although the data were generally consistent, shifts in gene expression were not as pronounced in either C. graminicola or C. orbiculare as they were in C. higginsianum, and this is likely because of the more synchronous development of the latter species. A previous LCM-enabled study of C. graminicola focused on analysis of biotrophic hyphae, but unfortunately did not provide a full account of the genes that were upregulated in those cells (Tang et al. 2006). Thus analyses of the C. higginsianum biotrophic phase, including cells colonizing living host cells, and also biotrophic hyphae isolated by the FACS technique, are likely to be the most informative on the transcriptional status of Colletotrichum biotrophic hyphae (Takahara et al. 2009; Kleemann et al. 2012; O’Connell et al. 2012).
As observed in appressoria , many of the genes expressed in C. higginsianum biotrophic hyphae encoded secreted effectors and SM enzymes (Kleemann et al. 2012; O’Connell et al. 2012). Several LysM protein genes were expressed specifically in biotrophic primary hyphae, suggesting a role in “masking” of the hyphal wall from host detection (O’Connell et al. 2012). Relatively few genes encoding lytic enzymes were expressed, and in this respect the biotrophic hyphae resemble the haustoria of obligate biotrophs (O’Connell et al. 2012). However, unlike haustoria, there was no specific induction of nutrient uptake transporters at this stage, which would be expected if the biotrophic hyphae of C. higginsianum function primarily as organs for nutrient uptake (O’Connell et al. 2012). Consistent with this view, recent evidence indicates that carbohydrate supply by the host is dispensable for the biotrophic growth of C. higginsianum (Engelsdorf et al. 2013). Thus, the primary role of biotrophic hyphae seems to be as organs for the secretion of effectors and SM that presumably modulate host responses and suppress host cell death. The SM enzymes and effectors of biotrophic hyphae differ from those expressed in appressoria, with distinct “waves” of these fungal modifiers produced over the course of pathogenic development (Gan et al. 2013; Kleemann et al. 2012; O’Connell et al. 2012).
The switch from biotrophy to necrotrophy is marked by the production of narrow secondary infection hyphae that are not separated from the host cell by a membrane (Fig. 3.4). Analysis of gene expression in all three species during necrotrophy revealed the induction of a large array of genes encoding secreted proteases and CAZymes, producing a cocktail of enzymes that is probably highly efficient for degrading plant cell walls (Gan et al. 2013; O’Connell et al. 2012). Between 23 and 25 % of the 100 most highly expressed genes upregulated by C. graminicola and C. higginsianum during the necrotrophic phase are CAZymes (O’Connell et al. 2012). However, each species apparently uses a different strategy to deconstruct host cell walls. More pectin-degrading enzymes (51) are induced during necrotrophy in C. higginsianum, while C. graminicola deploys more cellulases and hemicelluloses at this stage. An example is provided by the GH61 monooxygenases, which act in concert with classical cellulases to enhance lignocellulose hydrolysis (Beeson et al. 2012; Quinlan et al. 2011). Twenty-two of 28 GH61 monooxygenases were induced during C. graminicola necrotrophy, with 6 % of the most highly induced genes during necrotrophy belonging to this class (O’Connell et al. 2012). In contrast, only six out of 25 GH61 genes were expressed by C. higginsianum during necrotrophy, and none were highly induced (O’Connell et al. 2012).
Numerous nutrient uptake transporters are also induced at the switch to necrotrophy, suggesting that secondary hyphae provide the major organs for nutrient acquistion for Colletotrichum (Gan et al. 2013; O’Connell et al. 2012). In both C. higginsianum and C. orbiculare there was a general decrease in the number of SM gene clusters expressed during necrotrophy, but in C. graminicola a large percentage of the genes induced at this stage were SM genes. Notably, the SM genes expressed by C. graminicola during the necrotrophic phase differed from those expressed earlier in the interaction (Gan et al. 2013; O’Connell et al. 2012). Genes encoding secreted effectors, including putative necrosis-inducing proteins, were also induced at the necrotrophic stage of development in all three species (Gan et al. 2013; O’Connell et al. 2012).
3.7.3 How Can Genome Information Help Us to Better Manage and Exploit Colletotrichum?
We are now in the postgenomic era for Colletotrichum research. Numerous additional Colletotrichum genome-sequencing projects are underway as we write, and sequencing technology has advanced to the point that the genome or transcriptome of any strain of interest can be sequenced quickly and cheaply. An important consideration emerging from the comparative transcriptome analyses described here is the need to ensure that future analyses are performed at equivalent infection stages, and under conditions promoting synchronous development, if they are to be useful for a comparative analysis. For finer resolution of transcriptional differences, e.g., within intracellular primary hyphae occupying either living or dead host cells, it may be necessary to use single-cell sampling methods such as FACS or LCM (Kleemann et al. 2012; Takahara et al. 2009; Tang et al. 2006). Careful cytological studies and bioassay development are essential for each new species that is analyzed. Further considerations relate to aspects of bioinformatics and sequencing technologies. In particular, direct comparisons of genomes produced using different sequencing techniques and different assembly and annotation software must be made with extreme caution, and perceived differences must be confirmed by manual annotation before they can be fully accepted.
The genus Colletotrichum includes species displaying a broad spectrum of pathogenic lifestyles , providing many exciting opportunities for comparative genomics and transcriptomics to study the molecular and evolutionary basis of these lifestyles. Comparisons of species displaying “extreme” lifestyles, e.g., subcuticular necrotrophy or symptomless endophytism, should reveal how Colletotrichum fungi differ from the better-characterized hemibiotrophic species, e.g., in gene repertoires dedicated to host degradation (proteases, carbohydrate-active enzymes) or ‘stealth ’ (protein and secondary metabolite effectors), or in the timing with which those genes are deployed. Likewise, comparisons of species with contrasting lifestyles that infect the same host plant (endophytes vs. pathogens, necrotrophs versus hemibiotrophs, or species preferentially infecting different organs of the same host (shoots or roots) could be especially informative.
Several major conclusions can be drawn from the comparative genome analyses described herein, with implications for both applied and basic research disciplines. Remarkably, considering the extensive repertoire of pathogenicity-related genes encoded in the four sequenced Colletotrichum genomes and the patterns with which those ‘weapons’ are deployed during infection, it is clear that there is far more held in common by these phylogenetically diverse species than there is unique. Thus, one can anticipate that conserved components and mechanisms will be discovered that could provide potential targets for controlling many Colletotrichum diseases through chemical intervention or plant breeding. Strong evidence for host specialization was also revealed through genome-scale studies of Colletotrichum, including the apparent adaptation of the CAZyme repertoire and its expression to particular host cell wall composition, and the large degree of diversity in SM and secreted protein effectors. Many Colletotrichum effectors are species-specific , while others are shared within the genus, or even with other fungal genera, which may indicate conserved functions or host targets. The priority now will be to identify targets of both conserved and lineage-specific effectors, and to determine the mechanisms by which effectors manipulate host cells to induce compatibility.
Anthracnose diseases of field crops are generally managed by the use of resistant cultivars . But resistance failures occur frequently, due to the ability of pathogen populations to adapt rapidly to traditional R-gene-mediated resistance strategies. Ultimately, these boom-and-bust cycles lead to increased reliance on fungicides (Parlevliet 2002; Thakur 2007). In particular, management of postharvest diseases caused by Colletotrichum requires the use of expensive and toxic chemicals (Prusky 1996). Further genome/transcriptome-based insight into the conserved toolbox employed by members of the genus Colletotrichum could prove instrumental for the design of durable strategies for disease control through resistance breeding. For example, alternate breeding strategies, including the use of mutant susceptibility (S) genes identified using pathogen effectors as molecular probes, show promise in many pathosystems (Gawehns et al. 2013). S genes encode host proteins that are co-opted by plant pathogens, resulting in pathogen proliferation and ultimately leading to diseased host tissue. S gene inactivation reduces the pathogens’ ability to cause disease, providing a durable form of resistance (Pavan et al. 2010). In some cases, S gene-based immunity has provided broad spectrum resistance against pathogens for several decades (Gawehns et al. 2013). Although S genes provide highly effective sources of resistance, few have been identified to date and even fewer are commercially viable. But with the power of genomics, the discovery of new candidates may be accelerated through Colletotrichum effector-target screens.
3.7.4 The Genomics of Hemibiotrophy
Some of the most important results gleaned from genomics-enabled research of Colletotrichum are those that provide insight into the hemibiotrophic lifestyle , and the parallels between distinct life stages and the infection strategies of obligately biotrophic or necrotrophic plant pathogens.
The primary hyphae of pathogenic IH Colletotrichum are in some ways analogous to the haustoria of obligate intracellular biotrophs (Mendgen and Hahn 2002; O’Connell and Panstruga 2006; Perfect et al. 1999, 2001). These structures share certain morphological similarities, including the presence of a membrane that differs in composition from the normal plant plasma membrane, and serves to separate the fungus from the living host cell (Shimada et al. 2006). Obligate biotrophs cannot be cultured away from their plant hosts, and cannot be easily genetically manipulated, whereas hemibiotrophs are readily cultured and manipulated. This led to the idea that it might be possible to identify pathogen components required for biotrophic growth by studying the more experimentally tractable hemibiotrophs (Mendgen and Hahn 2002). However, genome analysis has suggested that the appressoria and biotrophic hyphae of Colletotrichum function primarily as organs for the synthesis and secretion of protein and secondary metabolite effectors to host cells, and not as organs of nutrient uptake like true haustoria.
Transcriptome analysis revealed that Colletotrichum fungi are highly responsive to unknown plant signals, and that gene expression can differ remarkably between morphologically similar structures formed in vitro and in planta. Understanding the role of plant signals in transcriptional reprogramming and the mechanisms by which those signals are sensed and transduced by the fungus could lead to new opportunities to alter those signals by manipulation of the plant genome or to interfere with their perception by the pathogen. Transcriptomics also demonstrated that massive shifts in gene expression underlie the developmental transitions that occur in planta, from spore germination to necrotrophy. Thus, the lifestyle switch to necrotrophy is characterized by a massive shift in fungal gene expression, with the activation of large numbers of genes encoding lytic enzymes and membrane transporters . It will be crucial to understand the signals that trigger this switch and the transcriptional/epigenetic regulators and signaling pathways that underlie it. It is possible that these differ between hemibiotrophic pathosystems, but comparative transcriptome analyses focused on the key transitional phase will help clarify this.
3.7.5 Genomics Applied to Elucidating the Systematics of Colletotrichum
Concurrent with genomics-enabled research of Colletotrichum pathology, the genus is undergoing a taxonomic renaissance, enabled by the application of molecular phylogenetic approaches (Crouch et al. 2006, 2009a, b, c; Damm et al. 2009, 2012a, b; Hyde et al. 2009; Rojas et al. 2010; Weir et al. 2012). Continued synergy between genomics, transcriptomics and molecular phylogenetic research is beginning to provide us with a long-awaited glimpse into the forces impacting the evolution of Colletotrichum species. Importantly, molecular phylogeny-based diagnosis of species boundaries will enable more accurate predictions about genome evolution, mechanisms of host adaptation, the evolution of key pathogenicity traits and other topics. One illustration of this point involves the host range of Colletotrichum species. Molecular phylogenetic studies are confirming long-held suspicions that twentieth century species concepts and host range assumptions made using classical morphology are overly broad and often inaccurate (Sutton 1980; Cannon et al. 2013). Increasingly well-resolved species diagnoses are now incorporated across the entire genus (Cannon et al. 2013). For instance, until recently, C. graminicola was recognized as a broad host range generalist pathogen of nearly every grass and cereal in the Poaceae family, despite considerable evidence of physiological specialization and distinctive appressorial structures (Sutton 1968; Sherriff et al. 1995; Hsiang and Goodwin 2001; Du et al. 2005; Crouch et al. 2006). Molecular data showed this broad circumscription as false, with C. graminicola limited to maize, while more than sixteen distinct Colletotrichum species are now described as pathogens and endophytes of the Poaceae (Crouch et al. 2006, 2009a, b; Crouch and Tomaso-Peterson 2012; Crouch 2013). Similar resolution has resulted from the study of C. gloeosporioides, C. acutatum and several other taxa (Damm et al. 2009, 2012a, b; Hyde et al. 2009; Weir et al. 2012). With increasingly precise demarcations of species boundaries, and the insight into host association that such data provides, accurate biological, epidemiological, and mechanistic interpretation of genome and transcriptome data become possible.
A very broad pattern of host association is evident across the Colletotrichum phylogeny, with older, basal lineages uniquely associated with dicots and non-graminicolous monocots (Fig. 3.1). Colletotrichum pathogenic to grasses form a cohesive, monophyletic group, the graminicola aggregate, originating from ancestral lines of non-graminicolous Colletotrichum. Thus, the pathogenic association of Colletotrichum with grass hosts appears to be a derived trait, of relatively recent origin. Taken together with data from genome and transcriptome analyses, this evolutionary trajectory suggests that the expanded gene cohorts held in common by dicot infecting Colletotrichum, particularly genes encoding pectinases, most closely reflect the ancestral state for the genus, and the reduced cohort characterized from C. graminicola is potentially due to the loss of these genes as they became unnecessary. Additional research is needed to investigate these possibilities.
Phylogenomic analysis and divergence dating from a sample of C. graminicola, C. higginsianum and seventeen other fungi estimated a recent divergence between these two species, just 47 million years ago (O’Connell et al. 2012). Notably, the divergence between C. graminicola and C. higginsianum occurred approximately 100 million years after the divergence between their respective host groups, monocots and dicots (Chaw et al. 2004). As such, it is unlikely that divergence in this part of the Colletotrichum phylogeny was temporally associated with host evolution. However, the divergence of species within the graminicola aggregate may have involved a coevolutionary process with the Poaceae family. The most basal species in the graminicola aggregate, C. cereale, is uniquely associated with cool-season (C3 physiology) Pooideae grasses such as wheat, oats, barley, bluegrass, etc. (Fig. 3.1). C. cereale is the progenitor of numerous Colletotrichum species adapted to warm-season (C4 physiology) cereals and grasses in the Panicoideae subfamily, including maize, sorghum and sugarcane. The divergence between wheat (Pooideae; C3) and maize (Panicoideae; C4) is estimated at 50–60 million years ago (Chaw et al. 2004), generally compatible with the estimated timing of the graminicola/higginsianum divergence, particularly given the margin for error associated with calibration of divergence times (O’Connell et al. 2012). This temporal correspondence suggests that speciation and host adaptation in the graminicola aggregate may have mirrored host diversification, or may even have been driven by the evolutionary radiation of the Poaceae family. Genome and transcriptome assessments of additional Colletotrichum species that are either members of, or closely related to, the graminicola and destructivum aggregates, especially those associated with non-graminicolous monocots or members of the Pooideae-infecting C. cereale, could be particularly informative.
3.7.6 Genomics Tools for Studying the Population Genetics of Colletotrichum
Phylogenetic and population genetic investigations are benefiting from the increased availability of genome-scale datasets. One of the major obstacles facing phylogenetic and population researchers is the identification of appropriate and informative molecular markers to gauge diversity, relationships, and evolutionary traits. Standard methodology uses “borrowed” markers: a handful of conserved loci that are used primarily because primers exist that are capable of amplifying a broad range of organisms with relative ease (e.g., Carbone and Kohn 1999; White et al. 1990). These markers have been in common use for almost two decades in some cases. However it is unknown whether such strategies yield a biased view of evolutionary relationships or gene-specific noise. There is an increasing body of work that shows many of these borrowed markers lack the power to fully resolve distinct organisms or provide incongruous results (Aguileta et al. 2008; Rokas et al. 2003; Townsend 2007; Townsend and Lopez-Giraldez 2010). To overcome these biases, researchers are increasingly developing datasets from larger numbers of orthologous genes, targeted genes or even whole genomes (e.g., Aguileta et al. 2008; Du et al. 2005; Crouch et al. 2009a, b, c; Fitzpatrick et al. 2006; Rokas et al. 2003; Wang et al. 2009). Comparative genome analysis of 52 Colletotrichum isolates from the graminicola and acutatum aggregates using Illumina sequenced restriction-associated DNA tagged (RAD-Seq) SNP datasets demonstrated that a subset of commonly used Sanger-sequenced PCR amplicon-derived single locus molecular markers—Apn2 and Sod2—individually provided reliable identification of Colletotrichum species comparable to a 1,723 locus genome-wide dataset (Crouch et al. 2013). These findings led to the development of real-time PCR diagnostic assay based on the Apn2 marker capable of population-specific detection of C. cereale from infected host tissue, and herbarium specimens up to 100 years old (Beirn et al. 2013). Similar development and application of genome data for the identification of other economically important Colletotrichum could provide pathologists with tools that help mitigate losses due to disease.
3.7.7 Genomics and the Commercial Exploitation of Colletotrichum
Colletotrichum have long been utilized for biotechnology applications , with many species yielding a diversity of compounds and secondary metabolites with commercially valuable biological activity (García-Pajón and Collado 2003). Recent notable applications include the purification of large quantities of the alkaloid compound huperzine A used for treatment of Alzheimer’s disease from C. gloeosporioides (Zhao et al. 2013), the use of lipid-accumulating Colletotrichum strains as a novel source of biodiesel feedstocks (Dey et al. 2011), and the purification of novel antimicrobial metabolites from C. gloeosporioides with effective antibiotic activity against multidrug resistant Staphylococcus aureus (Arivudainambi et al. 2011). The potential for identification of novel SM, including those with antimicrobial activity, from Colletotrichum appears virtually untapped. Colletotrichum may also provide a valuable source of novel enzymes for plant biomass transformation, notably the production of ‘second generation’ biofuels from lignocellulosic biomass such as residual crop waste or woody crops (Simmons et al. 2008). Genome analysis has revealed that Colletotrichum species possess extraordinarily large and complex repertoires of enzymes for lignocellulose degradation and that they tailor these enzymes for particular host substrates. There is thus significant potential for commercial exploitation of these enzyme systems to improve the efficiency of production of lignocellulose-derived biofuels. Transcriptomic information on which enzymes are co-regulated may suggest combinations of enzymes that act synergistically for efficient lignocellulose transformation.
References
Abang MM, Asiedu R, Hoffmann P, Wolf GA, Mignouna HD, Winter S (2006) Pathogenic and genetic variability among Colletotrichum gloeosporioides isolates from different yam hosts in the agroecological zones in Nigeria. J Phytopathol 154(1):51–61
Adegbite AA, Amusa NA (2008). The major economic field diseases of cowpea in the humid agro-ecologies of South-western Nigeria. Afr J Biotechnol. 7(25):4706–4712
Aguileta G, Hood ME, Refrégier G, Giraud T (2009) Genome evolution in plant pathogenic and symbiotic fungi. Adv Bot Res 49:151–193
Aguileta G, Marthey S, Chiapello H, Lebrun M, Rodolphe F, Fournier E, Gendrault-Jacquemard A, Giraud T (2008) Assessing the performance of single-copy genes for recovering robust phylogenies. Syst Biol 57:613–627
Akagi Y, Akamatsu H, Otani H, Kodama M (2009) Horizontal chromosome transfer, a mechanism for the evolution and differentiation of a plant-pathogenic fungus. Eukaryot Cell 8(11):1732–1738
Ammar MI, El-Naggar MA (2011) Date palm (Phoenix dactylifera L.) fungal diseases in Najran, Saudi Arabia. Int J Plant Pathol 2(3):126–135
Amselem J, Cuomo CA, van Kan JA, Viaud M, Benito EP, Couloux A, Levis C (2011) Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet 7(8):e1002230
Amyotte SG, Tan X, Pennerman K, Jimenez-Gasco M, Klosterman SJ, Ma LJ, Dobinson KF, Veronese P (2012) Transposable elements in phytopathogenic Verticillium spp.: insights into genome evolution and inter- and intra-specific diversification. BMC Genom 13:314
Arivudainambi US, Anand TD, Shanmugaiah V, Karunakaran C, Rajendran A (2011) Novel bioactive metabolites producing endophytic fungus Colletotrichum gloeosporioides against multidrug-resistant Staphylococcus aureus. FEMS Immunol Med Microbiol 61(3):340–345
Arnold AE, Mejía LC, Kyllo D, Rojas EI, Maynard Z, Robbins N, Herre EA (2003) Fungal endophytes limit pathogen damage in a tropical tree. Proc Natl Acad Sci 100(26):15649–15654
Barrus MF (1911) Variation of varieties of beans in their susceptibility to anthracnose. Phytopathology 1(6):190–195
Bastmeyer M, Deising HB, Bechinger C (2002) Force exertion in fungal infection. Annu Rev Biophys Biomol Struct 31(1):321–341
Bechinger C, Giebel KF, Schnell M, Leiderer P, Deising HB, Bastmeyer M (1999) Optical measurements of invasive forces exerted by appressoria of a plant pathogenic fungus. Science 285(5435):1896–1899
Beeson WT, Phillips CM, Cate JH, Marletta MA (2012) Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J Am Chem Soc 134(2):890–892
Beirn LA, Clarke BB, Crouch JA (2013) Influence of host and geographic locale on the distribution of Colletotrichum cereale lineages. PLoS ONE 9(5):e97706. doi: 10.1371/journal.pone.0097706
Bergstrom GC, Nicholson RL (1999) The biology of corn anthracnose: knowledge to exploit for improved management. Plant Dis 83(7):596–608
Bhadauria V, Banniza S, Vandenberg A, Selvaraj G, Wei Y (2013) Overexpression of a novel biotrophy-specific Colletotrichum truncatum effector, CtNUDIX, in hemibiotrophic fungal phytopathogens causes incompatibility with their host plants. Eukaryot Cell 12(1):2–11
Birker D, Heidrich K, Takahara H, Narusaka M, Deslandes L, Narusaka Y, O’Connell. R (2009) A locus conferring resistance to Colletotrichum higginsianum is shared by four geographically distinct Arabidopsis accessions. Plant J 60(4):602–613
Böhnert HU, Fudal I, Dioh W, Tharreau D, Notteghem JL, Lebrun MH (2004) A putative polyketide synthase/peptide synthetase from Magnaporthe grisea signals pathogen attack to resistant rice. The Plant Cell Online 16(9):2499–2513
Bölker M, Basse CW, Schirawski J (2008) Ustilago maydis secondary metabolism—from genomics to biochemistry. Fungal Genet Biol 45:S88–S93
Boyette CD, Jackson MA, Bryson CT, Hoagland RE, Connick WJ Jr, Daigle DJ (2007) Sesbania exaltata biocontrol with Colletotrichum truncatum microsclerotia formulated in ‘Pesta’granules. Biocontrol 52(3):413–426
Cambareri EB, Jensen BC, Schabtach E, Selker EU (1989) Repeat-induced G-C to A–T mutations in Neurospora. Science 244:1571–1575
Cannon P, Buddie A, Bridge P, de Neergaard E, Lübeck M, Askar M (2012a) Lectera, a new genus of the Plectosphaerellaceae for the legume pathogen Volutella colletotrichoides. MycoKeys 3:23–36
Cannon PF, Damm U, Johnston PR, Weir BS (2012b) Colletotrichum–current status and future directions. Stud Mycol 73(1):181–213
Cannon PF, Simmons CM (2002) Diversity and host preference of leaf endophytic fungi in the Iwokrama Forest Reserve, Guyana. Mycologia 94(2):210–220
Cano J, Guarro J, Gené J (2004) Molecular and morphological identification of Colletotrichum species of clinical interest. J Clin Microbiol 42(6):2450–2454
Carbone I, Kohn LM (1999) A method for designing primer sets for speciation studies in filamentous ascomyctes. Mycologia 91:553–556
Chaw SM, Chang CC, Chen HL, Li WH (2004) Dating the monocot-dicot divergence and the origin of core Eudicots using whole chloroplast genomes. J Mol Evol 58:424–441
Chen C, Dickman MB (2002) Colletotrichum trifolii TB3 kinase, a COT1 homolog, is light inducible and becomes localized in the nucleus during hyphal elongation. Eukaryot Cell 1(4):626–633
Chen C, Dickman MB (2004) Dominant active Rac and dominant negative Rac revert the dominant active Ras phenotype in Colletotrichum trifolii by distinct signalling pathways. Mol Microbiol 51(5):1493–1507
Chilton SJP, Lucas GB, Edgerton CW (1945) Genetics of Glomerella. III. Crosses with a conidial strain. Am J Bot 32:549–554
Chilton SJP, Wheeler HE (1949a) Genetics of Glomerella. VI. Linkage. Am J Bot 36:270–273
Chilton SJP, Wheeler HE (1949b) Genetics of Glomerella. VII. Mutation and segregation in plus cultures. Am J Bot 36:717–721
Choi J, Park J, Kim D, Jung K, Kang S, Lee YH (2010) Fungal secretome database: integrated platform for annotation of fungal secretomes. BMC Genom 11(1):105
Chona BL (1980) Red rot of sugarcane and sugar industry—a review. Indian Phytopathol 33(2):191–207
Chongo G, Gossen BD, Bernier CC (2002) Infection by Colletotrichum truncatum in resistant and susceptible lentil genotypes. Can J Plant Pathol 24(1):81–85
Chuma I, Tosa Y, Taga M, Nakayashiki H, Mayama S (2003) Meiotic behavior of a supernumerary chromosome in Magnaporthe oryzae. Curr Genet 43(3):191–198
Chung KR, Shilts T, Ertürk Ü, Timmer LW, Ueng PP (2003) Indole derivatives produced by the fungus Colletotrichum acutatum causing lime anthracnose and postbloom fruit drop of citrus. FEMS Microbiol Lett 226(1):23–30
Clutterbuck AJ (2011) Genomic evidence of repeat-induced point mutation in filamentous ascomycetes. Fungal Genet Biol 48:306–326
Coleman JJ, Rounsley SD, Rodriguez-Carres M, Kuo A, Wasmann CC, Grimwood J, VanEtten HD (2009) The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet 5(8):e1000618
Collemare J, Pianfetti M, Houlle AE, Morin D, Camborde L, Gagey MJ, Böhnert HU (2008) Magnaporthe grisea avirulence gene ACE1 belongs to an infection specific gene cluster involved in secondary metabolism. New Phytol 179(1):196–208
Cortese LM, Bonos SA (2012) Bioenergy traits of ten switchgrass populations grown in the Northeastern/Mid-Atlantic USA. Bioenergy Res 6:580–590
Covert SF (1998) Supernumerary chromosomes in filamentous fungi. Curr Genet 33(5):311–319
Crouch JA (2013) Colletotrichum caudatum is a species complex. IMA Fungus (in press)
Crouch JA, Beirn LA, Cortese LM, Bonos SA, Clarke BB (2009a) Anthracnose disease of switchgrass caused by the novel fungal species Colletotrichum navitas. Mycol Res 113(12):1411–1421
Crouch JA, Beirn LA, Ismaiel A, Oudemans PV, Clarke BB, Polashock JJ (2013) Phylogenomic RAD-Seq analysis of plant pathogenic Colletotrichum fungi. BMC Genomics (in review)
Crouch JA, Beirn LA (2009) Anthracnose of cereals and grasses. Fungal Divers 39:19
Crouch JA, Clarke BB, Hillman BI (2006) Unraveling evolutionary relationships among the divergent lineages of Colletotrichum causing anthracnose disease in turfgrass and corn. Phytopathology 96(1):46–60
Crouch JA, Clarke BB, White JF, Hillman BI (2009b) Systematic analysis of the falcate-spored graminicolous Colletotrichum and a description of six new species from warm-season grasses. Mycologia 101(5):717–732
Crouch JA, Glasheen BM, Giunta MA, Clarke BB, Hillman BI (2008) The evolution of transposon repeat-induced point mutation in the genome of Colletotrichum cereale: Reconciling sex, recombination and homoplasy in an ‘‘asexual” pathogen. Fungal Genet Biol 45(3):190–206
Crouch JA, Tomaso-Peterson M (2012) Anthracnose disease of centipedegrass turf caused by Colletotrichum eremochloa, a new fungal species closely related to Colletotrichum sublineola. Mycologia 104(5):1085–1096
Crouch JA, Tredway LP, Clarke BB, Hillman BI (2009c) Phylogenetic and population genetic divergence correspond with habitat for the pathogen Colletotrichum cereale and allied taxa across diverse grass communities. Mol Ecol 18(1):123–135
Cuomo CA, Birren BW (2010) The fungal genome initiative and lessons learned from genome sequencing. Methods Enzymol 470:833–855
Cuomo CA, Güldener U, Xu JR, Trail F, Turgeon BG, Di Pietro A, Kistler HC (2007) The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317(5843):1400–1402
Dahlberg J, Berenji J, Sikora V, Latkovic D (2011) Assessing sorghum (Sorghum bicolor (L) Moench) germplasm for new traits: food, fuels & unique uses. Maydica 56(1750):85–92
Damm U, Cannon PF, Woudenberg JHC, Crous PW (2012a) The Colletotrichum acutatum species complex. Stud Mycol 73(1):37–113
Damm U, Cannon PF, Woudenberg JHC, Johnston PR, Weir BS, Tan YP, Crous PW (2012b) The Colletotrichum boninense species complex. Stud Mycol 73(1):1–36
Damm U, Woudenberg JHC, Cannon PF, Crous PW (2009) Colletotrichum species with curved conidia from herbaceous hosts. Fungal Divers 39:45
Daub ME (1982) Cercosporin, a photosensitizing toxin from Cercospora species. Phytopathology 72(4):370–374
Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ, Birren BW (2005) The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434(7036):980–986
Dean R, Van Kan JA, Pretorius ZA, Hammond Kosack KE, Di Pietro A, Spanu PD, Foster GD (2012) The top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol 13(4):414–430
de Jonge R, Thomma BP (2009) Fungal LysM effectors: extinguishers of host immunity? Trends Microbiol 17(4):151–157
Dey P, Banerjee J, Maiti MK (2011) Comparative lipid profiling of two endophytic fungal isolates—Colletotrichum sp. and Alternaria sp. having potential utilities as biodiesel feedstock. Bioresour Technol 102:5815–5823
Dickman MB, Yarden O (1999) Serine/threonine protein kinases and phosphatases in filamentious fungi. Fungal Genet Biol 26(2):99–117
Dillard HR, Cobb AC (1998) Survival of Colletotrichum coccodes in infected tomato tissue and in soil. Plant Dis 82(2):235–238
Dingley JM, Gilmour JW (1972) Colletotrichum acutatum Simmds. f. sp. pinea associated with “terminal crook” disease of Pinus spp. NZ J Forest Sci 2(2):192–201
Driver CH, Wheeler HE (1955) A sexual hormone in Glomerella. Mycologia 47(3):311–316
Du Y, Chu H, Wang M, Chu IK, Lo C (2010) Identification of flavone phytoalexins and a pathogen-inducible flavone synthase II gene (SbFNSII) in sorghum. J Exp Bot 61(4):983–994
Du M, Schardl CL, Nuckles EM, Vaillancourt LJ (2005) Using mating-type gene sequences for improved phylogenetic resolution of Colletotrichum species complexes. Mycologia 97(3):641–658
Edgerton CW, Chilton SJP, Lucas GB (1945) Genetics of Glomerella. II. Fertilization between strains. Am J Bot 32:115–118
Engelsdorf T, Horst RJ, Pröls R, Pröschel M, Dietz F, Hückelhoven R, Voll LM (2013) Reduced carbohydrate availability enhances susceptibility of Arabidopsis towards Colletotrichum higginsianum. Plant Physiol 162(1):225–38
Fellbrich G, Romanski A, Varet A, Blume B, Brunner F, Engelhardt S, Nürnberger T (2002) NPP1, a Phytophthora associated trigger of plant defense in parsley and Arabidopsis. Plant J 32(3):375–390
Femenía-Ríos M, García-Pajón CM, Hernández-Galán R, Macías-Sánchez AJ, Collado IG (2006) Synthesis and free radical scavenging activity of a novel metabolite from the fungus Colletotrichum gloeosporioides. Bioorg Med Chem Lett 16(22):5836–5839
Finlay AR, Brown AE (1993) The relative importance of Colletotrichum musae as a crown rot pathogen on Windward Island bananas. Plant Pathol 42(1):67–74
Fitzpatrick DA, Logue ME, Stajich JE, Butler G (2006) A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol Biol 6:99
Forgey WM, Blanco MH, Loegering WQ (1978) Differences in pathological capabilities and host specificity of Colletotrichum graminicola on Zea mays (maize). Plant Dis Report 62:573–576
Freeman S, Horowitz S, Sharon A (2001) Pathogenic and nonpathogenic lifestyles in Colletotrichum acutatum from strawberry and other plants. Phytopathology 91(10):986–992
Freeman S, Shalev Z, Katan J (2002) Survival in soil of Colletotrichum acutatum and C. gloeosporioides pathogenic on strawberry. Plant Dis 86(9):965–970
Frey TJ, Weldekidan T, Colbert T, Wolters PJCC, Hawk JA (2011) Fitness evaluation of a locus that confers resistance to Colletotrichum graminicola (Ces.) GW Wils. using near-isogenic maize hybrids. Crop Sci 51(4):1551–1563
Galagan JE, Selker EU (2004) RIP: the evolutionary cost of genome defense. Trends Genet 20(9):417–423
Gan P, Ikeda K, Irieda H, Narusaka M, O’Connell RJ, Narusaka Y, Shirasu K (2013) Comparative genomic and transcriptomic analyses reveal the hemibiotrophic stage shift of Colletotrichum fungi. New Phytol 197(4):1236–1249
García E, Alonso Á, Platas G, Sacristán S (2013) The endophytic mycobiota of Arabidopsis thaliana. Fungal Divers 60(1):1–19
García-Pajón CM, Collado IG (2003) Secondary metabolites isolated from Colletotrichum species. Nat Prod Rep 20(4):426–431
García-Serrano M, Laguna EA, Simpson J, Rodríguez-Guerra R (2008) Analysis of the MATI-2-1 gene of Colletotrichum lindemuthianum. Mycoscience 49(5):312--317
Gardner MW (1918) Anthracnose of cucurbits (No. 727). US Dept. of Agriculture
Gawehns F, Cornelissen BJC, Takken FLW (2013) The potential of effector-target genes in breeding for plant innate immunity. Microb Biotechnol 6:223–229
Gazis R, Rehner S, Chaverri P (2011) Species delimitation in fungal endophyte diversity studies and its implications in ecological and biogeographic inferences. Mol Ecol 20(14):3001–3013
Geffroy V, Sicard D, de Oliveira JC, Sévignac M, Cohen S, Gepts P, Dron M (1999) Identification of an ancestral resistance gene cluster involved in the coevolution process between Phaseolus vulgaris and its fungal pathogen Colletotrichum lindemuthianum. Mol Plant Microbe Interact 12(9):774–784
Gengenbach BG, Miller RJ, Koeppe DE, Arntzen CJ (1973) The effect of toxin from Helminthosporium maydis (race T) on isolated corn mitochondria: swelling. Can J Bot 51(11):2119–2125
Gijzen M, Nürnberger T (2006) Nep1-like proteins from plant pathogens: recruitment and diversification of the NPP1 domain across taxa. Phytochemistry 67(16):1800–1807
Goddard R, Hatton IK, Howard JA, MacMillan J, Gilmore CJ (1976) X-Ray crystal and molecular structure of acetylcolletotrichin (colletotrichin), a metabolite of Colletotrichum capsici. J Chem Soc, Chem Commun 11:408–408
Göhre V, Robatzek S (2008) Breaking the barriers: microbial effector molecules subvert plant immunity. Annu Rev Phytopathol 46:189–215
Green KR, Simons SA (1994) ‘Dead skin’on yams (Dioscorea alata) caused by Colletotrichum gloeosporioides. Plant Pathol 43(6):1062–1065
Ha YS, Memmott SD, Dickman MB (2003) Functional analysis of Ras in Colletotrichum trifolii. FEMS Microbiol Lett 226(2):315–321
Hartman JC, Nippert JB, Orozco RA, Springer CJ (2011) Potential ecological impacts of switchgrass (Panicum virgatum L.) biofuel cultivation in the Central Great Plains, USA. Biomass Bioenergy 35(8):3415–3421
Hatta R, Ito K, Hosaki Y, Tanaka T, Tanaka A, Yamamoto M, Tsuge T (2002) A conditionally dispensable chromosome controls host-specific pathogenicity in the fungal plant pathogen Alternaria alternata. Genetics 161(1):59–70
He C, Nourse JP, Irwin JAG, Manners JM, Kelemu S (1996) CgT1: a non-LTR retrotransposon with restricted distribution in the fungal phytopathogen Colletotrichum gloeosporioides. Mol Gen Genet MGG 252:320–331
Horbach R, Graf A, Weihmann F, Antelo L, Mathea S, Liermann JC, Deising HB (2009) Sfp-type 4′-phosphopantetheinyl transferase is indispensable for fungal pathogenicity. The Plant Cell Online 21(10):3379–3396
Horie H, Sugata S, Abe Z (1988) Studies on anthracnose of komatsuna, Brassica rapa. Bull Tokyo Metrop Agr Exp Sta 21:189–237
Howard CM, Albregts EE (1983) Black leaf spot phase of strawberry anthracnose caused by Colletotrichum gloeosporioides (= C. fragariae). Plant Dis 67(10):1144–1146
Howard RJ, Valent B (1996) Breaking and entering: host penetration by the fungal rice blast pathogen Magnaporthe grisea. Annu Rev Microbiol 50:491–512
Hsiang T, Goodwin PH (2001) Ribosomal DNA sequence comparisons of Colletotrichum graminicola from turfgrasses and other hosts. Eur J Plant Pathol 107(6):593–599
Hua-Van A, Daviere J-M, Kaper F, Langin T, Daboussi M-J (2000) Genome organization in Fusarium oxysporum: clusters of class II transposons. Curr Genet 37:339–347
Hyde KD, Cai L, Cannon PF, Crouch JA, Crous PW, Damm U, Zhang JZ (2009) Colletotrichum—names in current use. Fungal Divers 39:147
Jaramillo VDA, Vargas WA, Sukno SA, Thon MR (2013) Horizontal transfer of a subtilisin gene from plants into an ancestor of the plant pathogenic fungal genus Colletotrichum. PLoS ONE 8(3):e59078
Kämper J, Kahmann R, Bölker M, Ma LJ, Brefort T, Saville BJ, Li W (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444(7115):97–101
Kanneganti TD, Huitema E, Cakir C, Kamoun S (2006) Synergistic interactions of the plant cell death pathways induced by Phytophthora infestans Nep1-like protein PiNPP1.1 and INF1 elicitin. Mol Plant Microbe Interact 19(8):854–863
Kelkar YD, Ochman H (2012) Causes and consequences of genome expansion in fungi. Genome Biol Evol 4(1):13–23
Kim YK, Kawano T, Li D, Kolattukudy PE (2000) A mitogen-activated protein kinase kinase required for induction of cytokinesis and appressorium formation by host signals in the conidia of Colletotrichum gloeosporioides. The Plant Cell Online 12(8):1331–1343
Kimura Y, Gohbara M, Suzuki A (1977) Assignment of 13C-nmr spectrum and biosynthesis of colletotrichin. Tetrahedron Lett 18(52):4615–4618
Kimura Y, Gohbara M, Suzuki A (1978) The biosynthesis of colletotrichins isolated from Colletotrichum nicotianae. Tetrahedron Lett 19(34):3115–3118
King BC, Waxman KD, Nenni NV, Walker LP, Bergstrom GC, Gibson DM (2011) Arsenal of plant cell wall degrading enzymes reflects host preference among plant pathogenic fungi. Biotechnol Biofuels, 4(4):1--14
Kleemann J, Takahara H, Stüber K, O’Connell R (2008) Identification of soluble secreted proteins from appressoria of Colletotrichum higginsianum by analysis of expressed sequence tags. Microbiology 154(4):1204–1217
Kleemann J, Rincon-Rivera LJ, Takahara H, Neumann U, van Themaat EVL, van der Does HC, O’Connell RJ (2012) Sequential delivery of host-induced virulence effectors by appressoria and intracellular hyphae of the phytopathogen Colletotrichum higginsianum. PLoS Pathog 8(4):e1002643
Kubo, Y (2012) Appressorium function in Colletotrichum orbiculare and prospect for genome based analysis. In: Pérez-Martín J, Di Pietro A (eds) Morphogenesis and pathogenicity in fungi, vol.22. Springer, Heidelberg, pp 115–131
Kubo Y, Nakamura H, Kobayashi K, Okuno T, Furusawa I (1991) Cloning of a melanin biosynthetic gene essential for appressorial penetration of Colletotrichum lagenarium. Mol Plant Microbe Interact 4:440–445
Kubo Y, Furusawa I (1991) Melanin biosynthesis: a prerequisite for successful invasion of the plant host by appressoria of Colletotrichum and Pyricularia. In: Cole GT, Hoch HC (eds) The fungal spore and disease initiation in plants. Plenum Press, New York, pp 205–218
Kubo Y, Takano Y (2013) Dynamics of infection-related morphogenesis and pathogenesis in Colletotrichum orbiculare. J Gen Plant Pathol. doi:10.1007/s10327-013-0451-9
Kuc J (1972) Phytoalexins. Annu Rev Phytopathol 10:207–232
Latunde-Dada AO, Lucas JA (2007) Localized hemibiotrophy in Colletotrichum: cytological and molecular taxonomic similarities among C. destructivum, C. linicola and C. truncatum. Plant Pathol 56(3):437–447
Lees AK, Hilton AJ (2003) Black dot (Colletotrichum coccodes): an increasingly important disease of potato. Plant Pathol 52(1):3–12
Legard DE (2000) Colletotrichum diseases of strawberry in Florida. In: Prusky D, Freeman S, Dickman M (eds) Colletotrichum: host specificity, pathogy, and host-pathogen interaction. APS Press, St. Paul MN, pp 292–299
Leite B, Nicholson RL (1992) Mycosporine-alanine: a self-inhibitor of germination from the conidial mucilage of Colletotrichum graminicola. Exp Mycol 16(1):76–86
Liao CY, Chen MY, Chen YK, Kuo KC, Chung KR, Lee MH (2012) Formation of highly branched hyphae by Colletotrichum acutatum within the fruit cuticles of Capsicum spp. Plant Pathol 61(2):262–270
Lin CL, Huang JW (2002) The occurrence of cruciferous vegetable anthracnose in Taiwan and identification of the pathogen. Plant Pathol Bull 11:173–178
López CE, Acosta IF, Jara C, Pedraza F, Gaitán-Solís E, Gallego G, Tohme J (2003) Identifying resistance gene analogs associated with resistances to different pathogens in common bean. Phytopathology 93(1):88–95
Lowe RG, Howlett BJ (2012) Indifferent, affectionate, or deceitful: lifestyles and secretomes of fungi. PLoS Pathog 8(3):e1002515
Lubbe CM, Denman S, Cannon PF, Groenewald JE, Lamprecht SC, Crous PW (2004) Characterization of Colletotrichum species associated with diseases of Proteaceae. Mycologia 96(6):1268–1279
Lucas GB (1946) Genetics of Glomerella. IV. Nuclear phenomena in the ascus. Am J Bot 33:802–806
Lucas GB, Chilton SJP, Edgerton CW (1944) Genetics of Glomerella. I. Studies on the behavior of certain strains. Am J Bot 31:233–239
Ma LJ, Van Der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, Di Pietro A, Sain D (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464(7287):367–373
Manire CA, Rhinehart HL, Sutton DA, Thompson EH, Rinaldi MG, Buck JD, Jacobson E (2002) Disseminated mycotic infection caused by Colletotrichum acutatum in a Kemp’s Ridley sea turtle (Lepidochelys kempi). J Clin Microbiol 40:4273–4280
Marcelino J, Giordano R, Gouli S, Gouli V, Parker BL, Skinner M, Cesnik R (2008) Colletotrichum acutatum var. fioriniae (teleomorph: Glomerella acutata var. fioriniae var. nov.) infection of a scale insect. Mycologia 100(3):353–374
Marcelino JA, Gouli S, Parker BL, Skinner M, Schwarzberg L, Giordano R (2009) Host plant associations of an entomopathogenic variety of the fungus, Collectotrichum acutatum, recovered from the elongate hemlock scale, Fiorinia externa. J Insect Sci 9(25):1--11
Masel AM, He C, Poplawski AM, Irwin JA, Manners JM (1996) Molecular evidence for chromosome transfer between biotypes of Colletotrichum gloeosporioides. Mol Plant Microbe Interact 9(5):339–348
Matthews DE, Gregory P, Gracen VE (1979) Helminthosporium maydis race T toxin induces leakage of NAD + from T cytoplasm corn mitochondria. Plant Physiol 63(6):1149–1153
Maynard DN, Hopkins DL (1999) Watermelon fruit disorders. HortTechnol 9(2):155–161
Mehrabi R, Bahkali AH, Abd-Elsalam KA, Moslem M, Ben M’Barek S, Gohari AM, de Wit PJ (2011) Horizontal gene and chromosome transfer in plant pathogenic fungi affecting host range. FEMS Microbiol Rev 35(3):542–554
Melotto M, Balardin RS, Kelly JD (2000) Host-pathogen interaction and variability of Colletotrichum lindemuthianum. In: PruskyD, Freeman S, Dickman M (eds) Colletotrichum host specificity, pathology, host–pathogen interaction. APS press, St. Paul, MN, pp 346–361
Melotto M, Kelly JD (2001) Fine mapping of the Co-4 locus of common bean reveals a resistance gene candidate, COK-4, that encodes for a protein kinase. Theor Appl Genet 103(4):508–517
Mendgen K, Hahn M (2002) Plant infection and the establishment of fungal biotrophy. Trends Plant Sci 7(8):352–356
Mendgen K, Hahn M, Diesing H (1996) Morphogenesis and mechanisms of penetration by plant pathogenic fungi. Annu Rev Phytopathol 34:367–386
Mims CW, Vaillancourt LJ (2002) Ultrastructural characterization of infection and colonization of maize leaves by Colletotrichum graminicola, and by a C. graminicola pathogenicity mutant. Phytopathology 92(7):803–812
Moses E, Nash C, Strange RN, Bailey JA (1996) Colletotrichum gloeosporioides as the cause of stem tip dieback of cassava. Plant Pathol 45(5):864–871
Mosquera G, Giraldo MC, Khang CH, Coughlan S, Valent B (2009) Interaction transcriptome analysis identifies Magnaporthe oryzae BAS1-4 as biotrophy-associated secreted proteins in rice blast disease. The Plant Cell Online 21(4):1273–1290
Moura-Costa PH, Kandasamy KI, Mantell SH (1993) Evaluation of in vitro screening methods for assessing anthracnose disease reactions in tropical yams (Dioscorea spp.). Tropical Agriculture- 70:147–147
Narusaka Y, Narusaka M, Park P, Kubo Y, Hirayama T, Seki M, Shinozaki K (2004) RCH1, a locus in Arabidopsis that confers resistance to the hemibiotrophic fungal pathogen Colletotrichum higginsianum. Mol Plant Microbe Interact 17(7):749–762
Narusaka M, Shirasu K, Noutoshi Y, Kubo Y, Shiraishi T, Iwabuchi M, Narusaka Y (2009) RRS1 and RPS4 provide a dual Resistance-gene system against fungal and bacterial pathogens. Plant J 60(2):218–226
Ni M, Feretzaki M, Sun S, Wang X, Heitman J (2011) Sex in Fungi. Annu Rev Genet 45:405–430
O’Connell R, Herbert C, Sreenivasaprasad S, Khatib M, Esquerré-Tugayé MT, Dumas B (2004) A novel Arabidopsis-Colletotrichum pathosystem for the molecular dissection of plant-fungal interactions. Mol Plant Microbe Interact 17(3):272–282
O’Connell RJ, Pain NA, Hutchison KA, Jones GL, Green JR (1996) Ultrastructure and composition of the cell surfaces of infection structures formed by the fungal plant pathogen Colletotrichum lindemuthianum. J Microsc 181(2):204–212
O’Connell RJ, Panstruga R (2006) Tete a tete inside a plant cell: establishing compatibility between plants and biotrophic fungi and oomycetes. New Phytol 171(4):699–718
O’Connell RJ, Perfect S, Hughes B, Carzaniga R, Bailey J, Green J (2000) Dissecting the cell biology of Colletotrichum infection processes. In: Prusky D, Freeman S, Dickman M (eds) Colletotrichum, host specificity, pathogenicity, and host-pathogen interaction. APS press, St. Paul, MN, pp 57–77
O’Connell RJ, Ride JP (1990) Chemical detection and ultrastructural localization of chitin in cell walls of Colletotrichum lindemuthianum. Physiol Mol Plant Pathol 37:39–53
O’Connell RJ, Thon MR, Hacquard S, Amyotte SG, Kleemann J, Torres MF, Vaillancourt LJ (2012) Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat Genet 44:1060–1065. doi:10.1038/ng.2372
Ohra J, Morita K, Tsujino Y, Tazaki H, Fujimori T, Goering M, Zorner P (1995) Production of the phytotoxic metabolite, ferricrocin, by the fungus Colletotrichum gloeosporioides. Biosci Biotechnol Biochem 59(1):113
Okayama K, Tsujimoto A (1994) Occurrence of strawberry anthracnose caused by Glomerella cingulata (Stoneman) Spaulding et Schrenk and pathogenicity of the fungus. Ann Phytopathological Soc Jpn 60(5):617--623
Olivieri F, Zanetti ME, Oliva CR, Covarrubias AA, Casalongué CA (2002) Characterization of an extracellular serine protease of Fusarium eumartii and its action on pathogenesis related proteins. Eur J Plant Pathol 108(1):63–72
O’Quinn RP, Hoffmann JL, Boyd AS (2001) Colletotrichum species as emerging opportunistic fungal pathogens: A report of 3 cases of phaeohyphomycosis and review. J Am Acad Dermatol 45(1):56–61
Osono T (2007) Endophytic and epiphytic phyllosphere fungi of red-osier dogwood (Cornus stolonifera) in British Columbia. Mycoscience 48(1):47–52
Osono T (2008) Endophytic and epiphytic phyllosphere fungi of Camellia japonica: seasonal and leaf age-dependent variations. Mycologia 100(3):387–391
Pain NA, O’Connell RJ, Mendgen K, Green JR (1994) Identification of glycoproteins specific to biotrophic intracellular hyphae formed in the Colletotrichum lindemuthianum bean interaction. New Phytol 127(2):233–242
Parlevliet JE (2002) Durability of resistance against fungal, bacterial and viral pathogens; present situation. Euphytica 124(2):147–156
Parra G, Bradnam K, Korf I (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23(9):1061–1067
Pavan S, Jacobsen E, Visser RGF, Bai Y (2010) Loss of susceptibility as a novel breeding strategy for durable and broad-spectrum resistance. Mol Breeding 25:1–12
Perfect SE, Green JR (2001) Infection structures of biotrophic and hemibiotrophic fungal plant pathogens. Mol Plant Pathol 2(2):101–108
Perfect SE, Green JR, O’Connell RJ (2001) Surface characteristics of necrotrophic secondary hyphae produced by the bean anthracnose fungus, Colletotrichum lindemuthianum. Eur J Plant Pathol 107(8):813–819
Perfect SE, Hughes HB, O’Connell RJ, Green JR (1999) Colletotrichum: A Model Genus for Studies on Pathology and Fungal-Plant Interactions. Fungal Genet Biol 27(2):186–198
Perfect SE, O’Connell RJ, Green EF, Doering-Saad C, Green JR (1998) Expression cloning of a fungal proline-rich glycoprotein specific to the biotrophic interface formed in the Colletotrichum–bean interaction. Plant J 15(2):273–279
Photita W, Lumyong S, Lumyong P, McKenzie EHC, Hyde KD (2004) Are some endophytes of Musa acuminata latent pathogens. Fungal Divers 16:131–140
Phoulivong S, Cai L, Chen H, McKenzie EH, Abdelsalam K, Chukeatirote E, Hyde KD (2010) Colletotrichum gloeosporioides is not a common pathogen on tropical fruits. Fungal Divers 44(1):33–43
Politis DJ (1975) The identity and perfect state of Colletotrichum graminicola. Mycologia 67:56–62
Prihastuti H, Cai L, Chen H, McKenzie EHC, Hyde KD (2009) Characterization of Colletotrichum species associated with coffee berries in northern Thailand. Fungal Divers 39:89
Promputtha I, Hyde KD, McKenzie EH, Peberdy JF, Lumyong S (2010) Can leaf degrading enzymes provide evidence that endophytic fungi becoming saprobes? Fungal Divers 41(1):89–99
Promputtha I, Lumyong S, Dhanasekaran V, McKenzie EHC, Hyde KD, Jeewon R (2007) A phylogenetic evaluation of whether endophytes become saprotrophs at host senescence. Microb Ecol 53(4):579–590
Prusky D (1996) Pathogen quiescence in postharvest diseases. Annu Rev Phytopathol 34(1):413–434
Prusky D, Freeman S, Rodriguez RJ, Keen NT (1994) A nonpathogenic mutant strain of Colletotrichum magna induces resistance to C. gloeosporioides in avocado fruits. MPMI-Mol Plant-Microbe Interact 7(3):326–333
Prusky D, McEvoy JL, Leverentz B, Conway WS (2001) Local modulation of host pH by Colletotrichum species as a mechanism to increase virulence. Mol Plant Microbe Interact 14(9):1105–1113
Quinlan RJ, Sweeney MD, Leggio LL, Otten H, Poulsen JCN, Johansen KS, Walton PH (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc Natl Acad Sci 108(37):15079–15084
Réblová M, Gams W, Seifert KA (2011) Monilochaetes and allied genera of the Glomerellales, and a reconsideration of families in the Microascales. Stud Mycol 68(1):163–191
Redman RS, Dunigan DD, Rodriguez RJ (2001) Fungal symbiosis from mutualism to parasitism: who controls the outcome, host or invader? New Phytol 151(3):705–716
Redman RS, Sheehan KB, Stout RG, Rodriguez RJ, Henson JM (2002) Thermotolerance generated by plant/fungal symbiosis. Science 298(5598):1581–1581
Ripoche A, Jacqua G, Bussière F, Guyader S, Sierra J (2008) Survival of Colletotrichum gloeosporioides (causal agent of yam anthracnose) on yam residues decomposing in soil. Applied Soil Ecology 38(3):270–278
Robinson M, Riov J, Sharon A (1998) Indole-3-Acetic Acid Biosynthesis in Colletotrichum gloeosporioides f. sp. aeschynomene. Appl Environ Microbiol 64(12):5030–5032
Rodriguez R, Redman R (2008) More than 400 million years of evolution and some plants still can’t make it on their own: plant stress tolerance via fungal symbiosis. J Exp Bot 59(5):1109–1114
Rodriguez RJ, White JF Jr, Arnold AE, Redman RS (2009) Fungal endophytes: diversity and functional roles. New Phytol 182(2):314–330
Rodríguez-Guerra R, Ramírez-Rueda MT, Cabral-Enciso M, García-Serrano M, Lira-Maldonado Z, Gerardo Guevara-González-Chavira M, simpson J (2005) Heterothallic mating observed between Mexican isolates of Glomerella lindemuthiana. Mycologia 97(4):793--803
Rojas EI, Rehner SA, Samuels GJ, Van Bael SA, Herre EA, Cannon P, Sha T (2010) Colletotrichum gloeosporioides sl associated with Theobroma cacao and other plants in Panama: multilocus phylogenies distinguish host-associated pathogens from asymptomatic endophytes. Mycologia 102(6):1318–1338
Rokas A, Williams BL, King N, Carroll SB (2003) Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 425:798–804
Rollins JA (1996) The characterization and inheritance of chromosomal variation in Glomerella graminicola. Ph.D. Dissertation, Purdue University, West Lafayette Indiana
Santamaría J, Bayman P (2005) Fungal epiphytes and endophytes of coffee leaves (Coffea arabica). Microb Ecol 50(1):1–8
Scott-Craig JS, Panaccione DG, Pocard JA, Walton JD (1992) The cyclic peptide synthetase catalyzing HC-toxin production in the filamentous fungus Cochliobolus carbonum is encoded by a 15.7-kilobase open reading frame. J Biol Chem 267(36):26044–26049
Sherriff C, Whelan MJ, Arnold GM, Bailey JA (1995) rDNA sequence analysis confirms the distinction between Colletotrichum graminicola and C. sublineolum. Mycol Res 99(4):475–478
Shwab EK, Keller NP (2008) Regulation of secondary metabolite production in filamentous ascomycetes. Mycol Res 112(2):225–230
Shen S, Goodwin PH, Hsiang T (2001) Infection of Nicotiana species by the anthracnose fungus, Colletotrichum orbiculare. Eur J Plant Pathol 107(8):767–773
Shimada C, Lipka V, O’Connell R, Okuno T, Schulze-Lefert P, Takano Y (2006) Nonhost resistance in Arabidopsis-Colletotrichum interactions acts at the cell periphery and requires actin filament function. Mol Plant Microbe Interact 19(3):270–279
Shivaprakash MR, Appannanavar SB, Dhaliwal M, Gupta A, Gupta S, Gupta A, Chakrabarti A (2011) Colletotrichum truncatum: an unusual pathogen causing mycotic keratitis and endophthalmitis. J Clin Microbiol 49(8):2894–2898
Simmons BA, Loque D, Blanch HW (2008) Next-generation biomass feedstocks for biofuel production. Genome Biol 9:242
Singh J, Quereshi S, Banerjee N, Pandey AK (2010) Production and extraction of phytotoxins from Colletotrichum dematium FGCC# 20 effective against Parthenium hysterophorus L. Braz Arch Biol Technol 53(3):669–678
Singh SP, Schwartz HF (2010) Breeding common bean for resistance to diseases: a review. Crop Sci 50(6):2199–2223
Soares DJ, Barreto RW, Braun U (2009) Brazilian mycobiota of the aquatic weed Sagittaria montevidensis. Mycologia 101(3):401–416
Spanu PD, Abbott JC, Amselem J, Burgis TA, Soanes DM, Stüber K, Reinhardt R (2010) Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science 330(6010):1543–1546
Sticher L, Mauch-Mani B, Métraux AJ (1997) Systemic acquired resistance. Annu Rev Phytopathol 35(1):235–270
Stephenson SA, Hatfield J, Rusu AG, Maclean DJ, Manners JM (2000) CgDN3: an essential pathogenicity gene of Colletotrichum gloeosporioides necessary to avert a hypersensitive-like response in the host Stylosanthes guianensis. Mol Plant Microbe Interact 13(9):929–941
Stergiopoulos I, de Wit PJ (2009) Fungal effector proteins. Annu Rev Phytopathol 47:233–263
Stukenbrock EH, Jorgensen FG, Zala M, Hansen TT, McDonald BA, Schierup MH (2010) Whole-genome and chromosome evolution associated with host adaptation and speciation of the wheat pathogen Mycosphaerella graminicola. PLos genetics 6(12):e1001189
Sukno SA, García VM, Shaw BD, Thon MR (2008) Root infection and systemic colonization of maize by Colletotrichum graminicola. Appl Environ Microbiol 74(3):823–832
Sutton BC (1968) The appressoria of Colletotrichum graminicola and C. falcatum. Can J Bot 46(7):873–876
Sutton BC (1980) The coelomycetes. Commonwealth Mycological Institute, Kew, Surrey, England, pp 522–537
Takahara H, Dolf A, Endl E, O’Connell R (2009) Flow cytometric purification of Colletotrichum higginsianum biotrophic hyphae from Arabidopsis leaves for stage-specific transcriptome analysis. Plant J 59(4):672–683
Takano Y, Kikuchi T, Kubo Y, Hamer JE, Mise K, Furusawa I (2000) The Colletotrichum lagenarium MAP kinase gene CMK1 regulates diverse aspects of fungal pathogenesis. Mol Plant Microbe Interact 13(4):374–383
Tang W, Coughlan S, Crane E, Beatty M, Duvick J (2006) The application of laser microdissection to in planta gene expression profiling of the maize anthracnose stalk rot fungus Colletotrichum graminicola. Mol Plant Microbe Interact 19(11):1240–1250
Thakur RP (2007) Host plant resistance to diseases: potential and limitations. Indian Journal of Plant Protection 35:17–21
Than PP, Jeewon R, Hyde KD, Pongsupasamit S, Mongkolporn O, Taylor PWJ (2008) Characterization and pathogenicity of Colletotrichum species associated with anthracnose on chilli (Capsicum spp.) in Thailand. Plant Pathol 57(3):562–572
Thon M, Pan H, Diener S, Papalas J, Taro A, Mitchell T, Dean R (2006) The role of transposable element clusters in genome evolution and loss of synteny in the rice blast fungus Magnaporthe oryzae. Genome Biol 7:R16
Townsend JP (2007) Profiling phylogenetic informativeness. Syst Biol 56:222–231
Townsend JP, Lopez-Giraldez F (2010) Optimal selection of gene and ingroup taxon sampling for resolving phylogenetic relationships. Syst Biol 59:446–457
Urefña-Padilla AR, MacKenzie SJ, Bowen BW, Legard DE (2002) Etiology and population genetics of Colletotrichum spp. Causing crown and fruit rot of strawberry. Phytopathology 92(11):1245--1252
Vaillancourt LJ, Hanau RM (1991) A method for genetic analysis of Glomerella graminicola (Colletotrichum graminicola) from maize. Phytopathology 81(5):530–534
Vaillancourt L, Du M, Wang J, Rollins J, Hanau R (2000a) Genetic analysis of cross fertility between two self-sterile strains of Glomerella graminicola. Mycologia 92:430–435
Vaillancourt L, Wang J, Hanau R (2000b) Genetic regulation of sexual compatibility in Glomerella graminicola. In: Prusky D, Freeman S, Dickman M (eds) Colletotrichum host specificity, pathology, and host-pathogen interaction. APS press, St. Paul MN, pp 29–44
Vargas WA, Martín JMS, Rech GE, Rivera LP, Benito EP, Díaz-Mínguez JM, Sukno SA (2012) Plant defense mechanisms are activated during biotrophic and necrotrophic development of Colletotricum graminicola in maize. Plant Physiol 158(3):1342–1358
Varzea VMP, Rodrigues CJ, Lewis BG (2002) Distinguishing characteristics and vegetative compatibility of Colletotrichum kahawe in comparison with other related species from coffee. Plant Pathol 51(2):202–207
Vega FE, Simpkins A, Aime MC, Posada F, Peterson SW, Rehner SA, Arnold AE (2010) Fungal endophyte diversity in coffee plants from Colombia, Hawai’i, Mexico and Puerto Rico. Fungal Ecol 3(3):122–138
Vogel J (2008) Unique aspects of the grass cell wall. Curr Opin Plant Biol 11(3):301–307
Voytas DF, Boeke JD (1993) Yeast retrotransposons and tRNAs. Trends Genet 9:421–427
Walker JC (1921) Onion smudge. University of Wisconsin, Madison, p 721
Waller JM (1992) Colletotrichum diseases of perennial and other cash crops. In: Bailey JA, Jeger MJ (eds) Colletotrichum: biology pathology and control. CAB International, Wallingford, pp 167–185
Wang H, Xu Z, Gao L, Hao B (2009) A fungal phylogeny based on 82 complete genomes using the composition vector method. BMC Evol Biol 9:195
Warwar V, Dickman MB (1996) Effects of calcium and calmodulin on spore germination and appressorium development in Colletotrichum trifolii. Appl Environ Microbiol 62(1):74–79
Wasilwa LA, Correll JC, Morelock TE, McNew RE (1993) Reexamination of races of the cucurbit anthracnose pathogen Colletotrichum orbiculare. Phytopathology 83(11):1190–1198
Waxman KD, Bergstrom GC (2011a) First report of anthracnose caused by Colletotrichum caudatum on indiangrass in New York. Plant Dis 95(9):1189–1189
Waxman KD, Bergstrom GC (2011b) First report of anthracnose caused by Colletotrichum navitas on switchgrass in New York. Plant Dis 95(8):1032–1032
Weir BS, Johnston PR, Damm U (2012) The Colletotrichum gloeosporioides species complex. Stud Mycol 73(1):115–180
Westcott C (2001) Westcott’s plant disease handbook. Kluwer Academic Pub, Boston
Wharton PS, Julian AM (1996) A cytological study of compatible and incompatible interactions between Sorghum bicolor and Colletotrichum sublineolum. New Phytol 134(1):25–34
Wharton PS, Julian AM, O’Connell RJ (2001) Ultrastructure of the infection of Sorghum bicolor by Colletotrichum sublineolum. Phytopathology 91:149–158
Wheeler HE (1950) Genetics of Glomerella. VIII. A genetic basis for the occurrence of minus mutants. Am J Bot 37:304–312
Wheeler HE (1954) Genetics and evolution of heterothallism in Glomerella. Phytopathology 44:342–345
Wheeler HE, Olive LS, Ernest CT, Edgerton CW (1948) Genetics of Glomerella. V. Crozier and ascus development. Am J Bot 35:722–728
Wheeler HE, McGahen JW (1952) Genetics of Glomerella. X. Genes affecting sexual reproduction. Am J Bot 39:110–119
White TJ, Bruns TD, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Glefand JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, San Diego, pp 315–322
Wicklow DT, Jordan AM, Gloer JB (2009) Antifungal metabolites (monorden, monocillins I, II, III) from Colletotrichum graminicola, a systemic vascular pathogen of maize. Mycol Res 113(12):1433–1442
Winter RL, Lawhon SD, Halbert ND, Levine GJ, Wilson HM, Daly MK (2010) Subcutaneous infection of a cat by Colletotrichum species. J Feline Med Surg 12(10):828–830
Xie L, Zhang JZ, Wan Y, Hu DW (2010) Identification of Colletotrichum spp. isolated from strawberry in Zhejiang Province and Shanghai City, China. J Zhejiang Univ Sci B, 11(1):61–70
Yamauchi J, Takayanagi N, Komeda K, Takano Y, Okuno T (2004) cAMP-PKA signaling regulates multiple steps of fungal infection cooperatively with Cmk1 MAP kinase in Colletotrichum lagenarium. Mol Plant Microbe Interact 17(12):1355–1365
Yang Z, Dickman MB (1997) Regulation of cAMP and cAMP dependent protein kinase during conidial germination and appressorium formation in Colletotrichum trifolii. Physiol Mol Plant Pathol 50(2):117–127
Yang Z, Dickman MB (1999a) Colletotrichum trifolii mutants disrupted in the catalytic subunit of cAMP-dependent protein kinase are nonpathogenic. Mol Plant Microbe Interact 12(5):430–439
Yang Z, Dickman MB (1999b) Molecular cloning and characterization of Ct-PKAR, a gene encoding the regulatory subunit of cAMP-dependent protein kinase in Colletotrichum trifolii. Arch Microbiol 171(4):249–256
Yoshino K, Irieda H, Sugimoto F, Yoshioka H, Okuno T, Takano Y (2012) Cell death of Nicotiana benthamiana is induced by secreted protein NIS1 of Colletotrichum orbiculare and is suppressed by a homologue of CgDN3. Mol Plant Microbe Interact 25(5):625–636
Zare R, Gams W, Culham A (2000) A revision of Verticillium sect. Prostrata. I. Phylogenetic studies using ITS sequences. Nova Hedwigia 71(3/4):465–480
Zeiders KE (1987) Leaf spot of indiangrass caused by Colletotrichum caudatum. Plant Dis 71(4):348–350
Zhang N, Castlebury LA, Miller AN, Huhndorf SM, Schoch CL, Seifert KA, Sung GH (2006) An overview of the systematics of the Sordariomycetes based on a four-gene phylogeny. Mycologia 98(6):1076–1087
Zhang M, Kadota Y, Prodromou C, Shirasu K, Pearl LH (2010) Structural basis for assembly of Hsp90-Sgt1-CHORD protein complexes: implications for chaperoning of NLR innate immunity receptors. Mol Cell 39(2):269–281
Zhao XM, Wang ZQ, Shu SH, Wang WJ, Xu HJ, Ahn YJ, Wang M, Hu X (2013) Ethanol and Methanol Can Improve Huperzine A Production from Endophytic Colletotrichum gloeosporioides ES026. PLoS ONE 8(4):e61777
Zhu P, Oudemans PV (2000) A long terminal repeat retrotransposon Cgret from the phytopathogenic fungus Colletotrichum gloeosporioides on cranberry. Curr Genet 38(5):241–247
Acknowledgements
We are grateful to Guillaume Robin for preparing Fig. 3.3, and to Adnan Ismaiel for the sequencing that contributed to the phylogenetic tree in Fig. 3.1. The work in the Shirasu lab is supported partly by the Programme for Promotion of Basic and Applied Researches for Innovations in Bio-oriented Industry and Grant-in-Aid for Scientific Research (KAKENHI; 24228008).
List of URLS
Broad Institute Colletotrichum Database: (www.broadinstitute.org/annotation/genome/colletotrichum_group/MultiHome.html)
Max Planck Institute for Plant Breeding Research Fungal Genomes: (www.mpipz.mpg.de/14157/fungal_genomes).
EnsemblFungi G. graminicola genome
(http://fungi.ensembl.org/Glomerella_graminicola/Info/Index)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Crouch, J. et al. (2014). The Genomics of Colletotrichum . In: Dean, R., Lichens-Park, A., Kole, C. (eds) Genomics of Plant-Associated Fungi: Monocot Pathogens. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-44053-7_3
Download citation
DOI: https://doi.org/10.1007/978-3-662-44053-7_3
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-44052-0
Online ISBN: 978-3-662-44053-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)