
Abstract
Filaggrin has become the major research focus within the field of skin barrier abnormality over the last 5–10 years. Null mutations in the (pro)-filaggrin gene (FLG) have been shown to be a major risk factor for the development of atopic dermatitis, a disease in which one of the hallmarks is a decreased skin barrier function. However, only between 30 and 40 % of the patients suffering from atopic dermatitis carry the mutation, and yet all of them have a barrier defect, and patients suffering from other skin diseases such as contact dermatitis and psoriasis also have a decreased skin barrier function. From the studies of keratinocyte cell cultures and animal models, it has become clear that inflammation in itself is actually able to induce downregulation of filaggrin expression on transcriptional and translational levels and decrease processing of pro-filaggrin into filaggrin. The literature on this subject is concentrated on the effect of inflammatory cytokines but is still very scarce. More research into this area may offer new insight into the pathogenesis of reduced skin barrier function in inflammatory skin diseases, and it may lead to new principles of treatment in these skin diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
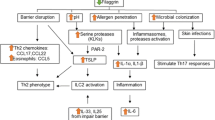
Since the milestone publication by Palmer et al. in 2006 [1], in which it was shown that mutations in the filaggrin gene (FLG), which codes for filaggrin “filament aggregating protein,” significantly increase the risk of atopic dermatitis (AD), filaggrin and barrier dysfunction have been the epicenter of AD research and other skin diseases with decreased skin barrier function. Filaggrin is important for the aggregation of keratin filaments forming the impenetrable keratin layer in the stratum corneum of the epidermis and also for the hydration of the skin through the “natural moisturizing factors” (NMFs) [2]. The decreased barrier function caused by the lack of filaggrin expression may increase the risk of allergic sensitization and thus offer a molecular background for the so-called atopic march, in which patients suffering from early childhood AD have a higher risk of developing asthma, rhinitis, and food allergies [3]. An interesting fact is that 44–85.8 % of AD patients do not carry an FLG mutation, but still all patients have a decreased skin barrier function [4], and that among patients carrying an FLG mutation, 40 % never show any signs of decreased skin barrier function [5].
These results indicate that regulatory mechanisms other than null mutations in the FLG are responsible for the decreased skin barrier function in AD patients. Mutations in other genes involved in the skin barrier function have been linked to the development of AD such as SPINK5 (LEKTI), a serine protease inhibitor, which inhibits the kallikrein’s breakdown of the corneodesmosomes [6] and very likely also filaggrin (KLK5) [7]. The inflammatory reaction taking place in the skin can modulate the expression of filaggrin on transcriptional, translational, and posttranslational levels, thus inducing a functional lack of filaggrin in the absence of an FLG null mutation [8, 9]. In this chapter, we will focus on the effect of inflammation on expression of filaggrin and the subsequent effect on skin barrier functions.
2 Production and Regulation of Filaggrin
Filaggrin has the property of aligning keratin intermediary filaments into a tight matrix in the keratinocytes in the upper layer of the epidermis leading to a collapse of the intracellular structure and the formation of the spindle-shaped and tightly packed cells of the stratum corneum. Filaggrin is thus one of the most important structural proteins in the establishment of an intact skin barrier [10–12].
Filaggrin is produced as the pre-protein pro-filaggrin containing 10–12 filaggrin repeats flanked by a C- and N-terminal region [13, 14]. The N-terminal region may act as a Ca2+-binding region and play a role for the terminal keratinocyte denucleation, whereas the exact role of the C-terminal region is unknown, although mice lacking this region are unable to process pro-filaggrin into filaggrin [15]. The gene for pro-filaggrin, FLG, is located on chromosome 1q21 in the epidermal differentiation complex that also contains other genes transcribed in the maturation process of the keratinocyte [14].
The interest in filaggrin was spurred by the finding that homozygous null mutations in the FLG lead to varying degrees of ichthyosis vulgaris [16] and that individuals heterozygous for the mutation had a higher risk of AD [1]. Until today, more than 40 different null mutations in the FLG have been identified, but the frequency of each mutation differs depending on the specific ancestral group [17].
The maturation process of pro-filaggrin is a very complex multistep process, and only an overview, aimed at describing possible points for regulation by inflammation, will be given here.
The expression of the FLG could be under control of the transcription factor AP-1 (fos and/or jun) as AP-1 responsive elements have been described in the promoter region of the FLG [18]. Furthermore, oct1, oct6, and skn1a/i bind in vitro to two responsive elements in the promotor region of the FLG [19]. Certain isoforms of p63 inhibit the transcription of FLG [20], and the FLG promotor region also contains glucocorticoid- and retinoic acid-responsive elements that suppress promotor activity [21].
Once pro-filaggrin is transcribed, it is heavily phosphorylated to prevent premature degradation to filaggrin [22]; however, when the keratinocytes reach the granular layer, filaggrin is dephosphorylated as one of the initial steps in the formation of the keratin matrix. The next step involves proteolytic cleavage of the pro-filaggrin molecule, primarily in the C- and N-terminal flanking regions, followed by cleavage of the linker region between the filaggrin repeats. The proteases involved in this process include matriptase [23] and prostasin [24]. In prostasin knockout mice, two- and three-domain filaggrin intermediates accumulate in the stratum corneum, but no filaggrin monomers, whereas pro-filaggrin accumulates in the stratum corneum in matriptase knockout mice [23–25]. LEKTI (lymphoepithelial Kazal-type-related inhibitor) is a serine protease inhibitor encoded by the SPINK5 gene. Mutations in this gene in humans are responsible for “Netherton’s syndrome,” in which the skin is very dry with abnormal maturation of the keratinocytes and a defect in skin barrier [26]. Under normal circumstances LEKTI inhibits the proteases that cleaves pro-filaggrin into filaggrin monomers, and in SPINK5 knockout mice, there is premature pro-filaggrin cleavage, but whether this is due to lack of inhibition of matriptase and prostasin is unknown [27, 28]. In mice lacking 12R-lipooxygenase, which catalyze dioxygenation of fatty acids, there is an aberrant pro-filaggrin cleavage, although the background for this is unclear [29].
The filaggrin monomers are broken down to hygroscopic amino acids that constitute the NMFs by caspase 14 [2]. Knockout mice lacking caspase 14 have dry skin and an increased susceptibility to ultraviolet (UV) B light. They do cleave filaggrin, but still there is an abnormal maturation of the skin and a decreased production of NMFs [30].
From this short overview of the maturation of pro-filaggrin into filaggrin, it is clear that there are several steps apart from null mutations in which the expression of filaggrin can be regulated.
3 Inflammation in the Skin and the Effect of Cytokines
The inflammatory reactions in the skin seen in inflammatory skin diseases are due to a complex interaction between the cells of the immune system, adaptive and innate; the keratinocytes; the endothelial cells; and the connective tissue. An offensive agent, such as bacteria, antigens, or local tissue damage, induces a cascade of events that attract the cells of the immune system such as polymorphnuclear leukocytes and lymphocytes. These cells are activated and differentiated in a manner specific for each inflammatory skin disease [31]. In AD, the dominating lymphocytes are of the Th2 subtype; in psoriasis, they are Th1; and in contact dermatitis, the lymphocytes are CD8+ rather than CD4+, although treatment may alter this dramatically [32]. The dermal dendritic cells also have a specific profile of surface receptors and cytokine production profile depending on the inflammatory skin disease [33]. The production and expression of cytokines in inflammatory skin diseases often take place in a specific spatial and timely sequence for each disease [34].
All the cells in the skin produce inflammatory mediators that attract inflammatory cells, and among these mediators are the cytokines that are small, biologically potent glycoproteins that regulate growth, migration, and differentiation of cells. The cytokines may be subgrouped in different, often overlapping, ways depending on function or molecular structure. The interleukins (ILs) are the cytokines initially described as produced by lymphocytes, monocytes, or macrophages, and the chemokines are a subgroup of these, with a highly conserved motif of cysteine residues in the N-terminal. For example, IL-8, which was originally termed so because it was produced by macrophages, was also found to have the structure of a CXC-chemokine; therefore, in accordance with the chemokine nomenclature subcommittee, it was renamed CXCL8 [35].
Other mediators of inflammation, besides the cytokines, are vasoactive amines such as histamine, 5-hydroxytryptamine, and serotonin; the reactive oxygen intermediates and nitrogen oxide; and the arachidonic acid products such as leukotrienes, prostaglandins, and thromboxanes.
4 Inflammatory Cytokines and Filaggrin Expression
4.1 Interleukin-4 and Interleukin-13
Patients suffering from AD have a decreased skin barrier function, but this is not confined to the group of patients carrying the FLG mutation. To address this question, the filaggrin expression on both mRNA and protein levels in the skin of 30 healthy patients was compared to the expression of lesional as well as non-lesional skin in 39 patients suffering from AD. In addition, a subgroup of patients was screened for the FLG null mutations 2282del4 and R501X, and 2 of 25 of the healthy controls had mutations, whereas 3 of 17 of the AD patients carried the 2282del4 mutation. None of the patients carried the R501X mutation. The results showed that filaggrin expression was significantly decreased in lesional as well as non-lesional skin of the AD patients not carrying the FLG mutation compared to the groups of healthy patients with or without FLG null mutation. The filaggrin expression in the skin of lesional AD in patients with FLG null mutation was significantly lower than that of AD patients without mutations. In the same study, it was shown that the typical Th2 cytokines IL-4 and IL-13, which are abundant in AD skin, could decrease the expression of filaggrin in primary keratinocyte cultures in which differentiation had been induced by increased calcium concentrations [8, 36]. From these studies it was clear that inflammation of the skin could induce a “functional filaggrin defect” in the skin and, thereby, a decreased skin barrier function.
Not only do IL-4 and IL-13 decrease filaggrin expression; they also decrease the expression of caspase 14 [9], which breaks down filaggrin to NMFs and contributes to the integrity of the skin barrier [2].
4.2 Interleukin-22 (IL-22)
IL-22 is a cytokine that was first described to be produced by Th17 cells, but later as a cytokine produced by a distinct subgroup of skin-homing T lymphocytes expressing the skin-homing chemokine receptor profile CCR4, CCR6, and CCR10, which do not express IL-17 [37]. This subgroup of lymphocytes, known as Th22 lymphocytes, are found in inflammatory skin disorders where they induce transcription of genes related to the innate and adaptive immune pathways in keratinocytes [38]. Dermal dendritic cells and Langerhans cells from the skin induce IL-22 production in T lymphocytes stressing the concept of the skin specificity of these cells [39]. Furthermore, exotoxins from Staphylococcus aureus that colonize the skin, especially in AD, induce IL-22 expression in CD4+ T lymphocytes [40]. In HaCaT cells (an immortalized keratinocytic cell line) cultured with high calcium concentrations to induce filaggrin expression, IL-22 downregulates pro-filaggrin and filaggrin expression on both mRNA and protein level [41].
4.3 Interleukin-17A (IL-17A) and Interleukin-17E (IL-17E)
Interleukin-17 is produced by Th17 lymphocytes after stimulation with IL-23. IL-17 was primarily associated with inflammatory diseases in the central nervous system and in the joints, because it was found to be the mediator of the IL-23-dependent models of collagen-induced arthritis and experimental autoimmune encephalitis. Later, important roles for IL-17 were described in autoimmune diseases, such as rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis, and in psoriasis. Interleukin-17 belongs to a family of at least five members – IL-17A–E. IL-17A is often described as a Th1-like cytokine, and it is the IL-17 member associated with psoriasis [42, 43]. IL-17E, also known as IL-25, is the opposite and is often described as a Th2-associated cytokine. IL-17E binds to the IL-17B-receptor, whereas IL-17A binds to the IL-17A receptor [44]. IL-17A has been shown to be able to downregulate not only pro-filaggrin and filaggrin expression in HaCaT cells cultured in high calcium concentrations but also proteins associated with cell adhesion between the keratinocytes, thereby affecting the skin barrier on at least two different levels [45]. The role of filaggrin downregulation in psoriasis in which IL-17A levels are high is not clear.
IL-17E (IL-25), on the other hand, is found in very high concentrations in Th2-mediated diseases such as asthma and AD. Il-17E is important in the response to helminthic infections in murine models [46], but also for the initiation and prolongation of the Th2 response. In asthma patients, Th2 cells, mast cells, and lung epithelial cells produce IL-17E [47], whereas in the skin of AD patients, dermal dendritic cells produce IL-25 [48]. Surprisingly, this production is downregulated by TSLP. The pro-filaggrin expression in primary keratinocytes cultured to 80 % confluency is reduced when co-cultured with IL-17E [48], a finding that could be reproduced in keratinocytes cultured with 1.3 mM calcium [36]. These results indicate that IL-17E is a key mediator in AD, as it has the ability both to induce the production of Th2 cytokines such as IL-4 and IL-13 and to downregulate filaggrin, thereby decreasing the skin barrier function.
4.4 Tumor Necrosis Factor-α (TNF-α)
Tumor necrosis factor-α (TNF-α) is a central mediator of inflammation. It inhibits viral replication and tumorigenesis, and in the context of skin diseases, it is a very central mediator of the inflammation in psoriasis, as proven by the high efficacy of TNF-α inhibitors in the treatment of psoriasis [49]. TNF-α is also expressed in AD, but since TNF-α inhibitors have almost no effect in the treatment of this disease, its role is very unclear [50]. In psoriasis a skin barrier defect also exists, although its role in the pathogenesis of psoriasis is not as clear as in AD. TNF-α acts on the filaggrin synthesis by downregulating the expression in primary keratinocytes cultured in 0.06 % calcium [51].
4.5 Interleukin 31 (IL-31)
IL-31 was originally described as a cytokine produced by Th2 lymphocytes, but other cells such as mast cells, dendritic cells, and monocytes may also produce the cytokine [52, 53]. It is increased in the skin and serum of AD patients, but also in a murine model of AD, the NC/Nga mouse, in which it induces pruritus through binding to IL-31 receptors in the dorsal ganglia. Blocking of IL-31 with monoclonal antibodies in this model decreases the itching behavior of the mice [54]. In a 3D model of the skin based on HaCaT cells, in which a doxycycline-inducible IL-31 receptor was inserted, IL-31 could repress the expression of pro-filaggrin. This effect was in part mediated through an IL-31-dependent induction of IL-20 and IL-24 production in the HaCaT cells, and these cytokines in turn downregulated pro-filaggrin [55] (Table 4.1).
5 The Effect of Anti-inflammatory Treatment on Filaggrin Expression in the Skin
5.1 Topical Steroids and Calcineurin Inhibitors
The most widely used therapy for inflammatory skin diseases of mild to moderate severity is topical corticosteroids, and in the case of AD also the topical calcineurin inhibitors tacrolimus and pimecrolimus. Topical corticosteroids exert their action partly through binding to steroid receptors in the cytosol of the cell, which then translocate into the nucleus, where it inhibits the transcription of inflammatory genes (transrepression) and induces expression of anti-inflammatory genes (transactivation), but also affects the synthesis of structural proteins [60]. Calcineurin inhibitors inhibit calcium-dependent dephosphorylation of the transcription factor nuclear factor of activated T cells (NFATs) that is required for the transcription of inflammatory cytokines such as IL-2 [61]. A study of genes expressed in the skin of AD patients treated with either betamethasone valerate or pimecrolimus showed that both reduced inflammation, betamethasone valerate more than pimecrolimus; both restored filaggrin and loricrin expression to normal levels; but that betamethasone valerate reduced expression of lipid synthesis rate-limiting enzymes and involucrin, which may be part of the explanation for the lack of effect of corticosteroids on the restoration of the skin barrier function [56].
5.2 Retinoic Acid
Retinoic acid is used in inflammatory skin diseases and has an anti-proliferative effect on the keratinocytes and also an anti-inflammatory effect; both effects are exerted through the RAR (retinoid acid receptors) and the RXR (retinoid X receptors). Filaggrin production is downregulated by retinoic acid as seen in keratinocytes cultured in multilayer on a collagen matrix at the air-liquid surface [57] and in oral keratinocytes in a lifted culture system [58]. The effect may be due to the presence of a retinoic acid-responsive element in the promoter region of the pro-FLG [21].
Chronic hand eczema may be treated with the retinoid alitretinoin (9-cis-retinoid) with good effect [62]. A side effect of the treatment is dry skin, and in this light, AD treated with alitretinoin (9-cis-retinoid) surprisingly shows good effect [63]. Larger studies, however, are needed.
5.3 Coal Tar
Coal tar is a treatment used for the treatment of skin diseases for more than two millennia. A recent publication has shown that the aryl hydrocarbon receptor (AHR) is involved not only in normalization of the Th2-dominated response by interfering with the STAT6-mediated response but also through an induction of filaggrin production along with other markers of epidermal differentiation in primary keratinocyte cell cultures and in an organotypic skin model of human keratinocytes both with and without filaggrin mutations, a finding that could be reproduced in skin biopsies from AD patients [59]. Thus, the AHR seems to be a very interesting and promising candidate for new filaggrin-targeted therapies for AD, as it both inhibits the inflammatory reaction and increases the differentiation of keratinocytes.
6 Discussion and Conclusion
The finding of the correlation between FLG null mutations, and AD and ichthyosis vulgaris spurred the interest in skin barrier defects, especially in AD but in also other eczematous diseases. The fact that AD patients have a skin barrier defect has long been recognized and has led to the discussion of the “inside-outside” hypothesis versus the “outside-inside” hypothesis. The “inside-outside” hypothesis favors the notion that the inflammatory reaction, initiated by allergens, infections, or autoimmune reactions, in the skin leads to skin barrier breakdown, whereas the “outside-inside” hypothesis favors the notion of the skin barrier defect as the initial event followed by an inflammatory/allergic reaction on the basis of noxious stimuli entering the body through the open barrier [64]. In this review, inflammatory stimuli leading to reduced filaggrin expression have been reviewed, and this could be read as a support of the “inside-outside” hypothesis. However, other studies have shown that a lack of filaggrin itself can enhance production of inflammatory stimuli such as IL-1 [65] and TSLP [66] (thymic stromal lymphopoietin). Thus, the two subelements of AD pathogenesis, inflammation and skin barrier defect, seem to be closely interconnected.
The downregulation of filaggrin expression by inflammation may also be of great importance in other inflammatory skin diseases such as contact dermatitis, in which inflammation leads to a decreased skin barrier, and this in turn increases the inflammatory reaction in a perpetual vicious circle, in which the risk of acquiring new allergies through the broken skin barrier is increased, as is the case in patients homozygous for the FLG mutations R501X and 2282del4 who have an odds Ration of 5.71 for sensitization to other allergens than nickel compared to healthy individuals [67].
Until now, the main focus on research in filaggrin and inflammation has been on the effect of various inflammatory mediators on the production of filaggrin in keratinocytes. However, inflammation also affects the posttranslational processing of both filaggrin and pro-filaggrin, as exemplified by the downregulation of caspase 14 by inflammatory cytokines [2, 9]. However, the effect of inflammation on posttranslational processes as well as transcriptional regulation is not well examined and should constitute a major focus area for research in skin barrier function in the next decade.
It is clear from the research so far that treatment of skin inflammation restores the skin barrier function with regard to filaggrin. The experiments performed so far indicate, for a large part, that this is due to the decreased production of inflammatory mediators by the inflammatory cells in the skin. However, the promoter region of pro-filaggrin contains retinoid as well as glucocorticoid responsive elements [21], and the effect of these immunosuppressants themselves on filaggrin production still remains to be clarified.
In conclusion, inflammation of all types, including Th1-, Th2-, Th17-, and Th22-dominated responses, induces skin barrier defects, and the current results suggest that this may, partly, be due to a downregulation of filaggrin.
References
Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38(4):441–6. PubMed PMID: 16550169.
Hoste E, Kemperman P, Devos M, Denecker G, Kezic S, Yau N, et al. Caspase-14 is required for filaggrin degradation to natural moisturizing factors in the skin. J Invest Dermatol. 2011;131(11):2233–41. PubMed PMID: 21654840.
Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242(1):233–46. PubMed PMID: 21682749. Pubmed Central PMCID: PMC3122139. Epub 2011/06/21. eng.
Irvine AD. Fleshing out filaggrin phenotypes. J Invest Dermatol. 2007;127(3):504–7. PubMed PMID: 17299430.
O’Regan GM, Sandilands A, McLean WH, Irvine AD. Filaggrin in atopic dermatitis. J Allergy Clin Immunol. 2008;122(4):689–93. PubMed PMID: 18774165.
Weidinger S, Baurecht H, Wagenpfeil S, Henderson J, Novak N, Sandilands A, et al. Analysis of the individual and aggregate genetic contributions of previously identified serine peptidase inhibitor Kazal type 5 (SPINK5), kallikrein-related peptidase 7 (KLK7), and filaggrin (FLG) polymorphisms to eczema risk. J Allergy Clin Immunol. 2008;122(3):560–8 e4. PubMed PMID: 18774391.
Sakabe JI, Yamamoto M, Hirakawa S, Motoyama A, Ohta I, Tatsuno K, et al. Kallikrein-related peptidase 5 functions in proteolytic processing of pro-filaggrin in cultured human keratinocytes. J Biol Chem. 2013;288(24):17179–89. PubMed PMID: 23629652.
Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, Debenedetto A, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. 2007;120(1):150–5. PubMed PMID: 17512043. Pubmed Central PMCID: PMC2669594. Epub 2007/05/22. eng.
Hvid M, Johansen C, Deleuran B, Kemp K, Deleuran M, Vestergaard C. Regulation of caspase 14 expression in keratinocytes by inflammatory cytokines – a possible link between reduced skin barrier function and inflammation? Exp Dermatol. 2011;20(8):633–6. PubMed PMID: 21539619. Epub 2011/05/05. eng.
Barker JN, Palmer CN, Zhao Y, Liao H, Hull PR, Lee SP, et al. Null mutations in the filaggrin gene (FLG) determine major susceptibility to early-onset atopic dermatitis that persists into adulthood. J Invest Dermatol. 2007;127(3):564–7. PubMed PMID: 16990802.
Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6(4):328–40. PubMed PMID: 15803139.
McGrath JA, Uitto J. The filaggrin story: novel insights into skin-barrier function and disease. Trends Mol Med. 2008;14(1):20–7. PubMed PMID: 18068483.
Gan SQ, McBride OW, Idler WW, Markova N, Steinert PM. Organization, structure, and polymorphisms of the human pro-filaggrin gene. Biochemistry. 1990;29(40):9432–40. PubMed PMID: 2248957.
McKinley-Grant LJ, Idler WW, Bernstein IA, Parry DA, Cannizzaro L, Croce CM, et al. Characterization of a cDNA clone encoding human filaggrin and localization of the gene to chromosome region 1q21. Proc Natl Acad Sci U S A. 1989;86(13):4848–52. PubMed PMID: 2740331. Pubmed Central PMCID: 297512.
Sandilands A, Terron-Kwiatkowski A, Hull PR, O’Regan GM, Clayton TH, Watson RM, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet. 2007;39(5):650–4. PubMed PMID: 17417636.
Smith FJ, Irvine AD, Terron-Kwiatkowski A, Sandilands A, Campbell LE, Zhao Y, et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet. 2006;38(3):337–42. PubMed PMID: 16444271.
Akiyama M. FLG mutations in ichthyosis vulgaris and atopic eczema: spectrum of mutations and population genetics. Br J Dermatol. 2010;162(3):472–7. PubMed PMID: 19958351.
Jang SI, Steinert PM, Markova NG. Activator protein 1 activity is involved in the regulation of the cell type-specific expression from the proximal promoter of the human pro-filaggrin gene. J Biol Chem. 1996;271(39):24105–14. PubMed PMID: 8798649.
Jang SI, Karaman-Jurukovska N, Morasso MI, Steinert PM, Markova NG. Complex interactions between epidermal POU domain and activator protein 1 transcription factors regulate the expression of the pro-filaggrin gene in normal human epidermal keratinocytes. J Biol Chem. 2000;275(20):15295–304. PubMed PMID: 10809764.
King KE, Ponnamperuma RM, Gerdes MJ, Tokino T, Yamashita T, Baker CC, et al. Unique domain functions of p63 isotypes that differentially regulate distinct aspects of epidermal homeostasis. Carcinogenesis. 2006;27(1):53–63. PubMed PMID: 16081516.
Presland RB, Tomic-Canic M, Lewis SP, Dale BA. Regulation of human pro-filaggrin promoter activity in cultured epithelial cells by retinoic acid and glucocorticoids. J Dermatol Sci. 2001;27(3):192–205. PubMed PMID: 11641059.
Resing KA, Johnson RS, Walsh KA. Characterization of protease processing sites during conversion of rat pro-filaggrin to filaggrin. Biochemistry. 1993;32(38):10036–45. PubMed PMID: 8399131.
List K, Szabo R, Wertz PW, Segre J, Haudenschild CC, Kim SY, et al. Loss of proteolytically processed filaggrin caused by epidermal deletion of Matriptase/MT-SP1. J Cell Biol. 2003;163(4):901–10. PubMed PMID: 14638864. Pubmed Central PMCID: 2173680.
Leyvraz C, Charles RP, Rubera I, Guitard M, Rotman S, Breiden B, et al. The epidermal barrier function is dependent on the serine protease CAP1/Prss8. J Cell Biol. 2005;170(3):487–96. PubMed PMID: 16061697. Pubmed Central PMCID: 2171460.
Netzel-Arnett S, Currie BM, Szabo R, Lin CY, Chen LM, Chai KX, et al. Evidence for a matriptase-prostasin proteolytic cascade regulating terminal epidermal differentiation. J Biol Chem. 2006;281(44):32941–5. PubMed PMID: 16980306.
Chavanas S, Bodemer C, Rochat A, Hamel-Teillac D, Ali M, Irvine AD, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25(2):141–2. PubMed PMID: 10835624.
Descargues P, Deraison C, Bonnart C, Kreft M, Kishibe M, Ishida-Yamamoto A, et al. Spink5-deficient mice mimic Netherton syndrome through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat Genet. 2005;37(1):56–65. PubMed PMID: 15619623.
Hewett DR, Simons AL, Mangan NE, Jolin HE, Green SM, Fallon PG, et al. Lethal, neonatal ichthyosis with increased proteolytic processing of filaggrin in a mouse model of Netherton syndrome. Hum Mol Genet. 2005;14(2):335–46. PubMed PMID: 15590704.
Epp N, Furstenberger G, Muller K, de Juanes S, Leitges M, Hausser I, et al. 12R-lipoxygenase deficiency disrupts epidermal barrier function. J Cell Biol. 2007;177(1):173–82. PubMed PMID: 17403930. Pubmed Central PMCID: 2064121.
Denecker G, Hoste E, Gilbert B, Hochepied T, Ovaere P, Lippens S, et al. Caspase-14 protects against epidermal UVB photodamage and water loss. Nat Cell Biol. 2007;9(6):666–74. PubMed PMID: 17515931.
Bieber T. Atopic dermatitis. N Engl J Med. 2008;358(14):1483–94. PubMed PMID: 18385500.
Vestergaard C, Deleuran M, Kragballe K. Two cases of atopic dermatitis-like conditions induced in psoriasis patients treated with infliximab. J Eur Acad Dermatol Venereol. 2007;21(9):1272–4. PubMed PMID: 17894730.
Zaba LC, Krueger JG, Lowes MA. Resident and “inflammatory” dendritic cells in human skin. J Invest Dermatol. 2009;129(2):302–8. PubMed PMID: 18685620. Pubmed Central PMCID: 2746703.
Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361(5):496–509. PubMed PMID: 19641206.
Homey B, Meller S. Chemokines and other mediators as therapeutic targets in psoriasis vulgaris. Clin Dermatol. 2008;26(5):539–45. PubMed PMID: 18755373.
Pellerin L, Henry J, Hsu CY, Balica S, Jean-Decoster C, Mechin MC, et al. Defects of filaggrin-like proteins in both lesional and nonlesional atopic skin. J Allergy Clin Immunol. 2013;131(4):1094–102. PubMed PMID: 23403047. Epub 2013/02/14. Eng.
Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. 2009;10(8):857–63. PubMed PMID: 19578369.
Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest. 2009;119(12):3573–85. PubMed PMID: 19920355. Pubmed Central PMCID: 2786807.
Fujita H, Nograles KE, Kikuchi T, Gonzalez J, Carucci JA, Krueger JG. Human Langerhans cells induce distinct IL-22-producing CD4+ T cells lacking IL-17 production. Proc Natl Acad Sci U S A. 2009;106(51):21795–800. PubMed PMID: 19996179. Pubmed Central PMCID: 2799849.
Niebuhr M, Scharonow H, Gathmann M, Mamerow D, Werfel T. Staphylococcal exotoxins are strong inducers of IL-22: a potential role in atopic dermatitis. J Allergy Clin Immunol. 2010;126(6):1176–83 e4. PubMed PMID: 20864149.
Gutowska-Owsiak D, Schaupp AL, Salimi M, Taylor S, Ogg GS. Interleukin-22 downregulates filaggrin expression and affects expression of pro-filaggrin processing enzymes. Br J Dermatol. 2011;165(3):492–8. PubMed PMID: 21564072. Epub 2011/05/14. eng.
Asarch A, Barak O, Loo DS, Gottlieb AB. Th17 cells: a new paradigm for cutaneous inflammation. J Dermatolog Treat. 2008;19(5):259–66. PubMed PMID: 18629676.
McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006;27(1):17–23. PubMed PMID: 16290228.
Lee J, Ho WH, Maruoka M, Corpuz RT, Baldwin DT, Foster JS, et al. IL-17E, a novel proinflammatory ligand for the IL-17 receptor homolog IL-17Rh1. J Biol Chem. 2001;276(2):1660–4. PubMed PMID: 11058597.
Gutowska-Owsiak D, Schaupp AL, Salimi M, Selvakumar TA, McPherson T, Taylor S, et al. IL-17 downregulates filaggrin and affects keratinocyte expression of genes associated with cellular adhesion. Exp Dermatol. 2012;21(2):104–10. PubMed PMID: 22229441.
Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med. 2006;203(4):1105–16. PubMed PMID: 16606668. Pubmed Central PMCID: 2118283.
Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, et al. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J Exp Med. 2007;204(8):1837–47. PubMed PMID: 17635955. Pubmed Central PMCID: 2118667.
Hvid M, Vestergaard C, Kemp K, Christensen GB, Deleuran B, Deleuran M. IL-25 in atopic dermatitis: a possible link between inflammation and skin barrier dysfunction? J Invest Dermatol. 2011;131(1):150–7. PubMed PMID: 20861853. Epub 2010/09/24. eng.
Richardson SK, Gelfand JM. Update on the natural history and systemic treatment of psoriasis. Adv Dermatol. 2008;24:171–96. PubMed PMID: 19256309. Pubmed Central PMCID: 2634854.
Deleuran MS, Vestergaard C. Therapy of severe atopic dermatitis in adults. J Dtsch Dermatol Ges. 2012;10(6):399–406. PubMed PMID: 22606964.
Kim BE, Howell MD, Guttman-Yassky E, Gilleaudeau PM, Cardinale IR, Boguniewicz M, et al. TNF-alpha downregulates filaggrin and loricrin through c-Jun N-terminal kinase: role for TNF-alpha antagonists to improve skin barrier. J Invest Dermatol. 2011;131(6):1272–9. PubMed PMID: 21346775. Epub 2011/02/25. eng.
Dillon SR, Sprecher C, Hammond A, Bilsborough J, Rosenfeld-Franklin M, Presnell SR, et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. 2004;5(7):752–60. PubMed PMID: 15184896.
Niyonsaba F, Ushio H, Hara M, Yokoi H, Tominaga M, Takamori K, et al. Antimicrobial peptides human beta-defensins and cathelicidin LL-37 induce the secretion of a pruritogenic cytokine IL-31 by human mast cells. J Immunol. 2010;184(7):3526–34. PubMed PMID: 20190140.
Grimstad O, Sawanobori Y, Vestergaard C, Bilsborough J, Olsen UB, Gronhoj-Larsen C, et al. Anti-interleukin-31-antibodies ameliorate scratching behaviour in NC/Nga mice: a model of atopic dermatitis. Exp Dermatol. 2009;18(1):35–43. PubMed PMID: 19054054.
Cornelissen C, Marquardt Y, Czaja K, Wenzel J, Frank J, Luscher-Firzlaff J, et al. IL-31 regulates differentiation and filaggrin expression in human organotypic skin models. J Allergy Clin Immunol. 2012;129(2):426–33, 33 e1–8. PubMed PMID: 22177328. Epub 2011/12/20. eng.
Jensen JM, Scherer A, Wanke C, Brautigam M, Bongiovanni S, Letzkus M, et al. Gene expression is differently affected by pimecrolimus and betamethasone in lesional skin of atopic dermatitis. Allergy. 2012;67(3):413–23. PubMed PMID: 22142306. Epub 2011/12/07. eng.
Asselineau D, Dale BA, Bernard BA. Filaggrin production by cultured human epidermal keratinocytes and its regulation by retinoic acid. Differentiation. 1990;45(3):221–9. PubMed PMID: 2090523.
Kautsky MB, Fleckman P, Dale BA. Retinoic acid regulates oral epithelial differentiation by two mechanisms. J Invest Dermatol. 1995;104(4):546–53. PubMed PMID: 7706775.
van den Bogaard EH, Bergboer JG, Vonk-Bergers M, van Vlijmen-Willems IM, Hato SV, van der Valk PG, et al. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J Clin Invest. 2013;123(2):917–27. PubMed PMID: 23348739. Pubmed Central PMCID: PMC3561798. Epub 2013/01/26. Eng.
Barnes PJ. Corticosteroids: the drugs to beat. Eur J Pharmacol. 2006;533(1–3):2–14. PubMed PMID: 16436275.
Liu J, Farmer Jr JD, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66(4):807–15. PubMed PMID: 1715244.
Ruzicka T, Lynde CW, Jemec GB, Diepgen T, Berth-Jones J, Coenraads PJ, et al. Efficacy and safety of oral alitretinoin (9-cis retinoic acid) in patients with severe chronic hand eczema refractory to topical corticosteroids: results of a randomized, double-blind, placebo-controlled, multicentre trial. Br J Dermatol. 2008;158(4):808–17. PubMed PMID: 18294310.
Grahovac M, Molin S, Prinz JC, Ruzicka T, Wollenberg A. Treatment of atopic eczema with oral alitretinoin. Br J Dermatol. 2010;162(1):217–8. PubMed PMID: 19886882.
Cork MJ, Danby SG, Vasilopoulos Y, Hadgraft J, Lane ME, Moustafa M, et al. Epidermal barrier dysfunction in atopic dermatitis. J Invest Dermatol. 2009;129(8):1892–908. PubMed PMID: 19494826.
Kezic S, O’Regan GM, Lutter R, Jakasa I, Koster ES, Saunders S, et al. Filaggrin loss-of-function mutations are associated with enhanced expression of IL-1 cytokines in the stratum corneum of patients with atopic dermatitis and in a murine model of filaggrin deficiency. J Allergy Clin Immunol. 2012;129(4):1031–9 e1. PubMed PMID: 22322004. Epub 2012/02/11. eng.
Lee KH, Cho KA, Kim JY, Kim JY, Baek JH, Woo SY, et al. Filaggrin knockdown and toll-like receptor 3 (TLR3) stimulation enhanced the production of thymic stromal lymphopoietin (TSLP) from epidermal layers. Exp Dermatol. 2011;20(2):149–51. PubMed PMID: 21255094. Epub 2011/01/25. eng.
Thyssen JP, Linneberg A, Ross-Hansen K, Carlsen BC, Meldgaard M, Szecsi PB, et al. Filaggrin mutations are strongly associated with contact sensitization in individuals with dermatitis. Contact Dermatitis. 2013;68(5):273–6. PubMed PMID: 23343419.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Vestergaard, C., Deleuran, M.S. (2014). Inflammatory-Driven Depletion of Filaggrin Proteins. In: Thyssen, J., Maibach, H. (eds) Filaggrin. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-54379-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-642-54379-1_4
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-54378-4
Online ISBN: 978-3-642-54379-1
eBook Packages: MedicineMedicine (R0)