
Abstract
Posterior uveitis (PU) represents a category of ocular inflammatory diseases, which can lead to a severe visual impairment. Many infectious agents can be the trigger of PU, such as Toxoplasma gondii, Toxocara canis, Mycobacterium tuberculosis, and syphilis. In addition, very recently, other infectious agents have been recognized as promoters of PU. Among these new entities, we recognize the West Nile fever virus, chikungunya virus, and spirochetes of the genus Leptospira.
Noninfectious uveitis is another subgroup, which is included in the family of PU. This category includes a wide spectrum of posterior uveitis which is characterized by a heterogeneous pattern of presentation. The therapeutic management is still a challenge for ocular immunologists and the hottest topic of such scientific area. In this chapter, we will show the clinical assessment and the therapeutic techniques used for the management of such diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
- Retinal Pigment Epithelium
- Cystoid Macular Edema
- Serous Retinal Detachment
- Posterior Uveitis
- Sympathetic Ophthalmia
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
10.1 Infectious Posterior Uveitis
10.1.1 Viral Posterior Uveitis
10.1.1.1 Acute Retinal Necrosis Syndrome
10.1.1.1.1 Definition
Acute retinal necrosis (ARN) is a condition that was initially described in 1971 by Urayama [115]. It is a fulminant viral infection caused by members of the herpesvirus family. ARN is characterized by peripheral full-thickness retinitis with discrete borders, occlusive vasculopathy with arteriolar involvement, rapid progression with circumferential spread in untreated eyes, and marked vitritis. Late retinal detachment remains a serious complication despite prophylactic laser photocoagulation and vitreoretinal surgery.
ARN is generally diagnosed on the basis of its clinical features, as summarized by the diagnostic criteria established by the executive committee of the American Uveitis Society (Table 10.1).
10.1.1.1.2 Epidemiology/Etiology
Necrotizing retinitis from alpha herpesviruses HSV and VZV is a rare disease. The age of onset has a bimodal distribution with peaks occurring at ages of 20 and 50. The disease is prevalently diffused in the elderly population and in immune-deficient patients, through the fifth to seventh decade. HSV infection seems to involve early adulthood, while VZV dermatitis seems to involve the older population. The etiology of ARN was clarified in 1982 when it was shown that almost every member of the herpesvirus family could be implicated as a causative agent.
10.1.1.1.3 Clinical Symptoms and Signs
ARN is characterized by acute peripheral necrotizing retinitis with well-demarcated borders and a tendency to rapidly spread towards the posterior pole. ARN is commonly associated with mild to severe vitritis and retinal arteriolitis in the context of an occlusive vasculopathy [121] (EBM:2+, C). Subsequent optic neuropathy is a frequent consequence of vasculitis. ARN is usually a unilateral disease, but in almost one-third of patients, the second eye becomes involved within 6 weeks.
ARN may begin with an anterior granulomatous uveitis. Usually within 21 days, the retinal necrosis reaches its maximum extension, and the macula is often spared. The regression of ARN leads to retinal atrophy in a Swiss cheese-like pattern. The massive cellular infiltration of the vitreous body (Fig. 10.1) creates the conditions for membrane proliferation with subsequent PVD and rhegmatogenous retinal detachment and a subsequent proliferative vitreoretinopathy. If there is a severe inflammatory response, an exudative retinal detachment can occur.

Acute retinal necrosis: color fundus photograph showing a dense vitritis with retinal vasculitis (black arrows). Fluorescein angiography (b) proves the occlusive nature of the retinal vasculitis (black arrows)
10.1.1.1.4 Differential Diagnosis of Acute Retinal Necrosis (Fig. 10.2)

Decision tree summarizing recommended approach to the patient with retinitis of unclear etiology [3]
CMV retinitis mostly affects immunocompromised patients. Toxoplasmic retinochoroiditis is the most frequent disease simulating necrotizing viral retinopathies; hemorrhage is not a characteristic of its lesions. Behcet’s disease is a systemic disease associated with an inconstant course of remissions and exacerbations. Intraocular lymphoma is characterized by a slow course as compared to ARN.
10.1.1.1.5 Treatment
The therapeutic strategy of alpha herpesvirus retinitis includes antivirals, anti-inflammatories, and antiglaucomatous medications. Intravenous administration of acyclovir remains the main approach, in the light of its efficacy against both HSV and VZV. It is given for 14–21 days; after that, it is switched to 4 g of acyclovir or 3 g of valacyclovir daily per os for a period of 1–3 months. Usually, lesions stabilize within 48 h, but in resistant cases, intravenous foscarnet or ganciclovir is employed ([128] (EBM: 1-, A)). Acyclovir significantly decreases contralateral disease as compared with those untreated ([121] (EBM:2++, C), [128] (EBM: 1-, A), [188] (EBM:2++, C)). Recently, oral valacyclovir has been successfully used for the treatment of ARN, even though its role has still to be discussed ([250] (EBM: 3, C)). Anti-inflammatory drugs are still being discussed in ARN syndrome: they are employed to minimize damages to the optic nerve and retinal vessels. On the other hand, they have to be used only in conjunction with antivirals. Steroids must be started at 1 mg/kg and progressively tapered ([275] (EBM: 1-, A)). The role of anticoagulants and aspirin remains controversial, in particular, their effect on the occlusive vasculopathy. Retinal detachment in ARN syndrome remains a major problem with an incidence of 75 % in the untreated patients. Prophylactic vitrectomy and laser photocoagulation ([196] (EBM: 4, D)) are associated with a variable visual function and are still controversial.
Very recently, Wong et al. ([274] (EBM: 2++, B)) have proposed to treat ARN with intravitreal foscarnet. The authors evaluated 33 eyes with HSV-ARN and 48 with VZV-ARN. Visual acuity on presentation was similar (p = 0.48), but a larger proportion had better vision (> or =20/60) in the HSV-ARN group (52 %) than the VZV-ARN group (35 %). A greater proportion of eyes with poor vision (< or =20/200) was found at the 12-month follow-up in the VZV-ARN group (60 %) compared with the HSV-ARN group (35 %). A greater degree of visual loss in the VZV-ARN group compared with the HSV-ARN group was detected. Retinal detachment was 2.5-fold more commonly observed in VZV-ARN (62 %) compared with HSV-ARN (24 %). The eyes treated with (n = 56) intravitreal foscarnet had 40 % lower rate in retinal detachment than those without (n = 25) intravitreal treatment for VZV-ARN (p = 0.23). Intravitreal foscarnet seemed to be a useful adjunct for the treatment of ARN in order to lower the rate of retinal detachment.
All commercially available antiviral drugs to date are virostatic, and this explains the frequent relapses, particularly, in absence of antiviral prophylaxis (Table 10.2).
10.1.1.2 Progressive Outer Retinal Necrosis
10.1.1.2.1 Definition
The designation of progressive outer retinal necrosis (PORN) generally describes patients with AIDS (CD4+ Tlymphocytes ≤50 cells/μl) or who are profoundly immunosuppressed.
PORN is a herpetic retinitis with less inflammation and a more aggressive clinical course than ARN. It is thought to be the second most frequent opportunistic retinal infection in patients with AIDS in North America.
10.1.1.2.2 Etiology
PORN is thought to be a variant necrotizing herpetic retinopathy in immunocompromised patients. There is sufficient evidence to identify VZV and HSV as causative factors of PORN. Very often, patients with PORN are infected with HIV (human immunodeficiency virus) and generally have advanced AIDS.
10.1.1.2.3 Clinical Symptoms and Signs
PORN is characterized by a sudden necrotizing retinitis of the deep retinal layers, starting at the posterior pole and arranged in a multifocal pattern. These inflammatory spots have a marked tendency towards peripheral spreading and confluence. Unlike ARN syndrome, retinal vasculitis, inflammatory reaction of the vitreous, and optic neuropathy are less common, particularly when associated with low Th-CD4+ cell counts. The frequent occurrence of retinal detachment and the marked resistance to antivirals make the visual prognosis extremely poor.
10.1.1.2.4 Differential Diagnosis
The differential diagnosis for PORN is similar to that of ARN. It is very important to differentiate these two disorders on the basis of precise criteria, such as the pattern of distribution of retinal lesions, as well as the depth of the necrosis in the retinal layers (outer vs. full thickness), involvement of posterior pole vs. midperiphery, and presence of vitritis and vasculitis.
10.1.1.2.5 Treatment
Since viral replication is less prone to be controlled in patients affected by PORN syndrome, several combinations of intravenous and intravitreal antivirals have been tried in order to stop the progression of the necrosis ([137] (EBM D 3) and [284] (EBM D 3)), with little or even lack of evident efficacy. However, aggressive therapy based on intravenous foscarnet or ganciclovir and intravitreal ganciclovir remains the mainstay of therapy ([82], (EBM:3, D)). Several papers have reported that combination of antiviral therapy and highly active antiretroviral therapy (HAART) may improve long-term visual outcomes for VZV-PORN (Ferente [137] (EBM D 4) and [284] (EBM D 4)). Corticosteroids generally must be avoided in order to prevent complications resulting from viral replication.
Core Message
-
The lytic reaction caused by herpetic ocular infection is accompanied by subsequent ocular inflammation.
-
The diagnosis is based on clinical typical findings; recently, molecular techniques such as PCR have been applied to ocular fluids.
-
Systemic antivirals are crucial in the control of viral replication. They should be used before corticosteroids.
-
Antiviral prophylaxis is very important in preventing relapses.
10.1.1.3 Cytomegalovirus Retinitis
10.1.1.3.1 Definition
Cytomegalovirus (CMV) is a beta herpesvirus and contains double-stranded DNA. Commonly, CMV retinitis tends to occur in patients whose immune system has been significantly depressed, such as HIV [23]. In 1997, Whitcup et al. [269] reported that CMV retinitis did not progress in patients receiving HAART, albeit they were not receiving any anti-CMV therapy.
10.1.1.3.2 Etiology
CMV reaches the retina via bloodstream and infects the vascular endothelium which then spreads to the retinal cells. Infected cells show the pathognomonic cytomegalic inclusions with intracellular, large, and eosinophilic bodies.
10.1.1.3.3 Clinical Symptoms and Signs
Patients present blurred vision with acute visual impairment.
Histopathology shows a full-thickness retinal necrosis, associated with coagulative vasculitis and choroiditis. The typical chorioretinal lesions observed in CMV retinitis include:
-
Hemorrhagic pattern which shows confluent area of full-thickness retinal necrosis with a yellow-white granular appearance, called “pizza pie” (Fig. 10.3)
Fig. 10.3 
Cytomegalovirus retinitis: “pizza pie” fundus (a) before the treatment. Note the improvement of the clinical picture after the treatment with ganciclovir and steroids (b)
-
“Brush-fire” pattern showing a rapid spreading of the CNV in the retinal tissue
-
“Granular pattern” which presents areas of retinal atrophy surrounded by whitish granular punctate lesions

Vitreous involvement can be variable in all the different retinal patterns.
One of the most severe complications of CMV retinitis is rhegmatogenous retina detachment. Persistent cystoid macular edema can occur.
10.1.1.3.4 Differential Diagnosis
Although the diagnosis of CMV retinitis is prevalently based on clinical criteria, the similarities between CMV retinitis and alpha herpesvirus retinitis cannot be easily distinguished. In order to make a correct diagnosis, both aqueous and vitreal polymerase chain reaction (PCR) analysis of intraocular antibody synthesis can confirm the diagnosis [245].
10.1.1.3.5 Treatment
The therapeutic approach to CMV retinitis is based on the patient’s immune status, which requires an interdisciplinary approach.
Up to date, ganciclovir represents the drug used as the first line. The standard dose of ganciclovir is 5 mg/kg intravenously every 12 h for 2 weeks followed by maintenance at 10 mg/kg/day ([269], EBM: 1 + A). Neutrophil count should be maintained higher than 500/μl.
Foscarnet is also used, particularly for those patients who have a low neutrophil count. The standard dose is 90 mg/kg twice daily, followed by maintenance therapy with 90–120 mg/kg [269]. Serum electrolytes should be regularly monitored.
Besides the traditional systemic approach, an intraocular ganciclovir implant seemed to be superior to intravenous ganciclovir in a large randomized controlled trials of HIV-associated CMV retinitis in the era before HAART ([174], EBM: B, 2++]). Unfortunately, the limitation of the intraocular ganciclovir implant to prevent CMV disease in the fellow eye represented its failure. As an alternative, oral valganciclovir has been proven as effective as initial intravenous ganciclovir for 4 weeks followed by oral valganciclovir. During the latter trial, most patients were also taking combination anti-HIV treatment. As the ocular penetration of systemically administered anti-CMV drugs is limited, current clinical guidelines include consideration of intraocular injection of anti-CMV drugs for patients who have sight-threatening CMV retinitis ([165], EBM: B, 2++).
Core Message
-
CMV is a highly adapted opportunistic agent, which can induce a severe sight-threatening retinitis.
-
Although the diagnosis of CMV retinitis is based on clinical criteria, PRC of ocular fluids is useful to detect the specific viral agent involved in the pathogenesis of the disease.
-
For the treatment of CMV retinitis, the sustained-release ganciclovir implant is more effective than intravenous ganciclovir, but patients treated with a ganciclovir implant alone remain at greater risk for the development of CMV disease in the fellow eye ([245], EBM: B, 2++).
-
Orally administered valganciclovir appears to be as effective as intravenous ganciclovir for induction treatment and is convenient and effective for the long-term management of cytomegalovirus retinitis in immunocompromised patients ([269], EBM: B, 2++).
10.1.2 Human Immunodeficiency Virus (HIV) Retinal Microvasculopathy
10.1.2.1 Definition
Human immunodeficiency virus (HIV)-1 is a single-strand RNA virus and represents the most widespread type of HIV within the retrovirus family.
Until now, HIV infection remains a worldwide diffused disease, with different prevalences depending on both socioeconomic and geographic factors [189].
10.1.2.2 Etiology
HIV can be sexually transmitted, even though intravenous and perinatal infections can occur. Incubation lasts approximately 3 weeks after which an acute retroviral syndrome can occur. Symptomatology includes fever, rash, myalgias, headaches, and gastrointestinal involvement. However, acute symptoms are not frequently observed.
Acquired immunodeficiency syndrome (AIDS) is the most severe manifestation of immunodepression, secondary to the progressive reduction of T-helper CD4+. AIDS typically leads to a significantly higher risk of developing opportunistic infections, such as CMV retinitis, which can occur as soon as Th-CD4+ count is <50 cells/μl ([145] (EBM: B2++)). Other ocular opportunistic infections include syphilis, toxoplasmosis, tuberculosis, candidosis, herpes simplex virus, and herpes zoster virus ([189] (EBM: C2+), [23] (EBM: B2++)).
10.1.2.3 Clinical Symptoms and Signs
The most common ocular finding is retinal microvasculopathy, which is characterized by small retinal hemorrhages and cotton-wool spots ([23] (EBM: B2++)). Up to date, pathophysiology has not been clearly demonstrated.
10.1.2.4 Differential Diagnosis
HIV retinopathy should be differentiated from the cotton-wool spots observed in the diabetic retinopathy, as well as in the hypertensive retinopathy.
10.1.2.5 Treatment
Up to date, there are no meta-analyses or systematic reviews of randomized clinical trials available about HIV-related retinal microvasculopathy. The clinical course of HIV infection has been dramatically reduced as soon as highly active antiretroviral therapy (HAART) has been introduced. Unfortunately, the availability of HAART is limited in developing countries. The treatment of HIV-related retinopathy is substantially not indicated ([23] (EBM: B2++)).
Core Message
-
Retinal microvasculopathy is the most common ocular manifestation that does not require treatment.
-
A significant higher risk of developing opportunistic infections when Th-CD4+ count is <50 cells/μl.
-
The clinical course of the disease has been dramatically improved since HAART was introduced.
10.1.3 Other Viral Uveitis
10.1.3.1 West Nile Virus
10.1.3.1.1 Definition
West Nile virus (WNV) is a zoonotic disease most often transmitted to humans by an infected Culex mosquito vector where wild birds serve as a vector. It is an enveloped single-stranded RNA flavivirus, member of the Japanese encephalitis virus serocomplex [87]. The disease has its peak in late summer.
10.1.3.1.2 Clinical Symptoms and Signs
About 80 % of human infections are apparently asymptomatic and the remaining 20 % become symptomatic manifesting almost always a self-limited febrile illness. The symptoms include high-grade fever, myalgia, arthralgia, malaise, nausea, headache, skin rash, weakness, and pharyngitis [13]. The acute illness typically lasts less than a week.
Since first described in 2002, several forms of ocular involvement have been recognized. Multifocal chorioretinitis [132], typically bilateral, with specific clinical and angiographic features is the most common finding, occurring in almost 80 % of patients with acute WNV infection. An associated mild to moderate vitreal inflammation is observed. Chorioretinal lesions involve the midzone and periphery in almost all eyes. The posterior pole is involved in nearly two-third of the eyes. Active lesions appear circular and creamy in ophthalmoscopy associated by early hypofluorescence and late staining in fluorescein angiography. Their size is variable. The linear cluster arrangement that the lesions take is a prominent feature. These streaks are typically oriented radially in the nasal and peripheral fundus or arranged in a curvilinear pattern in the temporal posterior fundus [131] (Fig. 10.4). ICGA tends to denote more choroidal lesions than those appreciated by fluorescein angiography. Diabetes mellitus appears being a risk factor for WNV-associated chorioretinitis [134].

Red-free fundus photograph (a) and fluorescein angiogram (b) of the left eye of a 64-year-old diabetic woman with West Nile virus infection show inactive multifocal chorioretinitis with a typical linear clustering of chorioretinal lesions. Note the presence of retinal arterial sheathing (a) and diabetic macular edema
Other ophthalmic manifestations include retinal hemorrhages, vascular sheathing and leakage, and occlusive vasculitis. Optic nerve involvement includes optic neuritis and optic disk swelling and staining [280].
10.1.3.1.3 Differential Diagnosis
The differential diagnosis of WNV systemic disease includes herpesvirus encephalitis, CNS involvement by legionella, rickettsioses, Epstein-Barr virus, hypertensive encephalopathy, and enteroviral aseptic meningitis. While the ocular involvement can enter in the differential diagnosis of syphilis, TBC, histoplasmosis, sarcoidosis, and idiopathic multifocal chorioretinitis.
10.1.3.1.4 Treatment
At present, there is no proven treatment for WNV infection ([87], EBM: C, 2-). Specific ophthalmic treatment such as topical steroids for anterior uveitis, peripheral retinal photocoagulation due to occlusive vasculitis, pars plana vitrectomy for vitreal hemorrhages or retinal detachment, and photodynamic therapy and anti-VEGF for choroidal neovascularization may be required in these specific situations ([226], EMB: 3, D).
Core Message
-
The most common intraocular finding of West Nile virus is the bilateral multifocal chorioretinitis which is frequently self-limited and asymptomatic in the majority of patients where the CNS is affected.
-
Multifocal chorioretinitis manifests a unique pattern which is helpful for diagnosis.
10.1.3.2 Dengue Fever
Dengue fever (DF) is caused by any of the four immunologically related serotypes of the dengue virus, which belong to the genus Flavivirus of the family Flaviviridae. It is transmitted through the bite of an infected female Aedes aegypti/Aedes albopictus mosquito.
DF is considered to be one of the most important arthropod borne disease in the tropical and subtropical regions, being endemic in more than 100 countries, including America, Southeast Asia, Western Pacific, Africa, and the Eastern Mediterranean [276].
10.1.3.2.1 Clinical Presentation
10.1.3.2.1.1 Systemic Disease
The incubation period for DF varies from 3 to 14 days. The initial infection may be asymptomatic, may result in a nonspecific febrile illness, or may produce features of classic DF including sudden onset of high fever, severe headache, myalgias, arthralgias, nausea, vomiting, and a maculopapular rash. The majority of DF cases are self-limiting. A small proportion of affected patients may develop life-threatening dengue hemorrhagic fever syndrome, which is characterized by increased capillary permeability and hemostatic disturbances, or dengue shock syndrome, which is characterized by severe systemic hypotension. DF is often associated with a bleeding tendency secondary to thrombocytopenia [53, 107].
10.1.3.2.1.2 Ocular Disease
The ocular involvement was found to occur in 10 % of patients hospitalized for serologically confirmed DF. It usually occurs within one month after onset of symptoms of DF and is often bilateral. A subconjunctival hemorrhage, petechial in type and associated with a platelet count of less than 50,000/μl, was the most common ocular manifestation in an East Indian population with DF [133]. Numerous posterior segment changes have been associated with DF including retinal hemorrhages, retinal vasculitis, yellow subretinal dots, retinal pigment epithelial mottling, and foveolitis, seen clinically as a round yellowish lesion at the fovea with corresponding focal outer neurosensory retina-retinal pigment epithelium thickening on OCT. Other findings include macular edema, serous retinal detachment, retinal vascular occlusion, choroidal changes, optic disk swelling, optic neuritis, and neuroretinitis [53, 58, 62, 67, 125, 127, 150, 237].
Dengue-associated ocular disease usually has a self-limited course, with a significant improvement of visual acuity in 2–4 weeks. However, persistent visual impairment may occur in a subset of patients with maculopathy or neuropathy [58, 62, 150].
10.1.3.2.2 Laboratory Diagnosis
Within the first 2 days of fever, diagnosis is possible only by detecting the virion, RNA, or dengue proteins, such as nonstructural protein 1 (NS1).
Detection of newly formed antibodies (IgM) usually is not possible until after viremia ends or after fever subsides [270]. MAC-ELISA has become a widely used assay but seems to have a high rate of false-positive results [260]. Other tests, including immunochromatographic assay [33], complement fixation, neutralization test, hemagglutination inhibition, and IgG enzyme-linked immunosorbent assay (ELISA), are also helpful to confirm the diagnosis of DF [260].
Apart from the dengue-specific parameters, platelet count should be performed.
10.1.3.2.3 Treatment
To date, there is no specific treatment available for dengue virus infection. Any medicine that decreases the platelet level should be avoided ([107] EBM:C4, [58] EBM:D3). In cases of dengue hemorrhagic fever, hospitalization, prompt treatment with intravenous fluids, and close monitoring of vital signs, as well as hematologic parameters, are indicated ([107] EBM; C4).
There is no established treatment for ocular manifestations of DF. Topical, periocular, oral, and intravenous steroids, as well as intravenous immunoglobulins, have been advocated for the management of dengue ocular complications, based on the postulated immune-mediated pathogenesis of the disease. Indications for treatment may include dengue-associated uveitis and optic neuritis, visual acuity worse than 20/40, and deterioration of vision ([133] EBM:D 4, 58). Preventive measures by avoiding contact with infected mosquitoes are required to decrease the infection incidence. Vaccines targeting all the four serotypes of dengue virus hopefully will be available in the near future ([57] EBM C 2+, [240] EBM C 2+).
Core Message
-
Dengue occurs in 10 % of patients hospitalized for serologically confirmed DF.
-
Ocular involvement can present different clinical patterns.
-
Treatment is still controversial.
10.1.3.3 Chikungunya
Chikungunya virus is a single-stranded RNA virus of the genus Alphavirus in the family Togaviridae which is transmitted to humans by the bite of infected Aedes mosquitoes (A. aegypti and A. albopictus). Since its first isolation in Tanzania in 1953, the virus has been associated with many epidemics in tropical regions of Africa, India, Southeast Asia, and South America. The infection which is endemoepidemic typically consists of an acute illness with fever, severe arthralgia, and skin rash [205].
10.1.3.3.1 Clinical Presentation
10.1.3.3.1.1 Systemic Disease
The incubation period ranges from 1 to 12 days, with an average of 2–4 days. Onset of the disease is abrupt and is characterized by high fever, severe arthralgia, and myalgia, along with headache and skin rash. Asymptomatic infections are rare (3–25 % of serologically proven infections) [47]. The debilitating polyarthralgia is very characteristic of chikungunya. Skin lesions may be seen in almost one-half of the patients. A pruriginous maculopapular rash, lasting for 2–3 days, is the most common feature [43, 202]. Rarely, severe infection associated with multiorgan failure, central neurological involvement, neonatal infection, and death occur [43, 202].
10.1.3.3.1.2 Ocular Disease
Ocular manifestations associated with chikungunya may be concomitant of the systemic disease or may follow its resolution [133]. Ocular involvement can be unilateral or bilateral. Acute anterior uveitis and retinitis are the most common ocular findings in chikungunya. The anterior uveitis is nongranulomatous or granulomatous and can be associated with increased intraocular pressure. Posterior synechiae are not common [133, 172]. The clinical course is typically benign.
Chikungunya retinitis presents in the form of areas of retinal whitening in the posterior pole with surrounding retinal and macular edema and associated mild vitritis [133]. FA usually shows early hypofluorescence and late hyperfluorescence of retinal lesions, along with focal areas of retinal vascular leakage and capillary non-perfusion [133]. OCT reveals increased reflectivity in the nerve fiber layer zone with aftershadowing corresponding to the areas of retinitis. It also helps in the detection and evaluation of associated retinal edema and exudative retinal detachment. Retinitis resolves gradually over a period of several weeks.
Other ophthalmic manifestations of chikungunya have been reported including conjunctivitis, episcleritis, keratitis, panuveitis, multifocal choroiditis, optic neuritis, neuroretinitis, central retinal artery occlusion, exudative retinal detachment, panophthalmitis, lagophthalmos, and sixth nerve palsy [133, 171].
Chikungunya-associated ocular disease is usually self-limiting, with most patients recovering good vision. However, permanent visual loss may occur mainly due to optic neuropathy.
10.1.3.3.2 Laboratory Diagnosis
In the acute phase of illness, diagnosis is based on the detection of viral nucleic acid in serum samples by RT-PCR, isolation of the virus, or detection of an antibody response. After resolution of the acute disease, the diagnosis is confirmed by the presence of an immune response. RT-PCR can detect viral nucleic acid from one day before onset of symptoms, up to day 7 after the beginning of the disease. Antigen capture ELISA may detect viral antigens as early as day 2 after onset. Indirect immunofluorescence and ELISA are rapid and sensitive techniques for the screening of IgM or IgG immune reaction. IgM antibody and IgG antibody response have been described to begin both by day 2 after onset [246].
10.1.3.3.3 Treatment
Nonsteroidal anti-inflammatory drugs are currently recommended for chikungunya-induced arthralgia. Ribavirin and interferon α may inhibit viral replication ([45] EBM C 2+), but further studies are needed to assess their efficacy in humans. Another potential treatment for chikungunya is chloroquine, but results of different studies have been inconclusive ([44] EBM C 2-, [69] EBM C 2 +).
Topical steroids and cycloplegic agents are used for anterior uveitis. Associated ocular hypertension is managed with topical beta-blockers and oral or topical carbonic anhydrase inhibitors. Systemic steroids may be used to control the inflammation in posterior uveitis, panuveitis, and optic neuritis ([133] EBM D 3). The use of acyclovir in association with corticosteroids has been described in some cases of chikungunya retinitis [133], but its efficacy remains doubtful. Efforts are to be made to prevent transmission of the virus and to develop efficient and safe vaccines against chikungunya ([47] EBM D 3).
Core Message
-
Ocular involvement in chikungunya can occur after the systemic disease.
-
Ocular chikungunya can present various clinical patterns.
-
No valid treatment is available at this time.
10.1.4 Posterior Uveitis: Bacterial Infections
10.1.4.1 Intraocular Tuberculosis
10.1.4.1.1 Definition
Intraocular inflammation associated with Mycobacterium tuberculosis [29].
10.1.4.1.2 Etiology
This condition is caused by dissemination of M. tuberculosis to ocular tissues, from the lung. The exact sequence of events is not known. However, there is histopathologic and molecular (polymerase chain reaction, PCR) evidence of the presence of this organism in the diseased eyes [258].
10.1.4.1.3 Clinical Symptoms and Signs
Intraocular tuberculosis can affect virtually every tissue in the eye. The clinical manifestations can be broadly classified as follows (Adapted from Ref. [29]):
-
1.
Anterior uveitis
-
Granulomatous, nongranulomatous, iris nodules, and ciliary body tuberculoma
-
-
2.
Intermediate uveitis granulomatous
-
3.
Posterior and panuveitis
-
Choroidal tubercle
-
Choroidal tuberculoma
-
Subretinal abscess
-
Multifocal serpiginoid choroiditis (previously called serpiginous-like choroiditis)
-
-
4.
Retinitis and retinal vasculitis
-
5.
Neuroretinitis and optic neuropathy
-
6.
Endophthalmitis and panophthalmitis
In a high endemic population, the following clinical signs were statistically found to be predictive of intraocular tuberculosis, in patients with latent or manifest systemic tuberculosis ([104], EBM: C, 2+):
-
Broad-based posterior synechiae
-
Retinal vasculitis with or without choroiditis patches overlying the blood vessels (Fig. 10.5)
Fig. 10.5 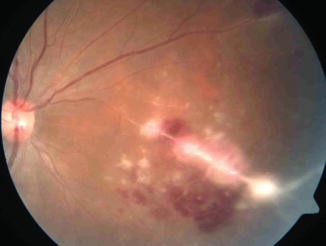
Tubercular retinal vasculitis showing perivascular exudation and hemorrhages, associated with active chorioretinitis patch overlying the blood vessel
-
Multifocal serpiginoid choroiditis

Infectious multifocal serpiginoid choroiditis: It is a form of superficial choroiditis characterized by multifocal lesions (Fig. 10.6) that are noncontiguous to the optic disk and show serpiginoid or ameboid spread ([26, 101], EBM:C, 2+). Lesions are often bilateral and have associated vitreous inflammation. The fovea is usually spared resulting in good final visual acuity. Fundus autofluorescence (FAF) and spectral domain optical coherence tomography (SD-OCT) are useful in following disease activity during the course of the disease. FAF usually shows hyperautofluorescence with ill-defined halo in acute stage with gradual stippling and progressive hypoautofluorescence as the lesions heal [102]. SD-OCT shows increased reflectivity from outer retinal layers in the acute stage followed by knobbly elevations in the outer retina and finally outer retinal atrophy and increased choroidal reflectivity [24]. Such lesions may also be observed in herpetic viral infections and syphilis in tuberculosis non-endemic regions [101].

Multifocal serpiginoid choroiditis showing multifocal areas of healed and active choroiditis
Infectious multifocal serpiginoid choroiditis needs to be distinguished from classical serpiginous choroiditis that is seen in non-endemic populations and is characterized by large, peripapillary lesions that are rarely multifocal or associated with vitritis [101, 175].
10.1.4.1.4 Diagnosis
Currently, intraocular tuberculosis is mostly diagnosed based on characteristic clinical signs (mentioned above), associated ancillary tests (immunologic and radiological), and exclusion of other disease entities – infectious and noninfectious – that may have similar clinical presentation, in a given geographic region [29].
Immunologic tests include the tuberculin skin test (TST) and interferon-gamma release assays like the QuantiFERON-TB Gold test (QFT) and the T-SPOT test. Current evidence shows that while TST is more sensitive [278], QFT and T-SPOT are more specific for the diagnosis of presumed ocular tuberculosis ([11, 12], EBM: C, 2+). TST and T-SPOT test should be the investigation of choice in high and low endemic populations, respectively ([12], EBM:C, 2+).
However, the absence of systemic evidence of tuberculosis (immunologic and radiological) needs to be interpreted with caution while diagnosing intraocular tuberculosis. In a large series of 42 cases of histopathologically proven cases of ocular tuberculosis, 40 % of tested patients had negative TST and 57 % had normal chest radiograph [258]. Thus, there is a need for definitive diagnosis of this condition. PCR (including its modifications – quantitative and multi-target) has shown promising results, but there is insufficient evidence regarding its role in clinical practice.
10.1.4.1.5 Differential Diagnosis
The key to diagnosis of ocular tuberculosis in the current scenario lies in exclusion of other disease entities (infectious and noninfectious), found in a given geographic region that can mimic ocular tuberculosis. Therefore, the list of differential diagnosis depends on the specific clinical presentation and geographic location. Since tuberculosis can affect virtually every ocular tissue, the differential diagnosis can include nearly every ocular inflammatory condition except morphologically distinct entities like toxoplasma retinochoroiditis or viral retinitis.
10.1.4.1.6 Treatment
Treatment of ocular tuberculosis requires a combination of antimicrobial/antituberculosis therapy (ATT) and anti-inflammatory therapy (usually corticosteroids). In a large series of 360 patients, those treated with ATT had a significantly reduced rate of recurrent inflammation (15.74 %) compared to those treated only with corticosteroids (46.53 %) ([22], EBM: C, 2+). ATT should be administered in consultation with a pulmonologist or infectious disease specialist. According to the Centers for Disease Control and Prevention (CDC) guidelines, ATT should be given for a minimum of 6 months in total – 2 months of four-drug therapy (isoniazid 5 mg/kg daily, rifampicin 450 mg daily, pyrazinamide 30 mg/kg daily, and ethambutol 15 mg/kg daily) followed by a 4-month continuation phase of isoniazid and rifampicin ([9], EBM: 1+, A). Many authors have suggested a longer duration for the continuation phase, citing slow response to the drug in intraocular tuberculosis [10, 29]. It was found that those receiving >9 months ATT were significantly less likely to develop recurrence compared to those not receiving ATT (p = 0.027). However, the reduction in recurrence compared to other ATT durations (<6 months, 6–9 months) was not statistically significant ([10], EBM: C, 2-). Patients on ATT need to be monitored for ocular and systemic side effects. Ocular side effects include optic neuritis (ethambutol, especially if used >15 mg/day for >2 months, and rarely, isoniazid) and anterior uveitis (rifabutin).
Concomitant corticosteroid therapy is vital to control the inflammatory tissue damage caused by delayed-type hypersensitivity to M. tuberculosis. The importance of corticosteroid therapy can be judged from its role in the management of continued progression or paradoxical worsening of ocular inflammation that is occasionally seen after initiation of ATT for intraocular tuberculosis [100]. Such paradoxical worsening usually occurs in the initial 4–6 weeks after initiation of ATT and needs to be differentiated from various causes of treatment failure like drug resistance, reinfection, or missed diagnosis [25]. The mode of corticosteroid therapy (topical, periocular, intraocular, or systemic) depends on the degree and primary site of inflammation.
10.1.4.1.7 Future Directions
-
1.
Definitive diagnosis based on PCR: Various modifications of PCR including quantitative PCR and multi-target PCR (targeting multiple gene sequences) are being applied to address the key challenge of low sensitivity of this technique [22].
10.1.4.2 Syphilis
10.1.4.2.1 Definition
Syphilis is a sexually transmitted disease caused by the spirochete Treponema pallidum. In acquired syphilis, the bacterium enters the body through small abrasions on the skin or the mucous membranes, mainly the genitals and mouth. If the condition is left untreated, it will progress through four stages with harmful effects in the major organs such as the heart and brain. Vertical transmission through the placental route is also possible but much less frequent, giving rise to congenital syphilis if the fetus survives.
10.1.4.2.2 Clinical Signs and Symptoms
10.1.4.2.2.1 Acquired Syphilis: Systemic Signs
10.1.4.2.2.1.1 Primary Syphilis
The main sign is the genital chancre which appears 3 weeks after infection (an ulcerated painless lesion) associated with regional lymphadenopathy.
10.1.4.2.2.1.2 Secondary Syphilis
It appears 6 weeks after the appearance of the chancre and is characterized by flu-like illness, by maculopapular skin rash (mainly on soles and palms), and rarely by symptomatic or asymptomatic meningitis.
10.1.4.2.2.1.3 Latent Stage
This stage is divided into early and late latent. During this stage, the clinical disease is not detectable.
10.1.4.2.2.1.4 Tertiary Syphilis
At this stage, syphilis may present with “gumma,” a small, rubbery granuloma with a necrotic center often located in the liver but also in the brain, heart, skin, and other tissues, leading to cardiovascular syphilis (aortitis and aortic aneurysms) and/or as neurosyphilis. In the late phase, neurosyphilis results in parenchymal lesions leading to encephalitis, stroke, tabes dorsalis, and Argyll-Robertson pupil, among other clinical findings.
Congenital syphilis can present during childhood and is divided into an early phase characterized by mucocutaneous lesions and osteochondritis and a late phase with the classic triad of Hutchinson keratitis, Hutchinson incisors, and eight nerve deafness.
Stages | Clinical findings |
|---|---|
Primary syphilis | The initial clinical manifestation is the primary chancre |
Usually painless, which distinguishes it from other causes of genital ulcers: herpes simplex (genital herpes) and Haemophilus ducreyi (chancroid) | |
Often heals without treatment over a period of a few weeks | |
Secondary syphilis | Untreated disease, approximately 25 % of patients will go on to develop systemic symptoms: rash, fever, headache, malaise, diffuse lymphadenopathy, alopecia |
Latent syphilis | Latent syphilis refers to patients without symptoms who have positive serologic testing for syphilis |
Early latent: <1 year | |
Late latent: >1 year | |
Tertiary syphilis | Clinical manifestations that may occur 1–30 years after infection when the infection is not treated |
10.1.4.2.2.2 Ocular Signs
10.1.4.2.2.2.1 Syphilitic Posterior Segment Involve ment
Ocular syphilis manifests itself in the secondary and tertiary stage of syphilis. It often affects the eye as anterior granulomatous uveitis (mutton-fat KPs at the corneal endothelium, iris nodules) but may involve any other ocular structure. Because of its very variable presentation, syphilis earned the label as “great mimicker.” Posterior pole manifestations vary ([122], EBM: 4, C). Unlike other infectious agents, treponemas have an affinity for all ocular layers including the posterior pole.
Deep chorioretinitis is the most common manifestation, with lesions, that can be divided into focal or multifocal lesions often located at the posterior pole. Focal lesions are often associated with serous retinal detachment and a significant degree of vitreous inflammation. Fluorescein angiography (FA) shows early hypofluorescence followed by late staining of the lesions.
Acute syphilitic posterior placoid chorioretinitis (ASPPC, Fig. 10.7) is a specific, uncommon entity described by Gass, associated with placoid subretinal lesions at the level of the RPE and not infrequently associated with a retinal detachment in which macular pseudohypopyon can be observed ([77], EBM: 2++, B). FA shows typical leopard-spot pattern in the cicatricial phase of the lesions.

Acute syphilitic posterior placoid chorioretinitis (ASPPC) characterized by placoid, yellowish subretinal lesions at the level of the RPE (a), associated with papillitis. Fluorescein angiography: note the corresponding staining of the dye at the late phases of the angiogram (b), with an evident hot disk
Syphilis may also manifest as a necrotizing retinitis, a severe condition associated with yellow-white patches of necrosis, retinal vasculitis, and vitritis, which can easily be confused with ARN.
Optic nerve involvement often occurs with minimal or no anterior segment inflammation. It often spills over into the retina being associated with either vasculitis, or focal areas of retinal edema. Untreated, it can lead to optic atrophy.
More peripheral involvement presenting as intermediate uveitis has been described, frequently associated with cystoid macular edema, vasculitis, and “hot disk.” Pars plana exudates are characteristically absent.
The diagnostic approach offers the nonspecific tests which suffer a low level of sensitivity as compared to the high level of sensitivity and specificity offered by the specific tests.
10.1.4.2.2.2.2 Syphilis and HIV
The association between syphilis and HIV is quite common as both are sexually transmitted diseases. The frequency is sufficiently high that in the presence of one infection, one should always consider the presence of the other. However, the clinical presentation of syphilis does not appear to be altered by the presence of HIV and is not correlated with the severity of immune compromise. While the presentation may be similar to that in immunocompetent patients, relapses are more frequent, as is bilaterality. Patients with HIV infection may have a higher prevalence of posterior uveitis ([256], EBM: 2++ B), which may present in as a more severe and atypical form. Treatment will require more prolonged therapy with higher doses of antibiotics. Analysis of cerebrospinal fluid (CSF) should be performed in all patients with ocular syphilis due to a high prevalence of neurosyphilis and a poor sensitivity of systemic antibody titers in this patient population [144].
10.1.4.2.3 Treatment
In the presence of ocular involvement, all ocular manifestations of the infection should be treated as neurosyphilis. Intravenous penicillin G or procaine penicillin G 18–24 (MU) daily plus probenecid for 10–14 days should be given, followed by IM procaine penicillin G, 2.4 MU weekly for 3 weeks ([144] (EBM:1++, A), [50] (EBM: 2++, B)). For patients with tertiary stage ocular syphilis, a three-week course of benzathine penicillin should be added to the above regimen. In patients allergic to penicillin, ceftriaxone can be used instead.
Systemic corticosteroids can be used in combination with antibiotic therapy. If not used initially, vision and the severity of the inflammatory response need to be carefully monitored as a Jarisch-Herxheimer reaction can occur in severe enough cases where the use of steroids would be the preferred course of action ([130] (EBM:3, D), [254] (EBM:3, D)).
Core Message
-
Syphilis is a sexually transmitted disease caused by the spirochete Treponema pallidum.
-
Ocular disease is characterized by a chronic granulomatous inflammation involving various ocular structures.
-
The most common eye manifestations are anterior uveitis, necrotizing retinitis, retinal vasculitis, panuveitis, and intermediate uveitis.
-
Because of its highly variable presentation and good treatment response, syphilis has always to be included in the differential diagnosis of any type of uveitis.
-
Syphilis is a treatable infection – especially in its primary and secondary stages – but is more difficult to manage in its tertiary stage.
10.1.4.3 Ocular Leptospirosis
10.1.4.3.1 Definition
Leptospirosis is a zoonosis caused by spirochetes of the genus Leptospira, whose natural reservoir is wild animals, mostly rodents and cattle. Initially, it was described by Adolf Weil in 1886 as a condition characterized by acute fever, malaise, and uveitis.
10.1.4.3.2 Etiology
Humans are accidental hosts, acquiring the disease by the contact with infected urine, tissues, or water. The disease can be considered occupational, infecting mostly farmers, veterinarians, and abattoir workers. It has the potential to occur both as epidemic outbreaks and as endemic disease, in tropical and temperate climates.
10.1.4.3.3 Clinical Symptoms and Signs
Leptospirosis is a multisystem disorder, characterized by a broad spectrum of illness ranging from subclinical illness to either a self-limited anicteric systemic illness (quasi 90 % of affected subjects) or a severe icteric septicemic illness associated with renal failure, liver failure, and pneumonitis with hemorrhagic diathesis. It is a biphasic disease with an initial septicemic phase followed by defervescence and the immune phase of illness. The most severe presentation that may develop after the initial leptospiremic phase is Weil’s disease, which is associated with impaired liver and kidney function. Mortality rates in these patients range from 5 to 40 % ([192] (EBM:2+)).
Ocular manifestations are seen in both the acute leptospiremic and immune phases of the illness. In the former phase, the most prominent findings are conjunctival chemosis and scleral icterus, while in the latter phase, there is a myriad of ocular signs such as interstitial keratitis iritis, hypopyon, cataract, membranous vitreous opacities, and retinal vasculitis; meanwhile, the most important systemic features of this immune phase are meningitis and leptospiruria. In leptospiral uveitis, hypopyon is the primary expression of the intraocular inflammation. Nongranulomatous uveitis is the hallmark of leptospiral uveitis.
10.1.4.3.4 Differential Diagnosis
It includes Behcet’s disease, HLA-B27-associated anterior uveitis, sarcoidosis, syphilis, toxoplasmosis, ARN, and endogenous endophthalmitis.
10.1.4.3.5 Treatment
Systemic leptospirosis has raised several controversies regarding antimicrobial treatment.
Despite a lack of evidence of the utility of antibiotic therapy for leptospirosis, penicillin, cephalosporins, and doxycycline are the commonly employed therapies in the management of leptospirosis. Despite its higher cost, interest in azithromycin against Leptospira spp. is increasing due to its broad activity against confounding pathogens, low mean inhibitory concentration (MIC), and fewer mild adverse events [286, 287].
For mild infection, doxycycline 100 mg po bid can be used for 7 days, amoxicillin 50 mg po q6 h for 7 days, and ampicillin 500–750 mg po q6 h for 7 days.
For moderate to severe infection, penicillin G 1.5 million UI IV q6 h can be used for 10 days, ampicillin 0.5–1 g q6 h for 10 days, and ceftriaxone 1 g IV qd for 10 days. For severe uveitis or neurological abnormalities or arthritis, ceftriaxone 2 g/day for 14–21 days ([78] (EBM:1+, B), [209] (EBM:2++, B), [42] (EBM:1-)) is given.
Corticosteroids are the mainstay of treatment for leptospiral uveitis. In unilateral panuveitis, sub-Tenon depot corticosteroids can be used, while in bilateral panuveitis, oral corticosteroids can be employed.
Core Message
-
Leptospirosis is a zoonotic waterborne infection commonly classified as a tropical disease that mainly affects young and middle-aged men.
-
It has a wide-ranging clinical and public health impact, in particular, in developing countries with a broad variety of clinical manifestation and significant mortality rate.
-
MAT is considered as a gold standard diagnostic test.
-
The most important intraocular clinical manifestations are nongranulomatous panuveitis, papillitis, and vitritis. Despite the lack of evidence, utility of antibiotic therapy is common, whereas corticosteroids are the mainstay of treatment for ocular involvement.
10.1.4.4 Lyme Disease
10.1.4.4.1 Definition
Lyme borreliosis is a bacterial infection caused by Borrelia burgdorferi. The spirochete is transmitted through the bite of infected ticks. The diagnosis is based on clinical history and examination and can be supplemented by laboratory investigations.
10.1.4.4.2 Etiology
The disease was described in 1977 by Steere et al., who described a group of children presenting with inflammatory arthropathy similar to that in juvenile rheumatoid arthritis. This entity was labeled “Lyme disease” after the town of Lyme, Connecticut [35, 170]. A characteristic rash was associated with the disease, labeled as erythema chronicum migrans often associated with severe headaches. Erythema migrans is the most common clinical presentation. Ocular involvement is uncommon and occurs mainly in the second and late stages of the disease. The causative agent was later identified by Burgdorfer, who described the spirochete in the midgut of the Ixodes tick. The hosts (deer and small rodents) and the Ixodes tick often thrive in the climates of the endemic regions such as northern Asia, Europe, and North America.
10.1.4.4.3 Clinical Signs and Symptoms
The disease is divided into stages ([118] (EBM: A+)). During early infection, it can be identified as the first (local) stage which appears a few days after the tick bite and includes erythema migrans (bull’s eye), fever, and arthralgias. However, 20–40 % of patients never develop a skin rash.
This is followed by the second (disseminated) stage during which the organism spreads to multiple organ systems. Particularly the skin, heart (associated with atrioventricular block), joints (associated with mono- or oligo-arthropathy), and nervous system are affected. Neurological involvement is frequently associated with palsies of the cranial nerves and meningitis. “Lymphocytoma benigna” is a bluish lesion occurring at the earlobes and nipple region.
The third or late (persistent) stage occurs after a disease-free period (months to years). Recurrent manifestations are the hallmark of this stage and include chronic relapsing arthritis mainly affecting the knee, acrodermatitis chronica atrophicans, chronic encephalopathy associated with cognitive dysfunction, and peripheral neuropathies.
10.1.4.4.4 Ocular Manifestations
Ocular manifestations are rare in patients with Lyme disease and can involve any of the ocular structures. Conjunctivitis is the most common finding, present in 11 % of patients with early disease. Episcleritis may occasionally be found with conjunctivitis during the local stage. Keratitis is one of the most common findings that appear during the late persistent stage.
Neuro-ophthalmic manifestations belong to the local and mainly disseminated stages, including optic neuritis, papillitis, papilledema, Bell’s palsy which is the most common cranial neuropathy, and Horner’s syndrome.
Intraocular inflammation has been reported to occur during the early and late stages of Lyme disease. Anterior uveitis, intermediate uveitis, posterior uveitis with choroiditis, and retinal vasculitis (Fig. 10.8) have been reported. Anterior uveitis associated with granulomatous KP-s and intermediate uveitis are the most common intraocular manifestations.

Retinal vasculitis complicated by “honeycomb” cystoid macular edema in a patient with Lyme disease
10.1.4.4.5 Differential Diagnosis
The most common infectious disorders are syphilis, TBC, viral keratitis, infectious arthritis, and viral encephalitis/meningitis. Noninfectious disorders that may have to be considered in the differential diagnosis from Lyme disease are sarcoidosis, VKH, multiple sclerosis, vasculitis, and collagen vascular disorders.
10.1.4.4.6 Treatment
Recommendations for the treatment of Lyme disease were reviewed by the Infectious Disease Society of America ([277] (EBM:1++, A)). In early infection, the adult doses are doxycycline 100 mg po bid for 14–21 days, amoxicillin 50 mg po tid for 14–21 days, and cefuroxime axetil 500 mg po bid for 14–21 days. In children, the doses are amoxicillin 50 mg/kg/day divided in three doses (maximum of 500 mg/dose) (Table 10.3).
In cases of severe ocular manifestations and neurological involvement, such as posterior uveitis, intravenous antibiotic therapy with ceftriaxone (2 g IV qd in adults for 2–4 weeks) is probably the treatment of choice ([277] (EBM:1++, A), [266] (EBM: 1+, A)). After systemic antibiotic treatment has been initiated, intraocular inflammation should be treated with topical corticosteroids and mydriatics. Systemic corticosteroids are proposed for severe posterior uveitis and neuro-ophthalmic involvement ([208] (EBM:4, D)). Attention should be directed at Jarisch-Herxheimer reaction after initiation of antibiotic therapy.
Core Message
-
Lyme disease is characterized by a wide variety of changes including rather nonspecific flu-like symptoms associated with tiredness, headaches, arthralgia, and skin manifestations. The characteristic skin rash “erythema migrans” is the most common clinical presentation appearing about 3–30 days after a tick bite. Left untreated, later symptoms may involve the joints, heart, and central nervous system.
-
The ocular manifestations of Lyme borreliosis most commonly occur during the late stages of this tick-transmitted disease.
-
A small proportion of patients who have had LB may go on to develop a post-infection syndrome resembling chronic fatigue syndrome or fibromyalgia, which has been termed “post-Lyme syndrome.”
-
Diagnostic strategies vary between early and late disease manifestations and usually include serologic methods. Erythema migrans is pathognomonic and does not require any further laboratory investigations. PCR has shown to be useful in the diagnosis of the disease, but serology should only be ordered in case of well-founded clinical suspicion for Lyme borreliosis, i.e., manifestations compatible with the diagnosis.
-
Antibiotics are the mainstay of therapy, with corticosteroids associated during severe intraocular inflammation.
10.1.4.5 Cat Scratch Disease
10.1.4.5.1 Definition
Cat scratch disease (CSD) is a self-limited, systemic disease caused by a gram-negative bacillus, Bartonella henselae [64]. The disease manifests itself as a mild lymphadenitis involving the lymph nodes draining the dermal/conjunctival sites. CSD manifests a clinical spectrum ranging from a mild self-limiting disease with neuroretinitis and macular star formation to retinal vasculitis with subsequent severe vision loss.
10.1.4.5.2 Etiology
Within different species of Bartonella, there are four recognized as human pathogens: B. bacilliformis (Carrion’s disease), B. elizabethae (endocarditis), B. quintana (trench fever), and B. henselae [271]. Bartonella species are gram-negative bacilli which have been associated with a clinical syndrome of self-limited lymphadenopathy associated with a transmission by cat scratch/bite. Human infections can be relatively asymptomatic or can produce symptoms such as fever, malaise, fatigue, and lymphadenopathy. On the other hand, severe systemic involvement can occur, characterized by splenomegaly, encephalopathy, pneumonia, granulomatous hepatitis, and osteomyelitis. Cat fleas are the major vector for CSD.
The eye can be involved either with the primary inoculation complex resulting in Parinaud’s oculoglandular syndrome, as is the most common presentation of Bartonella infection. The typical signs are unilateral granulomatous conjunctivitis and regional lymphadenopathy. Preauricular, submandibular, and cervical lymph nodes are typically affected. Vascular leakage from the optic nerve head (Fig. 10.9) ([56] (EBM:2, B)) and the “macular star” (Fig. 10.10) are the hallmark of the neuroretinitis, which may persist even after the resolution of posterior pole involvement. Typically, neuroretinitis is unilateral with a self-limited course.

(a) Color fundus photograph of the right eye of a patient with CSD shows a prominent optic disk edema associated with hemorrhage. (b) Fluorescein angiogram of the same eye shows optic disk leakage and masking effect by hemorrhages

(a) Photograph taken 15 days after initial presentation shows a complete macular star with partial resolution of optic disk edema. (b) Note the presence of optic disk telangiectatic vessels and associated preretinal hemorrhages
Both multifocal retinitis and choroiditis are typically seen in conjunction with disk swelling. These lesions are typically juxtavascular. The inner white retinal infiltrates may look similar to cotton-wool spots, but their distribution is not necessarily associated with the distribution of arterioles as in the case of cotton-wool spots. Other clinical findings can be observed, such as branch retinal artery and vein occlusions, local serous retinal detachments, and intraretinal bleeding.
10.1.4.5.3 Differential Diagnosis
Etiologies that must be differentiated include other causes of optic nerve swelling such as optic neuritis and sarcoid papillitis. Pseudotumor cerebri can mimic the rare occurrence of the bilateral CSD. Syphilitic perineuritis, TBC, Lyme disease, leptospirosis, and, rarely, toxoplasmosis can produce similar clinical appearance. Other causes of macular star formation include systemic hypertension.
Other causes of conjunctivitis associated with regional lymphadenopathy include tularemia, sporotrichosis, TBC, syphilis, lymphogranuloma venereum, and leprosy.
10.1.4.5.4 Treatment
Usually, no treatment is recommended for mild to moderate forms of systemic CSD, since the disease runs a self-limited course. Treatment is recommended for severe ocular/systemic complications of B. henselae infection, both in immunocompetent and immunocompromised patients ([207] (EBM D 3)). Currently, no controlled clinical trial has demonstrated efficacy in immunocompetent patients. Doxycycline (100 mg twice daily) has good intraocular and CNS penetration. For pediatric patients (8–12 years), erythromycin is recommended. The duration of treatment lasts 2–4 weeks in immunocompetent patients and 4 months in immunocompromised patients. Azithromycin, intramuscular gentamicin, ciprofloxacin, and trimethoprim/sulfamethoxazole are alternative antibiotics ([207] (EBM:4, D)). The role of corticosteroids in atypical CSD is somewhat controversial.
Core Message
-
B. henselae is a relatively common cause of neuroretinitis in CSD and probably underdiagnosed.
-
Mild to moderate forms of CSD run a self-limited course with no need for treatment. Patients with neuroretinitis, encephalopathy with or without hemiplegia, and acute solid organ transplant rejection have all been treated successfully with a combination of appropriate antibiotics and steroid therapy.
-
Patients with CSD have a good overall visual prognosis.
-
Good visual acuity at presentation was associated with a favorable visual outcome.
10.1.4.6 Rickettsial Diseases
Rickettsioses are worldwide distributed zoonoses due to obligate intracellular small gram-negative bacteria. Most of them are transmitted to humans by the bite of contaminated arthropods, such as ticks especially during spring or summer. Rickettsial agents are classified into three major categories: the spotted fever group, the typhus group, and the scrub typhus ([265] (EBM:2++, B)).The spotted fever group includes Mediterranean spotted fever (MSF), which is prevalent in Mediterranean countries and Central Asia; Rocky Mountain spotted fever, which is mainly encountered in the United States; and numerous other rickettsial agents.
10.1.4.6.1 Clinical Presentation
The initial clinical presentation includes high fever, myalgia, and headaches with a “tache noir” developing in the site of the bite. A maculopapular rash may be present at the time of presentation. Neurological signs ranging from small focal deficits to major neuropsychiatric disturbances have been reported.
10.1.4.6.1.1 Systemic Disease
The incubation period for rickettsial disease varies between 2 and 21 days. The initial presentation typically includes high fever with abrupt onset, headache, and myalgia. A maculopapular skin rash usually appears 3–5 days after the onset of fever. The skin rash, involving also the palms of the hands and the soles of the feet, is a hallmark of rickettsial infection. However, its absence should not rule out a possible rickettsial infection, especially during the first week of illness. A local skin lesion, termed tache noire (black spot), at the inoculating site may be seen in several rickettsial infections, including Mediterranean spotted fever, which is caused by Rickettsia conorii. Severe systemic complications may occur including interstitial pneumonitis, meningoencephalitic syndrome, acute renal failure, and disseminated intravascular coagulation [265].
10.1.4.6.1.2 Ocular Disease
Ocular involvement is common in patients with rickettsiosis, but since it is frequently asymptomatic and self-limited, it may be easily overlooked [265].
Bilateral or rarely unilateral non-necrotizing retinitis, with or without associated mild vitritis, is the most common ocular finding [265]. It typically presents in the form of white retinal lesions infiltrating the inner retinal layers (Fig. 10.11), located adjacent to retinal vessels, and varying in number, size, and location. Small retinal lesions in the posterior fundus may resemble cotton-wool spots, and large retinal lesions are usually associated with macular edema and exudative retinal detachment, which are accurately detectable by OCT. FA usually shows early hypofluorescence and late staining of large retinal lesions (Fig. 10.12) and slight hypofluorescence or isofluorescence of small retinal lesions [265]. Retinal vascular lesions are a prominent feature of rickettsial disease. They may include focal or diffuse vascular sheathing, vascular leakage on fluorescein angiography (FA), retinal hemorrhages, and retinal vascular occlusions, which mainly involve small branch retinal arteries [133]. A subclinical choroidal involvement only detectable by FA or indocyanine green angiography (ICGA) is also common [265].

Red-free fundus photograph of a patient with serologically proven rickettsial disease shows white retinal lesions of variable size, adjacent to retinal vessels

Fluorescein angiography shows early hypofluorescence (a) and late staining of retinal lesions associated with focal retinal vascular leakage and optic disk hyperfluorescence (b)
Other reported ocular manifestations of rickettsiosis include conjunctivitis, keratitis, nongranulomatous anterior uveitis, panuveitis, optic disk edema, optic disk staining, optic neuritis, neuroretinitis, anterior ischemic optic neuropathy, and endophthalmitis [265].
Ophthalmic involvement associated with rickettsial diseases often has a self-limited course. Areas of retinitis usually completely disappear without causing scarring in 3–10 weeks. Causes of persistent visual impairment include residual retinal changes due to resolved retinitis, macular edema, exudative retinal detachment, branch retinal artery or vein occlusion, and optic neuropathy [265].
10.1.4.6.2 Laboratory Diagnosis
Early diagnosis of rickettsial infection, primarily based on clinical features and epidemiologic data, is of utmost importance for early initiation of antibiotic therapy. Confirmation of diagnosis usually relies on positive indirect immunofluorescent antibody test results. Positive serologic criteria usually include either initial high antibody titer or a fourfold rise of the titer in the convalescent serum. Case confirmation with serology might take 2–3 weeks. Other laboratory tests, such as serologic testing using Western blot or detection of rickettsiae in the blood or tissue using PCR, may be useful in selected cases [265].
10.1.4.6.3 Management
Early treatment is required for a better outcome. Oral tetracyclines, particularly doxycycline (100 mg, twice a day for 7–10 days), are effective in the treatment of systemic rickettsial disease [265]. Fluoroquinolones are also effective. Macrolides, including clarithromycin, azithromycin, and particularly josamycin, can be used as alternative therapy in children and pregnant women.
Specific ophthalmic therapy may be needed in patients with ocular involvement. It includes topical antibiotics for conjunctivitis and keratitis, topical corticosteroids and mydriatics for anterior uveitis, and systemic steroids in association with antibiotics in cases of severe ophthalmic involvement such as extensive retinitis threatening the macula or the optic disk, macular edema, exudative retinal detachment, severe vitritis, optic neuropathy, and retinal vascular occlusions [265]. Prevention of rickettsial disease includes personal protection against tick bites in endemic areas and improvement of sanitary conditions.
Core Message
-
Ocular involvement in rickettsiosis is common but frequently asymptomatic.
-
Retinitis is a typical finding associated with vitritis and vascular lesions.
-
In order to diagnose this disease, a high index of suspicion is needed especially when associated with the specific clinical systemic symptoms and patients living or returning from endemic areas.
-
FA and ICGA are essential in subclinical cases.
-
Doxycycline is the mainstay of treatment.
10.1.5 Parasitic Uveitis
10.1.5.1 Ocular Toxoplasmosis
10.1.5.1.1 Definition
Ocular toxoplasmosis (OT) is considered as the most frequent infectious posterior uveitis. It is caused by the protozoan parasite Toxoplasma gondii, which exists in multiple clonal subpopulations, and in three stages, human seroprevalence of toxoplasmosis is high across the globe, but with remarkable geographic variation. A potential correlation of parasite genotype with disease is an area of current interest [160]. Ocular toxoplasmosis is more common in South America, Central America, and the Caribbean and parts of tropical Africa as compared to Europe and Northern America. Ocular disease in South America is more severe than in other continents due to the presence of extremely virulent genotypes of the parasite [200].
10.1.5.1.2 Etiology
The mode of T. gondii infection as either congenital or postnatally acquired is considered to be important. Although congenital infection frequently results in chronic recrudescent retinochoroiditis, most cases of OT are acquired after birth [249].
10.1.5.1.3 Clinical Symptoms and Signs
Symptoms vary but usually consist of unilateral floaters or blurred vision when the disease becomes active. Inactive disease rarely causes visual symptoms unless scarring is near the central retina or macula. Acute OT appears as a well-defined focus of retinal necrosis accompanied by a vitreous inflammatory reaction. In addition, there may be diffuse inflammation in the retina and choroid. OT typically runs a clinical course of 2–4 months of active intraocular inflammation followed by more or less long disease-free intervals, which may extend for several years. The reactivation of OT shows satellite lesions close to an old atrophic scar (Fig. 10.13). In the area surrounding the active retinitis, hemorrhage and vasculitis may be observed. Anterior uveitis may also be present. Atypical clinical findings may occur as well, such as vascular occlusive events and Kyrieleis’s arteritis [55]. Imaging can offer more information [97]. Bilateral involvement or atypical presentation can be observed in immune-compromised patients and often in congenital OT (Fig. 10.14).

Fundus photography of a 27-year-old patient presenting with recurrent paracentral retinochoroiditis caused by T. gondii.
Aqueous humor analysis: Goldmann-Witmer coefficient >5

Fundus photography of a 21-year-old patient affected by bilateral congenital ocular toxoplasmosis. The deep retinochoroidal lesion can be clearly seen
10.1.5.1.4 Differential Diagnosis
Necrotizing retinochoroiditis is considered as the typical presentation of OT and considered characteristic to such a degree that often further diagnostic workup is not needed. However, even when it is the most frequent manifestation of OT, there is considerable variation in the clinical features. Therefore, other necrotizing retinopathies, such as viral diseases, fungi, and other parasites, are important differential diagnoses. In such patients, analysis of intraocular antibody production and, therefore, Goldmann-Witmer coefficient plays a decisive role in the diagnosis of ocular toxoplasmosis, even more than PCR [48, 268] (Table 10.4).
10.1.5.1.5 Treatment
Despite the fact that OT continues to be a very common and sight-threatening cause of infectious posterior uveitis, treatment remains highly controversial (Fig. 10.15). This is related to a number of factors:

Histogram illustrating the preferred practice pattern derived from three surveys focusing on treatment of acquired ocular toxoplasmosis (Adapted from Basu et al. [28])
-
In many patients, T. gondii infection is a self-limited (often asymptomatic) disease that has been considered to need no treatment.
-
The parasites are able to form cysts that are impenetrable to medications and host enzymes; therefore, they cannot be eliminated from retinal tissue.
-
The persistence of retinal cysts also results in a very successful strategy for its survival and effectively avoids immunosurveillance by the host.
Despite the limited evidence of treatment effects, an increasing number of experienced ophthalmologists will treat patients with active OT [114].
Common clinical indications include:
-
Lesions within the vascular arcades threatening central vision
-
Active lesions in close proximity to the optic disk since substantial visual field defects may result
-
Large lesions >2 optic disk diameter which are often associated with dense vitreous haze
-
Immunosuppressed individuals because these patients very likely develop fulminant retinochoroiditis when left untreated
Several surveys of uveitis specialists indicate that even experts differ in their therapeutic approaches. Whereas some ophthalmologists will only care for sight-threatening lesions, others will treat all lesions independent on its location [28, 114, 255] (Fig. 10.16). Despite a lack of published evidence for effectiveness of current therapies, most ophthalmologists elect to treat patients with ocular toxoplasmosis that reduces or threatens visual acuity. Classic therapy consists of oral pyrimethamine and sulfadiazine, plus systemic corticosteroid. Substantial toxicity of this drug combination has spurred interest in alternative antimicrobials, as well as local forms of drug delivery ([217] [EBM C 3]). At this time, however, no therapeutic approach is curative of ocular toxoplasmosis.

Corresponding central scotoma of the patient of “Fig. 10.14” as findings in perimetry
In a Cochrane review, Gilbert et al. identified only three prospective, randomized, placebo-controlled clinical trials. Interestingly, two of these studies were conducted almost 40 years ago, using either eight weeks of pyrimethamine/trisulfapyrimidine vs. placebo or 4 weeks of pyrimethamine compared with placebo in acute OT [244] [EBM: B, 2++] [38] [EBM: 1+, B]. The third study determined the prophylactic effect of long-term (20 months) trimethoprim/sulfamethoxazole application vs. no treatment in patients with chronic relapsing OT [234] [EBM: B, 2++]. There was a lack of evidence in all three studies that antibiotics (short or long term) prevented vision loss. Only one study observing individuals infected with probably more aggressive South American strains of T. gondii demonstrated that long-term antibiotics (14 months) reduced the number of recurrences.
Because of toxicity and lack of effectivity, a number of alternative agents have been applied to OT patients. Clindamycin was a very promising substance when introduced in the 1980s [248] because it appeared to concentrate in ocular tissues and was considered to penetrate tissue cyst walls [248]. However, subsequent clinical experience showed no effect on disease recurrence [147] [EBM: C, 2+]. Recent treatment attempts focused on the use of clindamycin delivery as intravitreal injection [21, 140, 148] [EBM C 2+]. In a prospective randomized study comparing intravitreal clindamycin with 6 weeks of systemic clindamycin treatment, both appeared similarly effective [242] [EBM: B, 2++]. In a non-comparative, retrospective, multicentric interventional case series, 12 patients with active OT involving the posterior pole that were either intolerant to or contraindicated to oral medication received intravitreal injections of clindamycin (1.5 mg/0.1 ml) and dexamethasone (400 μg/0.1 ml) every 4 weeks (during pregnancy). During follow-up (24 months), resolution of OT was achieved in all cases and most eyes (83 %) improved, whereas two eyes (20 %) remained unchanged. No ocular or systemic adverse events were reported and furthermore no recurrences during 24 months of follow-up were observed [148] [EBM: C, 2+]. In particular, during pregnancy, sight-threatening lesions may be treated with intraocular injections of clindamycin and dexamethasone, combined with systemic sulfadiazine ([166] [EBM D 3]). Taken together, both studies demonstrated that IVI of clindamycin/dexamethasone might be an alternative to systemic treatment, offering a high drug availability and safer systemic adverse effect profile.
In addition, azithromycin [30] and atovaquone [197] were introduced into clinical use, but have not gained widespread acceptance (EBM: D, 3). There appears also an increasing use of the trimethoprim/sulfamethoxazole combination, offering a better option for compliance as does the standard combination of a dihydrofolate reductase inhibitor and sulphonamide. Small-scale uncontrolled studies showed apparently accelerated rates of resolution and improved acuities in patients on the combination [185] (EBM: D, 3). There remains, however, significant uncertainty with regard to proper medication by experts in the field. This is reflected by several surveys of uveitis specialists in the United States, Germany, and India, indicating that at least nine separate drugs in even more combinations are currently used in daily practice [28, 114, 255].
10.1.5.1.6 Management of Complications (CNV)
Several techniques have been proposed for CNV secondary to ocular toxoplasmosis (Fig. 10.17): PDT [EBM:C, 2+] [177], intravitreal anti-VEGF therapy [EBM: C, 2+] [164], and COMBO therapy [EBM: D, 3+] are the different therapeutic options [214].

Color picture (a) showing CNV in congenital ocular toxoplasmosis. Early (b), mid (c) and late (d) phases of FA showing an inactive CNV near the edge of an old, peripapillary toxoplasmic scar
10.1.5.1.6.1 Perioperative Prophylaxis
Perioperative prophylactic anti-toxoplasmic therapy may be warranted, in order to avoid reactivation of the disease [39] [EBM: C, 2+].
10.1.5.1.6.2 Management of Congenital Ocular Toxoplasmosis
Congenital OT is recognized as a major cause of child morbidity and mortality. Vertical transmission of toxoplasmosis occurs during primary infection in pregnant women, and generally, maternal disease goes unnoticed. Fetal infection occurs at up to 65–70 % and results in significant child morbidity with ocular lesions in up to 80 % of children as the most frequent manifestation [76, 169]. Therefore, prevention and treatment of congenital toxoplasmosis remains an important issue.
Worldwide remarkable differences exist regarding effective screening and treatment strategies. Reasons are related to the questionable benefit of early diagnosis and intervention since well-controlled studies are lacking and difficult to perform [83, 93, 94]. However, in most European centers, spiramycin remains the standard treatment and is immediately applied after diagnosis of maternal infection followed by pyrimethamine/sulphonamide as soon as a fetal infection is confirmed [93] [EBM: D, 2+]. Contrary, other treatment strategies rely initially on pyrimethamine/sulphonamide that will be changed to spiramycin if fetal diagnosis is negative [93] [EBM: C, 2+]. A meta-analysis investigating these different data in 2007 concluded that only weak evidence exists for an association between early treatment and reduced risk of congenital toxoplasmosis [252] [EBM: C, 2+]. However, it has been demonstrated by several studies that early treatment with spiramycin resulted in a significantly reduced rate (95 vs. 80 %) of placenta infection [61]. This led to a 50 % reduced incidence and lower severity of disease at birth of infected infants as compared to untreated individuals [135]. Recent observations confirm that it seems likely that more prompt diagnosis and treatment will result in better outcomes of congenital toxoplasmosis. New central chorioretinal lesions have been uncommon in children with congenital toxoplasmosis who are treated during their first year of life [201]. This contrasts markedly with previous observations for children left untreated or those treated for one month only (≥82 % retinal lesions).
10.1.5.1.6.3 The Role of Corticosteroids
As in many other ocular infections, the host immune response may have detrimental effects. Therefore, early intervention, e.g., by the use of corticosteroids, is often beneficial to reduce tissue damage. Interestingly, histopathologic specimen of the eyes from immunocompromised patients with OT showed no inflammatory cells in the infected tissue. These observations suggest that parasite proliferation, rather than inflammation, is the major cause of tissue damage in these individuals. Therefore, corticosteroid therapy is probably not necessary to control OT in immune-compromised individuals. On the contrary, it is commonly agreed upon that corticosteroid therapy without concurrent use of antimicrobial agents can lead to severe retina destruction and large lesions. A recent Cochrane review did not find supportive evidence for steroid treatment in either immune-competent or immune-compromised OT patients [120] [EBM: B, 2+].
10.1.5.1.6.4 Alternative Treatment Options
10.1.5.1.6.4.1 Surgical Options
Since the effect of medical treatment is uncertain, surgical options have been considered for OT. Argon laser photocoagulation has been applied with the intention to directly disrupt the organism or to reduce recurrence by surrounding old OT lesions with laser spots [215, 243] [EBM: D, 1+]. Unfortunately, even when the parasite was confirmed to be heat sensitive, neither direct destruction of the organisms nor reduced reactivation of tissue cysts could be obtained. Laser treatment has now been abandoned. Since active intraocular inflammation always carries a risk of complications, also any other intraocular surgery is not advised.
Taken together, treatment practices in OT are highly diverse. There is still no consensus regarding the choice of antiparasitic agents for treatment regimens. Despite this uncertainty, uveitis specialists appear to be more likely to treat patients with OT as compared to a decade ago [114].
Core Message
-
Ocular toxoplasmosis remains the most frequent infectious posterior uveitis in many parts of the world. Although congenital infection frequently results in retinochoroiditis, most ocular manifestations are acquired after birth (through nutrition).
-
Atypical clinical findings can be observed mainly in immunocompromised individuals or elderly people.
-
Although several treatment strategies have been proposed up to date, no therapeutic approach is curative of ocular toxoplasmosis.
-
Steroids are frequently applied to decrease intraocular inflammation but need careful monitoring and concurrent antimicrobial treatment.
10.1.5.2 Ocular Toxocariasis
10.1.5.2.1 Definition
Toxocara canis and Toxocara cati are nematodes, which live and mature in the dog or cat intestines, respectively.
10.1.5.2.2 Etiology
As a mature adult, the organism releases eggs which are passed in the stool. Contact with infected materials leads to human infection.
10.1.5.2.3 Clinical Symptoms and Signs
Toxocariasis is a rare infection and typically is observed in children. Toxocara infection can present fever; pulmonary symptoms such as a dry, hacking cough or asthma-like attacks; splenomegaly and hepatomegaly; skin lesions; neurological symptoms such as convulsions; and meningeal symptoms [129]. Ocular symptoms comprehend decreased vision, pain, photophobia, and floaters.
The most common eye manifestation is a granuloma, either in the posterior pole or in the periphery of the retina, with massive vitritis.
10.1.5.2.4 Differential Diagnosis
Differential diagnosis comprehends retinoblastoma, Coats’ disease, persistent fetal vasculature, retinopathy of prematurity, familial exudative vitreoretinopathy, idiopathic peripheral uveoretinitis, and toxoplasmosis. Retinoblastoma can be ruled out on the basis of B-scan echography, which typically finds calcifications, which are extremely uncommon in ocular toxocariasis. Non-inflamed eyes without cataract are also suggestive of retinoblastoma.
Laboratory tests are used to diagnose the disease: eosinophilia, hyperglobulinemia [232], and ELISA, even though a false-positive ELISA test can occur [161].
10.1.5.2.5 Treatment
The association of systemic prednisone (40 mg/day) and thiabendazole (2 g daily for 5 days) has been proposed ([220], EBM: D, 3). Albendazole (800 mg twice daily for adults or 400 mg twice daily for children, for 10 days to 2 weeks) can be considered as an alternative treatment option ([27], EBM: C, 2+).
Core Message
-
Ocular toxocariasis should be differentiated from several ocular diseases, particularly retinoblastoma.
-
Although different antihelminthic agents have been proposed, the role of such agents still remains unclear.
10.2 Noninfectious Posterior Uveitis
10.2.1 Multifocal Choroiditis (MFC)
MFC is part of the primary inflammatory choriocapillaropathies characterized by minimal signs of panuveitis mostly limited to cells in the posterior vitreous. ICGA made it clear that MFC typically involves primarily the choriocapillaris ([60, 238] (EBM:1+, A)) that shows inflammatory involvement even beyond the choriocapillaris in the choroidal stroma ([75] (EBM:3, D)). These findings may suggest an explanation for the more severe course and the propensity to develop choroidal neovascularization (CNV).
10.2.1.1 Clinical Symptoms and Signs
The majority of patients have bilateral disease that ranges from 66 to 79 % ([74, 213] (EBM:2+, D)), albeit asymmetric with the involved fellow eye. The most common symptoms are decreased central vision, photopsias, and subjective scotomas which are protracted in time. Signs of mild anterior uveitis including nongranulomatous keratic precipitates, cells, and flare can be seen associated with mild vitreous cellular activity when the disease is active. When keratic precipitates are granulomatous sarcoidosis, syphilis and tuberculosis have to be ruled out. Fundus examination reveals multiple round to oval, yellow-gray lesions, ranging in number from several to more than 100 scattered throughout the posterior pole and midperiphery. A distinct propensity for a peripapillary, nasal midperipheral, and peripheral distribution has been observed [213]. The active lesions evolve into round, atrophic chorioretinal scars with a punched-out appearance and varying degrees of hyperpigmentation. MFC shows a distinct peculiarity, that is, the high proportion of choroidal neovascular membranes complicating the disease, and typically, these almost always type II neovascular membranes seem to arise from atrophic scars or yellow nodular subretinal lesions frequently in association with active inflammation. Peripapillary atrophy similar to that seen in POHS is frequently seen on follow-up examination. Cystoid macular edema (CME) occurs in a range from 14 [74] to 41 % as reported by Nussenblatt et al. ([182] (EBM:4, D)). ICGA shows hypofluorescent areas during all the phases of the examination which are more numerous and involve a more extensive area than those appreciated on FA or clinical examination during the active stage of the disease. While during the active phase FA may show faint late hyperfluorescence reflecting the hypofluorescent lesion observed in ICGA, choroidal scars present a hyperfluorescent edge with a hypofluorescent center without leakage (Fig. 10.18). In a recent paper from Vance SK et al. ([257] (EBM:2++, B)) regarding the characteristic findings of MFC in SD-OCT, the authors concluded that the acute lesions of MFC include the presence of sub-RPE material, choroidal hyper-reflectivity below the lesions, and overlying vitreous cells in comparison to the findings of myopic degeneration. Visual field testing can show small scotomas corresponding to the chorioretinal scars. In the active phases, scotomas are larger reflecting the choriocapillaris non-perfusion. ERG suggests that MFC is a diffuse process with the degree of dysfunction relating to the severity and extent of chorioretinal involvement. MFC is a chronic disease that may persist for many years with the majority of patients experiencing multiple inflammatory recurrences in one or both eyes and the pathology affects predominantly young, healthy adults in their mid-30s with women predominantly affected [74, 213]. Inflammatory reactivation may manifest with swelling of the choroidal scars with surrounding subretinal fluid.

FA showing multiple spots of a MFC associated with a juxtafoveal CNV
10.2.1.2 Differential Diagnosis
ICGA is useful in ruling out the infectious causes such as West Nile virus choroiditis, Candida choroiditis, bacterial emboli, etc. However, in some instances, tuberculous choroiditis can also present with the same ICGA patterns as MFC. MFC exhibits anterior chamber and vitreous cells and a female predilection in contradistinction to POHS. MEWDS like MFC may present in young women with acute blind spot enlargement and vitreous inflammation; however, it is predominantly a unilateral disease. Although it has been suggested that SFU represents the terminal stage of MFC because of the multifocal lesions and the recurrent inflammatory episodes, SFU is limited to the posterior pole and eventuates in severe subretinal scarring. PIC is mostly related to MFC, with the high propensity in developing CNVM and its tendency towards bilaterality, but by definition, PIC is not associated with vitreous inflammation and the lesions tend to be smaller as compared to MFC. Other noninfectious entities to be considered in the differential diagnosis of MFC include sarcoidosis, APMPPE, and intraocular large cell lymphoma.
10.2.1.3 Treatment
Management, as for other entities under this group, is empirical. Although there are no controlled studies, clinical experience is probably sufficient to recommend corticosteroid therapy in cases with active disease; this can usually be diagnosed when patients complain of photopsia and is further evidenced by ICGA. If corticosteroids are insufficient, immunosuppressive agents may be added. The best follow-up parameter is ICGA which can show resolution of hypofluorescent areas. If inflammatory subretinal CNV is present, corticosteroids (sub-Tenon injections if the reactivation is unilateral or systemic if the reactivation is bilateral) should be tried first with concomitant or subsequent intravitreal anti-vascular endothelial growth factor (VEGF) therapy ([111] (EBM:4, D)). A recent paper published by Julian et al. reported the long-term results of 15 eyes (7 eyes with MFC) treated with intravitreal bevacizumab (IVB) as the first local treatment for CNV secondary to uveitis ([124] (EBM: 2++, C)). The intravitreal injections showed transient improvement in BCVA and CFT, in eyes under controlled inflammation, but further injections were needed in most cases with a mean number of 4.25 injections in 16 months. The peculiarity of this paper as compared to other works is the concomitant use of IVB in nine patients under systemic immunosuppression (corticosteroids and steroid-sparing agents). Another paper by Rouvas et al. ([219] (EBM:2+, C)) (three eyes) showed that ranibizumab resulted as a promising drug in maintaining stability or improving VA and OCT and FA findings in inflammatory choroidal neovascularization.
Core Message
-
MFC is part of the primary inflammatory choriocapillaropathies.
-
It is characterized by recurrent episodes of chorioretinal inflammation: photopsias, scotomata, and visual loss.
-
Vitreous cells during the active stage.
-
ICGA: active disease shows hypofluorescent zones.
-
Choroidal inflammatory neovascularization is a frequent sequela.
-
Therapy: the association of corticosteroids and immunosuppressive therapy has shown to be useful in the light of multiple recurrences, with associated anti-VEGFs in cases of inflammatory CNVM.
10.2.2 Punctate Inner Choroidopathy
Punctate inner choroidopathy (PIC) is a subset of MFC (primary inflammatory choriocapillaropathy) characterized by a similar clinical picture as far as symptoms, fundus signs, and neovascular complications are concerned, except that the lesions are smaller. PIC affects predominantly young myopic women. Although PIC in the majority of cases is a self-limited disease with good visual prognosis, permanent and severe visual loss can occur as a result of the development of choroidal neovascular membranes.
10.2.2.1 Clinical Symptoms and Signs
The predominant symptoms at presentation are scotomatas, followed by blurred vision, floaters, photophobia, and metamorphopsia, and these visual disturbances are usually unilateral ([92] (EBM:1+, B)). An analysis of refractive errors in inflammatory choriocapillaropathies revealed that PIC patients had the highest refractive errors. There is lack of inflammatory reaction in the anterior chamber and vitreous. Fundus examination reveals multiple, small, gray or yellow, round lesions concentrated in the posterior pole in a random or linear pattern that sometimes can be associated with serous detachment of the overlying neuroepithelium. They usually evolve into atrophic scars, and after 2–3 months, they may resemble old punched-out lesions seen in POHS. The scars involve the RPE and the choriocapillaris sparing the rest of the choroid. The fundus abnormalities are usually bilateral in 80 % ([46] (EBM:2+, D)) of cases. The most harmful clinical sequela of PIC is the formation of CNVM, and it is estimated that 17–40 % [46] of the eyes with PIC will develop it. The most frequent visual field defect is enlargement of the blind spot. During the active phases, ICGA shows hypofluorescent areas during all the phases of the examination which gradually resolve or remain hypofluorescent in case of scarring. FA shows early hypofluorescence followed by late hyperfluorescence and staining of the lesions (Fig. 10.19). While most PIC CNVs begin as multiple, yellow-green lesions, over time a tendency towards coalescence is seen to form larger CNVMs with bridging networks. FA of these membranes shows early hyperfluorescence followed by late leakage.

FA showing multiple hyperfluorescent macular spots typically observed in PIC
10.2.2.2 Differential Diagnosis
PIC can be differentiated from POHS by the lack of peripapillary atrophic changes and peripheral retinal lesions. MEWDS lesions are also concentrated in the posterior pole as in PIC, but these lesions have a less distinct border and do not have an associated serous detachment. In contrast to PIC lesions, APMPPE lesions are typically placoid and often confluent. ICGA findings are useful in excluding the infectious causes [164].
10.2.2.3 Treatment
No treatment is advised for the majority of patients with PIC where there is no evidence of CNV as the visual prognosis is excellent. The only exception to this would be those patients with inflammatory lesions, but no CNV, very close to fixation in whom medical treatment can be considered ([8] (EBM:2++, B)). Like multifocal choroiditis, new lesions respond to systemic or sub-Tenon corticosteroids, and additional immunosuppressive therapy is not always necessary. Corticosteroids are also thought to have a beneficial effect on the neovascular membrane and are the first line of treatment ([111] (EBM:4, D)). Corticosteroids can limit the extent of RPE disturbance and of scar formation following the insult of the acute PIC lesions. The treatment of inflammatory CNV is still a challenge, since no guidelines are available. Laser photocoagulation [184], periocular and systemic steroids ([80] (EBM:2+, C)), PDT ([112] (EBM:2+, D), [154] (EBM:2+, D)), immune suppression ([70] (EBM:2+, D)), and surgical removal ([157] (EBM:2+, D)) have been employed for management of inflammatory CNV in the pre-anti-VEGF era. Steroid-sparing agents such as cyclosporine A can be used for immune modulation; mycophenolate mofetil offers a more favorable safety and efficacy profile and is a promising drug for the long-term control of inflammatory CNV ([176] (EBM:2++, B)). Recent papers ([219] (EBM:2+, C), [14] (EBM:2++, C)) by Rouvas et al. (five eyes with PIC) and Arevalo et al.( eight eyes with PIC) showed that ranibizumab and bevacizumab, respectively, resulted as promising drugs in maintaining stability or improving VA and OCT and FA findings in inflammatory choroidal neovascularization [164].
Core Message
-
Punctate inner choroidopathy (PIC) is a subset of MFC primary inflammatory choriocapillaropathy, predominantly seen in healthy young myopic women.
-
PIC is characterized by recurrent episodes of chorioretinal inflammation.
-
The symptoms are scotomatas, followed by blurred vision, floaters, photophobia, and metamorphopsia.
-
Multiple, small, gray or yellow, round lesions concentrated in the posterior pole, which are usually bilateral in 80 % of cases.
-
ICGA: active disease shows hypofluorescent zones.
-
FA: scars show early hypofluorescence followed by late hyperfluorescence.
-
CNVM is estimated to occur in up to 40 % of eyes affected by PIC.
-
Therapy: the association of corticosteroids and immunosuppressive therapy has shown to be useful in the light of multiple recurrences, with associated anti-VEGFs in cases of inflammatory choroidal neovascularization.
10.2.3 Serpiginous Choroiditis
10.2.3.1 Definition
Serpiginous choroiditis (SC) is a rare, chronic, progressive, and recurrent bilateral inflammatory disease involving the RPE, the choriocapillaris, and the choroid of unknown etiology [5, 19]. SC affects mainly healthy young to middle-aged adults with a higher prevalence in men and with no racial or familial predilection. Because of the rarity and the variable course of the disease, the long-term management of patients with SC remains a challenging issue in ocular immunology.
10.2.3.2 Clinical Symptoms and Signs
Patients present typically with a painless unilateral decrease in central vision, metamorphopsia, or bilateral scotoma. Scotomata may be absolute or relative depending on the inflammatory activity. Usually, the anterior segment and the anterior vitreous do not present inflammation. The disease is characterized acutely by irregular, gray-white, or creamy-yellow subretinal infiltrates at the level of the choriocapillaris and the RPE. The overlying retina is usually edematous and an associated neurosensory retinal detachment may occur. Around 80 % of cases with SC have the classic peripapillary geographic pattern. The peripapillary serpentine lesions in the fundus are a characteristic feature of this type. Classically, lesions develop first in the peripapillary area and tend to spread centrifugally in a pseudopodial or serpentine fashion. Active lesions resolve spontaneously with or without treatment over 6–8 weeks leaving focal areas of atrophy. Recurrences usually, but not always, occur at the edges of previous atrophic scars, and they tend to be multiple at variable intervals ranging from months to years. Chronic cases are characterized by chorioretinal atrophy; subretinal fibrosis and extensive RPE clumping may be observed ([156] (EBM:2++, B)).
About two-thirds of patients with SC have scars in one or both eyes at the initial presentation 87 ([146] (EBM:2+, C)). Visual loss in one or both eyes with final VA of less than 20/200 is observed in up to 25 % of patients despite treatment ([59] (EBM:2+, C)). When SC begins in the macular area, it is termed “macular SC” [108]. Macular SC has a worse visual prognosis due to early foveal involvement and the higher risk of developing secondary choroidal neovascularization (CNV). This condition may be underdiagnosed. Occasionally, the lesions may occur in the periphery in an isolated or multifocal pattern termed “ampiginous choroiditis” [156] or “relentless placoid chorioretinitis” ([123] (EBM:4, D)). The evolution of APMPPE can mimic the clinical course of SC, with the difference that the multifocal nature of the lesions did not coincide to the extension of the old lesions. A paper by Gupta et al. reported that 20 out of 86 patients with SC presented with initially APMPPE which over time progressed to SC ([105] (EBM:1+, A)). Compared to patients with typical peripapillary SC, those with ampiginous choroiditis tend to have less central foveal involvement. ICGA findings are characterized by hypofluorescent areas during all the phases of the angiography. FA findings (Fig. 10.20) are the following: early serpiginous hypofluorescence followed by late diffuse staining and leakage at the edge of the retinal lesions ([40] (EBM:1+, A)). Visual field shows absolute scotomata associated with active lesions and relative ones associated with the resolution of the active lesions. ERG and EOG do not contribute to the evaluation of disease progression. The most common complication associated with SC is CNV, with a range from 13 to 35 % of patients [34, 59]. Other ocular conditions that may be considered as complications associated with SC are branch retinal vein occlusion (BRVO), periphlebitis, pigment epithelial detachment (PED), serous retinal detachment (SRD), cystoid macular edema (CME), optic disk neovascularization, and subretinal fibrosis.

Fluorescein angiography showing early hypofluorescence (a) and late hyperfluorescence with leakage at the edge of the retinal lesions (b) in a patient with noninfectious SC
10.2.3.3 Differential Diagnosis
The disease that most likely resembles the acute initial presentation of SC is APMPPE. The key difference is the clinical course where APMPPE lesions usually resolve spontaneously in two to three weeks, leaving a mottled RPE without significant choroidal involvement, and unlike SC, secondary CNV in APMPPE is rare and recurrence is uncommon. Patients with MFC differ from those with SC in that the latter do not show signs of vitreous inflammation and the lesions in MFC are smaller. Outer retinal toxoplasmosis may also mimic SC, but lesions do not coalesce and are virtually always unilateral. Tuberculosis infection, like SC, may affect the choroid and give rise to similar choroidal scars, but patients with ocular tuberculosis frequently present with vitritis, constitutional symptoms, and a positive tuberculin skin test. The angiographic features of choroidal ischemia and SC are similar, and conditions that result in occlusion of the posterior ciliary vessels, such as hypertension and systemic vasculitis, should be excluded. In older patients, metastatic tumors, non-Hodgkin’s lymphoma, and choroidal osteoma may mimic the appearance of the acute unilateral lesion of SC.
10.2.3.4 Treatment
Untreated active lesions typically resolve over months with a gradual extension of the primary lesion characterized by a variable natural history. The frequent recurrences also increase the risk of secondary CNV. Fundus photography and angiography are necessary to document the non-progression of SC in order to evaluate treatment. The rapid control of the active lesions during recurrences and prevention of further recurrences seem to represent the mainstay of a successful therapy. Based on the different etiologies proposed, many different treatments have been proposed such as antibiotics ([1, 156] (EBM:2++, B)), antivirals [1, 59], and immunosuppressive therapy [146]. Systemic corticosteroids and retrobulbar steroidal injections were effective in controlling the active lesions and in shortening the duration of active disease, but not in preventing recurrences [267]. Intravitreal steroids will likely be effective in the treatment of acute lesions, but probably will not prevent recurrences if not administered on a continuous basis. Treatment of SC with cyclosporine A (CsA) as a monotherapy has produced mixed results. Christmas et al. [59] reported successful results in 4 out of 6 patients with SC treated with immunosuppressive drugs such as CsA, mycophenolate mofetil, or azathioprine in a period ranging from 2 to 40 months, in terms of discontinuation of therapy without recurrences. Triple-agent therapy consisting of CsA, azathioprine, and prednisolone was reported to show satisfying results in terms of inflammation control ([1, 116] (EBM:2+, C)). Because of the potentially serious side effects, alkylating agents should be limited to patients with sight-threatening lesions that are unresponsive to conventional immunosuppressives. Nowadays, better visual acuity results are achievable through VEGF inhibitor injections with or without PDT. This is proven by a few publications with greater numbers of patients because of the rarity of the diseases and several case reports in the literature. In addition to CNV treatment, the control of intraocular inflammation should never be forgotten because it forms the leading CNV trigger ([273] (EBM:4, D)). Recent reports from Julian et al. ([124] (EBM: 2++, C)) (one eye with SC and two eyes with ampiginous choroiditis) and Arevalo et al. ([14] (EBM:2++, C)) (six eyes with SC) showed that intravitreal bevacizumab resulted as a promising drug in maintaining stability or improving VA and OCT and FA findings in inflammatory choroidal neovascularization. Based on the studies reported so far, the rapid control of any active lesion with aggressive immunosuppression and the maintenance on appropriate immunosuppression for at least 6 months to prevent eventual recurrences can be considered the initial management of patients with SC. Subsequent treatment will depend on the severity of the disease.
Core Message
-
SC is a rare, usually bilateral, chronic, progressive, recurrent inflammation of the choroid, RPE, and choriocapillaris.
-
SC can present into the peripapillary, macular, and ampiginous types.
-
SC is characterized by multiple recurrences.
-
Immunosuppressive treatment seems to be useful, but further clinical trials are required in order to achieve a gold standard of treatment.
10.2.4 Multiple Evanescent White Dot Syndrome (MEWDS) and Acute Idiopathic Blind Spot Enlargement (AIBSE)
10.2.4.1 Definition
MEWDS is a primary inflammatory choriocapillaritis of unknown etiology that results from inflammation at the level of the choriocapillaris causing areas of non-perfusion or hypoperfusion. The ischemic areas produce white lesions deep in the outer retina or at the level of the retinal pigment epithelium (RPE). AIBSE is most probably a variant of MEWDS ([72] (EBM:2+, C), [236] (EBM:2+, C)).
10.2.4.2 Clinical Symptoms and Signs
The majority of patients with MEWDS are within the younger age groups, and a definite female predominance is observed. In a large series, it was noted that a preceding flu-like episode or upper respiratory tract infection can occur in up to 50 % of patients ([36] (EBM:2++, B)). An autoimmune or immunologic mechanism is suspected after by reports of MEWDS developing after hepatitis B vaccination [20] or detection of HLA-B51 haplotype ([71] (EBM:2+, C)). Typically, patients with MEWDS present with unilateral visual impairment in the form of visual field defects including blind spot enlargement and central, cecocentral, and arcuate scotomas. Symptoms such as photopsias and scotomas are associated with ERG abnormalities in 80 % of cases and photoreceptor dysfunction. The lesions of MEWDS appear as multiple, small, round, yellow to white spots distributed over the posterior fundus especially at the perifoveal (around the vascular arcades) and peripapillary regions. Macular granularity is a uniform and distinguishing feature of MEWDS in the convalescent stage. Other common clinical features include anterior chamber cells, vitreous cells, an afferent papillary defect, and mild optic disk swelling. The disease usually hits once, and the evolution is spontaneously favorable with restoration of visual function within 6–12 weeks. Inflammation is usually moderate and limited to the vitreous, but the optic disk can be involved. Angiographic exams are essential in ascertaining the pathology. ICGA shows hypofluorescent dots and peripapillary hypofluorescence in the acute phases. FA is associated with early hypofluorescence and late hyperfluorescence (mild staining) and optic disk hyperfluorescence.
AIBSE manifests with peripapillary scotoma producing symptomatic enlargement of the blind spot that can be identified through visual field testing. ERG shows focal abnormalities around the optic disk. ICGA shows peripapillary hypofluorescence indicating choriocapillaris non-perfusion. AIBSE can be associated with several primary inflammatory choriocapillaropathies such as multifocal choroiditis and punctate inner choroidopathy.
10.2.4.3 Differential Diagnosis
The main differential diagnosis of MEWDS is retrobulbar optic neuritis, and the best way to ascertain the diagnosis is the performance of an ICGA.
10.2.4.4 Treatment
The lesions of MEWDS resolve spontaneously without treatment.
Core Message
-
MEDWS is the result of primary inflammatory involvement of the choriocapillaris that in up to 50 % of cases is preceded by a viral flu-like syndrome.
-
Usually unilateral and unique episode.
-
The characteristics of the fundus are discrete discolorations in the midperiphery and granularity of the macula.
-
Photoreceptors dysfunction.
-
Visual field changes and blind spot enlargement.
-
ICGA hypofluorescent dots and peripapillary hypofluorescence resolving in two months.
10.2.5 Birdshot Retinochoroiditis
10.2.5.1 Definition
Birdshot retinochoroiditis (BC) is a form of posterior uveitis, characterized by multiple, distinctive, hypopigmented choroidal lesions, that is strongly associated with the human leukocyte antigen (HLA)-A29 [153]. The term “birdshot retinochoroidopathy” was first used in 1980, by Ryan and Maumenee [223], even though very recently the term is turning into “birdshot retinochoroiditis.”
10.2.5.2 Etiology
The etiology of BC remains still receding. BC shows a strong association with the phenotype HLA-A29, which is present in 80–98 % of the white patients affected by this disease, compared to 7 % of controls [17, 18, 153]. The sensitivity (96 %) and specificity (93 %) of HLA-A29 phenotype can have a determining role in confirming the diagnosis [79]. BC has been diagnosed in 0.6–1.5 % of patients with uveitis who were referred to tertiary centers [168, 216]. BC is slightly predominant in females, with a mean age at onset of 53 years and with the majority of patients being Caucasians [229].
10.2.5.3 Clinical Symptoms and Signs
BC does not seem to be associated with systemic disorders, with the exception of a few studies showing associations with hypertension [206], hearing loss and cutaneous vitiligo [89], and loss of brightness and luster of colors. Involvement is always bilateral but can be asymmetric. The most common presentation of patients affected by BC is characterized by varying degrees of painless visual loss, blurred visual acuity, floaters, paracentral scotomas, photopsias, and nyctalopia [89, 126]. Visual acuity does not seem to be a specific marker for the disease severity [180]. The anterior segment usually remains quiet, but it can reveal a mild iritis associated with granulomatous keratic precipitates on the corneal endothelium in approximately 25 % of cases [142, 186]. Fundus examination reveals multiple, bilateral, ovoidal, cream-colored birdshot lesions, distributed throughout the post-equatorial retina. These spots can be best visualized in the inferotemporal quadrant [153]. Biomicroscopic examination points out that lesions are located at the level of the choroid and retinal pigment epithelium (RPE). The granulomatous nature of the inflammation in BC has been verified histopathologically in a recent autopsy case [91]. At the onset of disease, fundus examination shows papillitis, periphlebitis, few cream-colored lesions, and vitreous infiltration which is usually present. Chronic cystoid macular edema (CME) is the most common complication of BC, occurring in up to 50 % of cases [206, 262]. The late findings of BC consist in epiretinal membranes [206], optic atrophy [49], macular and peripapillary choroidal neovascularization (CNV) in 6 % of cases [206], and RPE atrophy. The fundus fluoroangiographic (FFA) findings of the early and active phase show sectorial vasculitis mainly of the retinal veins, diffuse vasculitis of the retinal capillaries, pseudo-delay in arteriovenous circulation, optic disk hyperfluorescence, and CME, while the long-lasting cases “old lesions” show window defects of atrophic areas with early hypofluorescence and late hyperfluorescence and vessel attenuation [191]. The angiographic findings of indocyanine green angiography (ICGA) in the early and non-treated phase of the disease are numerous hypofluorescent dark dots corresponding to stromal granulomas (Fig. 10.21), regularly distributed along the posterior pole and midperiphery [190]. These dots may remain hypofluorescent or become isofluorescent in the late frames of ICGA; another characteristic angiographic sign pointed out by ICGA is the vasculitis of the larger choroidal vessels. Ocular coherence tomography (OCT) is a complementary examination to FFA. It evaluates macular thickness in case of vascular leakage, the photoreceptors inner/outer segment junction in cases of blurred VA, abnormal color, etc. The fundus autofluorescence (FAF) examination reveals hypoautofluorescence due to RPE atrophy, nonuniform correspondence between hypoautofluorescent areas and birdshot lesions, linear hypoautofluorescent streaks along retinal blood vessels which correspond to the visible changes at the level of the RPE, placoid hypoautofluorescent area at the macula which is correlated to a best corrected VA ≤ 20/50, macular RPE atrophy which predicts a low central VA, and non-correspondence of the RPE atrophic areas to hypopigmented lesions suggesting an independent affection of the RPE and choroid [143]. Visual field testing is a routine follow-up examination, and the occurrence or progression of visual field changes is considered an indication to introduce therapy [68]. Visual filed loss is much more indicative of the retinal dysfunction than VA alone. There are a variety of abnormalities despite normal best corrected VA such as multiple foci of scotomas, arcuate defects, and loss of the third highly reflective band on OCT that is associated with retinal damage. The introduction of immunomodulatory therapy (IMT) can reverse the visual field loss related to retinal dysfunction [253]. Full-field electroretinogram (ERG) becomes abnormal as the disease progresses, a fact that indicates relentless retinal deterioration. ERG shows decrease of the rod a- and b-wave amplitudes with an increase of their implicit times [85]. The most sensitive and prevalent abnormality is a delay of the cone system-derived 30 Hz flicker ERG [113].

Patient with Birdshot retinochoroiditis. Note the hot disk with few hyperfluorescent spots at the FA (a); a different clinical assessment can be done at the ICGA (b), where the distribution of the choroidal granulomata is better appreciated
10.2.5.4 Differential Diagnosis
The diagnosis of BC is mainly a clinical one, based on a careful ophthalmic examination and review of systems. The required diagnostic criteria are bilateral disease, birdshot lesions ≥3 inferior or nasal to optic nerve, low-grade anterior chamber inflammation, and low-grade vitreous inflammation. The supportive criteria are HLA-A29 positivity, retinal vasculitis, and CME. The exclusion criteria are posterior synechiae and other infectious, neoplastic, inflammatory entity [155]. Diagnosing birdshot retinochoroiditis is most challenging at the onset of disease, particularly if the typical birdshot lesions are subtle. The presence of the following signs such as mutton-fat keratic precipitates, hypopyon, or posterior synechiae in the early phase of the disease, in the majority of cases, is compatible with other pathologic entities such as Vogt-Koyanagi-Harada (VKH) disease, sarcoidosis, syphilis, and tuberculosis. The presence of concurrent systemic inflammatory disease suggests an alternative diagnosis, such as posterior scleritis, sarcoidosis, syphilis, tuberculosis, VKH disease, or intraocular lymphoma. The short and usually self-limited course of both acute posterior multifocal placoid pigment epitheliopathy (APMPPE) and multiple evanescent white dot syndrome (MEWDS) will distinguish these entities from the chronic and protracted course of birdshot retinochoroiditis. Pars planitis can almost always be distinguished from birdshot retinochoroiditis either by the presence of snowballs or snowbanking or by the absence of birdshot lesions in later phases of the disease. Sarcoidosis is the disease that is most difficult to distinguish from birdshot retinochoroiditis, because it shares a chronic course, and fundus lesions can occur in patients with sarcoidosis that mimics birdshot lesions [211, 264]. Patients with documented sarcoidosis who are HLA-A29-positive have been reported.
10.2.5.5 Treatment
The mainstay of treatment has been the employment of systemic and periocular corticosteroids (CS). CS are very important in the short-term management of vitritis and CME, but they show an inconsistent and transient efficacy in the long-term management because of the high maintenance dose of >15 mg/day to prevent CME and the severe adverse effects [85]. The use of regional CS is mainly adjunctive, employed in the inflammatory relapses in patients with systemic therapy or in asymmetrical disease. The early introduction of IMT shows an inherent anti-inflammatory effect and also a steroid-sparing effect of 10 mg/day of CS. The main treatment outcomes should include the reduction of inflammation and recurrences, preservation or reversal of a possible visual field loss, reduction of the risk of CME, and induction of long-term remission. The therapeutic modalities utilized in the treatment of BC include antimetabolites such as methotrexate (MTX), azathioprine, and mycophenolate mophetil (MMF); T-cell transduction inhibitors such as cyclosporine A (CSA), tacrolimus, and combination of MTX and MMF; biologics such as adalimumab, infliximab, and daclizumab; fluocinolone acetonide implant; emerging therapies such as voclosporin, anti-interleukin-17 monoclonal, and interferon α-2a; and intravenous immunoglobulins (IVIG). The indications for initiating therapy are symptomatic patients with photopsias, floaters and nyctalopia, vitritis, retinal vasculitis, CME, and peripheral retinal dysfunction revealed by visual field examination and ERG. A randomized, double-masked study comparing CSA to prednisolone in the treatment of endogenous uveitis by Nussenblatt RB et al. reported the efficacy of CSA in the treatment of two patients affected by BC at a dose of 10 mg/kg/day [179]. These findings were supported by Le Hoang et al., who treated 21 patients (42 eyes) affected by BC. There was a marked reduction of vitritis, improved visual acuity in 54.8 % of eyes, stabilization of VA in 26.2 % of eyes, and marked reduction of retinal vasculitis at the dose of 10 mg/kg/day of CSA [151]. This dose of CSA is now known to be associated with a high rate of nephrotoxic and hypertensive effects. Vitale et al. reported favorable outcomes in 19 patients (19 eyes) treated with CSA at a dosage of 2.5–5.0 mg/kg/day alone in 8 patients or in combination with AZA 1.5–2.0 mg/kg/day in 5 patients. The remaining patients were on systemic/periocular CS. Vitritis was controlled in 88.5 % of the cases; VA improved or stabilized in 83.8 % of the cases; there was a reduction of recurrences; and no nephrotoxic effects were observed [262]. Kiss et al. reported on a long-term follow-up of 81.2 months, in 28 patients affected by BC. All patients were treated with CS-sparing systemic IMT at some point during their follow-up: 92.9 % were treated with CSA, 67.9 % with MMF, 17.9 % with AZA, 10.7 % with oral MTX, and 7.1 % with daclizumab. VA remained stable or improved in 78.6 % OD, 89.3 % OS. The 30-Hz flicker implicit time was prolonged in 58.3 % of initial ERGs and in 62.5 % of final ERGs. The bright scotopic amplitude was abnormal in 45.5 % of initial and final ERGs [141]. Rothova et al. reported on the efficacy of low-dose MTX in 76 patients affected by BC, 46 of whom were followed for ≥ 5 years. The mean visual acuity underwent a statistically significant increase over time in the MTX-treated patients, remained unchanged in patients on systemic CS, and decreased in the patients without systemic treatment [218]. In a retrospective cohort study by Cervantes-Castaneda RA et al., there were 40 reported patients (80 eyes) affected by BC and treated with a combination therapy of CSA/MMF in a median time of 25.6 months with a median total patient follow-up of 52.6 months. At the 12-month point, a statistically significant reduction of vitritis and CME was achieved. Inflammatory control off systemic CS in 92.5 % of cases, long-term remission followed by absence of relapses in 64.9 % of cases, no reduction under 30 Hz of the amplitude/implicit times OD/OS, at least one relapse requiring change of IMT in 35.1 % of the cases, and a mean LogMAR VA not statistically different in both eyes were also reported [51]. In a report by Sorbin et al., there is satisfactory evidence of daclizumab employment in eight patients affected by BC and refractory to conventional IMT. The dose of daclizumab used was 1 mg/kg IV every 2 weeks with a mean follow-up of 25.6 months. Seven patients had either stabilization or improvement in visual acuity of both eyes and complete resolution of vitreous inflammation. Six patients had resolution of vasculitis on fluorescein angiography. The ERG 30-Hz implicit times and the bright scotopic amplitudes worsened in some patients despite abolition of clinically evident inflammation. Four patients were able to discontinue all other IMT and remain inflammation-free while receiving only daclizumab treatment. Two patients developed adverse effects that led to daclizumab treatment discontinuation [241], while Yeh et al. bring another modality of high doses of daclizumab in two patients with BC. They used 8 mg/kg at day 0 and 4 mg/kg at day 14. These treatment modality resulted in a mean visual acuity (10 eyes in 5 patients) that was 69.2 ETDRS letters and following treatment was 78.2 letters (p < 0.12). Anterior chamber cell, vitreous cell, and vitreous haze also improved in the majority of eyes. Adverse events were generally mild except for one episode of left-lower lobe pneumonia requiring hospitalization and treatment [283]. In a recent paper by Artornsombudh et al., there are reports on infliximab treatment of 22 patients with BC refractory to conventional IMT. The mean duration of the disease prior to infliximab was 58.62 months and the mean duration of infliximab therapy 13.55 months. All patients received 4–5 mg/kg infliximab at 4- to 8-week intervals. The main outcome measures were abolition of all evidence of active inflammation, visual acuity (VA), and presence of CME at 6 months and 1 year. Control of inflammation was achieved in 81.8 % at 6 months and in 88.9 % at the 1-year follow-up. The rate of CME decreased from 22.7 % at baseline to 13.9 % at 6 months and 6.7 % at 1 year after receiving the drug. Initial VA of 20/40 or better was found in 34 eyes (84.1 %). At 6 months and 1 year, 91.7 and 94.4 % of eyes, respectively, had VA of 20/40 or better. Three patients had active inflammation during therapy. Six patients developed adverse events requiring drug discontinuation [15]. Le Hoang et al. reported in a clinical study the tolerance and efficiency of IVIG treatment in 18 patients (36 eyes) with active BC with a mean follow-up of 39 months. IVIG was given as sole treatment at 1.6 g/kg every four weeks for six months, followed by injections of 1.2–1.6 g/kg at 6- to 8-week intervals. The results showed that the final VA of the 26 eyes with an initial VA of < or =20/30 was increased by two or more lines in 14 eyes (53.8 %) and decreased in two (7.7 %). When present, macular edema was improved in 17/23 eyes on fluorescein angiography and the visual field improved in 20/26 eyes. Benign side effects were observed in 12 patients: moderate transient arterial hypertension (7), headache (6), eczematous lesions (6), and hyperthermia (4) [152]. In a retrospective, multicenter, interventional case study, Rush et al., report on the outcomes in 22 patients (36 eyes) affected by BC and HLA-A29 positive following intravitreal implantation of a fluocinolone acetonide-containing drug delivery device. Nineteen of 22 patients (32 eyes) completed 12 months of follow-up with improvement in median visual acuity (p = 0.015). Eyes with zero vitreous haze increased from 7 of 27 scored eyes (26 %) at baseline to 30 of 30 eyes (100 %) by 12 months (p < 0.001). CME decreased from 13 of 36 eyes (36 %) at baseline to 2 of 32 eyes (6 %) at 12 months (p = 0.006). Prior to implantation, 18 of 22 patients (82 %) received immunosuppressive therapy vs. 1 of 19 (5 %) by 12 months (p < 0.001). Nineteen of twenty-two patients (32 eyes) completed 12 months of follow-up with improvement in median visual acuity (p = 0.015). Nineteen patients underwent cataract surgery, and all of the 22 patients had ocular hypertension, while 33 % of the cases required glaucoma surgery or pressure-lowering therapy [222].
Core Message
-
BC is a chronic progressive, sight-threatening disease.
-
Early and aggressive IMT limits ocular structural damage, preserves global visual function, and establishes long-term remission.
-
The treatment threshold is suggested by the markers of progressive disease such as the clinical indices of intraocular inflammation, visual field evaluation, ERG, and the various imaging modalities (Table 10.5).
Table 10.5 Treatment protocol for birdshot chorioretinopathy
10.2.6 Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE)
10.2.6.1 Definition
APMPPE is a primary inflammatory choriocapillaropathy characterized by sudden loss of vision caused by the sudden appearance of deep multiple yellow-white, flat inflammatory lesions.
10.2.6.2 Clinical Symptoms and Signs
APMPPE has a predilection for young adults with peak occurrence between the ages of 20 and 40 years and a range of 8–66 years [224] and both sexes are equally affected. APMPPE is most commonly bilateral and involvement may however be asymmetric and sequential in time. Patients present with sudden complaints of visual disturbance, photopsias, and scotomas without external evidence of ocular inflammation. During the active phase, fundus examination discloses multiple round, circumscribed, flat, yellow-white, subretinal lesions involving the RPE. The lesions may be multiple and confluent, forming large patches, and are localized mainly at the post-equatorial area. After several days to weeks, in the convalescent phase, the lesions begin to disappear leaving behind scattered areas of chorioretinal scars and mottling of the pigment epithelium in the zones of maximal involvement. Visual loss varies from minimal to severe and depends on the location of lesions. Visual field testing identifies the scotomas that are localized to the areas of fundus involvement. Other ocular findings include minimal anterior segment inflammation and cells in the vitreous and serous retinal detachment that can be seen in severe and hyperacute cases. Most of the patients with APMPPE have a history of a preceding flu-like syndrome before the onset of ocular symptoms, ([16, 60] (EBM: 2++, C)) and even preceding infectious episodes such as mumps and streptococcal group A infection ([37] (EBM:3, D), [158] (EBM:3, D)). Most commonly, the disease has occurred once, but in rare instances, it may recur. ICGA shows geographic hypofluorescent areas during all the course of the exam, while FA shows early hypofluorescence followed by late hyperfluorescence that has a geographic aspect. Electroretinography shows moderate and transient abnormalities in APMPPE ([195] (EBM:4, D)). In general, the visual prognosis in patients with APMPPE is good, and the time between the onset of visual loss and improvement may take as long as 6 months.
10.2.6.3 Differential Diagnosis
The most important disease to exclude is the early stage of serpiginous choroiditis which can mimic APMPPE in the beginning. In the case of strong suspects of serpiginous choroiditis, the tubercular etiology must be ruled out. Vogt-Koyanagi-Harada is another pathology to be excluded in the hyperacute forms of APMPPE, and ICGA is essential.
10.2.6.4 Treatment
Corticosteroids and/or immunosuppressive therapy has not been proved to be useful in APMPPE. If the tubercular etiology has been ruled out, a systemic corticosteroid therapy can be considered in patients with macular involvement given the vasculitic component in APMPPE ([198] (EBM:4, D)).
Core Message
-
Primary inflammatory choriocapilla-ropathy.
-
APMPPE usually has self-limiting course and is characterized by bilateral discolorations at the posterior pole.
-
The main symptoms are visual loss, scotomas, and photopsias.
-
FA: early hypofluorescence late hyperfluorescence.
-
ICGA: hypofluorescent areas.
-
ERG shows no abnormalities.
-
Most patients do not require therapy, but in cases of macular involvement, systemic steroid therapy can be considered because of the inflammatory etiology.
10.2.7 Vogt-Koyanagi-Harada (VKH) Disease
10.2.7.1 Definition
VKH disease is a severe granulomatous bilateral panuveitis and multisystem disorder affecting the eyes, auditory system, meninges, and skin [173, 239].
10.2.7.2 Etiology
Although the exact etiology of VKH remains unknown, the underlying immunopathological mechanism in VKH disease is believed to involve a T-cell-mediated autoimmune reaction against a melanocyte-related antigen, which is a member of the tyrosinase family of proteins [281]. VKH has been linked to human leukocyte antigen DR4 (HLA-DR4) and HLA-Dw53 [66], with the strongest associated risk for HLA-DRB1*0405 haplotype [233]. In the United States, Nussenblatt et al. reported that 44 % of the patients in their series were blacks [181]. VKH is a common cause of endogenous uveitis in Japan, with at least 8 % of the total cases [247]. Most patients develop VKH in the second to fifth decades of life, showing a slight female predominance [173, 247].
10.2.7.3 Clinical Symptoms and Signs
The clinical course of VKH has been divided into four clinical stages [247]. The prodromal stage which mimics a systemic viral infection whose symptoms include fever, headache, nausea, vertigo, orbital pain, meningismus, and tinnitus that represents a typical clinical symptom. The symptoms of prodromal stage normally last for a few days and are followed by the acute uveitis phase that lasts for several weeks. Patients in this stage present with acute blurring of vision and bilateral uveitis in both eyes in up to 70 % of patients [247], while the remaining 30 % may show a delay of 1–3 days regarding the involvement of the second eye. The early findings of the posterior segment consist in thickening of the posterior choroid, manifested as an elevation of the peripapillary retinochoroidal complex, and swelling of the optic nerve head [98]. Subsequent retinal pigment epithelium (RPE) breakdown causes multifocal exudative non-rhegmatogenous retinal detachment which can give rise to frank bullous exudative retinal detachment. The swelling of the optic nerve head is a marker of severe inflammation and is noted in 87 % of the patients with evolving disease [183]. The uveitis in the anterior segment initially may manifest as a nongranulomatous nature, which transforms into granulomatous at the later stages, causing mutton-fat keratic precipitates (KP) and iris nodules. In the early stages, the anterior chamber may be shallow [138] because of ciliary edema, serous detachment of the ciliary body [96], and forward displacement of the lens-iris diaphragm. These findings may cause a rise of the intraocular pressure (IOP) and acute angle closure glaucoma [139]. Harada’s disease represents the condition in which posterior uveitis, serous retinal detachment, and cerebrospinal fluid (CSF) pleocytosis are the only manifestations of VKH disease, while the Vogt-Koyanagi disease represents the form associated with bilateral iridocyclitis, vitiligo, poliosis, and auditory problems. The convalescent stage that follows may last for several months and is associated with depigmentation of the skin and uveal tract. Sugiura’s sign is a typical finding of early perilimbal depigmentation highly occurring in Japanese patients [84, 183]. The sunset glow fundus or depigmentation of the choroid typically occurs 2–3 months after the uveitic phase [98]. Dalen-Fuchs-like nodules, similar to those described in sympathetic ophthalmia, are frequently found in the midperiphery. The chronic recurrent phase, which abruptly interrupts the convalescent stage, is characterized by smoldering panuveitis with exacerbations of, typically, acute episodes of granulomatous anterior uveitis that are often resistant to systemic steroid therapy. Iris nodules are a characteristic finding of these recurrent episodes of disease. Being that VKH is a systemic disease, the presence of extraocular manifestations has an important role regarding the diagnosis. In the integumentary system, poliosis and vitiligo usually occur during the convalescent stage. Headache is the most common neurological complaint [30]. CSF pleocytosis has been found in 80 % of VKH patients [247]. Hearing loss usually involves the high frequencies, but all the frequencies can be affected in the early stage [117]. In the acute stage of the VKH, fundus fluorescein angiography (FFA) shows multiple punctate hyperfluorescent dots at the RPE level, which gradually enlarge and pool in the subretinal fluid underlying areas of exudative retinal detachment [90]. Optic nerve leakage is usually seen. In the chronic stage, FFA shows multiple window defects of the RPE or blocked fluorescence corresponding to damage of RPE. In some eyes, choroidal neovascularization (CNV) and subretinal fibrosis can occur as late complications [279]. The indocyanine green angiography (ICGA) findings in the acute phase are represented by filling delay of larger choroidal artery, fewer choroidal vessels in the posterior and peripheral fundus, patchy filling delay of choriocapillaris, ICG dye leakage, and multiple hypofluorescent spots, while in the convalescent phase, ICGA shows improvement of all the signs mentioned superiorly. The main ICGA signs for the evaluation of the inflammation and follow-up are hypofluorescent dark dots (Fig. 10.22), early hyperfluorescent choroidal vessels, fuzzy choroidal stromal vessels, and ICGA optic disk hyperfluorescence [40, 41]. Recently, optical coherence tomography (OCT) has shown its importance in evaluating and monitoring serous retinal detachment during the acute and chronic phases of VKH disease [167, 193]. OCT can discover small serous detachments, otherwise not detectable by slit-lamp biomicroscopy [193]. Ultrasonography (USG) has shown its utility as a diagnostic tool in the presence of obscured fundus view or in atypical presentations of VKH [81]. Regarding the diagnosis of VKH disease, new criteria, taking into account the multisystem nature of Vogt-Koyanagi-Harada disease, with allowance for the different ocular findings present in the early and late stages of the disease, were formulated and agreed at the First International Workshop on VKH Disease on October 19–21, 1999, at the University of California, Los Angeles, Conference Center [210].

FA of a patient with VKH disease. Note the widespread hypofluorescent spots at the early phase (a), followed by the hyperfluorescent spots at the late phase (b). Early (c) and late (d) phases of ICGA show a more extended involvement compromising the retina
10.2.7.4 Differential Diagnosis
The differential diagnosis of VKH disease includes other causes that manifest with granulomatous inflammation, exudative retinal detachment, and white dot syndromes such as sympathetic ophthalmia, Lyme disease with ocular involvement, multiple evanescent white dot syndrome (MEWDS), posterior scleritis, acute posterior multifocal placoid pigment epitheliopathy (APMPPE), and uveal effusion syndrome.
10.2.7.5 Treatment
The typical treatment for VKH disease is high-dose corticosteroids (CS) in the range of 1–2 mg/kg/day followed by a slow tapering of the drug for at least 3–6 months in order to prevent further recurrence. Sasamoto et al. evaluated the significance of corticosteroid therapy on 47 new patients with VKH disease in a follow-up period of 6 months. Eighteen patients received systemic CS as pulse therapy, 20 patients received high-dose CS starting with prednisolone 200 mg, 2 patients received conventional-dose CS, and 7 patients received no systemic CS therapy. After 6 months, anterior chamber inflammation was significantly less in patients with pulse and high-dose CS therapy than in those without systemic corticosteroid therapy, final visual acuity was significantly better in patients with pulse and high-dose CS than in those without them, while there was no significant difference between patients with pulse therapy and those with high-dose CS therapy [228]. In a paper of Rubsamen et al., we can find a review of 26 patients (44 eyes) affected by VKH disease treated with systemic CS, for a median period of 6 months, that was prolonged (48 months) in patients who developed chronic uveitis. The disease recurred in nine (43 %) of 21 patients in the first 3 months, usually in association with a rapid tapering of steroid dosage, and a final visual acuity of better than 20/30 in 29 (66 %) of 44 eyes and of worse than 20/400 in only 3 (7 %) of 44 eyes [221]. Yamanaka et al. evaluated through OCT the rapid effects of pulse CS therapy on the serous retinal detachment found at the acute phase of VKH disease on nine Japanese patients. OCT images showed a marked decrease in the retinal detachment immediately after the first intravenous injection of CS and subsequent resolution [282]. A multicenter study has shown an equal efficacy between the intravenous pulse steroid therapy and oral therapy with CS in improving the visual outcome [212]. Jaffe et al. investigated the safety and efficacy of a fluocinolone acetonide intravitreal implant in the treatment of 32 patients with a history of recurrent noninfectious posterior uveitis. None of these eyes experienced a recurrence for the first 2 years after implantation. There was a reduction in systemic and local therapy used in the device-implanted eyes. Inflammation was effectively controlled over the follow-up period. The posterior sub-Tenon capsule injection rate significantly decreased from a mean of 2.2 injections per eye per year to 0.07 injections per eye per year. Mean baseline visual acuity for the device-implanted eyes improved significantly from +1.1 logarithm of the minimum angle of resolution (logMAR) units to +0.81 logMAR units (20/125) at 30 months. The most common adverse event was intraocular pressure (IOP) rise [119]. In a case report by Perente et al., sub-Tenon triamcinolone acetonide injection is recommended in addition to systemic CS and cyclosporine (CSA) treatments if systemic medications fail to stop the progression of the VKH disease activity [199]. Despite proper treatment with CS, several studies reported the development of chronic recurrent granulomatous inflammation and sunset glow fundus with peripapillary atrophy and depigmented small atrophic lesions at the level of RPE [3, 7, 194]. In the article of Paredes et al., the main focus was on the use of immunomodulatory therapy (IMT) in a group of patients with VKH disease and to compare the outcomes with those of another group of patients with VKH who were treated for prolonged periods with CS. Their results suggest that IMT as first-line therapy for VKH is associated with a superior visual outcome when compared to CS as monotherapy or with delayed addition of IMT [194]. Recently, several studies suggested that the use of nonsteroid immunomodulatory therapy with CSA, azathioprine (AZA), methotrexate (MTX), and mycophenolate mofetil (MMF) as first-line therapy in addition to corticosteroids is associated with good clinical results. In a retrospective chart review by Sachdev et al., the clinical profile, management with AZA in association with CS, and outcome in seven patients with posterior segment recurrence in Vogt-Koyanagi-Harada (VKH) disease were reported. All the recurrent episodes of VKH were bilateral and were characterized by vitritis (8 eyes), papillitis (14 eyes), multiple yellow-white oval subretinal lesions6 eyes), and exudative retinal detachment (10 eyes). The first episodes of recurrence were managed with oral CS (1.0–1.5 mg/day) and AZA (2.0–2.5 mg/day). Three patients experienced a second episode of posterior segment recurrence, which also responded to the CS-AZA combination [225]. In a recent retrospective analysis of 87 patients, of whom 53 have initial-onset acute VKH disease and 34 have chronic recurrent VKH disease, Abu El-Asrar et al. recommend the use of CSA and MMF as first-line therapy. The results pointed out that this treatment modality significantly reduced the development of complications in the whole study group and in the initial-onset acute group, while the visual outcomes improved in the whole study group and in the chronic recurrent group [2]. A recent paper by Cuchacovich et al. reported a prospective comparison between 2 immunosuppressive regimens in patients with active VKH disease in spite of systemic glucocorticoid treatment, in 44 patients. Twenty-one patients developed chronic intraocular inflammation in spite of glucocorticoid treatment and were randomized to receive either prednisone and AZA (n = 12) or prednisone and CSA (n = 9). The results suggested that both regimens showed a good clinical efficacy, but CSA seemed to be a better glucocorticoid-sparing agent than AZA [63]. Another prospective study by Abu El-Asrar et al. deals with the effectiveness of MMF as first-line therapy combined with systemic CS in 19 patients with acute uveitis related to VKH disease, with mean follow-up period of 27.0 ± 11.1 months. The results showed a statistically significant reduction of recurrent inflammation (p = 0.0383) in the CS + MMF group (3 %) as compared to CS group (18 %). Development of all complications was significantly higher in the CS group (43 %) compared with the CS + MMF group (8 %) (p < 0.001). None of the eyes in the CS + MMF group developed sunset glow fundus [3]. A case report by Dolz-Marco et al. reports the employment of rituximab in a patient with chronic recurrent VKH, refractory to conventional IMT treatment [73]. Wu et al. reported in their paper the usefulness of intravitreal bevacizumab in two patients who had developed subfoveal and extrafoveal CNV due to VKH disease.
Core Message
-
VKH is a multisystem autoimmune disorder selectively targeting tissues containing melanocytes.
-
Ocular symptoms are preceded by headache, dysacousia, or tinnitus.
-
The ocular findings in the acute stage include bilateral and multifocal serous retinal detachment and swelling of the optic disk, while in the convalescent phase, the main findings are sunset glow fundus and irregular and linear pigmentation, which may result long after the onset of the disease.
-
The main FFA findings of the acute phase include multiple punctate hyperfluorescent dots at the RPE level, pooling in the subretinal fluid, and optic nerve leakage. The convalescent stage results in window defects due to RPE damage. The ICGA findings of the acute stage consist in by filling delay of larger choroidal artery, fewer choroidal vessels in the posterior and peripheral fundus, patchy filling delay of choriocapillaris, ICG dye leakage, and multiple hypofluorescent spots, while in the convalescent phase ICGA shows improvement of all the signs mentioned superiorly.
-
Though ocular inflammation responds to CS therapy, there is progressive depigmentation of the fundus. The damage to melanocyte-containing tissues goes on resulting in vitiligo, alopecia, and poliosis.
-
The principles of therapy in VKH disease are directed towards the suppression of the initial intraocular inflammation in the acute posterior uveitis stage with early and high-dose systemic CS followed by slow tapering, but despite proper treatment with corticosteroids, several studies have reported the development of chronic recurrent granulomatous inflammation and sunset glow fundus with peripapillary atrophy and depigmented small atrophic lesions at the level of retinal pigment epithelium. Recently published evidence is suggesting that the employment of nonsteroid immunomodulatory therapy with CSA, AZA, MTX, and MMF as first-line therapy in addition to CS is associated with good clinical results.
10.2.8 Sympathetic Ophthalmia (SO)
10.2.8.1 Definition
SO is a bilateral diffuse granulomatous panuveitis occurring either after surgery or penetrating trauma to one eye. The eye responsible for initiation of the inflammation is called exciting eye while the noninjured eye is known as the sympathizing eye. Penetrating or surgical injury to the exciting eye leads to an inflammatory response in both the exciting and the sympathizing eye [6].
10.2.8.2 Etiology
Trauma was considered as the most common precipitating event [6], while the recent papers tend to consider ocular surgery as a major risk factor, particularly vitreoretinal surgery [88, 136, 204]. Kilmartin et al. [136] calculated the risk of developing SO in retinal surgical procedures, which resulted to be higher more than twice the risk of developing endophthalmitis after vitrectomy. Continuous advances in the management of traumatized eyes associated with less invasive microsurgical techniques may be held responsible for the observed etiologic and epidemiologic changes from penetrating injuries to surgical traumas. Other etiologic factors involved with the development of SO are laser and surgical procedures such as glaucoma filtration surgery, peripheral iridectomy, cataract surgery, scleral buckling, evisceration, Nd-YAG laser cyclotherapy, and cyclocryotherapy [95, 99, 109, 163, 230, 235].
10.2.8.3 Clinical Symptoms and Signs
SO is a bilateral granulomatous uveitis occurring either after intentional or unintentional penetrating trauma to one eye. The latent period is usually between 2 weeks and 3 months; however, there are reports of cases presenting as early as 5 days and as late as 66 years after the incident [261, 285]. Approximately 80 % of cases present within the first 3 months and 90 % of cases present by 1 year of the penetrating trauma [159]. The inflammatory response in the anterior chamber is a granulomatous one, with mutton-fat keratic precipitates (KPs) on the corneal endothelium and the typical findings of acute anterior uveitis such as ciliary flush, pain, and photophobia in the sympathizing eye. Iritis may manifest with posterior synechiae. In the early stages, the inflammation may be nongranulomatous, associated with cells in the retrolental space. Intraocular pressure (IOP) may be high or low as a result of inflammatory cells crowding the trabecular meshwork or ciliary shutdown, respectively. The posterior segment findings typically consist of moderate to severe vitritis associated with papillitis and multiple peripheral white to yellow choroidal lesions, which later show a tendency towards confluency. These represent the clinical appearance of Dalen-Fuchs nodules. Papillitis is an important marker of disease activity and progression. The clinical appearance of SO varies within a mild to severe range. In a long-term study of Gupta et al., the most important posterior segment manifestations were exudative retinal detachment in the majority of patients, Dalen-Fuchs nodules, papillitis, and vasculitis as secondary events [103]. The complications of chronic inflammation include secondary glaucoma, cataract, and chronic maculopathy. Misdiagnosis and inappropriate treatment of this condition may result in severe inflammatory sequela such as retinal and optic atrophy, inflammatory choroidal neovascularization (CNV), choroidal atrophy, and phthisis bulbi [103]. Rarely, SO may be associated with the typical extraocular findings accompanying Vogt-Koyanagi-Harada syndrome, such as alopecia, vitiligo, cells in the cerebrospinal fluid, and dysacousia. In the acute phase of SO, fundus fluorescein angiography (FFA) shows multiple hyperfluorescent sites of leakage at the retinal pigment epithelium (RPE) in the transit stage, while in the late phase, FFA shows late leakage. In severe cases, the sites of leakage may coalesce, with pooling of dye and consequent exudative neurosensory detachment. Late staining of the optic nerve head may sometimes be seen. Dalen-Fuchs lesions may appear hyper- or hypofluorescent, depending on the RPE condition [231]. During the intermediate phase of indocyanine green angiography (ICGA), the examiner observes hypofluorescent areas [31]. During the late phase of ICGA, the hypofluorescent areas seen in the intermediate phase may persist or may fade to isofluorescent ones, reflecting the behavior of full-thickness and partial-thickness choroidal granulomas, respectively. Late atrophic lesions of SO appear as hypofluorescent areas, not changing their behavior in ICGA even in the presence of systemic corticosteroids (CS).
10.2.8.4 Differential Diagnosis
SO represents a clinical diagnosis, relying essentially on a history of ocular trauma or surgery, which evolves in a bilateral granulomatous uveitis. The clinical findings of SO may be difficult to distinguish from those of VKH [98]. Patients with VKH do not have history of trauma. They typically show bilateral localized exudative neurosensory detachments, a sign which is absent in SO patients. VKH patients frequently present with auditory, integumentary, and meningeal signs, which are very rare in SO patients. VKH has a predilection for darkly pigmented races such as blacks and Asians, and most patients are affected during the 2nd–5th decades of life, while SO is not associated with these epidemiologic factors. VKH shows a tendency to involve choriocapillaris during its course, while SO does not. Other important differential diagnoses include granulomatous conditions such as tuberculosis, sarcoidosis, infective endophthalmitis, intraocular lymphoma, and lens-induced uveitis.
10.2.8.5 Treatment
Management of SO patients includes surgical and medical treatment. Enucleation of the injured eye is generally recommended within 14 days after ocular injury [159]. Because of the decreasing incidence of injured eyes developing SO, this approach is no longer advised. Nowadays, a lot of controversy exists regarding the value of enucleation once the inflammatory process has begun, as the exciting eye may actually present with better visual acuity (VA) than the sympathizing one. Lubin et al. reported that early enucleation of the exciting eye after onset of symptoms in the fellow eye was found to improve visual prognosis ([32] EBM C 2 +), while in another review of Winter, there are reports that show no benefits from enucleation of the exciting eye ([272] EBM D 4). Corticosteroids have served as the mainstay of treatment following onset ([52] EBM B 2+), which are used as intravenous, oral, topical, or regional injections ([54] EBM D 3, [110] EBM C3, [187] EBM D3). Nussenblatt et al. are recommending 3 months of high daily dose of 1–2 mg/kg CS, tapering in three to six months and considering uveitis quiescent with a maintenance dose of ≤15 mg of CS ([178] EBM D 4). In severe cases of SO, short courses of intravenous CS may be considered [110]. In cases that seem to be refractory to corticosteroids and in patients who show significant systemic side effects, steroid-sparing therapy that combines systemic corticosteroids and other immunosuppressive agents such as cyclosporine or azathioprine can improve prognosis, particularly in patients that have initial response to steroid but exhibit rebound activity when steroid is tapered to lower doses. Combining CS with other steroid-sparing agents such as cyclosporine A (CSA) and azathioprine (AZA) has shown benefits in patients that are refractory to the association of CS with a single immunosuppressive agent ([106] EBM C 3, [263] EBM C3). Nussenblatt suggests the employment of triple immunosuppressive therapy in patients where SO is difficult to control, such as CS, CSA, immunosuppressive agents, and biologics ([178] EBM C 4). Being that in attendance to recent studies, evidence suggests that sympathetic ophthalmia represents an autoimmune inflammatory response against choroidal melanocytes mediated by T cells ([65] EBM C 3, [203]); CSA represents the steroid-sparing agent of choice in CS refractory SO. Therapies including immunosuppressive agents such as mycophenolate mofetil (MMF) and chlorambucil have shown efficacy in patients refractory to “conventional” treatment ([149] EBM D3, [251] EBM D 3). Disruption of leukocyte recruitment by targeting gelatinase B (matrix metalloproteinase-9), CCL2, and CXCL12 may hold promise for future treatments ([4] EBM D 3). Furusato et al. reported that M1 macrophages, IL-23, CCL19, CXCL11, and IL-17 predominate within the granulomatous infiltrates of SO, findings which suggest the targeting of M1 macrophages and their cytokines and chemokines, Th17, or Th1 lymphocytes ([86] EBM D 3). Treatment with anti-TNFα agents has been used for uveitis suggesting potential benefit for sympathetic ophthalmia ([259] EBM D 3). Mahajan et al. examined the results of fluocinolone acetonide implantation (Retisert) in eight patients with active SO reporting satisfactory control of inflammation and a decrease in the dependence on systemic immunosuppression ([162] EBM D 3).
Key Points
-
1.
SO is a bilateral, diffuse granulomatous panuveitis that occurs after eye injury (trauma or surgery).
-
2.
Other etiologic factors involved with the development of SO are laser and surgical procedures.
-
3.
Histopathologic findings include granulomatous inflammation of the uvea, sparing the choriocapillaris.
-
4.
The most common clinical findings include mutton-fat KPs, papillitis, Dalen-Fuchs nodules, and vitritis.
-
5.
Chronic inflammation develops sequelae such as chronic maculopathy, CNV, optic atrophy, and secondary glaucoma.
-
6.
The most important diagnostic tools include FFA and ICGA.
-
7.
Management involves long-term CS, CSA, and other immunosuppressive agents, while enucleation is controversial.
References
Abrez H, Biswas J, Sudharshan S. Clinical profile, treatment, and visual outcome of serpiginous choroiditis. Ocul Immunol Inflamm. 2007;15(4):325–35.
Abu El-Asrar AM, Al Tamimi M, Hemachandran S, Al-Mezaine HS, Al-Muammar A, Kangave D. Prognostic factors for clinical outcomes in patients with Vogt-Koyanagi-Harada disease treated with high-dose corticosteroids. Acta Ophthalmol. 2013;91(6):e486–93.
Abu El-Asrar AM, Hemachandran S, Al-Mezaine HS, Kangave D, Al-Muammar AM. The outcomes of mycophenolate mofetil therapy combined with systemic corticosteroids in acute uveitis associated with Vogt-Koyanagi-Harada disease. Acta Ophthalmol. 2012;90(8):e603–8.
Abu El-Asrar AM, Struyf S, Van den Broeck C, Van Damme J, Opdenakker G, Geboes K, Kestelyn P. Expression of chemokines and gelatinase B in sympathetic ophthalmia. Eye (Lond). 2007;21(5):649–57 [3 D].
Abu el-Asrar AM. Serpiginous (geographical) choroiditis. Int Ophthalmol Clin. 1995;35:87–91.
Albert DM, Diaz-Rohena R. A historical review of sympathetic ophthalmia and its epidemiology. Surv Ophthalmol. 1989;34(1):1–14.
Al-Kharashi AS, Aldibhi H, Al-Fraykh H, Kangave D, Abu El-Asrar AM. Prognostic factors in Vogt-Koyanagi-Harada disease. Int Ophthalmol. 2007;27(2–3):201–10.
Amer R, Lois N. Punctate inner choroidopathy. Surv Ophthalmol. 2011;56(1):36–53.
American Thoracic Society. Targeted tuberculin testing and treatment of latent tuberculosis infection. MMWR Recomm Rep. 2000;49:1e51 (A).
Ang M, Hedayatfar A, Wong W, Chee SP. Duration of anti-tubercular therapy in uveitis associated with latent tuberculosis: a case-control study. Br J Ophthalmol. 2012;96(3):332–6 (C).
Ang M, Htoon HM, Chee SP. Diagnosis of tuberculous uveitis: clinical application of an interferon-gamma release assay. Ophthalmology. 2009;116(7):1391–6 (C).
Ang M, Wong WL, Li X, Chee SP. Interferon ϒ release assay for the diagnosis of uveitis associated with tuberculosis: a Bayesian evaluation in the absence of a gold standard. Br J Ophthalmol. 2013;97(8):1062–7. (C)s.
Anninger W, Lubow M. Visual loss with West Nile virus infection: a wider spectrum of a “new” disease. Clin Infect Dis. 2004;38:e55–6.
Arevalo JF, Adan A, Berrocal MH, Espinoza JV, Maia M, Wu L, Roca JA, Quiroz-Mercado H, Ruiz-Moreno JM, Serrano MA, Pan-American Collaborative Retina Study Group. Intravitreal bevacizumab for inflammatory choroidal neovascularization: results from the pan-American collaborative retina study group at 24 months. Retina. 2011;31(2):353–63.
Artornsombudh P, Gevorgyan O, Payal A, Siddique SS, Foster CS. Infliximab treatment of patients with birdshot retinochoroidopathy. Ophthalmology. 2013;120(3):588–92.
Azar P, Gohd RS, Waltman D, Gitter KA. Acute posterior multifocal placoid pigment epitheliopathy associated with an adenovirus type 5 infection. Am J Ophthalmol. 1975;80:1003–5.
Baarsma GS, Kijlstra A, Oosterhuis JA, et al. Association of birdshot retinochoroidopathy and HLA-A29 antigen. Doc Ophthalmol. 1986;61:267–9.
Baarsma GS, Priem HA, Kijlstra A. Association of birdshot retinochoroidopathy and HLA-A29 antigen. Curr Eye Res. 1990;9(Suppl):63–8.
Bacin F, Larmande J, Boulmier A, et al. Serpiginous choroiditis and placoid epitheliopathy. Bull Soc Ophthalmol Fr. 1983;83:1153–62.
Baglivo E, Safran AB, Borruat FX. Multiple evanescent white dot syndrome after hepatitis B vaccine. Am J Ophthalmol. 1996;122:431.
Baharivand N, Mahdavifard A, Fouladi RF. Intravitreal clindamycin plus dexamethasone versus classic oral therapy in toxoplasmic retinochoroiditis: a prospective randomized clinical trial. Int Ophthalmol. 2013;33(1):39–46.
Balne PK, Barik MR, Sharma S, Basu S. Development of a loop-mediated isothermal amplification assay targeting the mpb64 gene for diagnosis of intraocular tuberculosis. J Clin Microbiol. 2013;51(11):3839–40.
Banker A. Posterior segment manifestations of human immunodeficiency virus/acquired immune deficiency syndrome. Indian J Ophthalmol. 2008;56(5):377–83.
Bansal R, Gupta A, Gupta V, Dogra MR, Bambery P, Arora SK. Role of anti-tubercular therapy in uveitis associated with latent tuberculosis. Am J Ophthalmol. 2008;146(5):772–9. C.
Bansal R, Gupta A, Gupta V, Dogra MR, Sharma A, Bambery P. Tubercular serpiginous-like choroiditis presenting as multifocal serpiginoid choroiditis. Ophthalmology. 2012;119(11):2334–42.
Bansal R, Kulkarni P, Gupta A, Gupta V, Dogra MR. High-resolution spectral domain optical coherence tomography and fundus autofluorescence correlation in tubercular serpiginous-like choroiditis. J Ophthalmic Inflamm Infect. 2011;1(4):157–63.
Barisani-Asenbauer T, Maca SM, Hauff W, et al. Treatment of ocular toxocariasis with albendazole. J Ocul Pharmacol Ther. 2001;17:287–94.
Basu S, Biswas J, Pleyer U, Pathangay A, Manohar BB. An ophthalmologist survey-based study of the atypical presentations and current treatment practices of ocular toxoplasmosis in India. J Parasit Dis. 2011;35:148–54.
Basu S, Nayak S, Padhi TR, Das T. Progressive ocular inflammation following anti-tubercular therapy for presumed ocular tuberculosis in a high-endemic setting. Eye. 2013;27(5):657–62.
Beniz J, Forster DJ, Lean JS, Smith RE, Rao NA. Variations in clinical features of the Vogt-Koyanagi-Harada syndrome. Retina. 1991;11(3):275–80.
Bernasconi O, Auer C, Zografos L, Herbort CP. Indocyanine green angiographic findings in sympathetic ophthalmia. Graefes Arch Clin Exp Ophthalmol. 1998;236(8):635–8.
Bilyk JR. Enucleation, evisceration, and sympathetic ophthalmia. Curr Opin Ophthalmol. 2000;11(5):372–86 [2+ C].
Blacksell SD, Newton PN, Bell D, et al. The comparative accuracy of 8 commercial rapid immunochromatographic assays for the diagnosis of acute dengue virus infection. Clin Infect Dis. 2006;42:1127–34.
Blumenkranz MS, Gass JD, Clarkson JG. Atypical serpiginous choroiditis. Arch Ophthalmol. 1982;100:1773–5.
Bodaghi B. Ocular manifestations of Lyme disease. Med Mal Infect. 2007;37(7–8):518–22.
Borruat FX, Otheni-Girard P, Safran AB. Multiple evanescent white dot syndrome. Klin Monatsbl Augenheilkd. 1991;198:453–6.
Borruat FX, Piguet B, Herbort CP. Acute posterior multifocal placoid pigment epitheliopathy following mumps. Ocul Immunol Inflamm. 1998;6:189–93.
Bosch-Driessen LE, Berendschot TT, Ongkosuwito JV, Rothova A. Ocular toxoplasmosis: clinical features and prognosis of 154 patients. Ophthalmology. 2002;109:869–78.
Bosch-Driessen LH, Plaisier MB, Stilma JS, Van der Lelij A, Rothova A. Reactivations of ocular toxoplasmosis after cataract extraction. Ophthalmology. 2002;109:41–5.
Bouchenaki N, Cimino L, Auer C, et al. Assessment and classification of choroidal vasculitis in posterior uveitis using indocyanine green angiography. Klin Monatsbl Augenheilkd. 2002;219:243–9.
Bouchenaki N, Herbort CP. The contribution of indocyanine green angiography to the appraisal and management of Vogt-Koyanagi-Harada disease. Ophthalmology. 2001;108(1):54–64.
Brett-Major DM, Coldren R. Antibiotics for leptospirosis. Cochrane Database Syst Rev. 2012;2:CD008264. EbM 1-.
Brighton SW, Prozesky OW, de la Harpe AL. Chikungunya virus infection. A retrospective study of 107 cases. S Afr Med J. 1983;63:313–5.
Brighton SW. Chloroquine phosphate treatment of chronic Chikungunya arthritis. An open pilot study. S Afr Med J. 1984;66:217–8.
Briolant S, Garin D, Scaramizzino N, Jouan A, Crance JM. In vitro inhibition of Chikungunya and Semliki Forest viruses replication by antiviral compounds: synergistic effect of interferon-alpha and ribavirin combination. Antiviral Res. 2004;61:111–7.
Brown Jr J, Folk JC, Reddy CV, Kimura AE. Visual prognosis of multifocal choroiditis, punctate inner choroidopathy, and diffuse subretinal fibrosis syndrome. Ophthalmology. 1996;103:1100–5.
Burt FJ, Rolph MS, Rulli NE, Mahalingam S, Heise MT. Chikungunya: a re-emerging virus. Lancet. 2012;379(9816):662–7.
Butler NJ, Furtado JM, Winthrop KL, Smith JR. Ocular toxoplasmosis II: clinical features, pathology and management. Clin Experiment Ophthalmol. 2013;41:95–108.
Caballero-Presencia A, Diaz-Guia E, Lopez-Lopez JM. Acute anterior ischemic optic neuropathy in birdshot retinochoroidopathy. Ophthalmologica. 1988;196:87–91.
Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2006. MMWR Morb Mortal Wkly Rep. 2006;55(No.RR-11):1–96.
Cervantes-Castaneda RA, Bhat P, Fortuna E, Acevedo S, Foster CS. Induction of durable remission in ocular inflammatory diseases. Eur J Ophthalmol. 2009;19(1):118–23.
Chan CC, Roberge RG, Whitcup SM, Nussenblatt RB. 32 cases of sympathetic ophthalmia. A retrospective study at the National Eye Institute, Bethesda, Md., from 1982 to 1992. Arch Ophthalmol. 1995;113(5):597–600.
Chan DP, Teoh SC, Tan CS, et al. Ophthalmic complications of dengue. Emerg Infect Dis. 2006;12:285–9.
Chan RV, Seiff BD, Lincoff HA, Coleman DJ. Rapid recovery of sympathetic ophthalmia with treatment augmented by intravitreal steroids. Retina. 2006;26(2):243–7 [3 D].
Chazalon E, Conrath J, Ridings B, Matonti F. Kyrieleis arteritis: report of two cases and literature review. J Fr Ophtalmol. 2013;36:191–6.
Chi SL, Stinnett S, Eggenberger E, Foroozan R, Golnik K, Lee MS, Bhatti MT. Clinical characteristics in 53 patients with cat scratch optic neuropathy. Ophthalmology. 2012;119(1):183–7. EbM 2b.
Chia A, Luu CD, Mathur R, Cheng B, Chee SP. Electrophysiological findings in patients with dengue-related maculopathy. Arch Ophthalmol. 2006;124(10):1421–6.
Chlebicki MP, Ang B, Barkham T, Lande A. Retinal hemorrhages in 4 patients with dengue fever. Emerg Infect Dis. 2005;11:770–1.
Christmas NJ, Oh KT, Oh DM, et al. Long-term follow up of patients with serpiginous choroiditis. Retina. 2002;22:550–6.
Cimino L, Auer C, Herbort CP. Sensitivity of indocyanine green angiography for the follow-up of active inflammatory choriocapillaropathies. Ocul Immunol Inflamm. 2000;8:275–83.
Couvreur J, Thulliez P, Daffos F, et al. In utero treatment of toxoplasmic fetopathy with the combination pyrimethamine-sulfadiazine. Fetal Diagn Ther. 1993;8:45–50.
Cruz-Villegas V, Berrocal AM, Davis JL. Bilateral choroidal effusions associated with dengue fever. Retina. 2003;23:576–8.
Cuchacovich M, Solanes F, Díaz G, Cermenati T, Avila S, Verdaguer J, Verdaguer JI, Carpentier C, Stopel J, Rojas B, Traipe L, Gallardo P, Sabugo F, Zanoli M, Merino G, Villarroel F. Comparison of the clinical efficacy of two different immunosuppressive regimens in patients with chronic Vogt-Koyanagi-Harada disease. Ocul Immunol Inflamm. 2010;18(3):200–7.
Cunningham ET, Koehler JE. Ocular bartonellosis. Am J Ophthalmol. 2000;130(3):340–9.
Damico FM, Kiss S, Young LH. Sympathetic ophthalmia. Semin Ophthalmol. 2005;20(3):191–7 [3C].
Davis JL, Mittal KK, Freidlin V, Mellow SR, Optican DC, Palestine AG, Nussenblatt RB. HLA associations and ancestry in Vogt-Koyanagi-Harada disease and sympathetic ophthalmia. Ophthalmology. 1990;97(9):1137–42.
De Amorim Garcia CA, Gomes AH, De Oliveira AG. Bilateral stellar neuroretinitis in a patient with dengue fever. Eye. 2006;20(12):1382–3.
De Courten C, Herbort CP. The potential role of computerized field testing for the appraisal and follow-up of birdshot chorioretinopathy. Arch Ophthalmol. 1998;116:1389–91.
De Lamballerie X, Boisson V, Reynier JC, et al. On chikungunya acute infection and chloroquine treatment. Vector Borne Zoonotic Dis. 2008;8:837–9.
Dees C, Arnold JJ, Forrester JV, Dick AD. Immunosuppressive treatment of choroidal neovascularization associated with endogenous posterior uveitis. Arch Ophthalmol. 1998;116:1456–61.
Desarnaulds AB, Borruat FX, Herbort CP, Spertini F. Multiple evanescent white dot syndrome: a genetic disorder? Klin Monatsbl Augenheilkd. 1996;208:301–2.
Dodwell DD, Jampol LM, Rosenberg M, et al. Optic nerve involvement associated with multiple evanescent white dot syndrome. Ophthalmology. 1990;97:862.
Dolz-Marco R, Gallego-Pinazo R, Díaz-Llopis M. Rituximab in refractory Vogt-Koyanagi-Harada disease. J Ophthalmic Inflamm Infect. 2011;1(4):177–80.
Dreyer RF, Gass JDM. Multifocal choroiditis and panuveitis: a syndrome that mimics ocular histoplasmosis. Arch Ophthalmol. 1984;102:1776.
Dunlop AA, Cree IA, Hagee S, Luthert PJ, Lightman S. Multifocal choroiditis, clinicopathologic correlation. Arch Ophthalmol. 1998;116:801–3.
Dunn D, Wallon M, Peyron F, Petersen E, et al. Mother-to-child transmission of toxoplasmosis: risk estimates for clinical counselling. Lancet. 1999;353:1829–33.
Eandi CM, Neri P, Adelman RA, Yannuzzi LA, Cunningham Jr ET, International Syphilis Study Group. Acute syphilitic posterior placoid chorioretinitis: report of a case series and comprehensive review of the literature. Retina. 2012;32(9):1915–41.
Faine S, Adler B, Bolin C, Perolat P. Leptospira and leptospirosis. 2nd ed. Melbourne: Med Sci; 1999. p. 1–230.
Feltkamp TE. Ophthalmological significance of HLA associated uveitis. Eye. 1990;4:839–44.
Flaxel CJ, Owens SL, Mulholland B, Schwartz SD, Gregor ZJ. The use of corticosteroids for choroidal neovascularization in young patients. Eye (Lond). 1998;12:266–72.
Forster DJ, Cano MR, Green RL, Rao NA. Echographic features of the Vogt-Koyanagi-Harada syndrome. Arch Ophthalmol. 1990;108(10):1421–6.
Forster DJ, Dugel PU, Frangieh GT, Liggett PE, Rao NA. Rapidly progressive outer retinal necrosis in the acquired immunodeficiency syndrome. Am J Ophthalmol. 1990;110(4):341–8.
Freeman K, Tan HK, Prusa A, Petersen E, et al. Predictors of retinochoroiditis in children with congenital toxoplasmosis: European, prospective cohort study. Pediatrics. 2008;121:e1215–22.
Friedman AH, Deutsch-Sokol RH. Sugiura’s sign. Perilimbal vitiligo in the Vogt-Koyanagi-Harada syndrome. Ophthalmology. 1981;88(11):1159–65.
Fuerst DJ, Tessler HH, Fishman GA, et al. Birdshot retinochoroidopathy. Arch Ophthalmol. 1984;102:214–9.
Furusato E, Shen D, Cao X, Furusato B, Nussenblatt RB, Rushing EJ, Chan CC. Inflammatory cytokine and chemokine expression in sympathetic ophthalmia: a pilot study. Histol Histopathol. 2011;26(9):1145–51 [3 D].
Garg S, Jampol LM. Systemic and intraocular manifestations of West Nile virus infection. Surv Ophthalmol. 2005;50:3–13.
Gass JD. Sympathetic ophthalmia following vitrectomy. Am J Ophthalmol. 1982;93(5):552–8.
Gass JD. Vitiliginous chorioretinitis. Arch Ophthalmol. 1981;99:1778–87.
Gass JDM. Harada’s disease. In: Gass JDM, editor. Stereoscopic atlas of macular disease. Diagnosis and treatment. St Louis: Mosby; 1987. p. 150–3.
Gaudio PA, Kaye DB, Brooks CJ. Histopathology of birdshot retinochoroidopathy. Br J Ophthalmol. 2002;86:1439–41.
Gerstenblith AT, Thorne JE, Sobrin I, et al. Punctate inner choroidopathy: a survey analysis of 77 persons. Ophthalmology. 2007;114:1201–4.
Gilber RE, Freeman K, Lago EG, Bahia-Oliveira LM, et al. Ocular sequelae of congenital toxoplasmosis in Brazil compared with Europe. PLoS Negl Trop Dis. 2008;2:e277.
Gilbert R, Dezateux C. Newborn screening for congenital toxoplasmosis: feasible, but benefits are not established. Arch Dis Child. 2006;91:629–31.
Glavici M. Sympathetic ophthalmia after a cataract operation. Oftalmologia. 1992;36(4):397–401.
Gohdo T, Tsukahara S. Ultrasound biomicroscopy of shallow anterior chamber in Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol. 1996;122(1):112–4.
Goldenberg D, Goldstein M, Loewenstein A, Habot-Wilner Z. Vitreal, retinal, and choroidal findings in active and scarred toxoplasmosis lesions: a prospective study by spectral-domain optical coherence tomography. Graefes Arch Clin Exp Ophthalmol. 2013;251:2037–45.
Goto H, Rao NA. Sympathetic ophthalmia and Vogt-Koyanagi-Harada syndrome. Int Ophthalmol Clin. 1990;30(4):279–85.
Green WR, Maumenee AE, Sanders TE, Smith ME. Sympathetic uveitis following evisceration. Trans Am Acad Ophthalmol Otolaryngol. 1972;76(3):625–44.
Gupta A, Bansal R, Gupta V, Sharma A, Bambery P. Ocular signs predictive of tubercular uveitis. Am J Ophthalmol. 2010;149:562–70. C.
Gupta A, Bansal R, Gupta V, Sharma A. Fundus autofluorescence in serpiginous-like choroiditis. Retina. 2012;32(4):814–25.
Gupta V, Bansal R, Gupta A. Continued progression of tubercular serpiginous-like choroiditis after initiating antituberculosis treatment. Am J Ophthalmol. 2011;152:857–63.
Gupta V, Gupta A, Dogra MR. Posterior sympathetic ophthalmia: a single centre long-term study of 40 patients from North India. Eye (Lond). 2008;22(12):1459–64.
Gupta V, Gupta A, Rao NA. Intra-ocular tuberculosis – an update. Surv Ophthalmol. 2007;52:561–87.
Gupta V, Agarwal A, Gupta A, Bambery P, Narang S. Clinical characteristics of serpiginous choroidopathy in North India. Am J Ophthalmol. 2002;134(1):47–56.
Hakin KN, Pearson RV, Lightman SL. Sympathetic ophthalmia: visual results with modern immunosuppressive therapy. Eye (Lond). 1992;6(Pt 5):453–5 [3C].
Halstead SB. Dengue. Curr Opinion Infect Dis. 2002;15:471–6.
Hardy RA, Schatz H. Macular geographic helicoid choroidopathy. Arch Ophthalmol. 1987;105:1237–42.
Harrison TJ. Sympathetic ophthalmia after cyclocryotherapy of neovascular glaucoma without ocular penetration. Ophthalmic Surg. 1993;24(1):44–6.
Hebestreit H, Huppertz HI, Sold JE, Dämmrich J. Steroid-pulse therapy may suppress inflammation in severe sympathetic ophthalmia. J Pediatr Ophthalmol Strabismus. 1997;34(2):124–6 [3C].
Herbort CP, Papadia M, Neri P. Myopia and inflammation. J Ophthalmic Vis Res. 2011;6(4):270–83. EBM (4).
Hogan A, Behan U, Kilmartin DJ. Outcomes after combination photodynamic therapy and immunosuppression for inflammatory subfoveal choroidal neovascularization. Br J Ophthalmol. 2005;89:1109–11.
Holder GE, Ag R, Pavesio CP, Graham EM. Electrophysiological characterization and monitoring in the management of birdshot chorioretinopathy. Br J Ophthalmol. 2005;89:709–18.
Holland GN, Lewis KG. An update on current practices in the management of ocular toxoplasmosis. Am J Ophthalmol. 2002;134:102–14.
Holland GN. Standard diagnostic criteria for the acute retinal necrosis syndrome. Executive Committee of the American Uveitis Society. Am J Ophthalmol. 1994;117:663–7.
Hooper PL, Kaplan HJ. Triple agent immunosuppression in serpiginous choroiditis. Ophthalmology. 1991;98(6):944–51.
Hoshi H, Tamada Y, Murada Y, et al. Changes in audiogram in the course of Harada’s disease. Jpn J Clin Ophthalmol. 1977;31:23–30.
Huppertz HI, Bartmann P, Heininger U, Fingerle V, Kinet M, Klein R, Korenke GC, Nentwich HJ, Committee for Infectious Diseases and Vaccinations of the German Academy for Pediatrics and Adolescent Health. Rational diagnostic strategies for Lyme borreliosis in children and adolescents: recommendations by the Committee for Infectious Diseases and Vaccinations of the German Academy for Pediatrics and Adolescent Health. Eur J Pediatr. 2012;171(11):1619–24.
Jaffe GJ, McCallum RM, Branchaud B, Skalak C, Butuner Z, Ashton P. Long-term follow-up results of a pilot trial of a fluocinolone acetonide implant to treat posterior uveitis. Ophthalmology. 2005;112(7):1192–8.
Jasper S, Vedula SS, John SS, Horo S, Sepah YJ, Nguyen QD. Corticosteroids for ocular toxoplasmosis. Cochrane Database Syst Rev. 2013;4:CD007417.
Jeon S, Kakizaki H, Lee WK, et al. Effect of prolonged oral acyclovir treatment in acute retinal necrosis. Ocul Immunol Inflamm. 2012;20(4):288–92.
John M, Samson CM, Foster CS. Syphilis. In: Foster CS, Vitale AT, editors. Diagnosis & treatment of uveitis. New Delhi: Jaypee Medical Publishers; 2013. p. 337–45.
Jones BE, Jampol LM, Yannuzzi LA, et al. Relentless placoid chorioretinitis: a new entity or an unusual variant of serpiginous choroiretinitis? Am J Ophthalmol. 2000;118:931–8.
Julián K, Terrada C, Fardeau C, Cassoux N, Français C, LeHoang P, Bodaghi B. Intravitreal bevacizumab as first local treatment for uveitis-related choroidal neovascularization: long-term results. Acta Ophthalmol. 2011;89(2):179–84.
Kanungo S, Shukla D, Kim R. Branch retinal artery occlusion secondary to dengue fever. Indian J Ophthalmol. 2008;56(1):73–4.
Kaplan HJ, Aaberg TM. Birdshot retinochoroidopathy. Am J Ophthalmol. 1980;90:773–82.
Kapoor HK, Bhai S, John M, Xavier J. Ocular manifestations of dengue fever in an East Indian epidemic. Can J Ophthalmol. 2006;41:741–6.
Kawaguchi T, Spencer DB, Mochizuki M. Therapy for acute retinal necrosis. Semin Ophthalmol. 2008;23(4):285–90.
Kennedy JJ, Defeo E. Ocular toxocariasis demonstrated by ultrasound. Ann Ophthalmol. 1981;13:1357–8.
Kent ME, Romanelli F. Reexamining syphilis: an update on epidemiology, clinical manifestations and management. Ann Pharmacother. 2008;42:226–36.
Khairallah M, Ben Yahia S, Attia S, Zaouali S, Ladjimi A, Messaoud R. Linear pattern of West Nile virus-associated chorioretinitis is related to retinal nerve fibres organization. Eye (Lond). 2007;21(7):952–5.
Khairallah M, Ben Yahia S, Ladjimi A, Zeghidi H, Ben Romdhane F, Besbes L, Zaouali S, Messaoud R. Chorioretinal involvement in patients with West Nile virus infection. Ophthalmology. 2004;111(11):2065–70.
Khairallah M, Kahloun R, Ben Yahia S, Jelliti B, Messaoud R. New infectious etiologies for posterior uveitis. Ophthalmic Res. 2013;49(2):66–72.
Khairallah M, Yahia SB, Letaief M, Attia S, Kahloun R, Jelliti B, Zaouali S, Messaoud R. A prospective evaluation of factors associated with chorioretinitis in patients with West Nile virus infection. Ocul Immunol Inflamm. 2007;15(6):435–9.
Kieffer F, Wallon M, Garcia P, Thulliez P, Peyron F, Franck J. Risk factors for retinochoroiditis during the first 2 years of life in infants with treated congenital toxoplasmosis. Pediatr Infect Dis J. 2008;27:27–32.
Kilmartin DJ, Dick AD, Forrester JV. Sympathetic ophthalmia risk following vitrectomy: should we counsel patients? Br J Ophthalmol. 2000;84(5):448–9.
Kim SJ, Equi R, Belair ML, Fine HF, Dunn JP. Long-term preservation of vision in progressive outer retinal necrosis treated with combination antiviral drugs and highly active antiretroviral therapy. Ocul Immunol Inflamm. 2007;15:425–7.
Kimura R, Sakai M, Okabe H. Transient shallow anterior chamber as initial symptom in Harada’s syndrome. Arch Ophthalmol. 1981;99(9):1604–6.
Kishi A, Nao-i N, Sawada A. Ultrasound biomicroscopic findings of acute angle-closure glaucoma in Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol. 1996;122(5):735–7.
Kishore K, Conway MD, Peyman GA. Intravitreal clindamycin and dexamethasone for toxoplasmic retinochoroiditis. Ophthalmic Surg Lasers. 2001;32:183–92.
Kiss S, Ahmed M, Letko E, Foster CS. Long-term follow-up of patients with birdshot retinochoroidopathy treated with corticosteroid-sparing systemic immunomodulatory therapy. Ophthalmology. 2005;112(6):1066–71.
Knecht PB, Papadia M, Herbort CP. Granulomatous keratic precipitates in birdshot retinochoroiditis. Int Ophthalmol. 2013;33(2):133–7.
Koizumi H, Pozzoni MC, Spaide RF. Fundus autofluorescence in birdshot chorioretinopathy. Ophthalmology. 2008;115(5):e15–20.
Kunkel J, Schürmann D, Pleyer U, Rüther K, Kneifel C, Krause L, Reichert M, Ignatius R, Schneider T. Ocular syphilis-indicator of previously unknown HIV-infection. J Infect. 2009;58:32–6.
Kupperman BD, Petty JG, Richman DD, et al. Correlation between CD4+ counts and the prevalence of cytomegalovirus retinitis and human immunodeficiency virus-related noninfectious retinal vasculopathy in patients with acquired immunodeficiency syndrome. Am J Ophthalmol. 1993;115:575–82.
Laatikanen L, Erkkilä H. A follow-up study on serpiginous choroiditis. Acta Ophthalmol (Copenh). 1981;59:707–18.
Lakhanpal V, Schocket SS, Nirankari VS. Clindamycin in the treatment of toxoplasmic retinochoroiditis. Am J Ophthalmol. 1983;95:605–13.
Lasave AF, Diaz-Llopis M, Muccioli C, Belfort Jr R, Arevalo JF. Intravitreal clindamycin and dexamethasone for zone 1 toxoplasmic retinochoroiditis at twenty-four months. Ophthalmology. 2010;117:1831–8.
Lau CH, Comer M, Lightman S. Long-term efficacy of mycophenolate mofetil in the control of severe intraocular inflammation. Clin Exp Ophthalmol. 2003;31(6):487–91 [3 D].
Laurence B, Harold M, Thierry D. Ocular complications of dengue fever. Ophthalmology. 2008;115:1100–1.
Le Hoang P, Girard B, Deray G, Le Minh H, De Kozak Y, Thillaye B, Faure JP, Rousselie F. Cyclosporine in the treatment of birdshot retinochoroidopathy. Transplant Proc. 1988;20(3 Suppl 4):128–30.
LeHoang P, Cassoux N, George F, Kullmann N, Kazatchkine MD. Intravenous immunoglobulin (IVIg) for the treatment of birdshot retinochoroidopathy. Ocul Immunol Inflamm. 2000;8(1):49–57.
LeHoang P, Ryan SJ. Birdshot retinochoroidopathy. In: Pepose JS, Holland GN, Wilhelmus KR, editors. Ocular infection & immunity. St. Louis: Mosby-Year Book Inc; 1996. p. 570–8.
Leslie T, Lois N, Christopoulou D, Olson JA, Forrester JV. Photodynamic therapy for inflammatory choroidal neovascularization unresponsive to immunosuppression. Br J Ophthalmol. 2005;89:147–50.
Levinson RD, Brezin A, Rothova A, Accorinti M, Holland GN. Research criteria for the diagnosis of birdshot chorioretinopathy: results of an international consensus conference. Am J Ophthalmol. 2006;141(1):185–7.
Lim WK, Buggage RR, Nussenblatt RB. Serpiginous choroiditis. Surv Ophthalmol. 2005;50(3):231–44.
Lit ES, Kim RY, Damico DJ. Surgical removal of subfoveal choroidal neovascularization without removal of posterior hyaloid: a consecutive series in younger patients. Retina. 2001;21:317–23.
Lowder CY, Foster RE, Gordon SM, Gutman FA. Acute posterior multifocal placoid pigment epitheliopathy after acute group A streptococcal infection. Am J Ophthalmol. 1996;122:115–7.
Lubin JR, Albert DM, Weinstein M. Sixty-five years of sympathetic ophthalmia. A clinicopathologic review of 105 cases (1913–1978). Ophthalmology. 1980;87(2):109–21.
Maenz M, Schlüter D, Liesenfeld O, Schares G, Gross U, Pleyer U. Ocular toxoplasmosis: past, present and new aspects of an old disease. Prog Retin Eye Res. 2014;39c:77–106.
Magnaval JF, Fabre R, Maurières P, et al. Application of the western blotting procedure for the immunodiagnosis of human toxocariasis. Parasitol Res. 1991;77:697–702.
Mahajan VB, Gehrs KM, Goldstein DA, Fischer DH, Lopez JS, Folk JC. Management of sympathetic ophthalmia with the fluocinolone acetonide implant. Ophthalmology. 2009;116(3):552–7 [3 D].
Maisel JM, Vorwerk PA. Sympathetic uveitis after giant tear repair. Retina. 1989;9(2):122–6.
Mansour AM, Arevalo JF, Fardeau C, Hrisomalos EN, et al. Three-year visual and anatomic results of administrating intravitreal bevacizumab in inflammatory ocular neovascularization. Can J Ophthalmol. 2012;47:269–74.
Martin DF, Sierra-Madero J, Walmsley S, Wolitz RA, Macey K, Georgiou P. A controlled trial of valganciclovir as induction therapy for cytomegalovirus retinitis. N Engl J Med. 2002;346:1119–26.
Martinez CE, Zhang D, Conway MD, et al. Successful management of ocular toxoplasmosis during pregnancy using combined intraocular clindamycin and dexamethasone with systemic sulfadiazine. Int Ophthalmol. 1999;22:85–8.
Maruyama Y, Kishi S. Tomographic features of serous retinal detachment in Vogt-Koyanagi-Harada syndrome. Ophthalmic Surg Lasers Imaging. 2004;35(3):239–42.
McCannel CA, Holland GN, Helm CJ, et al. Causes of uveitis in the general practice of ophthalmology. UCLA Community-Based Uveitis Study Group. Am J Ophthalmol. 1996;121:35–46.
Melamed J, Eckert GU, Spadoni VS, Lago EG, Uberti F. Ocular manifestations of congenital toxoplasmosis. Eye. 2010;24:528–34.
Mikkilä HO, Seppälä IJ, Viljanen MK, Peltomaa MP, Karma A. The expanding clinical spectrum of ocular lyme borreliosis. Ophthalmology. 2000;107(3):581–7.
Mittal A, Mittal S, Bharathi JM, Ramakrishnan R, Saravanan S, Sathe PS. Optic neuritis associated with chikungunya virus infection in South India. Arch Ophthalmol. 2007;125:1381–6.
Mittal A, Mittal S, Bharathi JM, Ramakrishnan R, Sathe PS. Uveitis during outbreak of chikungunya fever. Ophthalmology. 2007;114:1798.
Moorthy RS, Inomata H, Rao NA. Vogt-Koyanagi-Harada syndrome. Surv Ophthalmol. 1995;39:265–92.
Musch DC, Martin DF, Gordon JF, Davis MD, Kuppermann BD, Ganciclovir Implant Study Group. Treatment of cytomegalovirus retinitis with a sustained-release ganciclovir implant. N Engl J Med. 1997;337:83–90. 90.
Nazari Khanamiri H, Rao NA. Serpiginous choroiditis and infectious multifocal serpiginoid choroiditis. Surv Ophthalmol. 2013;58(3):203–32.
Neri P, Manoni M, Fortuna C, Lettieri M, Mariotti C, Giovannini A. Association of systemic steroids and mycophenolate mofetil as rescue therapy for uveitic choroidal neovascularization unresponsive to the traditional immunosuppressants: interventional case series. Int Ophthalmol. 2009;30:583–90.
Neri P, Mercanti L, Mariotti C, Salvolini S, Giovannini A. Long-term control of choroidal neovascularization in quiescent congenital toxoplasma retinochoroiditis with photodynamic therapy: 4-year results. Int Ophthalmol. 2010;30:51–6.
Nussenblatt R. Sympathetic ophthalmia. In: Nussenblatt RB SW, Whitcup SW, editors. Uveitis: fundamentals and clinical practice. 4th ed. St. Louis: Mosby Elsevier; 2010. p. 289–302 [4 D].
Nussenblatt RB, Palestine AG, Chan CC, Stevens Jr G, Mellow SD, Green SB. Randomized, double-masked study of cyclosporine compared to prednisolone in the treatment of endogenous uveitis. Am J Ophthalmol. 1991;112(2):138–46.
Nussenblatt RB, Whitcup SM, Palestine AG. Birdshot retinochoroidopathy. In: Nussenblatt RB, Whitcup SM, Palestine AG, editors. Uveitis: fundamentals and clinical practice. 2nd ed. St Louis: Mosby-Year Book; 1996. p. 325.
Nussenblatt RB, Whitcup SM, Palestine AG. Vogt-Koyanagi-Harada syndrome. In: Nussenblatt RB, Whitcup SM, Palestine AG, editors. Uveitis: fundamental and clinical practice. 2nd ed. St Louis: Mosby; 1996. p. 312–24.
Nussenblatt RB, Whitcup SM, Palestine AG. White dot syndromes. In: Nussenblatt RB, Whitcup SM, Palestine AG, editors. Fundamentals and clinical practice. 2nd ed. St. Louis: Mosby-Year Book, Inc; 1996. p. 373.
Ohno S, Minakawa R, Matsuda H. Clinical studies of Vogt-Koyanagi-Harada’s disease. Jpn J Ophthalmol. 1988;32(3):334–43.
Olk RJ, Burgess DB. Treatment of recurrent juxtafoveal subretinal neovascular membranes with krypton red laser photocoagulation. Ophthalmology. 1985;92:1035–46.
Opremcak EM, Scales DK, Sharpe MR. Trimethoprim-sulfamethoxazole therapy for ocular toxoplasmosis. Ophthalmology. 1992;99:920–5.
Opremcak EM. Birdshot retinochoroiditis. In: Albert DM, Jakobiec FA, editors. Principles and practice of ophthalmology, vol. 1. Philadelphia: W.B. Saunders; 1994. p. 475.
Ozdemir H, Karacorlu M, Karacorlu S. Intravitreal triamcinolone acetonide in sympathetic ophthalmia. Graefes Arch Clin Exp Ophthalmol. 2005;243(7):734–6 [3 D].
Palay D, Sternberg P, Davis J. Decrease in the risk of bilateral acute retinal necrosis by acyclovir therapy. Ophthalmology. 1991;112:250–5.
Palestine AG, Rodrigues MM, Macher AM, et al. Ophthalmic involvement in the acquired immune deficiency syndrome. Ophthalmology. 1984;91:1092–9.
Papadia M, Herbort CP. Indocyanine green angiography (ICGA) is essential for the early diagnosis of birdshot chorioretinopathy. Klin Monbl Augenheilkd. 2012;229(4):348–52.
Papadia M, Herbort CP. Reappraisal of birdshot retinochoroiditis (BRC): a global approach. Graefes Arch Clin Exp Ophthalmol. 2013;251(3):861–9.
Pappachan JM, Mathew S, Thomas B, Renjini K, Scaria CK, Shukla J. The incidence and clinical characteristics of the immune phase eye disease in treated cases of human leptospirosis. Indian J Med Sci. 2007;61(8):441–7. EbM 2+.
Parc C, Guenoun JM, Dhote R, Brézin A. Optical coherence tomography in the acute and chronic phases of Vogt-Koyanagi-Harada disease. Ocul Immunol Inflamm. 2005;13(2–3):225–7.
Paredes I, Ahmed M, Foster CS. Immunomodulatory therapy for Vogt-Koyanagi-Harada patients as first-line therapy. Ocul Immunol Inflamm. 2006;14(2):87–90.
Park D, Shatz H, McDonald R, Johnson RN. Indocyanine green angiography of acute posterior multifocal placoid pigment epitheliopathy. Ophthalmology. 1995;102:1877–83.
Park JJ, Pavesio C. Prophylactic laser photocoagulation for acute retinal necrosis: does it raise more questions than answers? Br J Ophthalmol. 2008;92(9):1161–2.
Pearson PA, Piracha AR, Sen HA, Jaffe GJ. Atovaquone for the treatment of toxoplasma retinochoroiditis in immunocompetent patients. Ophthalmology. 1999;106:148–53.
Pedroza Seres M. Acute posterior multifocal placoid pigment epitheliopathy. In: Foster CS, Vitale AT, editors. Diagnosis and treatments of uveatis. Philadelphia: W.B. Saunders Company; 2002. p. 772–7.
Perente I, Utine CA, Cakir H, Kaya V, Tutkun IT, Yilmaz OF. Management of ocular complications of Vogt-Koyanagi-Harada syndrome. Int Ophthalmol. 2009;29(1):33–7.
Petersen E, Kiilstra A, Stanford M. Epidemiology of ocular toxoplasmosis. Ocul Immunol Inflamm. 2012;20:68–75.
Phan L, Kasza K, Jalbrzikowski J, et al. Longitudinal study of new eye lesions in treated congenital toxoplasmosis. Ophthalmology. 2008;115:553–9. e558.
Pialoux G, Gaüzère BA, Jauréguiberry S, Strobel M. Chikungunya, an epidemic arbovirosis. Lancet Infect Dis. 2007;7:319–27.
Pleyer U, Dutescu M. Sympathetic ophthalmia. Ophthalmologe. 2009;106(2):167–75 [4C].
Pollack AL, McDonald HR, Ai E, Green WR, Halpern LS, Jampol LM, Leahy JM, Johnson RN, Spencer WH, Stern WH, Weinberg DV, Werner JC, Williams GA. Sympathetic ophthalmia associated with pars plana vitrectomy without antecedent penetrating trauma. Retina. 2001;21(2):146–54.
Powers AM, Brault AC, Tesh RB, Weaver SC. Re-emergence of chikungunya and O’nyong-nyong viruses: evidence for distinct geographical lineages and distant evolutionary relationships. J Gen Virol. 2000;81:471–9.
Priem HA, Oosterhuis JA. Birdshot chorioretinopathy: clinical characteristics and evolution. Br J Ophthalmol. 1988;72:646–59.
Prutsky G, Domecq JP, Mori L, Bebko S, Matzumura M, Sabouni A, Shahrour A, Erwin PJ, Boyce TG, Montori VM, Malaga G, Murad MH. Treatment outcomes of human bartonellosis: a systematic review and meta-analysis. Int J Infect Dis. 2013;17(10):e811–9. pii: S1201-9712(13)00107-0. (EbM 1-).
Rahn DW, Felz MW. Lyme disease: current approach to early, disseminated and late disease. Postgrad Med. 1998;103(5):51–4. 57–9: 63–4.
Rathinam SR, Ratnam S, Selvaraj S, et al. Uveitis associated with an epidemic outbreak of leptospirosis. Am J Ophthalmol. 1997;124:71–9.
Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tessler HH, Usui M. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol. 2001;131(5):647–52.
Read RW, Rao NA, Sharma OP. Sarcoid choroiditis initially diagnosed as birdshot choroidopathy. Sarcoidosis Vasc Diffuse Lung Dis. 2000;17:85–6.
Read RW, Yu F, Accorinti M, Bodaghi B, Chee SP, Fardeau C, Goto H, Holland GN, Kawashima H, Kojima E, Lehoang P, Lemaitre C, Okada AA, Pivetti-Pezzi P, Secchi A, See RF, Tabbara KF, Usui M, Rao NA. Evaluation of the effect on outcomes of the route of administration of corticosteroids in acute Vogt-Koyanagi-Harada disease. Am J Ophthalmol. 2006;142(1):119–24.
Reddy CV, Brown J, Folk JC, et al. Enlarged blind spots in chorioretinal inflammatory disorders. Ophthalmology. 1996;103:606.
Rishi P, Venkataraman A, Rishi E. Combination photodynamic therapy and bevacizumab for choroidal neovascularization associated with toxoplasmosis. Indian J Ophthalmol. 2011;59:62–4.
Rodriguez A. Photocoagulation in toxoplasmic retinochoroiditis. Am J Ophthalmol. 1981;91:417–8.
Rodriguez A, Calonge M, Pedroza-Seres M, et al. Referral patterns of uveitis in a tertiary eye care center. Arch Ophthalmol. 1996;114:593–9.
Rothova A, Bosch-Driessen LE, van Loon NH, Treffers WF. Azithromycin for ocular toxoplasmosis. Br J Ophthalmol. 1998;82:1306–8.
Rothova A, Ossewaarde-van Norel A, Los LI, Berendschot TT. Efficacy of low-dose methotrexate treatment in birdshot chorioretinopathy. Retina. 2011;31(6):1150–5.
Rouvas A, Petrou P, Douvali M, Ntouraki A, Vergados I, Georgalas I, Markomichelakis N. Intravitreal ranibizumab for the treatment of inflammatory choroidal neovascularization. Retina. 2011;31(5):871–9.
Rubin ML, Kaufman HE, Tierney JP, et al. An intraretinal nematode. Trans Am Acad Ophthalmol Otolaryngol. 1968;72:855–66.
Rubsamen PE, Gass JD. Vogt-Koyanagi-Harada syndrome. Clinical course, therapy, and long-term visual outcome. Arch Ophthalmol. 1991;109(5):682–7.
Rush RB, Goldstein DA, Callanan DG, Meghpara B, Feuer WJ, Davis JL. Outcomes of birdshot chorioretinopathy treated with an intravitreal sustained-release fluocinolone acetonide-containing device. Am J Ophthalmol. 2011;151(4):630–6.
Ryan SJ, Maumenee AE. Birdshot retinochoroidopathy. Am J Ophthalmol. 1980;89:31–45.
Ryan SJ, Maumenee AE. Acute posterior multifocal placoid pigment epitheliopathy. Am J Ophthalmol. 1972;74:1066–74.
Sachdev N, Gupta V, Gupta A, Singh R. Posterior segment recurrences in Vogt-Koyanagi-Harada disease. Int Ophthalmol. 2008;28(5):339–45.
Samuel MA, Diamond MS. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol. 2005;79:13350–61.
Sanjay S, Wagle AM, Au Eong KG. Dengue optic neuropathy. Ophthalmology. 2009;116(1):170.
Sasamoto Y, Ohno S, Matsuda H. Studies on corticosteroid therapy in Vogt-Koyanagi-Harada disease. Ophthalmologica. 1990;201(3):162–7.
Shah KH, Levinson RD, Yu F, Goldhardt R, Gordon LK, Gonzales CR, Heckenlively JR, Kappel PJ, Holland GN. Birdshot chorioretinopathy. Surv Ophthalmol. 2005;50(6):519–41.
Shammas HF, Zubyk NA, Stanfield TF. Sympathetic uveitis following glaucoma surgery. Arch Ophthalmol. 1977;95(4):638–41.
Sharp DC, Bell RA, Patterson E, Pinkerton RM. Sympathetic ophthalmia. Histopathologic and fluorescein angiographic correlation. Arch Ophthalmol. 1984;102(2):232–5.
Shields JA. Ocular toxocariasis: a review. Surv Ophthalmol. 1984;28:361–81.
Shindo Y, Inoko H, Yamamoto T, Ohno S. HLA-DRB1 typing of Vogt-Koyanagi-Harada’s disease by PCR-RFLP and the strong association with DRB1*0405 and DRB1*0410. Br J Ophthalmol. 1994;78(3):223–6.
Silveira C, Belfort Jr R, Muccioli C, et al. The effect of long-term intermittent trimethoprim/sulfamethoxazole treatment on recurrences of toxoplasmic retinochoroiditis. Am J Ophthalmol. 2002;134:41–6.
Singh G. Sympathetic ophthalmia after Nd:YAG cyclotherapy. Ophthalmology. 1993;100(6):798–9.
Singh K, de Frank MP, Shults WT, Watzke RC. Acute idiopathic blind spot enlargement. A spectrum of disease. Ophthalmology. 1991;98:497.
Siqueira RC, Vitral NP, Campos WR, Orefice F, de Moraes Figueiredo LT. Ocular manifestations in dengue fever. Ocul Immunol Inflamm. 2004;12:323–7.
Slakter JS, Giovannini A, Yannuzzi LA, et al. Indocyanine green angiography of multifocal choroiditis. Ophthalmology. 1997;104:1813.
Snyder DA, Tessler HH. Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol. 1980;90(1):69–75.
Sobia S, Ashfaq UA. A brief review on dengue molecular virology, diagnosis, treatment and prevalence in Pakistan. Genet Vaccines Ther. 2012;10(1):6.
Sobrin L, Huang JJ, Christen W, Kafkala C, Choopong P, Foster CS. Daclizumab for treatment of birdshot chorioretinopathy. Arch Ophthalmol. 2008;126(2):186–91.
Soheilian M, Ramezani A, Azimzadeh A, et al. Randomized trial of intravitreal clindamycin and dexamethasone versus pyrimethamine, sulfadiazine, and prednisolone in treatment of ocular toxoplasmosis. Ophthalmology. 2011;118:134–41.
Spalter HF, Campbell CJ, Noyori KS, Rittler MC, Koester CJ. Prophylactic photocoagulation of recurrent toxoplasmic retinochoroiditis. A preliminary report. Arch Ophthalmol. 1966;75:21–31.
Stanford MR, See SE, Jones LV, et al. Antibiotics for toxoplasmic retinochoroiditis: an evidence-based systematic review. Ophthalmology. 2003;110:926–31.
Stewart JF, Croxson MC, Powell KF, Polkinghorne PJ. Identification of cytomegalovirus in vitreous using the polymerase chain reaction. Aust N Z J Ophthalmol. 1993;21(3):165–9.
Sudeep AB, Parashar D. Chikungunya: an overview. J Biosci. 2008;33:443–9.
Sugiura S. Vogt-Koyanagi-Harada disease. Jpn J Ophthalmol. 1978;22:9–35.
Tabbara KF, O'Connor GR. Treatment of ocular toxoplasmosis with clindamycin and sulfadiazine. Ophthalmology. 1980;87:129–34.
Talabani H, Mergey T, Yera H, Delair E, Brézin AP, Langsley G, Dupouy-Camet J. Factors of occurrence of ocular toxoplasmosis. A review. Parasite. 2010;17(3):177–82.
Taylor SRJ, Hamilton R, Hooper CY, et al. Valacyclovir in the treatment of acute retinal necrosis. BMC Ophthalmol. 2012;12:48.
Tessler HH, Jennings T. High-dose short-term chlorambucil for intractable sympathetic ophthalmia and Behçet’s disease. Br J Ophthalmol. 1990;74(6):353–7 [3 D].
Thiébaut R, Leproust S, Chêne G, Gilbert R. Effectiveness of prenatal treatment for congenital toxoplasmosis: a meta-analysis of individual patients’ data. Lancet. 2007;369:115–22.
Thorne JE, Jabs DA, Kedhar SR, Peters GB, Dunn JP. Loss of visual field among patients with birdshot chorioretinopathy. Am J Ophthalmol. 2008;145(1):23–8.
Tomsak RL, Lystad LD, Katirji MB, Brassel TC. Rapid response of syphilitic optic neuritis to posterior subtenon’s steroid injection. J Clin Neuroophthalmol. 1992;12:6–7.
Torun N, Sherif Z, Garweg J, Pleyer U. Diagnosis and treatment of ocular toxoplasmosis: a survey of German-speaking ophthalmologists. Ophthalmologe. 2008;105:1023–8.
Tucker JD, Li JZ, Robbins GK, Davis BT, Lobo AM, Kunkel J, Papaliodis GN, Durand ML, Felsenstein D. Ocular syphilis among HIV-infected patients: a systematic analysis of the literature. Sex Transm Infect. 2011;87(1):4–8. doi:10.1136/sti.2010.043042. Epub 26 Aug 2010.
Vance SK, Khan S, Klancnik JM, Freund KB. Characteristic spectral-domain optical coherence tomography findings of multifocal choroiditis. Retina. 2011;31:717–23.
Vasconcelos-Santos DV, Rao PK, Davies JB, Sohn EH, Rao NA. Clinical features of tuberculous serpiginouslike choroiditis in contrast to classic serpiginous choroiditis. Arch Ophthalmol. 2010;128(7):853–8.
Vazquez-Cobian LB, Flynn T, Lehman TJ. Adalimumab therapy for childhood uveitis. J Pediatr. 2006;149(4):572–5 [3 D].
Venkatramani J, Lim WK. Bilateral vitreous haemorrhage associated with dengue fever. Eye. 2006;20(12):1405–6.
Verhoeff F. An effective treatment for sympathetic uveitis. Arch Ophthalmol. 1927;56:28.
Vitale AT, Rodriguez A, Foster CS. Low-dose cyclosporine therapy in the treatment of birdshot retinochoroidopathy. Ophthalmology. 1994;101:822–31.
Vote BJ, Hall A, Cairns J, Buttery R. Changing trends in sympathetic ophthalmia. Clin Experiment Ophthalmol. 2004;[3C](5):542–5 [3C].
Vrabec TR, Augsburger JJ, Fischer DH, et al. Taches de bougie. Ophthalmology. 1995;102:1712–21.
Walker DH, Raoult D. Rickettsia rickettsii and other spotted fever group rickettsiae (Rocky Mountain spotted fever and other spotted fevers). In: Mandell GL, Douglas JE, Bennett R, editors. Principles and practice of infectious disease. New York: Churchill Livingstone; 1995. p. 1721–7.
Warshafsky S, Lee DH, Francois LK, Nowakowski J, Nadelman RB, Wormser GP. Efficacy of antibiotic prophylaxis for the prevention of Lyme disease: an updated systematic review and meta-analysis. J Antimicrob Chemother. 2010;65(6):1137–44.
Weiss H, Annesley WH, Shields JA, et al. The clinical course of serpiginous choroidopathy. Am J Ophthalmol. 1979;87:133–42.
Westeneng AC, Rothova A, de Boer JH, et al. Infectious uveitis in immunocompromised patients and the diagnostic value of polymerase chain reaction and Goldmann-Witmer coefficient in aqueous analysis. Am J Ophthalmol. 2007;144:781–5.
Whitcup SM, Fortin E, Nussenblatt RB, et al. Therapeutic effect of combination antiretroviral therapy on cytomegalovirus retinitis. JAMA. 1997;277:1519–20.
Wichmann O, Lauschke A, Frank C, et al. Dengue antibody prevalence in German travelers. Emerg Infect Dis. 2005;11:762–5.
Windsor JJ. Cat scratch disease: epidemiology, etiology and treatment. Br J Biomed Sci. 2001;58:101–10.
Winter FC. Sympathetic uveitis; a clinical and pathologic study of the visual result. Am J Ophthalmol. 1955;39(3):340–7 [4 D].
Winterhalter S, Joussen AM, Pleyer U, Stübiger N. Inflammatory choroidal neovascularizations. Klin Monbl Augenheilkd. 2012;229(9):897–904.
Wong R, Pavesio CE, Laidlaw DAH, et al. The effects of intravitreal foscarnet and virus type on outcome. Ophthalmology. 2010;117(3):556–60.
Wong RW, Jumper JM, McDonald HR, Johnson RN, Fu A, Lujan BJ, Cunningham Jr ET. Emerging concepts in the management of acute retinal necrosis. Br J Ophthalmol. 2013;97(5):545–52. doi:10.1136/bjophthalmol-2012-301983. Epub 12 Dec 2012.
World Health Organization. Dengue hemorrhagic fever: diagnosis, treatment and control. 2nd ed. Geneva: WHO; 1997.
Wormser GP, Dattwyler RJ, Shapiro ED, Halperin JJ, Steere AC, Klempner MS, Krause PJ, Bakken JS, Strle F, Stanek G, Bockenstedt L, Fish D, Dumler JS, Nadelman RB. The clinical assessment, treatment, and prevention of lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43(9):1089–134.
Wroblewski KJ, Hidayat AA, Neafie RC, Rao NA, Zapor M. Ocular tuberculosis: a clinicopathologic and molecular study. Ophthalmology. 2011;118:772–7.
Wu L, Evans T, Saravia M, Schlaen A, Couto C. Intravitreal bevacizumab for choroidal neovascularization secondary to Vogt-Koyanagi-Harada syndrome. Jpn J Ophthalmol. 2009;53(1):57–60.
Yahia SB, Khairallah M. Ocular manifestations of West Nile virus infection. Int J Med Sci. 2009;6(3):114–5.
Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. Tyrosinase family proteins are antigens specific to Vogt-Koyanagi-Harada disease. J Immunol. 2000;165:7323–9.
Yamanaka E, Ohguro N, Yamamoto S, Nakagawa Y, Imoto Y, Tano Y. Evaluation of pulse corticosteroid therapy for Vogt-Koyanagi-Harada disease assessed by optical coherence tomography. Am J Ophthalmol. 2002;134(3):454–6.
Yeh S, Wroblewski K, Buggage R, Li Z, Kurup SK, Sen HN, Dahr S, Sran P, Reed GF, Robinson R, Ragheb JA, Waldmann TA, Nussenblatt RB. High-dose humanized anti-IL-2 receptor alpha antibody (daclizumab) for the treatment of active, non-infectious uveitis. J Autoimmun. 2008;31(2):91–7.
Yin PD, Kurup SK, Fischer SH, Rhee HH, Byrnes GA, Levy-Clarke GA, Buggage RR, Nussenblatt RB, Mican JM, Wright ME. Progressive outer retinal necrosis in the era of highly active antiretroviral therapy: successful management with intravitreal injections and monitoring with quantitative PCR. J Clin Virol. 2007;38(3):254–9.
Zaharia MA, Lamarche J, Laurin M. Sympathetic uveitis 66 years after injury. Can J Ophthalmol. 1984;19(5):240–3.
Phimda K, Hoontrakul S, Suttinont C, Chareonwat S, Losuwanaluk K, Chueasuwanchai S, Chierakul W, Suwancharoen D, Silpasakorn S, Saisongkorh W, Peacock SJ, Day NP, Suputtamongkol Y. Doxycycline versus azithromycin for treatment of leptospirosis and scrub typhus. Antimicrob Agents Chemother. 2007;51(9):3259–63. Epub 2007 July 16.
Ressner RA, Griffith ME, Beckius ML, Pimentel G, Miller RS, Mende K, Fraser SL, Galloway RL, Hospenthal DR, Murray CK. Antimicrobial susceptibilities of geographically diverse clinical human isolates of Leptospira. Antimicrob Agents Chemother. 2008;52(8):2750–4. doi: 10.1128/AAC.00044-08. Epub 2008 Apr 14.
Holland GN, Lewis KG. An update on current practices in the management of ocular toxoplasmosis. Am J Ophthalmol. 2002;134(1):102–14.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Neri, P. et al. (2014). Posterior Uveitis. In: Pleyer, U., Alió, J., Barisani-Asenbauer, T., Le Hoang, P., Rao, N. (eds) Immune Modulation and Anti-Inflammatory Therapy in Ocular Disorders. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-54350-0_10
Download citation
DOI: https://doi.org/10.1007/978-3-642-54350-0_10
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-54349-4
Online ISBN: 978-3-642-54350-0
eBook Packages: MedicineMedicine (R0)