
Abstract
In vivo imaging of β-amyloid (Αβ) has transformed the assessment of Αβ pathology and its changes over time, extending our insight into Aβ deposition by providing highly accurate, reliable and reproducible quantitative statements of regional or global Aβ burden in the brain, essential for therapeutic trial recruitment and for the evaluation of anti-Αβ treatments. Although cross-sectional evaluation of Αβ burden does not strongly correlate with cognitive impairment in AD, it does correlate with memory impairment and a higher risk for cognitive decline in the aging population and MCI subjects. This correlation with memory impairment, one of the earliest symptoms of AD, suggests that Αβ deposition is not part of normal aging, supporting the hypothesis that Αβ deposition occurs well before the onset of symptoms. Longitudinal observations, coupled with different disease-specific biomarkers to assess potential downstream effects of Aβ, are required to confirm this hypothesis and further elucidate the role of Αβ deposition in the course of Alzheimer’s disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Positron Emission Tomography
- Mild Cognitive Impairment
- Dementia With Lewy Body
- Cerebral Amyloid Angiopathy
- Mild Cognitive Impairment Subject
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Alzheimer’s disease (AD) is a progressive and irreversible neurodegenerative disorder clinically characterised by memory loss and cognitive decline that severely affect the activities of daily living (Masters et al. 2006). AD is the principal cause of dementia in the elderly, leading invariably to death, usually within 7–10 years after diagnosis (Khachaturian 1985). The progressive nature of the neurodegeneration suggests an age-dependent process that ultimately leads to synaptic failure and neuronal damage in cortical areas of the brain essential for memory and other cognitive domains (Isacson et al. 2002). AD not only has devastating effects on the sufferers and their caregivers, but it also has a tremendous socioeconomic impact on families and the health system, a burden which will only increase in the upcoming years as the population of most countries ages (Johnson et al. 2000). In the absence of reliable biomarkers, direct pathological examination of brain tissue derived from either biopsy or autopsy remains the only definitive method for establishing a diagnosis of AD (O’Brien et al. 2000). The typical macroscopic picture is gross cortical atrophy, while microscopically, there is widespread cellular degeneration and diffuse synaptic and neuronal loss, accompanied by reactive gliosis and the presence of the pathological hallmarks of the disease: intracellular neurofibrillary tangles (NFT) and extracellular amyloid plaques (Jellinger 1990; Masters 2005; Masters and Beyreuther 2005). While NFT are intraneuronal bundles of paired helical filaments mainly composed of the aggregates of an abnormally phosphorylated form of tau protein (Jellinger and Bancher 1998; Michaelis et al. 2002), neuritic plaques consist of dense extracellular aggregates of β-amyloid (Αβ) (Masters et al. 1985), surrounded by reactive gliosis and dystrophic neurites. Αβ is a 4 kDa 39–43 amino acid metalloprotein derived from the proteolytic cleavage of the amyloid precursor protein (APP), by β- and γ-secretases (Cappai and White 1999). To date, all available genetic, pathological, biochemical and cellular evidence strongly supports the notion that an imbalance between the production and removal of Αβ leading to its progressive accumulation is central to the pathogenesis of AD (Villemagne et al. 2006). The ‘Aβcentric theory’ (Masters et al. 2006) postulates that Aβ plaque deposition is the primary event in a cascade of effects that lead to neurofibrillary degeneration and dementia (Hardy 1997).
Despite the proposed new diagnostic criteria (McKhann et al. 2011), the clinical diagnosis of AD is still largely based on progressive impairment of memory and decline in at least one other cognitive domain and by excluding other diseases that might also present with dementia such as frontotemporal lobar degeneration (FTLD), dementia with Lewy bodies (DLB), stroke, brain tumour, normal pressure hydrocephalus or depression (Larson et al. 1996; Cummings et al. 1998). A variable period of up to 15 years of prodromal decline in cognition characterised by a relatively isolated impairment in short-term memory that may also be accompanied by impairments of working memory, known as amnestic mild cognitive impairment (MCI), usually precedes the formal diagnosis of AD (Petersen et al. 1999; Petersen 2000). At this point, there is no cure for AD nor proven way to slow the rate of neurodegeneration. Symptomatic treatment with an acetylcholinesterase inhibitor or a glutamatergic moderator provides modest benefit in some patients usually by temporary stabilisation rather than a noticeable improvement in memory function (Masters and Beyreuther 2006).
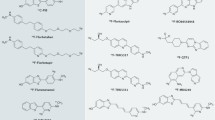
2 Aβ Imaging Radiotracers
Αβ plaques and NFT are the hallmark brain lesions of AD. These microscopic aggregates are still well beyond the resolution of conventional neuroimaging techniques used for the clinical evaluation of patients with AD. Positron emission tomography (PET) is a sensitive molecular imaging technique that allows in vivo quantification of radiotracer concentrations in the picomolar range, allowing the noninvasive assessment of molecular processes at their sites of action, detecting disease processes at asymptomatic stages when there is no evidence of anatomic changes on computed tomography (CT) and magnetic resonance imaging (MRI) (Phelps 2000). Since Αβ is at the centre of AD pathogenesis and given that several pharmacological agents aimed at reducing Αβ levels in the brain are being developed and tested, many efforts have been focused on generating radiotracers for imaging Αβ in vivo (Sair et al. 2004; Mathis et al. 2005; Villemagne et al. 2005). Several applications of Aβ imaging have been proposed (Table 10.1).
As a quantitative neuroimaging probe, the Aβ radiotracer must possess a number of key general properties: it should be a lipophilic, nontoxic small molecule with a high specificity and selectivity for Αβ, amenable for high specific activity labelling with 18F or other long lived radioisotope, with no radiolabelled metabolites that enter the brain, while reversibly binding to Αβ in a specific and selective fashion (Laruelle et al. 2003; Nordberg 2004; Pike 2009). Overall, binding affinity and lipophilicity are the most crucial properties for in vivo neuroimaging tracers. While high affinity for Αβ is desirable to provide an adequate signal-to-noise ratio, this high affinity might also delay reaching binding equilibrium requiring an extended scanning time. And while lipophilicity is necessary for the tracer to cross the blood-brain barrier (BBB), if the radiotracer is too lipophilic, its nonspecific binding might be too high (Laruelle et al. 2003; Pike 2009).
Through the years, several compounds have been evaluated with different fortune as potential Aβ probes: monoclonal and anti-Aβ antibodies fragments, serum amyloid P, basic fibroblast growth factor, Aβ fragments, derivatives of histopathological dyes such as Congo red, chrysamine G and thioflavin S and T (for review, see (Villemagne and Rowe 2010)). While selective tau imaging for in vivo NFT quantification is still in the early stages of development (Okamura et al. 2005; Maruyama et al. 2009; Ojida et al. 2009; Fodero-Tavoletti et al. 2011), several Αβ radiotracers found their way into human clinical trials.
Almost a decade after unsuccessful trials with anti-Αβ antibodies (Majocha et al. 1992), Αβ imaging came to fruition with the first report of successful imaging in an AD patient with 18F-FDDNP, a tracer claimed to bind both plaques and NFT (Shoghi-Jadid et al. 2002). Since then, human Αβ imaging studies have been conducted in AD patients, normal controls and patients with other dementias using 11C-PiB (Klunk et al. 2004), 11C-SB13 (Verhoeff et al. 2004), 11C-ST1859 (Bauer et al. 2006), 11C-BF-227 (Kudo et al. 2007), 11C-AZD2138 (Nyberg et al. 2009), 18F-florbetaben (Rowe et al. 2008), 18F-flutemetamol (Serdons et al. 2009a), 18F-florbetapir (Wong et al. 2010) and 18F-AZD4694 (Cselenyi et al. 2012) with PET.
In order to test compounds that could have more widespread application, preliminary studies with SPECT Aβ radiotracers 123I-CQ (Opazo et al. 2006) and 123I-IMPY (Newberg et al. 2006) showed limited utility for the evaluation of Aβ burden in AD, although 123I-IMPY might be useful in the evaluation of transmissible spongiform encephalopathies (Song et al. 2008). New SPECT radiotracers labelled with 123I or those that could be potentially labelled with 99mTc are being evaluated (Qu et al. 2007a, b; Serdons et al. 2007; Lin et al. 2009).
While all of the aforementioned tracers bind with varying degrees of success to Αβ fibrils and brain homogenates of AD patients, Congo red and thioflavin T—and some of their derivatives—have recently been shown to also bind to soluble oligomeric forms of Αβ (Maezawa et al. 2008). On the other hand, Αβ soluble species represent less than 1 % of the total brain Αβ (McLean et al. 1999), and the reported affinity of PiB for these soluble oligomers seems to be significantly lower than for Αβ fibrils (Maezawa et al. 2008). Until highly selective radiotracers are developed to bind the Αβ soluble species, the contribution of these oligomers to the PET signal in sporadic AD from tracers such as 11C-PiB is considered to be negligible (Mathis et al. 2007).
2.1 11C-Labelled Radiotracers
2.1.1 11C-PiB
11C-PiB (PiB), the most successful and widely used of the currently available Αβ tracers, has been shown to possess high affinity and high selectivity for fibrillar Αβ in plaques and other Αβ-containing lesions (Klunk et al. 2001; Mathis et al. 2002, 2005; Price et al. 2005; Ye et al. 2005; Cohen et al. 2012). In vitro studies with high specific activity 3H-PiB demonstrated two binding sites for PiB in brain homogenates and Aβ1–42 fibrils (Klunk et al. 2005). Recent studies have shown that 11C-PiB binds with high affinity to the N-terminally truncated and modified Αβ and AβN3-pyroglutamate species in senile plaques (Maeda et al. 2007). In vitro assessment of 3H-PiB binding to white matter homogenates failed to show any specific binding (Fodero-Tavoletti et al. 2009b). PiB is a derivative of thioflavin T, a fluorescent dye commonly used to assess fibrillisation into β-sheet conformation (LeVine 1999), and as such PiB has been shown to bind to a range of additional Αβ-containing lesions including diffuse plaques and cerebral amyloid angiopathy (CAA) (Lockhart et al. 2007), as well as to Αβ oligomers—although with lower affinity (Maezawa et al. 2008). PiB also displayed lower affinity towards other misfolded proteins with a similar β-sheet secondary structure such as α-synuclein (Fodero-Tavoletti et al. 2007; Ye et al. 2008) and tau (Lockhart et al. 2007; Ikonomovic et al. 2008). This is relevant particularly since AD has been described as a ‘triple brain amyloidosis’ (Trojanowski 2002). Most importantly, these studies have shown that, at the concentrations achieved during a PET scan, 11C-PiB cortical retention in AD or DLB primarily reflects Αβ-related cerebral amyloidosis and not binding to Lewy bodies (LB) or NFT (Klunk et al. 2003; Fodero-Tavoletti et al. 2007; Lockhart et al. 2007; Ikonomovic et al. 2008). 11C-PiB has consistently provided quantitative information on Αβ burden in vivo, contributing new insights into Αβ deposition in the brain, allowing earlier detection of AD pathology (Klunk et al. 2004; Mintun et al. 2006; Rowe et al. 2007; Cohen et al. 2012) and accurate differential diagnosis of the dementias (Ng et al. 2007b; Rabinovici et al. 2007; Rowe et al. 2007).
2.1.2 11C-BF-227
11C-BF-227 (BF-227), a benzoxazole derivative, is also a promising tool as an Aβ imaging tracer (Okamura et al. 2004). In vitro binding studies with BF-227 demonstrated a Ki value of 4.3 nM for the compound to Aβ1–42 fibrils (Kudo 2006). A PET study using BF-227 showed AD patients were clearly distinguishable from age-matched controls with AD patients displaying significantly higher tracer retention in the cerebral cortex than controls (Kudo et al. 2007). Regional parametric analysis of the images further demonstrated a higher BF-227 retention in the posterior association cortex in AD patients. Unlike PiB, BF-227 presents with similar binding affinities for Aβ and α-synuclein (Fodero-Tavoletti et al. 2009a).
A few other C-11 Aβ radiotracers have been tested in humans. The evaluation of the stilbene derivative, 11C-SB13, showed that it could differentiate between five AD and six controls but had a lower effect size values when compared with 11C-PiB (Verhoeff et al. 2004). A preliminary study with the anti-Aβ agent 11C-ST1859 revealed small differences in radiotracer retention between nine AD patients and three healthy controls, but showing that the biodistribution and specificity of therapeutic agents can be assessed with PET (Bauer et al. 2006). Another Aβ radiotracer, 11C-AZD2138, showed not only reversible binding but also displayed very low nonspecific binding to white matter (Johnson et al. 2009; Nyberg et al. 2009).
2.2 18F-Labelled Radiotracers
Unfortunately, the 20-min radioactive decay half-life of carbon-11 (11C) limits the use of 11C-PiB to centres with an on-site cyclotron and 11C radiochemistry expertise, making the cost of studies prohibitive for routine clinical use. To overcome these limitations, several Aβ tracers labelled with fluorine-18 (18F; half-life of 110 min) that permits centralised production and regional distribution have been developed and tested. Recent studies with the newly introduced 8F-labelled Aβ-specific radiotracers, 18F-florbetapir (Wong et al. 2010; Clark et al. 2011), 18F-florbetaben (Rowe et al. 2008; Barthel et al. 2011; Villemagne et al. 2011a), 18F-flutemetamol (Nelissen et al. 2009; Vandenberghe et al. 2010b) and 18F-AZD4694 (Cselenyi et al. 2012), have been successfully replicating the results obtained with 11C-PiB. The latest entry into the list of fluorinated Aβ radiotracers is 18F-MK-3328, developed by Merck, that has the disadvantage that >17 % of its cortical signal was attributed to binding to monoamine oxidase-B (MAO-B) (Sur et al. 2010).
2.2.1 18F-FDDNP
A marked progression in the development of Aβ imaging tracers was the synthesis and characterisation by Barrio and colleagues of a very lipophilic radiofluorinated 6-dialkylamino-2-naphthyethylidene derivative that presents nanomolar affinity to Aβ fibrils (Barrio et al. 1999; Agdeppa et al. 2001a, b). 18F-FDDNP is reported to bind both the extracellular Aβ plaques and the intracellular NFT in AD (Shoghi-Jadid et al. 2002) while also binding to prion plaques in Creutzfeldt-Jakob disease (CJD) brain tissue (Bresjanac et al. 2003). However, in vitro evaluation of FDDNP in concentrations similar to those achieved during a PET scan showed limited binding to both NFT and Aβ plaques (Thompson et al. 2009). 18F-FDDNP was used to obtain the first human PET images of Aβ in an 82-year-old woman with AD. AD patients present with higher accumulation and slower clearance of 18F-FDDNP than controls in brain areas such as the hippocampus (Shoghi-Jadid et al. 2002). Retention time of 18F-FDDNP in these brain regions was correlated with lower memory performance scores, regional glucose hypometabolism and brain atrophy (Small et al. 2002, 2006). However, the dynamic range of 18F-FDDNP cortical uptake values between HC and AD patients is small (9 %). Direct comparison of 18F-FDDNP with 11C-PIB in monkeys (Noda et al. 2008) and in human subjects showed very limited dynamic range of 18F-FDDNP (Shih et al. 1987; Tolboom et al. 2009b) and, in a longitudinal study, was found to be less useful than 11C-PiB and 18F-fluorodeoxyglucose (FDG) for examining disease progression (Ossenkoppele et al. 2012). Interestingly, 18F-FDDNP still remains the only Aβ tracer showing retention in the medial temporal cortex of AD patients (Shih et al. 1987) and correlation with cerebrospinal fluid (CSF) tau (Tolboom et al. 2009b).
2.2.2 18F-AZD4694
Developed by AstraZeneca, the most salient features of 18F-AZD4694 are, as with 11C-AZD2138, fast tracer kinetics and low nonspecific binding to white matter, similar to that one observed with 11C-PiB, suggesting it might be helpful for the detection of small Aβ cortical deposits at very early stages of the disease process (Sundgren-Andersson et al. 2009; Jureus et al. 2010). Initial clinical studies with 18F-AZD4694 showed a clear distinction in tracer retention between six healthy controls and ten AD patients (Cselenyi et al. 2012).
2.2.3 18F-Florbetaben
18F-Florbetaben (a.k.a. AV1; BAY94-9172), synthesised by Kung and colleagues (2005b) and developed by Bayer Healthcare, has been shown to bind with high affinity to Aβ in brain homogenates and selectively labelled Aβ plaques and CAA in AD tissue sections (Zhang et al. 2005b). After injection into Tg2576 transgenic mice, ex vivo brain sections showed localisation of 18F-florbetaben in regions with Aβ plaques as confirmed by thioflavin binding (Zhang et al. 2005a). At tracer concentrations achieved during human PET studies, florbetaben did not show binding to LB or NFT tau in postmortem cortices from DLB or FTLD patients (Fodero-Tavoletti et al. 2012). In human studies, cortical retention of 18F-florbetaben was higher at 90-min postinjection in all AD subjects compared to age-matched controls and FTLD patients, with binding matching the reported postmortem distribution of Aβ plaques (Rowe et al. 2008). Recently completed phase II clinical studies further confirmed these results (Barthel et al. 2011), and an ongoing longitudinal study in MCI subjects is helping to establish the usefulness of FBB PET in predicting progression to AD (Ong et al. 2010). 18F-Florbetaben is highly correlated with 11C-PiB (r = 0.97 with a slope of 0.71) (Villemagne et al. 2012c) and was used to detect the presence or absence of AD pathology in the brain in subjects from a wide spectrum of neurodegenerative diseases (Villemagne et al. 2011a). Phase III studies for 18F-florbetaben have reached completion (Sabbagh et al. 2012).
2.2.4 18F-Florbetapir
As 18F-florbetaben, 18F-florbetapir is a stilbene derivative that was also synthesised by Kung and colleagues at the University of Pennsylvania (Zhang et al. 2005a) and has been developed by Avid Radiopharmaceuticals. Initial in vitro evaluation showed binding to Aβ plaques in AD brain sections (Choi et al. 2009; Lin et al. 2009). The most salient feature of this tracer is its rapid reversible binding characteristics allowing scanning only at 45–50 min after injection, similar to 11C-PiB (Wong et al. 2010). 18F-Florbetapir (Lister-James et al. 2011) has become the most widely used Aβ radiotracer after 11C-PiB and has been adopted as the Aβ radiotracer for ADNI 2 and also used in patient selection and to evaluate treatment response in a large number of multicenter therapeutic trials around the world. Several multicenter phase I and II studies in AD, MCI and HC showed the ability of 18F-florbetapir to discriminate between AD and age-matched controls, with ~50 % of the MCI subjects presenting with high 18F-florbetapir retention (Fleisher et al. 2011). Multicenter studies using 18F-florbetapir to assess the relationship between Aβ burden and cognition showed that in clinically normal older individuals, Aβ burden in the brain is associated with poorer memory performance (Sperling et al. 2013), while MCI subjects with high Aβ burden in the brain are at a significantly higher risk of cognitive decline over 18 months (Doraiswamy et al. 2012). An initial phase III study in 35 volunteers demonstrated a 96 % agreement between 18F-florbetapir and neuropathology for the detection of brain Aβ in vivo and no retention in young controls (Clark et al. 2011). An extension phase III in 59 volunteers establishes that 18F-florbetapir has a sensitivity of 92 % and a specificity of 100 % for the detection of Aβ pathology (Clark et al. 2012). Using a semiquantitative approach, it has been shown that 18F-florbetapir is >90 % sensitive and specific to detect Aβ pathology in the brain (Camus et al. 2012) and that it is correlated with 11C-PiB (r = 0.77 with a slope of 0.33) (Wolk et al. 2012b). 18F-Florbetapir (as Amyvid®) is the first radiotracer approved by the Food and Drug Administration (FDA) for detection of Aβ in vivo and the first 18F-labelled radiotracer approved by the FDA since FDG.
2.2.5 18F-Flutemetamol
Another fluorinated tracer, developed by GE Healthcare, that also recently completed a phase III study (Buckley et al. 2012) is 18F-flutemetamol (a.k.a. GE067) (Serdons et al. 2009a, b). Phase I and II studies demonstrated that 18F-flutemetamol can differentiate between AD and HC (Nelissen et al. 2009; Vandenberghe et al. 2010b) and that when combined with brain atrophy it could be predictive of disease progression in MCI subjects (Thurfjell et al. 2012). 18F-Flutemetamol brain retention is also highly correlated with 11C-PIB (r = 0.91 with a slope of 0.90) (Vandenberghe et al. 2010b). Furthermore, there was a high correlation between Aβ burden as measured by 18F-flutemetamol and by immunohistochemical assessment of brain biopsy tissue (Wolk et al. 2011; Wong et al. 2013).
While the cortical retention of 18F-florbetapir, 18F-florbetaben and 18F-flutemetamol provides a clear separation of AD patients from HC subjects, the degree of cortical retention with 18F-florbetapir and 18F-florbetaben is lower than with 11C-PiB (Villemagne et al. 2012c; Wolk et al. 2012b), showing a narrower dynamic range of SUVR values, that visually appears as a relatively higher degree of nonspecific binding to white matter. While the cortical retention of 18F-flutemetamol is similar to that of 11C-PiB, the nonspecific retention in white matter is much higher (Vandenberghe et al. 2010b). Due to this lower signal-to-noise ratio, visual readouts of the images are more challenging than with 11C-PiB (Rowe and Villemagne 2011). While 11C-PiB PET images in subjects with AD pathology usually clearly show high radiotracer retention in the grey matter in excess of that in subjacent white matter, these novel 18F tracers frequently show loss of the normal grey-white matter demarcation as the predominant evidence of cortical Aβ deposition (Rowe and Villemagne 2011).
To date, the vast majority of the Aβ imaging literature is based on 11C-PiB studies, with studies using the first generation of fluorinated Aβ radiotracers only recently being reported. The following survey of the state of the field is a reflection of this, and, unless otherwise specified, the reported results are from 11C-PiB studies.
3 Aβ Imaging in Alzheimer’s Disease
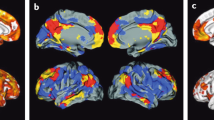
On visual inspection, cortical 11C-PiB retention is higher in AD, with a regional brain distribution that is highest in frontal, cingulate, precuneus, striatum, parietal and lateral temporal cortices, while occipital, sensorimotor and mesial temporal cortices are much less affected (Fig. 10.1). Both quantitative and visual assessments of PET images present a pattern of radiotracer retention that seems to replicate the sequence of Αβ deposition found at autopsy (Braak and Braak 1997), with initial deposition in the orbitofrontal cortex, inferior temporal, cingulate gyrus and precuneus, followed by the remaining prefrontal cortex and lateral temporal and parietal cortices (Fig.10.2). Multimodality studies in early AD have shown that the regional pattern of Αβ deposition is similar to the anatomy of the ‘default network’ (Buckner et al. 2005; Sperling et al. 2009), a specific, anatomically defined brain system responsible for internal modes of cognition, such as self-reflection processes, conscious resting state or episodic memory retrieval (Buckner et al. 2008; Wermke et al. 2008), an association that is present even in non-demented subjects (Drzezga et al. 2011).

Representative sagittal (top row), transaxial (middle row) and coronal (bottom row) PET images overlaid on MRI from Alzheimer’s disease patients obtained with different Aβ imaging radiotracers. From left to right, 11C-PiB, 18F-florbetaben, 18F-flutemetamol, 18F-florbetapir and 18F-AZD4694. The images show the typical pattern of tracer retention in AD, with the highest retention in frontal, temporal and posterior cingulate cortices, reflecting Aβ plaque burden

Top row. Schematics showing the stages of Aβ deposition in the human brain as proposed by Braak and Braak (1997). Bottom row. Representative sagittal PET images showing the regional brain distribution of 11C-PIB in two asymptomatic healthy age-matched controls, one with low (PiB- HC) and one with high Aβ burden (PiB + HC), a subject classified as mild cognitive impairment (MCI) with significant Aβ deposition in the brain and an Alzheimer’s disease (AD) patient with even a higher Aβ burden in the brain. The pattern of PiB retention replicates the sequence of Αβ deposition described from postmortem studies (Braak and Braak 1997)
The regional distribution of 11C-PiB retention is not always the same and varies with the specific Αβ distribution characteristic of the underlying pathology. For example, carriers of mutations associated with familial AD (Klunk et al. 2007; Koivunen et al. 2008b; Villemagne et al. 2009a), subjects with posterior cortical atrophy (Ng et al. 2007b; Tenovuo et al. 2008; Kambe et al. 2010; Formaglio et al. 2011) or CAA (Johnson et al. 2007; Dierksen et al. 2010) each presenting a different regional 11C-PiB distribution of retention to the one usually observed in sporadic AD (Klunk et al. 2004; Rowe et al. 2007).
Quantitative PET studies using diverse Αβ radiotracers have shown a robust difference in retention between AD patients and age-matched controls (Klunk et al. 2004; Price et al. 2005; Mintun et al. 2006; Kudo et al. 2007; Pike et al. 2007; Rabinovici et al. 2007; Rowe et al. 2007, 2010; Jack et al. 2008a; Morris et al. 2009; Furst et al. 2012; Barthel et al. 2011; Fleisher et al. 2011; Villemagne et al. 2011a; Camus et al. 2012; Cselenyi et al. 2012).
Longitudinal studies with 11C-PiB are showing that significant, although small, increases in Αβ deposition can be measured but also that these increases in Αβ deposition are present in the whole spectrum of cognitive stages, from cognitively unimpaired individuals to AD dementia patients (Engler et al. 2006; Jack et al. 2009; Okello et al. 2009b; Resnick et al. 2010; Rinne et al. 2010; Sojkova et al. 2011b; Villemagne et al. 2011b; Villain et al. 2012). Αβ accumulation is observed in individuals considered to have low Αβ loads with about 7 % of them progressing above the threshold in about 2.5 years (Vlassenko et al. 2011). Αβ accumulation is a slow process, and the evidence suggests that it remains constant in the preclinical and prodromal stages of the disease in those individuals deemed to have high Αβ burdens while being slightly higher at the early stages of AD (Villemagne et al. 2011b). In individuals with the highest Aβ burdens independent of clinical classification and also as Alzheimer’s dementia progresses, the rates of Aβ deposition start to slow down, although not completely reaching a plateau (Villain et al. 2012). This slower rate of Αβ deposition associated with disease progression might be attributed to a saturation of the process or, more likely, extensive neuronal death that precludes Αβ production.
4 Antemortem and Postmortem Correlations
While clinicopathological studies have shown that the accuracy of clinical assessments for the diagnosis of AD ranges between 70 and 90 %, depending on the specialisation of the centre, the regional retention of 11C-PiB and the novel 18F radiotracers is highly correlated with regional Αβ plaques as reported at autopsy or biopsy (Bacskai et al. 2007; Ikonomovic et al. 2008; Leinonen et al. 2008b; Villemagne et al. 2009b; Burack et al. 2010; Clark et al. 2011; Kadir et al. 2011; Sabbagh et al. 2011; Sojkova et al. 2011a; Wolk et al. 2011; Wong et al. 2013), with a higher Aβ concentration in the frontal cortex than in hippocampus, consistent with previous reports (Arnold et al. 2000; Naslund et al. 2000). As mentioned before and in contrast with CSF studies, Αβ imaging studies can assess both the quantity and regional brain distribution of Αβ, as observed in the different patterns of regional distribution of Αβ observed in sporadic AD (Klunk et al. 2004; Nordberg 2007), familial AD (Klunk et al. 2007; Koivunen et al. 2008b; Villemagne et al. 2009a), familial British dementia (Villemagne et al. 2010) and CAA subjects (Johnson et al. 2007; Dierksen et al. 2010). The results from Αβ imaging might be considered for the brain regions to be sampled and stained for the neuropathological diagnosis of AD (Montine et al. 2012).
There have been a handful of cases reported with a discrepancy between a positive neuropathological finding or a strong biomarker profile of AD and a low PET signal. To date, only seven discrepant cases have been reported, three of them in mutation carriers (Tomiyama et al. 2008; Schöll et al. 2011) and four in sporadic cases (Leinonen et al. 2008a; Cairns et al. 2009; Sojkova et al. 2011a; Ikonomovic et al. 2012). No ‘false-positive’ cases have been reported. Tomiyama and colleagues reported a case of a novel APP mutation (E693Delta) where the mutant Αβ does not fibrillise in the same way as wild-type Aβ (Inayathullah and Teplow 2011) in which the 11C-PiB study showed low cortical retention (Tomiyama et al. 2008). Schöll and colleagues report on two subject carriers of the Arctic APP mutation (Schöll et al. 2011). These two mutation carriers, while presenting with low 11C-PiB retention, had a clear ‘AD pattern’ of brain hypometabolism as measured by FDG, low Aβ1–42 and elevated t-tau and p-tau in CSF (Schöll et al. 2011). While neuropathology was not available in these two subjects, a family member carrier of the same mutation presented with atypical plaque morphology—with ringlike plaques lacking a congophilic core—at autopsy (Basun et al. 2008). In regard to sporadic cases, Leinonen et al., while correlating 11C-PiB retention and frontal lobe Αβ burden from biopsies on subjects undergoing intraventricular monitoring for normal pressure hydrocephalus, described a non-demented case with plaques that had a negative 11C-PiB scan (Leinonen et al. 2008a). Cairns et al. reported a longitudinally characterised subject with a low 11C-PiB scan that presented with evidence of cognitive decline and a CSF profile indicative of AD a year later (Cairns et al. 2009). At the time of autopsy, two and a half years after the 11C-PiB PET, diffuse Αβ plaques were observed with only minimal neuritic plaques and NFT pathology. Sojkova et al. reported imaging-neuropathological correlations in five participants from the Baltimore Longitudinal Study of Aging (Sojkova et al. 2011a). While there was agreement in four subjects, even in one that had only sparse plaques at autopsy, a fifth presented with low global and regional 11C-PiB retention despite presenting moderate plaques at neuropathological examination and meeting CERAD criteria for ‘possible AD’ (Mirra et al. 1991). Ikonomovic et al. (2012) reported autopsy findings in a subject with a clinical diagnosis of DLB and possible AD that presented with low 11C-PiB retention 17 months before death. At autopsy, focally frequent neocortical neuritic plaques fulfilled CERAD criteria of ‘definite AD’ (Mirra et al. 1991). Cortical Αβ1–40 concentration levels were on the same range to those found in a typical AD case with high 11C-PiB retention, but Αβ1–42 concentrations were lower. Furthermore, the majority of plaques labelled weakly with 11C-PiB and 3H-PiB binding were low, all findings suggesting low fibrillar Αβ.
On the one hand, these cases illustrate the possibility that different conformations of Aβ deposits (Levine and Walker 2010b) may affect the binding of tracers and that Aβ imaging may not recognise all types of Aβ pathologies with equal sensitivity (Walker et al. 2008; Rosen et al. 2011). On the other, it raises issues not only in regard to the sensitivity of Aβ imaging (e.g. What is the minimal amount of Aβ detected by PET?) but also in regard to the evaluation of brain tissue (e.g. type of antibodies used for immunohistochemistry and ELISA, fluorescent dyes, diagnostic criteria).
5 Aβ Deposition in Non-Demented Individuals and Its Relation with Cognition
Αbout 25–35 % of elderly subjects performing within normal limits on cognitive tests present with high cortical 11C-PiB retention, predominantly in the prefrontal and posterior cingulate/precuneus regions (Mintun et al. 2006; Rowe et al. 2007, 2010; Aizenstein et al. 2008; Villemagne et al. 2008b; Mormino et al. 2009; Reiman et al. 2009b). These findings are in perfect agreement with postmortem reports that ~25 % of non-demented older individuals over the age of 75 have Aβ plaques (Davies et al. 1988; Morris and Price 2001; Forman et al. 2007). Aβ deposition in these non-demented individuals (Mintun et al. 2006; Rowe et al. 2007; Aizenstein et al. 2008; Villemagne et al. 2008b) might reflect preclinical AD (Price and Morris 1999). Furthermore, the prevalence of subjects with high 11C-PiB retention increases each decade at the same rate as the reported prevalence of plaques in non-demented subjects in autopsy studies (Rowe et al. 2010). While cross-sectional examination of these controls essentially shows no significant differences in cognition (Mintun et al. 2006; Aizenstein et al. 2008; Jack et al. 2008a; Villemagne et al. 2008b; Rowe et al. 2010), significant cognitive differences in females were reported (Pike et al. 2011). More recently, Sperling and colleagues reported that high Aβ deposition in the brain was correlated with lower immediate and delayed memory scores in otherwise healthy controls (Sperling et al. 2013). The detection of Aβ pathology at the presymptomatic stage is of crucial importance because if this group truly represents preclinical AD (Price and Morris 1999; Thal et al. 2004; Backman et al. 2005; Small et al. 2007), it is precisely the group that may benefit the most from therapies aimed at reducing or eliminating Αβ from the brain before irreversible neuronal or synaptic loss occurs (Sperling et al. 2011b).
Individuals fulfilling criteria for MCI are a heterogeneous group with a wide spectrum of underlying pathologies (Petersen 2000; Winblad et al. 2004). About 40–60 % of carefully characterised subjects with MCI will subsequently progress to meet criteria for AD over a 3–4-year period (Petersen et al. 1995, 1999; Petersen 2000). Αβ imaging has proven useful in identifying MCI individuals with and without AD pathology. Approximately 50–70 % of the subjects classified as MCI present with high cortical 11C-PiB retention (Price et al. 2005; Kemppainen et al. 2007; Pike et al. 2007; Forsberg et al. 2008; Lowe et al. 2009; Mormino et al. 2009). While most studies found that subjects classified as non-amnestic MCI show low Αβ levels in the brain (Pike et al. 2007; Lowe et al. 2009), other reports did not (Wolk et al. 2009). More specifically, subjects classified as non-amnestic single-domain MCI usually do not show Αβ deposition, consistent with a non-AD underlying pathological process (Pike et al. 2007; Villemagne et al. 2011b).
To date, the relationship between Αβ deposition and the development of clinical symptoms is not fully understood. Although Aβ burden as assessed by PET does not correlate with measures of memory impairment in established clinical AD, it does correlate with memory impairment in MCI and healthy older subjects (Pike et al. 2007; Villemagne et al. 2008b). In subjects with MCI, this Aβ-related impairment is thought to be mediated through hippocampal atrophy (Mormino et al. 2009) while modulated by cognitive reserve (Roe et al. 2008; Rentz et al. 2010) and glucose metabolism (Cohen et al. 2009), although at the AD stage there is no longer correlation between glucose metabolism, cognition and Αβ burden (Furst et al. 2012).
Longitudinal studies are helping clarify the relationship between Aβ burden and cognitive decline in healthy individuals and MCI subjects. High 11C-PiB cortical retention in non-demented elderly subjects is associated with a greater risk of cognitive decline (Villemagne et al. 2008b, 2011b; Resnick et al. 2010). For example, the evaluation of either the cognitive trajectories (Villemagne et al. 2008b; Resnick et al. 2010) or changes in cognition prospectively (Morris et al. 2009; Villemagne et al. 2011b; Doraiswamy et al. 2012) shows that individuals with substantial Αβ deposition are more likely to present with cognitive decline. Aggregating the results from recently reported longitudinal studies reveals that 65 % of MCI subjects with marked 11C-PiB retention progressed to AD over 2–3 years, showing that MCI subjects with high Aβ deposition in the brain are >10 times more likely to progress to AD than those with low Aβ deposition, highlighting the clinical relevance and potential impact in patient management of Aβ imaging (Forsberg et al. 2008; Okello et al. 2009b; Wolk et al. 2009; Villemagne et al. 2011b). The observations that 11C-PiB retention in non-demented individuals relates to episodic memory impairment, one of the earliest clinical symptoms of AD, and that extensive Aβ deposition is associated with a significant higher risk of cognitive decline in HC and with progression from MCI to AD emphasise the non-benign nature of Aβ deposition and support the hypothesis that Aβ deposition occurs well before the onset of symptoms, further suggesting that early disease-specific therapeutic intervention at the presymptomatic stage might be the most promising approach to either delay onset or halt disease progression (Sperling et al. 2011b).
The high prevalence of Αβ deposition in non-demented individuals raises questions in regard to Αβ toxicity and deposition. The evidence points to the fact that Αβ toxicity in the form of oligomers and Αβ deposition in the form of aggregates precede by many years the appearance of clinical symptoms (Price and Morris 1999). These neurotoxic processes eventually lead to synaptic and neuronal dysfunction, manifested as cognitive impairment, grey matter atrophy and glucose hypometabolism (Eckert et al. 2003; Suo et al. 2004; Leuner et al. 2007). Therefore, it is highly likely that 11C-PiB retention in non-demented individuals reflects preclinical AD (Mintun et al. 2006; Rowe et al. 2007; Aizenstein et al. 2008; Villemagne et al. 2008b). This may be associated or attributed to a different susceptibility/vulnerability to Αβ, at either a cellular (frontal neurons seems to be more resistant to Αβ than hippocampal neurons) (Roder et al. 2003; Resende et al. 2007), a regional (compensatory upregulation of choline acetyltransferase (ChAT) activity in the frontal cortex and hippocampi of MCI and early AD subjects) (DeKosky et al. 2002) as well as at an individual or personal level, either due to a particular cognitive reserve (Mortimer 1997; Stern 2002; Kemppainen et al. 2008; Roe et al. 2008), differences in Aβ conformation affecting toxicity and/or aggregation (Lockhart et al. 2005; Deshpande et al. 2006; Levine and Walker 2010a; Walker et al. 2008), or because an idiosyncratic threshold must be exceeded for synaptic failure and neuronal death to ensue (Suo et al. 2004). These factors would help explain why some older individuals with a significant Αβ burden are cognitively unimpaired, while others with lower Αβ burden and no genetic predisposing factors have already developed the full clinical AD phenotype. Ongoing longitudinal studies will help identify those subjects that despite substantial Aβ deposition do no develop the AD phenotype, thus allowing to isolate genetic or environmental factors that may convey resistance to Aβ (Sojkova and Resnick 2011).
The lack of a strong association between Aβ deposition and measures of cognition, synaptic activity and neurodegeneration in AD, in addition to the evidence of Αβ deposition in a high percentage of MCI and asymptomatic HC, suggests that Αβ is an early and necessary, though not sufficient, cause for cognitive decline in AD (Villemagne et al. 2008b; Rabinovici et al. 2010; Sojkova and Resnick 2011), indicating the involvement of other downstream mechanisms, likely triggered by Αβ such as NFT formation, synaptic failure and eventually neuronal loss. Also at play are factors as age, years of education, occupational level and brain volume among others that, under the umbrella of ‘cognitive reserve’, appear to have a modulatory role between cognition and Αβ deposition (Roe et al. 2008; Chetelat et al. 2010b; Tucker and Stern 2011). Perhaps specific in vivo tau imaging will help elucidate or bridge the gap between Aβ deposition and the persistent decline in cognitive function observed in AD (Fodero-Tavoletti et al. 2011; Villemagne et al. 2012a).
6 Relationship of Aβ Imaging with Other Biomarkers
Another rapidly growing area is the exploration of the potential association between biomarkers of Aβ deposition or neurodegeneration (Wahlund and Blennow 2003; Morris et al. 2005; Sunderland et al. 2005; de Leon et al. 2006; Thal et al. 2006; Blennow et al. 2007; Shaw et al. 2007; Clark et al. 2008; Jack et al. 2010; Albert et al. 2011; McKhann et al. 2011; Storandt et al. 2012) and Aβ burden as measured by PET (Jack et al. 2008a, 2010).
6.1 FDG
While the typical pattern of Aβ deposition in sporadic AD is highest in frontal, cingulate, precuneus, striatum, parietal and lateral temporal cortices (Klunk et al. 2004), in FDG studies, the typical ‘AD pattern’ is of temporoparietal and posterior cingulate hypometabolism with sparing of the basal ganglia, thalamus, cerebellum and primary sensorimotor cortex (Salmon et al. 1994; Devanand et al. 1997; Coleman 2005; Jagust et al. 2007). Due to its high sensitivity (>90 %) for detecting temporoparietal and posterior cingulate hypometabolism, FDG-PET has improved diagnostic and prognostic accuracy in patients with probable AD (Salmon et al. 1994; Kennedy et al. 1995; Small et al. 1995; Silverman et al. 2001, 2002). A similar pattern of hypometabolism has been reported in normal elderly ApoE ε4 carriers (Reiman et al. 1996), MCI (Chetelat et al. 2003, 2005; Mosconi et al. 2006a), asymptomatic subjects with mutations associated with familial AD (Kennedy et al. 1995; Rossor et al. 1996) and subjects with a strong family history of AD (Mosconi et al. 2006b). FDG hypometabolism is correlated with cognition (Landau et al. 2011; Furst et al. 2012) and is predictive of future cognitive decline (Drzezga et al. 2003, 2005; Mosconi et al. 2004).
Visual assessment of PET images by clinicians blinded to the clinical status has demonstrated that 11C-PiB was more accurate than FDG to distinguish AD from HC (Ng et al. 2007a) and slightly better than FDG in differentiating AD from FTLD (Rabinovici et al. 2011). Similarly, Aβ imaging outperforms FDG at identifying MCI subtypes (Lowe et al. 2009).
While some reports found no association between FDG and 11C-PiB in AD (Furst et al. 2012), others found an inverse correlation between them in temporal and parietal cortices (Cohen et al. 2009), but no correlation has been shown in the frontal lobe (Klunk et al. 2004; Edison et al. 2007). A possible explanation for this dissociation is that there is a compensatory mechanism such as upregulation of ChAT in the frontal lobe of MCI individuals and mild AD patients (DeKosky et al. 2002; Ikonomovic et al. 2007) that might delay the manifestation of synaptic dysfunction; therefore, no hypometabolism is observed on FDG in the frontal lobe. Glucose metabolism in the frontal lobe eventually decreases as the disease progresses, and the characteristic temporoparietal pattern of AD becomes less apparent (Mielke et al. 1992; Herholz 1995). Conversely to what is observed in the frontal area, in AD patients, ChAT is significantly decreased in the posterior cingulate gyrus (Ikonomovic et al. 2011), which is characterised by being one of the regions where marked and early Aβ deposition is associated with substantial hypometabolism (Drzezga et al. 2011; Yokokura et al. 2011).
6.2 CSF
Analysis of CSF allows simultaneous measurement of the concentration of the two main hallmarks of AD, Aβ and tau. It has been reported to be highly accurate in the diagnosis of AD as well of predicting cognitive decline (Blennow et al. 2007; de Leon et al. 2007; Hansson et al. 2007; Mattsson et al. 2009, 2011). The typical CSF profile in AD is low Aβ1–42 and high total (t-tau) and phosphorylated tau (p-tau) (Strozyk et al. 2003; Hampel et al. 2010). Slower Aβ clearance from the brain (Bateman et al. 2006; Mawuenyega et al. 2010) is likely to lead to Aβ deposition in the brain and lower CSF Aβ1−42 concentrations. While there is no correlation between 11C-PiB retention and CSF t-tau or p-tau (Tolboom et al. 2009a; Forsberg et al. 2010), several studies have reported a strong inverse correlation between Aβ deposition in the brain as measured by 11C-PiB and CSF Aβ1–42 (Fagan et al. 2006, 2007, 2009; Forsberg et al. 2008; Koivunen et al. 2008a; Grimmer et al. 2009). Both high 11C-PiB retention and low CSF Aβ1–42 have been observed in cognitively unimpaired individuals probably reflecting Aβ deposition years before the manifestation of the AD phenotype (Mintun et al. 2006; Fagan et al. 2007, 2009; Rowe et al. 2007; Aizenstein et al. 2008). Large population studies such as ADNI (Shaw et al. 2007; Apostolova et al. 2010) will further validate the value of Aβ imaging and CSF assessments as antecedent biomarkers of AD.
6.3 MRI
While Aβ imaging or CSF Aβ provides information in regard to Aβ pathology, CSF tau quantification, FDG and structural MRI provide information related to the neurodegenerative process (Jack et al. 2010). Hippocampal and cortical grey matter atrophy along with ventricular enlargement are typical MRI findings in AD and MCI (Frisoni et al. 2010; Drago et al. 2011). Furthermore, it has been shown that the rates of hippocampal atrophy might be predictive of conversion from MCI to AD (Jack et al. 2008b).
Aβ deposition is associated with regional cerebral atrophy as measured by MRI (Archer et al. 2006; Bourgeat et al. 2010; Chetelat et al. 2010a; Becker et al. 2011) and correlates with the rates of cerebral atrophy (Becker et al. 2011; Tosun et al. 2011). Moreover, the data suggest that the relationship between Aβ deposition and cortical atrophy is sequential, where Aβ deposition precedes synaptic dysfunction and neuronal loss (Chetelat et al. 2010a; Drzezga et al. 2011; Forster et al. 2012), then manifested as structural changes (Becker et al. 2011). While one report did not find a correlation between atrophy trajectories and 11C-PiB retention in cognitively unimpaired elderly individuals (Driscoll et al. 2011), another reported that those with high Aβ deposition showed a significantly higher rate of atrophy in the temporal and posterior cingulate cortices compared to those with low Aβ burden (Chetelat et al. 2012).
6.4 Neuroinflammation
Microglia are the primary resident immune surveillance cells in the brain and are thought to play a significant role in the pathogenesis of several neurodegenerative disorders including AD (Venneti et al. 2006). Activated microglia can be measured using radioligands for the translocator protein (TSPO), formerly known as the peripheral benzodiazepine receptor. The TSPO is upregulated on activated macrophages and microglia and has been established as a biomarker of neuroinflammation in the central nervous system. Most reports found that in AD there is no association between Aβ deposition and microglial activation measured by [11C]PK11195 (Edison et al. 2008a; Okello et al. 2009a; Wiley et al. 2009) despite being an association between [11C]PK11195 binding and both glucose hypometabolism and severity of dementia (Edison et al. 2008a; Yokokura et al. 2011). More recently, PET studies assessing the relationship between Aβ deposition and neuroinflammation have been proposed using the MAO-B inhibitor L-deprenyl as a marker for astrocytosis (Carter et al. 2012).
7 Relationship of Aβ Deposition with Genetic Risks and Predisposing Factors
Age is the strongest risk factor in sporadic AD, with the prevalence of the disease increasing exponentially with age. These risks are increased in the presence of the ApoE ε4 allele (Farrer et al. 1997). Both risk factors have been directly associated with Aβ burden as measured by PET (Reiman et al. 2009b; Morris et al. 2010; Rowe et al. 2010).
To date, ApoE ε4 is the most consistent genetic risk factor associated with sporadic AD, and its presence has been associated with an earlier age of onset and a dose-dependent—either one or two ApoE ε4 alleles—higher risk of developing AD (Farrer et al. 1997). Examination of ApoE ε4 allele status revealed that, independent of clinical classification, ε4 carriers present with significantly higher 11C-PiB retention than non-ε4 carriers, further emphasising the crucial role that ApoE plays in the metabolism of Αβ (Reiman et al. 2009b; Morris et al. 2010; Rowe et al. 2010). When specific clinical groups are examined, the results—from both autopsy and Αβ imaging studies—are not that clear cut. While some studies found significant differences in Αβ burden between AD ε4 carriers and non-ε4 carriers (Gomez-Isla et al. 1996; Drzezga et al. 2008; Grimmer et al. 2010), others did not (Berg et al. 1998; Klunk et al. 2004; Rowe et al. 2007; Rabinovici et al. 2010).
Both symptomatic and asymptomatic individual carriers of mutations within the APP or presenilin 1 or 2 genes associated with familial AD present with very high and early 11C-PiB retention in the caudate nuclei, retention that seems to be independent of mutation type or disease severity (Klunk et al. 2007; Koivunen et al. 2008b; Remes et al. 2008; Villemagne et al. 2009a). Larger multicenter studies are helping elucidate the value of several biomarkers in these autosomal mutation carriers, assessment that might lead to better targeted disease-specific therapeutic strategies (Bateman et al. 2012).
While a similar pattern of Aβ deposition as seen in AD has been reported in older adults with Down syndrome (Landt et al. 2011), a completely different pattern of 11C-PiB retention was observed in a mutation carrier of familial British dementia (Villemagne et al. 2010).
There are several other factors that have been associated with AD. Among these, the most relevant are vascular risk factors (VRF), such as diabetes, hypercholesterolemia, hypertension and hyperhomocysteinemia. The influence of these VRF on AD pathology is an area that, despite the long and intense scientific debate, has not been clearly established (Viswanathan et al. 2009). While some studies support a direct role of VRF in the development of AD (Qiu et al. 2010), others suggest that they may accelerate its clinical presentation (Riekse et al. 2004). Recent studies have used MRI white matter hyperintense lesions as surrogate markers of vascular disease in association with assessment of Aβ deposition and cognition (Kuczynski et al. 2008; Marchant et al. 2012).
As with AD, VRF are strongly age-dependent factors that share similar pathogenic pathways, such as oxidative stress or inflammation, so it is not easy to extricate one from the other in order to ascertain a causal, casual or co-morbid association between them (Rosendorff et al. 2007). On the other hand, in light of the absence of disease-modifying therapies for AD, the relevance of these risk factors rests in the fact that early intervention over them, especially at middle age, might have an impact on either the onset or progression of AD.
8 Aβ Imaging in Other Neurodegenerative Conditions
Aβ imaging in a wide spectrum of neurodegenerative conditions has yielded diverse results. Although lower than in AD, similar patterns of 11C-PiB retention are usually observed in DLB (Rowe et al. 2007; Gomperts et al. 2008; Maetzler et al. 2009). Cortical 11C-PiB retention is also higher in subjects diagnosed with CAA (Johnson et al. 2007), while there is usually no cortical 11C-PiB retention in patients with FTLD (Rabinovici et al. 2007; Rowe et al. 2007; Drzezga et al. 2008; Engler et al. 2008). Aβ imaging has also facilitated differential diagnosis in cases of patients with atypical presentations of dementia (Ng et al. 2007b; Wolk et al. 2012a).
8.1 Cerebral Amyloid Angiopathy
CAA, characterised by Aβ deposits in and around the media of small arteries and arterioles of the cerebral cortex and leptomeninges, has been found to be present in most patients with AD (Jellinger and Attems 2005). To this date, neuropathological examination of the brain remains the only definitive method for diagnostic confirmation of CAA. However, the combination of Aβ imaging with 11C-PiB PET and T2* susceptibility-weighted (SWI) MR imaging (Knudsen et al. 2001) allows for the concomitant assessment of molecular and structural changes in vivo. The combination of cerebral microhaemorrhages (MH) and superficial siderosis (SS) has been suggested as radiological markers for CAA (Feldman et al. 2008). Aβ imaging studies have shown both processes may be intimately associated with Aβ deposition (Dierksen et al. 2010; Dhollander et al. 2011), preferentially in posterior areas of the brain, showing a distinct pattern of 11C-PiB retention than the one observed in sporadic AD (Johnson et al. 2007). Furthermore, focal Aβ deposition might be useful in predicting new MH in CAA patients (Gurol et al. 2012). While deep subcortical MH are generally associated with VRF, lobar MH (particularly posterior) (Pettersen et al. 2008) are usually attributed to CAA (Knudsen et al. 2001; Vernooij et al. 2008). Lobar MH are a frequent finding in AD patients or even in cognitively normal older individuals, and they are strongly associated with increasing age and Aβ deposition (Yates et al. 2011). This association between Aβ and vascular lesions has crucial implications not only for the selection and risk stratification of individuals undergoing anticoagulant therapies but also in those enrolled in anti-Aβ therapeutic trials (Weller et al. 2009).
8.2 Lewy Body Diseases
While AD is the most common cause of dementia in the elderly, DLB accounts for 20 % of the cases (McKeith et al. 2005). The pathological hallmark of DLB is the presence of α-synuclein-containing LB within the neocortical and limbic regions (McKeith et al. 2005), as well as substantial loss of pigmented dopaminergic neurons in the substantia nigra, reflected in a marked dopaminergic terminal denervation in the striatum (McKeith and Mosimann 2004). More refined neuropathological techniques have revealed extensive synaptic deposits of α-synuclein, more concordant with the DLB clinical phenotype than the usually sparse cortical LB (Schulz-Schaeffer 2010). Overlap of cognitive symptoms early in the disease course offers a challenge for clinicians making it difficult to distinguish between patients that will develop AD versus those that will ultimately develop DLB. Postmortem histopathological studies have identified that 50–80 % of DLB cases often have cortical Aβ deposits with characteristics and a distribution similar to AD patients (McKeith et al. 2005; Rowe et al. 2007; Edison et al. 2008b). This has also been described as ‘mixed’ DLB/AD or ‘Lewy body variant of AD’. The so-called ‘pure’ DLB is much less common, and it is not clear if the clinical characteristics and prognosis differ from the mixed-pathology cases. On visual inspection, cortical 11C-PiB retention in DLB is generally lower and more variable than in AD, probably reflecting a larger spectrum of Αβ deposition (Rowe et al. 2007; Gomperts et al. 2008; Maetzler et al. 2009). Therefore, Aβ imaging is not a useful tool in differentiating between AD and DLB but might assist in the differential diagnosis of those cases (‘pure’ DLB) without Aβ deposition in the brain (Armstrong et al. 2000).
Taking advantage of its high affinity for α-synuclein (Fodero-Tavoletti et al. 2009a), 11C-BF-227 has been used for the assessment of multiple system atrophy (MSA), a disease characterised by the glial accumulation of α-synuclein in the absence of Aβ plaques, showing a higher retention in MSA patients compared to age-matched controls (Kikuchi et al. 2010). Interestingly, while 11C-BF-227 retention in DLB patients was lower compared to AD in most cortical regions, DLB presented with significantly higher 11C-BF-227 retention in the amygdala (Furukawa et al. 2011).
In contrast to Aβ imaging, assessing the integrity of dopaminergic terminals via dopamine transporter (DAT) or vesicular monoamine transporter type 2 (VMAT2) imaging can robustly detect reductions of dopaminergic nigrostriatal afferents in DLB patients, assisting in the differential diagnosis from AD (O’Brien et al. 2004; Koeppe et al. 2008; Villemagne et al. 2012d).
Cortical Aβ is not usually present in cognitively intact Parkinson’s disease (PD) patients, and no significant 11C-PiB cortical retention is usually observed in these patients (Johansson et al. 2008). Aβ deposits, both vascular and parenchymal, are frequent, however, in PD patients who develop dementia (PDD) (Edison et al. 2008b; Gomperts et al. 2008; Maetzler et al. 2008, 2009; Burack et al. 2010; Foster et al. 2010; Kalaitzakis et al. 2011).
The contribution of Aβ to the development of Lewy body diseases remains unclear, but cortical Aβ deposits are associated with extensive α-synuclein lesions and higher levels of insoluble α-synuclein lesions (Pletnikova et al. 2005), as well as exacerbation of neuronal injury (Masliah et al. 2001). Furthermore, it has been shown that in PD, CSF Aβ is predictive of cognitive decline (Siderowf et al. 2010). Along these lines, while Aβ imaging cannot contribute to the differential diagnosis between AD and DLB, it may have prognostic relevance in these Lewy body diseases. For example, similar to what has been reported for PDD, where higher Αβ burden was associated with a shorter prodromal phase before dementia (Halliday et al. 2008), Αβ burden in DLB patients was inversely correlated with the interval from onset of cognitive impairment to the full development of the DLB phenotype (Rowe et al. 2007).
8.3 Frontotemporal Lobar Degeneration
FTLD is a syndrome that can also be clinically difficult to distinguish from early-onset AD, especially at the initial stages of the disease. Based on clinical phenotype, FTLD has been categorised mainly into two classes: behavioural variant of FTLD (bvFTLD) with distinct changes in behaviour and personality with little effect on language functions and progressive aphasias (comprised by semantic dementia (SD), progressive non-fluent aphasia (PNFA) and logopenic aphasia (LPA)) showing progressive language deficits, with less obvious personality and behavioural changes (Rabinovici and Miller 2010).
Aβ deposition is not a pathological trait of FTLD, and definitive differential diagnosis can only be established after postmortem examination of the human brain. Neuropathological examination of the brain in FTLD shows variable frontal and temporal atrophy (Cairns et al. 2007; Hodges and Patterson 2007; Snowden et al. 2007), microvacuolation and neuronal loss, with white matter myelin loss, astrocytic gliosis as well as neuronal and glial inclusions (Cairns et al. 2007). Three intraneuronal inclusion types have been identified in FTLD: (1) hyperphosphorylated tau (FTLD tau), (2) ubiquitin hyperphosphorylated and proteolysed TAR-DNA-binding protein 43 (FTLD-TDP-43) and (3) fused in sarcoma (FUS) positive (Taniguchi et al. 2004; Mott et al. 2005; Neumann et al. 2006, 2009). Tau-positive inclusions account for approximately 40 % of all FTLD cases, the remainder being tau negative and TDP-43/ubiquitin positive (Josephs et al. 2004). In bvFTLD, both tau and TDP-43 are equally prevalent; and while 90 % of the SD cases show TDP-43 pathology, PNFA presents predominantly (70 %) tau pathology (Josephs et al. 2004; Mackenzie et al. 2008). In contrast, LPA is thought to be a language presentation of AD, with typical Aβ and NFT (Leyton et al. 2011).
Aβ imaging has been helpful in the differential diagnosis between FTLD and AD (Rabinovici et al. 2007; Rowe et al. 2007; Drzezga et al. 2008; Engler et al. 2008). Furthermore, Aβ imaging has been used to ascertain the absence of AD pathology in the different FTLD aphasias (Drzezga et al. 2008; Rabinovici et al. 2008; Leyton et al. 2011). Despite displaying similar specificities, the diagnostic performance of Aβ imaging proved to be more sensitive for the diagnosis of FTLD than FDG (Rabinovici et al. 2011).
8.4 Prion Diseases
No cortical 11C-PiB retention was observed in sporadic Creutzfeldt-Jakob disease (CJD) cases (Villemagne et al. 2009b). Due to the rapid progression and relatively short duration of symptomatic illness, sporadic CJD patients do not demonstrate appreciable PrP-plaque deposition at autopsy (Liberski 2004). Although some sporadic CJD molecular subtypes can manifest larger PrP plaques (Hill et al. 2003), the most common subtype usually demonstrates only very fine ‘synaptic’ and small perivacuolar deposits (Hill et al. 2003; Liberski 2004). A preliminary study in transmissible spongiform encephalopathies showed that Aβ imaging with 11C-BF-227 might be able to distinguish between Gerstmann-Straussler-Scheinker disease (GSS) and sporadic CJD disease with clear tracer retention in brain regions of PrP deposition (Okamura et al. 2010).
9 Aβ Imaging in the Development of Disease-Specific Therapeutics
Aβ imaging with PET is also contributing to the development of more effective therapies by allowing better selection of patients for anti-Aβ therapy trials and providing a means to measure their impact on Aβ burden (Rinne et al. 2010; Ostrowitzki et al. 2012). While these studies represent one of the principal applications of Aβ imaging, they have brought forward an issue that needs to be addressed, namely, that the advent of novel Aβ tracers with differing pharmacological and pharmacokinetics properties, summed to the diverse arsenal of quantitative approaches, and contrasting criteria for the selection of internal scaling region pose a challenge when trying to compare results from therapeutic trials that use different Aβ tracers or quantitative tools.
The contribution of Aβ imaging to the selection of patients for therapeutic trials needs to be complemented with adequate MRI sequences such as SWI or GRE for the detection of vascular pathology to rule out the possibility of vascular complications such as amyloid-related imaging abnormalities (ARIA), either as ‘vasogenic edema’ and/or sulcal effusion (ARIA-E) or as hemosiderin deposits including MH and SS (ARIA-H) (Cordonnier 2010; Sperling et al. 2011c). The presence of MH is impacting on patient selection in therapeutic trials, and some anti-Aβ immunotherapy trials are already excluding individuals with MH. To make matters worse, about one third of non-demented individuals with high Aβ burden and no prior history of cerebrovascular disease showed MH (Yates et al. 2011). While focal Aβ deposition might be a better predictor of a vascular event (Gurol et al. 2012), only longitudinal follow-up of large cohorts of patients will allow elucidation of the relevant risk factors for lobar MH.
10 Pending Issues
There are still some pending issues in regard to Aβ imaging; some are clinically relevant, while others remain in the realm of research settings.
One of the issues is in regard to the utility of a cut-off given the continuous spectrum of Aβ deposition (Jagust 2011; Mormino et al. 2012). In order to adopt a specific threshold, it needs to be shown that this threshold is relevant and accurate from both a diagnostic and prognostic point of view. From a clinical point of view, a visual binary read will help separate those with a significant Aβ burden in the brain from those with a null or low Aβ burden. But some questions arise: What is really a ‘negative’ scan? What is the clinical relevance of focal Aβ deposition? What is the weight to be attributed to this focal Aβ deposition given the known functional and/or structural regional susceptibility such as the one observed in the posterior cingulate (Drzezga et al. 2011) and orbitofrontal cortices (Resnick et al. 2007)? Similar dilemmas arise in research settings. For example, is someone with a 11C-PiB SUVR of 1.44 really ‘negative’ for Aβ because it is below a 1.50 SUVR threshold? In order to avoid being trapped in this dichotomy, some researchers use tertiles (Resnick et al. 2010), while others, like Prof. Klunk, are proposing to distinguish two thresholds for ‘positivity’, one more sensitive to detect significant Aβ deposition usually in still cognitively unimpaired elderly individuals and another higher one that can be more specifically ascribe the clinical phenotype of dementia to Aβ pathology (Klunk et al. 2012). In addition to establishing a clinically relevant threshold, there is a need to change the nomenclature employed to distinguish this binary partition. The terms ‘positive’ and ‘negative’ have denotations, connotations and implications that might be, in the long run, prejudicial for the field. Given the potentially misleading positive connotations of a ‘negative’ scan, it might be more appropriate to use the term ‘low’, while instead of using ‘positive’ it might be more accurate to use ‘high’, the same way other risk factors such as blood pressure or cholesterol are described in clinical settings.
Other issues are related to the choice of reference region. This matter is further complicated by the introduction of novel Aβ radiotracers with physicochemical and pharmacokinetic features somewhat different from 11C-PiB that benefit from different approaches in the sampling and selection of the reference region. For example, studies with 18F-florbetapir tend to use the whole cerebellum (Clark et al. 2011), while using the pons as reference region seems to reduce variability for 18F-flutemetamol (Vandenberghe et al. 2010a). Closely related to the choice and sampling of the reference region are issues referring to the quantitative or semiquantitative approaches used to determine Aβ burden. Given that the studies are performed in individuals or populations who might not tolerate long scanning sessions, the majority of studies use a short 20- or 30-min PET scan at apparent steady state (Lopresti et al. 2005). While all the novel F-18 Aβ tracers have a somewhat fixed time window for optimal scanning (40–50 min postinjection for 18F-florbetapir and 18F-AZD4694 and about 90 min for 18F-florbetaben and 18F-flutemetamol), several intervals (40–60, 40–70, 50–70 or 60–90 min postinjection) have been used with 11C-PiB (Lopresti et al. 2005; Rowe et al. 2007; Yee et al. 2007; Edison et al. 2008b). While these image acquisition intervals are more than enough to obtain clinically useful images, are they adequate to reliably establish Aβ burden in longitudinal studies or therapeutic trials? (Rinne et al. 2010; Ostrowitzki et al. 2012; Ossenkoppele et al. 2012). Is arterial blood sampling required? Is a simple ratio enough, or is a full compartmental analysis needed? (Lopresti et al. 2005; Price et al. 2005). If different Aβ imaging tracers are used and/or different quantification approaches are applied (use of either BP or SUVR, as well as choice of reference region), in between tracer equivalence or standards must be established for the interpretation and comparison of efficacy in therapeutic trials. Finally, the issue of partial volume correction (PVC) raises several other questions: How much of the intrinsic noise of the PET signal is augmented by PVC? Can longitudinal studies be evaluated without PVC (Jack et al. 2009; Villemagne et al. 2011b)? How to correct for spillover radioactivity from white matter? All these issues remain contentious.
Finally, and given that there is no cure for AD, diagnosing the presence or absence of AD pathology in vivo raises ethical issues. While the presence of AD pathology has undoubtedly been associated with a significantly greater risk of cognitive decline, there is still not enough longitudinal data available to attach a firm prognostic clinical significance to a ‘positive’ scan. While this is clearer in subjects classified as MCI, it is still controversial in cognitively unimpaired individuals. At this stage, Aβ imaging, in combination with other modalities, is probably the best diagnostic tool to rule out Aβ pathology in cognitively impaired individuals being evaluated for cognitive decline. The available data clearly show that Aβ production and deposition are not benign processes with a more prominent role at the early, mostly asymptomatic, stages of the disease (Hyman 2011; Karran et al. 2011); therefore, anti-Aβ therapy at these stages seems a logical approach (Sperling et al. 2011b). On the other hand, we still do not have the full grasp of the long-term cognitive consequences of Aβ deposition. Longitudinal Aβ imaging, especially of cognitively unimpaired elderly individuals, is allowing the assessment of pathology almost simultaneously with cognition and other biomarkers of neurodegeneration. This will provide not only a better picture of how these different factors interact but also the weight or relevance this complementary and/or supplementary information plays at different stages of the disease.
11 CODA
The clinical diagnosis of AD is typically based on progressive cognitive impairments while excluding other diseases. Clinical diagnosis of sporadic disease is challenging, however, often presenting mild and nonspecific symptoms attributable to diverse and overlapping pathology presenting similar phenotypes. Overall, the accuracy of clinical diagnosis of AD compared to neuropathological examination ranges between 70 and 90 % (Beach et al. 2012). While clinical criteria, together with current structural neuroimaging techniques (CT or MRI), are sufficiently sensitive and specific for the diagnosis of AD at the mid to late stages of the disease, they are focused on relatively nonspecific features such as memory and functional activity decline or brain atrophy that are late features in the course of the disease. Confirmation of diagnosis of AD still relies on autopsy. But how reliable is pathology? How much of a ‘gold standard’ is autopsy? (Jellinger 2010; Scheltens and Rockwood 2011). A more sensible paradigm, adopting a more fluid model that integrates all cognitive, biochemical and imaging biomarkers that can provide a better predictive framework, has been proposed in the new diagnostic criteria for AD (Dubois et al. 2010; Sperling and Johnson 2010; McKhann et al. 2011), MCI (Albert et al. 2011) and preclinical AD (Sperling et al. 2011a). Along the same lines, Scheltens and Rockwood suggest that ‘a shift in focus from why people with AD have plaques and tangles to why not all people with plaques and tangles have AD would be welcome’ (Scheltens and Rockwood 2011). While most people with AD pathology develop AD dementia, there are some that while having the pathology do not. What makes them different? What conveys resistance to some or, conversely, what makes others more vulnerable to the same pathological burden? Identifying these factors will allow a more discerning selection of individuals that will benefit from early intervention with anti-Aβ medication while also providing insights into potential preventive strategies or even new therapeutic approaches (Sojkova and Resnick 2011; Sperling et al. 2011b). While the new diagnostic criteria have blurred once again the boundaries between MCI and AD (Morris 2012), it also allows for earlier diagnosis and therapeutic intervention, arguing that when there is a clear history of progressive cognitive decline, objective evidence from psychometric tests of episodic memory impairment and characteristic abnormalities in the CSF and/or neuroimaging studies such as Aβ imaging, dementia might not be required for the diagnosis of probable AD (Dubois et al. 2010; Sperling and Johnson 2010; McKhann et al. 2011). Thus, as the criteria for the diagnosis of AD change, Aβ imaging is likely to play an increasingly important role in clinical practice provided it is accessible and affordable (Villemagne and Rowe 2010).
The pathological process usually begins decades before symptoms are evident, making early identification difficult. This in turn precludes early intervention with disease-modifying medications during the presymptomatic period, which by arresting neuronal loss would presumably achieve the maximum benefits of such therapies (Villemagne et al. 2008a). Therefore, a change in the diagnostic paradigm is warranted, where diagnosis moves away from identification of signs and symptoms of neuronal failure—indicating that central compensatory mechanisms have been exhausted and extensive synaptic and neuronal damage is present—to the noninvasive detection of specific biomarkers for particular traits underlying the pathological process (Clark et al. 2008). Given the complexity and sometimes overlapping characteristics of these disorders and despite recent advances in molecular neurosciences, it is unlikely that a single biomarker will be able to provide the diagnostic certainty required for the early detection of neurodegenerative diseases like AD, especially for the identification of at-risk individuals before the development of the typical phenotype. Therefore, a multimodality approach combining biochemical and neuroimaging methods is warranted (Shaw et al. 2007).
Biomarkers are important as they can be used to identify the risk of developing a disease (antecedent biomarkers), aid in identifying disease (diagnostic biomarkers), assess disease evolution (progression biomarkers), predict future disease course (prognostic biomarkers) or evaluate and/or customise therapy (theranostic biomarkers). While CSF Aβ and tau, structural and molecular neuroimaging (both FDG and Aβ imaging) are all good—especially if combined—diagnostic biomarkers for AD, evaluation of the data available to date suggests that Aβ imaging and CSF assessments of Aβ are good antecedent biomarkers at the preclinical and prodromal stages of the disease (Jack et al. 2010; Ossenkoppele et al. 2011; Villemagne et al. 2011b). Conversely, CSF assessments of tau and p-tau, structural neuroimaging and FDG seem to be excellent biomarkers of disease progression (Jack et al. 2010). Despite the fact that Aβ accumulation is still ongoing at the AD stage (Villemagne et al. 2011b; Villain et al. 2012) and in contrast to what is observed at the preclinical and prodromal phases (Pike et al. 2007; Villemagne et al. 2008a; Cohen et al. 2012), Aβ burden at this stage is not correlated with markers of neurodegeneration, disease severity or cognitive impairment (Hyman 2011; Karran et al. 2011). A review of the literature shows that quantification of Aβ1–42, total tau and p-tau in CSF, FDG and a long list of assessments of global or regional brain atrophy as well as white matter hyperintensities with MRI have all been proposed as predictors of conversion to AD (Chang and Silverman 2004; Smith et al. 2008; Jack et al. 2010). While larger longitudinal studies will further clarify the role of Aβ imaging in predicting conversion from MCI to AD, one of the most important conclusions to be drawn from these studies is that the likelihood of developing AD in a cognitively unimpaired individual with a low Aβ burden scan is extremely small (Villemagne et al. 2011b).
Another aspect to be considered is prediction of therapeutic response. As new therapies enter clinical trials, the role of Aβ imaging in vivo is becoming increasingly crucial. Αβ imaging allows the in vivo assessment of brain Αβ pathology and its changes over time, providing highly accurate, reliable and reproducible quantitative statements of regional or global Aβ burden in the brain. As mentioned before, therapies, especially those targeting irreversible neurodegenerative processes, have a better chance to succeed if applied early, making early detection of the underlying pathological process so important. Therefore, Aβ imaging is an ideal tool for the selection of adequate candidates for anti-Aβ therapeutic trials while also monitoring—and potentially predicting—treatment response, thus aiding in reducing sample size, minimising cost while maximising outcomes. Preliminary studies are already showing the utility of this technique (Rinne et al. 2010; Ostrowitzki et al. 2012). These preliminary results, taken together with the results from longitudinal studies, support the growing consensus that anti-Aβ therapy to be effective may need to be given early in the course of the disease, perhaps even before symptoms appear (Sperling et al. 2011b), and that downstream mechanisms may also need to be addressed to successfully prevent the development of AD. Furthermore, the slow rate of Aβ deposition indicates that the time window for altering Aβ accumulation prior to the full manifestation of the clinical phenotype may be very wide (Villemagne et al. 2011b).
The introduction of radiotracers for the noninvasive in vivo quantification of Αβ burden in the brain has revolutionised the approach to the evaluation of AD. Αβ imaging has extended our insight into Aβ deposition in the brain, where Aβ burden as measured by PET matches histopathological reports of Αβ distribution in aging and dementia, and it appears more accurate than FDG for the diagnosis of AD and is an excellent aid in the differential diagnosis of AD from FTLD (Mathis et al. 2005, 2007; Johnson 2006; Villemagne et al. 2008a, 2012b; Jagust 2009; Rabinovici and Jagust 2009; Drzezga 2010; Klunk 2011). ApoE ε4 carriers, independent of diagnosis or disease severity, present with higher Aβ burden than non-ε4 carriers, and high retention of Aβ radiotracers closely corresponds with low CSF Aβ levels and increasing age (Reiman et al. 2009a; Fagan and Holtzman 2010; Morris et al. 2010; Rowe et al. 2010). Although Αβ burden as assessed by PET does not strongly correlate with cognitive impairment in AD, it does correlate with memory impairment and a higher risk for cognitive decline in the aging population and MCI subjects. This correlation with memory impairment, one of the earliest symptoms of AD, suggests that Αβ deposition is not part of normal aging, supporting the hypothesis that Αβ deposition occurs well before the onset of symptoms and it likely represents preclinical AD in asymptomatic individuals and prodromal AD in MCI. Further longitudinal observations, coupled with different disease-specific biomarkers to assess potential downstream effects of Aβ, are required to confirm this hypothesis and further elucidate the role of Αβ deposition in the course of Alzheimer’s disease.
References
Agdeppa ED, Kepe V, Liu J et al (2001a) Binding characteristics of radiofluorinated 6-dialkylamino-2-naphthylethylidene derivatives as positron emission tomography imaging probes for beta-amyloid plaques in Alzheimer’s disease. J Neurosci 21:RC189
Agdeppa ED, Kepe V, Shoghi-Jadid K et al (2001b) In vivo and in vitro labeling of plaques and tangles in the brain of an Alzheimer’s disease patient: a case study. J Nucl Med 42:65P
Aizenstein HJ, Nebes RD, Saxton JA et al (2008) Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol 65:1509–1517
Albert MS, Dekosky ST, Dickson D et al (2011) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:270–279
Apostolova LG, Hwang KS, Andrawis JP et al (2010) 3D PIB and CSF biomarker associations with hippocampal atrophy in ADNI subjects. Neurobiol Aging 31:1284–1303
Archer HA, Edison P, Brooks DJ et al (2006) Amyloid load and cerebral atrophy in Alzheimer’s disease: an 11C-PIB positron emission tomography study. Ann Neurol 60:145–147
Armstrong RA, Cairns NJ, Lantos PL (2000) Beta-amyloid deposition in the temporal lobe of patients with dementia with Lewy bodies: comparison with non-demented cases and Alzheimer’s disease. Dement Geriatr Cogn Disord 11:187–192
Arnold SE, Han LY, Clark CM et al (2000) Quantitative neurohistological features of frontotemporal degeneration. Neurobiol Aging 21:913–919
Backman L, Jones S, Berger AK et al (2005) Cognitive impairment in preclinical Alzheimer’s disease: a meta-analysis. Neuropsychology 19:520–531
Bacskai BJ, Frosch MP, Freeman SH et al (2007) Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Arch Neurol 64:431–434
Barrio JR, Huang SC, Cole G et al (1999) PET imaging of tangles and plaques in Alzheimer disease with a highly hydrophobic probe. J Label Compd Radiopharm 42:S194–S195
Barthel H, Gertz HJ, Dresel S et al (2011) Cerebral amyloid-beta PET with florbetaben ((18)F) in patients with Alzheimer’s disease and healthy controls: a multicentre phase 2 diagnostic study. Lancet Neurol 10:424–435
Basun H, Bogdanovic N, Ingelsson M et al (2008) Clinical and neuropathological features of the arctic APP gene mutation causing early-onset alzheimer disease. Arch Neurol 65:499–505
Bateman RJ, Munsell LY, Morris JC et al (2006) Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med 12:856–861
Bateman RJ, Xiong C, Benzinger TL et al (2012) Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 367:795–804
Bauer M, Langer O, Dal-Bianco P et al (2006) A positron emission tomography microdosing study with a potential antiamyloid drug in healthy volunteers and patients with Alzheimer’s disease. Clin Pharmacol Ther 80:216–227
Beach TG, Monsell SE, Phillips LE et al (2012) Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J Neuropathol Exp Neurol 71:266–273
Becker JA, Hedden T, Carmasin J et al (2011) Amyloid-beta associated cortical thinning in clinically normal elderly. Ann Neurol 69:1032–1042
Berg L, McKeel DW Jr, Miller JP et al (1998) Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol 55:326–335
Blennow K, Zetterberg H, Minthon L et al (2007) Longitudinal stability of CSF biomarkers in Alzheimer’s disease. Neurosci Lett 419:18–22
Bourgeat P, Chetelat G, Villemagne VL et al (2010) Beta-amyloid burden in the temporal neocortex is related to hippocampal atrophy in elderly subjects without dementia. Neurology 74:121–127
Braak H, Braak E (1997) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18:351–357
Bresjanac M, Smid LM, Vovko TD et al (2003) Molecular-imaging probe 2-(1-[6-[(2-fluoroethyl)(methyl) amino]-2-naphthyl]ethylidene) malononitrile labels prion plaques in vitro. J Neurosci 23:8029–8033
Buckley C, Ikonomovic M, Smith A et al (2012) Flutemetamol F 18 injection PET images reflect brain beta-amyloid levels. Alzheimers Dement 8:P90
Buckner RL, Snyder AZ, Shannon BJ et al (2005) Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci 25:7709–7717
Buckner RL, Andrews-Hanna JR, Schacter DL (2008) The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci 1124:1–38
Burack MA, Hartlein J, Flores HP et al (2010) In vivo amyloid imaging in autopsy-confirmed Parkinson disease with dementia. Neurology 74:77–84
Cairns NJ, Bigio EH, Mackenzie IR et al (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the consortium for frontotemporal lobar degeneration. Acta Neuropathol 114:5–22
Cairns NJ, Ikonomovic MD, Benzinger T et al (2009) Absence of Pittsburgh Compound B detection of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: a case report. Arch Neurol 66:1557–1562
Camus V, Payoux P, Barre L et al (2012) Using PET with (18)F-AV-45 (florbetapir) to quantify brain amyloid load in a clinical environment. Eur J Nucl Med Mol Imaging 39:621–631
Cappai R, White AR (1999) Amyloid beta. Int J Biochem Cell Biol 31:885–889
Carter SF, Scholl M, Almkvist O et al (2012) Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh Compound B and 18F-FDG. J Nucl Med 53:37–46
Chang CY, Silverman DH (2004) Accuracy of early diagnosis and its impact on the management and course of Alzheimer’s disease. Expert Rev Mol Diagn 4:63–69
Chetelat G, Desgranges B, de la Sayette V et al (2003) Dissociating atrophy and hypometabolism impact on episodic memory in mild cognitive impairment. Brain 126:1955–1967
Chetelat G, Eustache F, Viader F et al (2005) FDG-PET measurement is more accurate than neuropsychological assessments to predict global cognitive deterioration in patients with mild cognitive impairment. Neurocase 11:14–25
Chetelat G, Villemagne VL, Bourgeat P et al (2010a) Relationship between atrophy and beta-amyloid deposition in Alzheimer disease. Ann Neurol 67:317–324
Chetelat G, Villemagne VL, Pike KE et al (2010b) Larger temporal volume in elderly with high versus low beta-amyloid deposition. Brain 133:3349–3358
Chetelat G, Villemagne VL, Villain N et al (2012) Accelerated cortical atrophy in cognitively normal elderly with high beta-amyloid deposition. Neurology 78:477–484
Choi SR, Golding G, Zhuang Z et al (2009) Preclinical properties of 18F-AV-45: a PET agent for Abeta plaques in the brain. J Nucl Med 50:1887–1894
Clark CM, Davatzikos C, Borthakur A et al (2008) Biomarkers for early detection of Alzheimer pathology. Neurosignals 16:11–18
Clark CM, Schneider JA, Bedell BJ et al (2011) Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA 305:275–283
Clark CM, Pontecorvo MJ, Beach TG et al (2012) Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-beta plaques: a prospective cohort study. Lancet Neurol 11:669–678
Cohen AD, Price JC, Weissfeld LA et al (2009) Basal cerebral metabolism may modulate the cognitive effects of Abeta in mild cognitive impairment: an example of brain reserve. J Neurosci 29:14770–14778
Cohen AD, Rabinovici GD, Mathis CA et al (2012) Using Pittsburgh Compound B for in vivo PET imaging of fibrillar amyloid-beta. Adv Pharmacol 64:27–81
Coleman RE (2005) Positron emission tomography diagnosis of Alzheimer’s disease. Neuroimaging Clin N Am 15:837–846, x
Cordonnier C (2010) Brain microbleeds. Pract Neurol 10:94–100
Cselenyi Z, Jonhagen ME, Forsberg A et al (2012) Clinical validation of 18F-AZD4694, an amyloid-beta-specific PET radioligand. J Nucl Med 53:415–424
Cummings JL, Vinters HV, Cole GM et al (1998) Alzheimer’s disease: etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology 51:S2–S17; discussion S65–S17
Davies L, Wolska B, Hilbich C et al (1988) A4 amyloid protein deposition and the diagnosis of Alzheimer’s disease: prevalence in aged brains determined by immunocytochemistry compared with conventional neuropathologic techniques. Neurology 38:1688–1693
de Leon MJ, DeSanti S, Zinkowski R et al (2006) Longitudinal CSF and MRI biomarkers improve the diagnosis of mild cognitive impairment. Neurobiol Aging 27:394–401
de Leon MJ, Mosconi L, Blennow K et al (2007) Imaging and CSF studies in the preclinical diagnosis of Alzheimer’s disease. Ann N Y Acad Sci 1097:114–145
DeKosky ST, Ikonomovic MD, Styren SD et al (2002) Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol 51:145–155
Deshpande A, Mina E, Glabe C et al (2006) Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 26:6011–6018
Devanand DP, Jacobs DM, Tang MX et al (1997) The course of psychopathologic features in mild to moderate Alzheimer disease. Arch Gen Psychiatry 54:257–263
Dhollander I, Nelissen N, Van Laere K et al (2011) In vivo amyloid imaging in cortical superficial siderosis. J Neurol Neurosurg Psychiatry 82:469–471
Dierksen GA, Skehan ME, Khan MA et al (2010) Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Ann Neurol 68:545–548
Doraiswamy PM, Sperling RA, Coleman RE et al (2012) Amyloid-beta assessed by florbetapir F 18 PET and 18-month cognitive decline: a multicenter study. Neurology 79:1636–1644
Drago V, Babiloni C, Bartres-Faz D et al (2011) Disease tracking markers for Alzheimer’s disease at the prodromal (MCI) stage. J Alzheimers Dis 26(Suppl 3):159–199
Driscoll I, Zhou Y, An Y et al (2011) Lack of association between (11)C-PiB and longitudinal brain atrophy in non-demented older individuals. Neurobiol Aging 32:2123–2130
Drzezga A (2010) Amyloid-plaque imaging in early and differential diagnosis of dementia. Ann Nucl Med 24:55–66
Drzezga A, Lautenschlager N, Siebner H et al (2003) Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer’s disease: a PET follow-up study. Eur J Nucl Med Mol Imaging 30:1104–1113
Drzezga A, Grimmer T, Riemenschneider M et al (2005) Prediction of individual clinical outcome in MCI by means of genetic assessment and (18)F-FDG PET. J Nucl Med 46:1625–1632
Drzezga A, Grimmer T, Henriksen G et al (2008) Imaging of amyloid plaques and cerebral glucose metabolism in semantic dementia and Alzheimer’s disease. Neuroimage 39:619–633
Drzezga A, Becker JA, Van Dijk KR et al (2011) Neuronal dysfunction and disconnection of cortical hubs in non-demented subjects with elevated amyloid burden. Brain 134:1635–1646
Dubois B, Feldman HH, Jacova C et al (2010) Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 9:1118–1127
Eckert A, Keil U, Marques CA et al (2003) Mitochondrial dysfunction, apoptotic cell death, and Alzheimer’s disease. Biochem Pharmacol 66:1627–1634
Edison P, Archer HA, Hinz R et al (2007) Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology 68:501–508
Edison P, Archer HA, Gerhard A et al (2008a) Microglia, amyloid, and cognition in Alzheimer’s disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis 32:412–419
Edison P, Rowe CC, Rinne JO et al (2008b) Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry 79:1331–1338
Engler H, Forsberg A, Almkvist O et al (2006) Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain 129:2856–2866
Engler H, Santillo AF, Wang SX et al (2008) In vivo amyloid imaging with PET in frontotemporal dementia. Eur J Nucl Med Mol Imaging 35:100–106
Fagan AM, Holtzman DM (2010) Cerebrospinal fluid biomarkers of Alzheimer’s disease. Biomark Med 4:51–63
Fagan AM, Mintun MA, Mach RH et al (2006) Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta(42) in humans. Ann Neurol 59:512–519
Fagan AM, Roe CM, Xiong C et al (2007) Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 64:343–349
Fagan AM, Mintun MA, Shah AR et al (2009) Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer’s disease. EMBO Mol Med 1:371–380
Farrer LA, Cupples LA, Haines JL et al (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease meta analysis consortium. JAMA 278:1349–1356
Feldman HH, Maia LF, Mackenzie IR et al (2008) Superficial siderosis: a potential diagnostic marker of cerebral amyloid angiopathy in Alzheimer disease. Stroke 39:2894–2897
Fleisher AS, Chen K, Liu X et al (2011) Using positron emission tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Arch Neurol 68:1404–1411
Fodero-Tavoletti MT, Smith DP, McLean CA et al (2007) In vitro characterization of Pittsburgh Compound-B binding to Lewy bodies. J Neurosci 27:10365–10371
Fodero-Tavoletti MT, Mulligan RS, Okamura N et al (2009a) In vitro characterisation of BF227 binding to alpha-synuclein/Lewy bodies. Eur J Pharmacol 617:54–58
Fodero-Tavoletti MT, Rowe CC, McLean CA et al (2009b) Characterization of PiB binding to white matter in Alzheimer disease and other dementias. J Nucl Med 50:198–204
Fodero-Tavoletti MT, Okamura N, Furumoto S et al (2011) 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer’s disease. Brain 134:1089–1100
Fodero-Tavoletti MT, Brockschnieder D, Villemagne VL et al (2012) In vitro characterisation of [18F]–florbetaben, an Aβ imaging radiotracer. Nucl Med Biol 39:1042–1048. doi:10.1016/j.nucmedbio.2012.1003.1001
Formaglio M, Costes N, Seguin J et al (2011) In vivo demonstration of amyloid burden in posterior cortical atrophy: a case series with PET and CSF findings. J Neurol 258:1841–1851
Forman MS, Mufson EJ, Leurgans S et al (2007) Cortical biochemistry in MCI and Alzheimer disease: lack of correlation with clinical diagnosis. Neurology 68:757–763
Forsberg A, Engler H, Almkvist O et al (2008) PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging 29:1456–1465
Forsberg A, Almkvist O, Engler H et al (2010) High PIB retention in Alzheimer’s disease is an early event with complex relationship with CSF biomarkers and functional parameters. Curr Alzheimer Res 7:56–66
Forster S, Grimmer T, Miederer I et al (2012) Regional expansion of hypometabolism in Alzheimer’s disease follows amyloid deposition with temporal delay. Biol Psychiatry 71:792–797
Foster ER, Campbell MC, Burack MA et al (2010) Amyloid imaging of Lewy body-associated disorders. Mov Disord 25:2516–2523
Frisoni GB, Fox NC, Jack CR Jr et al (2010) The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol 6:67–77
Furst AJ, Rabinovici GD, Rostomian AH et al (2012) Cognition, glucose metabolism and amyloid burden in Alzheimer’s disease. Neurobiol Aging 33(2):215–225
Furukawa K, Okamura N, Tashiro K et al (2011) PET imaging with BF-227 in dementia with Lewy bodies. Alzheimers Dement 7:S12–S13
Gomez-Isla T, West HL, Rebeck GW et al (1996) Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer’s disease. Ann Neurol 39:62–70
Gomperts SN, Rentz DM, Moran E et al (2008) Imaging amyloid deposition in Lewy body diseases. Neurology 71:903–910
Grimmer T, Riemenschneider M, Forstl H et al (2009) Beta amyloid in Alzheimer’s disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol Psychiatry 65:927–934
Grimmer T, Tholen S, Yousefi BH et al (2010) Progression of cerebral amyloid load is associated with the apolipoprotein E epsilon4 genotype in Alzheimer’s disease. Biol Psychiatry 68:879–884
Gurol ME, Dierksen G, Betensky R et al (2012) Predicting sites of new hemorrhage with amyloid imaging in cerebral amyloid angiopathy. Neurology 79:320–326
Halliday G, Hely M, Reid W et al (2008) The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol 115:409–415
Hampel H, Blennow K, Shaw LM et al (2010) Total and phosphorylated tau protein as biological markers of Alzheimer’s disease. Exp Gerontol 45:30–40
Hansson O, Zetterberg H, Buchhave P et al (2007) Prediction of Alzheimer’s disease using the CSF Abeta42/Abeta40 ratio in patients with mild cognitive impairment. Dement Geriatr Cogn Disord 23:316–320
Hardy J (1997) Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci 20:154–159
Herholz K (1995) FDG PET and differential diagnosis of dementia. Alzheimer Dis Assoc Disord 9:6–16
Hill AF, Joiner S, Wadsworth JD et al (2003) Molecular classification of sporadic Creutzfeldt-Jakob disease. Brain 126:1333–1346
Hodges JR, Patterson K (2007) Semantic dementia: a unique clinicopathological syndrome. Lancet Neurol 6:1004–1014
Hyman BT (2011) Amyloid-dependent and amyloid-independent stages of Alzheimer disease. Arch Neurol 68:1062–1064
Ikonomovic MD, Abrahamson EE, Isanski BA et al (2007) Superior frontal cortex cholinergic axon density in mild cognitive impairment and early Alzheimer disease. Arch Neurol 64:1312–1317
Ikonomovic MD, Klunk WE, Abrahamson EE et al (2008) Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 131:1630–1645
Ikonomovic MD, Klunk WE, Abrahamson EE et al (2011) Precuneus amyloid burden is associated with reduced cholinergic activity in Alzheimer disease. Neurology 77:39–47
Ikonomovic MD, Abrahamson EE, Price JC et al (2012) Early AD pathology in a [C-11]PiB-negative case: a PiB-amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathol 123:433–447
Inayathullah M, Teplow DB (2011) Structural dynamics of the DeltaE22 (Osaka) familial Alzheimer’s disease-linked amyloid beta-protein. Amyloid 18:98–107
Isacson O, Seo H, Lin L et al (2002) Alzheimer’s disease and Down’s syndrome: roles of APP, trophic factors and ACh. Trends Neurosci 25:79–84
Jack CR Jr, Lowe VJ, Senjem ML et al (2008a) 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain 131:665–680
Jack CR Jr, Weigand SD, Shiung MM et al (2008b) Atrophy rates accelerate in amnestic mild cognitive impairment. Neurology 70:1740–1752
Jack CR Jr, Lowe VJ, Weigand SD et al (2009) Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain 132:1355–1365
Jack CR Jr, Knopman DS, Jagust WJ et al (2010) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9:119–128
Jagust W (2009) Mapping brain beta-amyloid. Curr Opin Neurol 22:356–361
Jagust WJ (2011) Amyloid imaging: liberal or conservative? Let the data decide. Arch Neurol 68:1377–1378
Jagust W, Reed B, Mungas D et al (2007) What does fluorodeoxyglucose PET imaging add to a clinical diagnosis of dementia? Neurology 69:871–877
Jellinger K (1990) Morphology of Alzheimer disease and related disorders. In: Maurer K et al (eds) Alzheimer disease: epidemiology, neuropathology, neurochemistry, and clinics. Springer, Berlin, pp 61–77
Jellinger KA (2010) Con: can neuropathology really confirm the exact diagnosis? Alzheimers Res Ther 2:11
Jellinger KA, Attems J (2005) Prevalence and pathogenic role of cerebrovascular lesions in Alzheimer disease. J Neurol Sci 229–230:37–41
Jellinger KA, Bancher C (1998) Neuropathology of Alzheimer’s disease: a critical update. J Neural Transm Suppl 54:77–95
Johansson A, Savitcheva I, Forsberg A et al (2008) [(11)C]-PIB imaging in patients with Parkinson’s disease: preliminary results. Parkinsonism Relat Disord 14:345–347
Johnson KA (2006) Amyloid imaging of Alzheimer’s disease using Pittsburgh Compound B. Curr Neurol Neurosci Rep 6:496–503
Johnson N, Davis T, Bosanquet N (2000) The epidemic of Alzheimer’s disease. How can we manage the costs? Pharmacoeconomics 18:215–223
Johnson KA, Gregas M, Becker JA et al (2007) Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol 62:229–234
Johnson AE, Jeppsson F, Sandell J et al (2009) AZD2184: a radioligand for sensitive detection of beta-amyloid deposits. J Neurochem 108:1177–1186
Josephs KA, Holton JL, Rossor MN et al (2004) Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol Appl Neurobiol 30:369–373
Jureus A, Swahn BM, Sandell J et al (2010) Characterization of AZD4694, a novel fluorinated Abeta plaque neuroimaging PET radioligand. J Neurochem 114:784–794
Kadir A, Marutle A, Gonzalez D et al (2011) Positron emission tomography imaging and clinical progression in relation to molecular pathology in the first Pittsburgh Compound B positron emission tomography patient with Alzheimer’s disease. Brain 134:301–317
Kalaitzakis ME, Walls AJ, Pearce RK et al (2011) Striatal Abeta peptide deposition mirrors dementia and differentiates DLB and PDD from other parkinsonian syndromes. Neurobiol Dis 41:377–384
Kambe T, Motoi Y, Ishii K et al (2010) Posterior cortical atrophy with [11C] Pittsburgh Compound B accumulation in the primary visual cortex. J Neurol 257:469–471
Karran E, Mercken M, De Strooper B (2011) The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10:698–712
Kemppainen NM, Aalto S, Wilson IA et al (2007) PET amyloid ligand [11C]PIB uptake is increased in mild cognitive impairment. Neurology 68:1603–1606
Kemppainen NM, Aalto S, Karrasch M et al (2008) Cognitive reserve hypothesis: Pittsburgh Compound B and fluorodeoxyglucose positron emission tomography in relation to education in mild Alzheimer’s disease. Ann Neurol 63:112–118
Kennedy AM, Frackowiak RS, Newman SK et al (1995) Deficits in cerebral glucose metabolism demonstrated by positron emission tomography in individuals at risk of familial Alzheimer’s disease. Neurosci Lett 186:17–20
Khachaturian ZS (1985) Diagnosis of Alzheimer’s disease. Arch Neurol 42:1097–1105
Kikuchi A, Takeda A, Okamura N et al (2010) In vivo visualization of alpha-synuclein deposition by carbon-11-labelled 2-[2-(2-dimethylaminothiazol-5-yl)ethenyl]-6-[2-(fluoro)ethoxy]benzoxazole positron emission tomography in multiple system atrophy. Brain 133:1772–1778
Klunk WE (2011) Amyloid imaging as a biomarker for cerebral beta-amyloidosis and risk prediction for Alzheimer dementia. Neurobiol Aging 32(Suppl 1):S20–S36
Klunk WE, Wang Y, Huang GF et al (2001) Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life Sci 69:1471–1484
Klunk WE, Wang Y, Huang GF et al (2003) The binding of 2-(4'-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci 23:2086–2092
Klunk WE, Engler H, Nordberg A et al (2004) Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol 55:306–319
Klunk WE, Lopresti BJ, Ikonomovic MD et al (2005) Binding of the positron emission tomography tracer Pittsburgh Compound-B reflects the amount of amyloid-beta in Alzheimer’s disease brain but not in transgenic mouse brain. J Neurosci 25:10598–10606
Klunk WE, Price JC, Mathis CA et al (2007) Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci 27:6174–6184
Klunk W, Cohen AD, Bi W et al (2012) Why we need two cutoffs for amyloid imaging: early versus Alzheimer’s-like amyloid-positivity. Alzheimers Dement 8:P453–P454
Knudsen KA, Rosand J, Karluk D et al (2001) Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 56:537–539
Koeppe RA, Gilman S, Junck L et al (2008) Differentiating Alzheimer’s disease from dementia with Lewy bodies and Parkinson’s disease with (+)-[11C]dihydrotetrabenazine positron emission tomography. Alzheimers Dement 4:S67–S76
Koivunen J, Pirttila T, Kemppainen N et al (2008a) PET amyloid ligand [C]PIB uptake and cerebrospinal fluid beta-amyloid in mild cognitive impairment. Dement Geriatr Cogn Disord 26:378–383
Koivunen J, Verkkoniemi A, Aalto S et al (2008b) PET amyloid ligand [11C]PIB uptake shows predominantly striatal increase in variant Alzheimer’s disease. Brain 131:1845–1853
Kuczynski B, Reed B, Mungas D et al (2008) Cognitive and anatomic contributions of metabolic decline in Alzheimer disease and cerebrovascular disease. Arch Neurol 65:650–655
Kudo Y (2006) Development of amyloid imaging PET probes for an early diagnosis of Alzheimer’s disease. Minim Invasive Ther Allied Technol 15:209–213
Kudo Y, Okamura N, Furumoto S et al (2007) 2-(2-[2-Dimethylaminothiazol-5-yl]Ethenyl)-6- (2-[Fluoro]Ethoxy)Benzoxazole: a novel PET agent for in vivo detection of dense amyloid plaques in Alzheimer’s disease patients. J Nucl Med 48:553–561
Landau SM, Harvey D, Madison CM et al (2011) Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging 32(7):1207–1218
Landt J, D’Abrera JC, Holland AJ et al (2011) Using positron emission tomography and carbon 11-labeled Pittsburgh Compound B to image brain fibrillar beta-amyloid in adults with Down syndrome: safety, acceptability, and feasibility. Arch Neurol 68:890–896
Larson EB, Edwards JK, O’Meara E et al (1996) Neuropathologic diagnostic outcomes from a cohort of outpatients with suspected dementia. J Gerontol A Biol Sci Med Sci 51(suppl 6):M313–M318
Laruelle M, Slifstein M, Huang Y (2003) Relationships between radiotracer properties and image quality in molecular imaging of the brain with positron emission tomography. Mol Imaging Biol 5:363–375
Leinonen V, Alafuzoff I, Aalto S et al (2008) Assessment of beta-amyloid in a frontal cortical brain biopsy specimen and by positron emission tomography with carbon 11-labeled Pittsburgh Compound B. Arch Neurol 65:1304–1309
Leuner K, Hauptmann S, Abdel-Kader R et al (2007) Mitochondrial dysfunction: the first domino in brain aging and Alzheimer’s disease? Antioxid Redox Signal 9:1659–1675
LeVine H 3rd (1999) Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol 309:274–284
Levine H 3rd, Walker LC (2010a) Molecular polymorphism of Abeta in Alzheimer’s disease. Neurobiol Aging 31(4):542–548
Levine H 3rd, Walker LC (2010b) Molecular polymorphism of Abeta in Alzheimer’s disease. Neurobiol Aging 31:542–548
Leyton CE, Villemagne VL, Savage S et al (2011) Subtypes of progressive aphasia: application of the international consensus criteria and validation using {beta}-amyloid imaging. Brain 134:3030–3043
Liberski PP (2004) Amyloid plaques in transmissible spongiform encephalopathies (prion diseases). Folia Neuropathol 42(Suppl B):109–119
Lin KS, Debnath ML, Mathis CA et al (2009) Synthesis and beta-amyloid binding properties of rhenium 2-phenylbenzothiazoles. Bioorg Med Chem Lett 19:2258–2262
Lister-James J, Pontecorvo MJ, Clark C et al (2011) Florbetapir f-18: a histopathologically validated beta-amyloid positron emission tomography imaging agent. Semin Nucl Med 41:300–304
Lockhart A, Ye L, Judd DB et al (2005) Evidence for the presence of three distinct binding sites for the thioflavin T class of Alzheimer’s disease PET imaging agents on beta-amyloid peptide fibrils. J Biol Chem 280:7677–7684
Lockhart A, Lamb JR, Osredkar T et al (2007) PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain 130:2607–2615
Lopresti BJ, Klunk WE, Mathis CA et al (2005) Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med 46:1959–1972
Lowe VJ, Kemp BJ, Jack CR Jr et al (2009) Comparison of 18F-FDG and PiB PET in cognitive impairment. J Nucl Med 50:878–886
Mackenzie IR, Foti D, Woulfe J et al (2008) Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain 131:1282–1293
Maeda J, Ji B, Irie T et al (2007) Longitudinal, quantitative assessment of amyloid, neuroinflammation, and anti-amyloid treatment in a living mouse model of Alzheimer’s disease enabled by positron emission tomography. J Neurosci 27:10957–10968
Maetzler W, Reimold M, Liepelt I et al (2008) [11C]PIB binding in Parkinson’s disease dementia. Neuroimage 39:1027–1033
Maetzler W, Liepelt I, Reimold M et al (2009) Cortical PIB binding in Lewy body disease is associated with Alzheimer-like characteristics. Neurobiol Dis 34:107–112
Maezawa I, Hong HS, Liu R et al (2008) Congo red and thioflavin-T analogs detect Abeta oligomers. J Neurochem 104:457–468
Majocha RE, Reno JM, Friedland RP et al (1992) Development of a monoclonal antibody specific for ß/A4 amyloid in Alzheimer’s disease brain for application to in vivo imaging of amyloid angiopathy. J Nucl Med 33:2184–2189
Marchant NL, Reed BR, Decarli CS et al (2012) Cerebrovascular disease, beta-amyloid, and cognition in aging. Neurobiol Aging 33(5):1006.e25–36
Maruyama M, Maeda J, Ji B et al (2009) In-vivo optical and PET detections of fibrillar tau lesions in a mouse model of tauopathies. Alzheimers Dement 55:P209–P210 [abstract]
Masliah E, Rockenstein E, Veinbergs I et al (2001) Beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci U S A 98:12245–12250
Masters CL (2005) Neuropathology of Alzheimer’s disease. In: Burns A et al (eds) Dementia, 3rd edn. Hodder Arnold, London, pp 393–407
Masters CL, Beyreuther K (2005) The neuropathology of Alzheimer’s disease in the year 2005. In: Beal MF et al (eds) Neurodegenerative diseases: neurobiology, pathogenesis and therapeutics. Cambridge University Press, Cambridge, pp 433–440
Masters CL, Beyreuther K (2006) Alzheimer’s centennial legacy: prospects for rational therapeutic intervention targeting the Abeta amyloid pathway. Brain 129:2823–2839
Masters CL, Simms G, Weinman NA et al (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A 82:4245–4249
Masters CL, Cappai R, Barnham KJ et al (2006) Molecular mechanisms for Alzheimer’s disease: implications for neuroimaging and therapeutics. J Neurochem 97:1700–1725
Mathis CA, Bacskai BJ, Kajdasz ST et al (2002) A lipophilic thioflavin-T derivative for positron emission tomography (PET) imaging of amyloid in brain. Bioorg Med Chem Lett 12:295–298
Mathis CA, Klunk WE, Price JC et al (2005) Imaging technology for neurodegenerative diseases: progress toward detection of specific pathologies. Arch Neurol 62:196–200
Mathis CA, Lopresti BJ, Klunk WE (2007) Impact of amyloid imaging on drug development in Alzheimer’s disease. Nucl Med Biol 34:809–822
Mattsson N, Zetterberg H, Hansson O et al (2009) CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 302:385–393
Mattsson N, Andreasson U, Persson S et al (2011) The Alzheimer’s Association external quality control program for cerebrospinal fluid biomarkers. Alzheimers Dement 7:386–395, e386
Mawuenyega KG, Sigurdson W, Ovod V et al (2010) Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330:1774
McKeith IG, Mosimann UP (2004) Dementia with Lewy bodies and Parkinson’s disease. Parkinsonism Relat Disord 10(Suppl 1):S15–S18
McKeith IG, Dickson DW, Lowe J et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872
McKhann GM, Knopman DS, Chertkow H et al (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:263–269
McLean CA, Cherny RA, Fraser FW et al (1999) Soluble pool of Aß amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol 46:860–866
Michaelis ML, Dobrowsky RT, Li G (2002) Tau neurofibrillary pathology and microtubule stability. J Mol Neurosci 19:289–293
Mielke R, Herholz K, Grond M et al (1992) Differences of regional cerebral glucose metabolism between presenile and senile dementia of Alzheimer type. Neurobiol Aging 13:93–98
Mintun MA, Larossa GN, Sheline YI et al (2006) [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology 67:446–452
Mirra SS, Heyman A, McKeel D et al (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41:479–486
Montine TJ, Phelps CH, Beach TG et al (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123:1–11
Mormino EC, Kluth JT, Madison CM et al (2009) Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain 132:1310–1323
Mormino EC, Brandel MG, Madison CM et al (2012) Not quite PIB-positive, not quite PIB-negative: slight PIB elevations in elderly normal control subjects are biologically relevant. Neuroimage 59:1152–1160
Morris JC (2012) Revised criteria for mild cognitive impairment may compromise the diagnosis of Alzheimer disease dementia. Arch Neurol 69:700–708. doi:10.1001/archneurol.2011.3152
Morris JC, Price AL (2001) Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Mol Neurosci 17:101–118
Morris JC, Kimberly A, Quaid K et al (2005) Role of biomarkers in studies of presymptomatic Alzheimer’s disease. Alzheimers Dement 1:145–151
Morris JC, Roe CM, Grant EA et al (2009) Pittsburgh Compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol 66:1469–1475
Morris JC, Roe CM, Xiong C et al (2010) APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 67:122–131
Mortimer JA (1997) Brain reserve and the clinical expression of Alzheimer’s disease. Geriatrics 52(Suppl 2):S50–S53
Mosconi L, Perani D, Sorbi S et al (2004) MCI conversion to dementia and the APOE genotype: a prediction study with FDG-PET. Neurology 63:2332–2340
Mosconi L, De Santi S, Li Y et al (2006a) Visual rating of medial temporal lobe metabolism in mild cognitive impairment and Alzheimer’s disease using FDG-PET. Eur J Nucl Med Mol Imaging 33:210–221
Mosconi L, Sorbi S, de Leon MJ et al (2006b) Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. J Nucl Med 47:1778–1786
Mott RT, Dickson DW, Trojanowski JQ et al (2005) Neuropathologic, biochemical, and molecular characterization of the frontotemporal dementias. J Neuropathol Exp Neurol 64:420–428
Naslund J, Haroutunian V, Mohs R et al (2000) Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 283:1571–1577
Nelissen N, Van Laere K, Thurfjell L et al (2009) Phase 1 study of the Pittsburgh Compound B derivative 18F-flutemetamol in healthy volunteers and patients with probable Alzheimer disease. J Nucl Med 50:1251–1259
Neumann M, Sampathu DM, Kwong LK et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Neumann M, Rademakers R, Roeber S et al (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132:2922–2931
Newberg AB, Wintering NA, Plossl K et al (2006) Safety, biodistribution, and dosimetry of 123I-IMPY: a novel amyloid plaque-imaging agent for the diagnosis of Alzheimer’s disease. J Nucl Med 47:748–754
Ng S, Villemagne VL, Berlangieri S et al (2007a) Visual assessment versus quantitative assessment of 11C-PIB PET and 18F-FDG PET for detection of Alzheimer’s disease. J Nucl Med 48:547–552
Ng SY, Villemagne VL, Masters CL et al (2007b) Evaluating atypical dementia syndromes using positron emission tomography with carbon 11 labeled Pittsburgh Compound B. Arch Neurol 64:1140–1144
Noda A, Murakami Y, Nishiyama S et al (2008) Amyloid imaging in aged and young macaques with [11C]PIB and [18F]FDDNP. Synapse 62:472–475
Nordberg A (2004) PET imaging of amyloid in Alzheimer’s disease. Lancet Neurol 3:519–527
Nordberg A (2007) Amyloid imaging in Alzheimer’s disease. Curr Opin Neurol 20:398–402
Nyberg S, Jonhagen ME, Cselenyi Z et al (2009) Detection of amyloid in Alzheimer’s disease with positron emission tomography using [11C]AZD2184. Eur J Nucl Med Mol Imaging 36:1859–1863
O’Brien J, Ames D, Burns A (2000) Dementia, 2nd edn. Arnold, London
O’Brien JT, Colloby S, Fenwick J et al (2004) Dopamine transporter loss visualized with FP-CIT SPECT in the differential diagnosis of dementia with Lewy bodies. Arch Neurol 61:919–925
Ojida A, Sakamoto T, Inoue MA et al (2009) Fluorescent BODIPY-based Zn(II) complex as a molecular probe for selective detection of neurofibrillary tangles in the brains of Alzheimer’s disease patients. J Am Chem Soc 131:6543–6548
Okamura N, Suemoto T, Shimadzu H et al (2004) Styrylbenzoxazole derivatives for in vivo imaging of amyloid plaques in the brain. J Neurosci 24:2535–2541
Okamura N, Suemoto T, Furumoto S et al (2005) Quinoline and benzimidazole derivatives: candidate probes for in vivo imaging of tau pathology in Alzheimer’s disease. J Neurosci 25:10857–10862
Okamura N, Shiga Y, Furumoto S et al (2010) In vivo detection of prion amyloid plaques using [(11)C]BF-227 PET. Eur J Nucl Med Mol Imaging 37:934–941
Okello A, Edison P, Archer HA et al (2009a) Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology 72:56–62
Okello A, Koivunen J, Edison P et al (2009b) Conversion of amyloid positive and negative MCI to AD over 3 years: an 11C-PIB PET study. Neurology 73:754–760
Ong K, Villemagne VL, Lamngon N et al (2010) Assessment of Aβ deposition in mild cognitive impairment with 18F-Florbetaben. Alzheimer Dement 6:S26
Opazo C, Luza S, Villemagne VL et al (2006) Radioiodinated clioquinol as a biomarker for beta-amyloid: Zn complexes in Alzheimer’s disease. Aging Cell 5:69–79
Ossenkoppele R, van Berckel BN, Prins ND (2011) Amyloid imaging in prodromal Alzheimer’s disease. Alzheimers Res Ther 3:26
Ossenkoppele R, Tolboom N, Foster-Dingley JC et al (2012) Longitudinal imaging of Alzheimer pathology using [11C]PIB, [18F]FDDNP and [18F]FDG PET. Eur J Nucl Med Mol Imaging 39:990–1000
Ostrowitzki S, Deptula D, Thurfjell L et al (2012) Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol 69(2):198–207
Petersen RC (2000) Mild cognitive impairment: transition between aging and Alzheimer’s disease. Neurologia 15:93–101
Petersen RC, Smith GE, Ivnik RJ et al (1995) Apolipoprotein E status as a predictor of the development of Alzheimer’s disease in memory-impaired individuals. JAMA 273:1274–1278
Petersen RC, Smith GE, Waring SC et al (1999) Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 56:303–308
Pettersen JA, Sathiyamoorthy G, Gao FQ et al (2008) Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Arch Neurol 65:790–795
Phelps ME (2000) PET: the merging of biology and imaging into molecular imaging. J Nucl Med 41:661–681
Pike VW (2009) PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol Sci 30:431–440
Pike KE, Savage G, Villemagne VL et al (2007) Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain 130:2837–2844
Pike KE, Ellis KA, Villemagne VL et al (2011) Cognition and beta-amyloid in preclinical Alzheimer’s disease: data from the AIBL study. Neuropsychologia 49:2384–2390
Pletnikova O, West N, Lee MK et al (2005) Abeta deposition is associated with enhanced cortical a-synuclein lesions in Lewy body diseases. Neurobiol Aging 26:1183–1192
Price JL, Morris JC (1999) Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol 45:358–368
Price JC, Klunk WE, Lopresti BJ et al (2005) Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab 25:1528–1547
Qiu C, Xu W, Fratiglioni L (2010) Vascular and psychosocial factors in Alzheimer’s disease: epidemiological evidence toward intervention. J Alzheimers Dis 20:689–697
Qu W, Kung MP, Hou C et al (2007a) Novel styrylpyridines as probes for SPECT imaging of amyloid plaques. J Med Chem 50:2157–2165
Qu W, Kung MP, Hou C et al (2007b) Radioiodinated aza-diphenylacetylenes as potential SPECT imaging agents for beta-amyloid plaque detection. Bioorg Med Chem Lett 17:3581–3584
Rabinovici GD, Jagust WJ (2009) Amyloid imaging in aging and dementia: testing the amyloid hypothesis in vivo. Behav Neurol 21:117–128
Rabinovici GD, Miller BL (2010) Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs 24:375–398
Rabinovici GD, Furst AJ, O’Neil JP et al (2007) 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology 68:1205–1212
Rabinovici GD, Jagust WJ, Furst AJ et al (2008) Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol 64:388–401
Rabinovici GD, Furst AJ, Alkalay A et al (2010) Increased metabolic vulnerability in early-onset alzheimer’s disease is not related to amyloid burden. Brain 133:512–528
Rabinovici GD, Rosen HJ, Alkalay A et al (2011) Amyloid vs FDG-PET in the differential diagnosis of AD and FTLD. Neurology 77:2034–2042
Reiman EM, Caselli RJ, Yun LS et al (1996) Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med 334:752–758
Reiman EM, Chen K, Liu X et al (2009) Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc Natl Acad Sci U S A 106:6820–6825
Remes AM, Laru L, Tuominen H et al (2008) Carbon 11-labeled Pittsburgh Compound B positron emission tomographic amyloid imaging in patients with APP locus duplication. Arch Neurol 65:540–544
Rentz DM, Locascio JJ, Becker JA et al (2010) Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol 67:353–364
Resende R, Pereira C, Agostinho P et al (2007) Susceptibility of hippocampal neurons to Abeta peptide toxicity is associated with perturbation of Ca2+ homeostasis. Brain Res 1143:11–21
Resnick SM, Lamar M, Driscoll I (2007) Vulnerability of the orbitofrontal cortex to age-associated structural and functional brain changes. Ann N Y Acad Sci 1121:562–575
Resnick SM, Sojkova J, Zhou Y et al (2010) Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology 74:807–815
Riekse RG, Leverenz JB, McCormick W et al (2004) Effect of vascular lesions on cognition in Alzheimer’s disease: a community-based study. J Am Geriatr Soc 52:1442–1448
Rinne JO, Brooks DJ, Rossor MN et al (2010) 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol 9:363–372
Roder S, Danober L, Pozza MF et al (2003) Electrophysiological studies on the hippocampus and prefrontal cortex assessing the effects of amyloidosis in amyloid precursor protein 23 transgenic mice. Neuroscience 120:705–720
Roe CM, Mintun MA, D’Angelo G et al (2008) Alzheimer disease and cognitive reserve: variation of education effect with carbon 11-labeled Pittsburgh Compound B uptake. Arch Neurol 65:1467–1471
Rosen RF, Walker LC, Levine H, 3rd (2011) PIB binding in aged primate brain: enrichment of high-affinity sites in humans with Alzheimer’s disease. Neurobiol Aging 32:223–234
Rosendorff C, Beeri MS, Silverman JM (2007) Cardiovascular risk factors for Alzheimer’s disease. Am J Geriatr Cardiol 16:143–149
Rossor MN, Kennedy AM, Frackowiak RS (1996) Clinical and neuroimaging features of familial Alzheimer’s disease. Ann N Y Acad Sci 777:49–56
Rowe CC, Villemagne VL (2011) Brain amyloid imaging. J Nucl Med 52:1733–1740
Rowe CC, Ng S, Ackermann U et al (2007) Imaging beta-amyloid burden in aging and dementia. Neurology 68:1718–1725
Rowe CC, Ackerman U, Browne W et al (2008) Imaging of amyloid beta in Alzheimer’s disease with (18)F-BAY94-9172, a novel PET tracer: proof of mechanism. Lancet Neurol 7:129–135
Rowe CC, Ellis KA, Rimajova M et al (2010) Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 31:1275–1283
Sabbagh MN, Fleisher A, Chen K et al (2011) Positron emission tomography and neuropathologic estimates of fibrillar amyloid-beta in a patient with Down syndrome and Alzheimer disease. Arch Neurol 68:1461–1466
Sabbagh M, Seibyl J, Akatsu H et al (2012) Multicentre phase 3 trial on florbetaben for beta-amyloid brain PET in Alzheimer’s disease. Alzheimers Dement 8:P90
Sair HI, Doraiswamy PM, Petrella JR (2004) In vivo amyloid imaging in Alzheimer’s disease. Neuroradiology 46:93–104
Salmon E, Sadzot B, Maquet P et al (1994) Differential diagnosis of Alzheimer’s disease with PET. J Nucl Med 35:391–398
Scheltens P, Rockwood K (2011) How golden is the gold standard of neuropathology in dementia? Alzheimers Dement 7:486–489
Schöll M, Almqvist O, Graff C et al (2011) Amyloid imaging in members of a family harbouring the Arctic mutation. Alzheimers Dement 7(Suppl 1):303
Schulz-Schaeffer WJ (2010) The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol 120:131–143
Serdons K, Verduyckt T, Cleynhens J et al (2007) Synthesis and evaluation of a (99m)Tc-BAT-phenylbenzothiazole conjugate as a potential in vivo tracer for visualization of amyloid beta. Bioorg Med Chem Lett 17:6086–6090
Serdons K, Terwinghe C, Vermaelen P et al (2009a) Synthesis and evaluation of (18)F-Labeled 2-Phenylbenzothiazoles as positron emission tomography imaging agents for amyloid plaques in Alzheimer’s disease. J Med Chem 52:1428–1437
Serdons K, Verduyckt T, Vanderghinste D et al (2009b) Synthesis of 18F-labelled 2-(4'-fluorophenyl)-1,3-benzothiazole and evaluation as amyloid imaging agent in comparison with [11C]PIB. Bioorg Med Chem Lett 19:602–605
Shaw LM, Korecka M, Clark CM et al (2007) Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat Rev Drug Discov 6:295–303
Shih WJ, Markesbery WR, Clark DB et al (1987) Iodine-123 HIPDM brain imaging findings in subacute spongiform encephalopathy (Creutzfeldt-Jakob disease). J Nucl Med 28:1484–1487
Shoghi-Jadid K, Small GW, Agdeppa ED et al (2002) Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry 10:24–35
Siderowf A, Xie SX, Hurtig H et al (2010) CSF amyloid beta 1–42 predicts cognitive decline in Parkinson disease. Neurology 75:1055–1061
Silverman DH, Small GW, Chang CY et al (2001) Positron emission tomography in evaluation of dementia: regional brain metabolism and long-term outcome. JAMA 286:2120–2127
Silverman DH, Cummings JL, Small GW et al (2002) Added clinical benefit of incorporating 2-deoxy-2-[18F]fluoro-D-glucose with positron emission tomography into the clinical evaluation of patients with cognitive impairment. Mol Imaging Biol 4:283–293
Small GW, Mazziotta JC, Collins MT et al (1995) Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 273:942–947
Small GW, Agdeppa ED, Kepe V et al (2002) In vivo brain imaging of tangle burden in humans. J Mol Neurosci 19:323–327
Small GW, Kepe V, Ercoli LM et al (2006) PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med 355:2652–2663
Small BJ, Gagnon E, Robinson B (2007) Early identification of cognitive deficits: preclinical Alzheimer’s disease and mild cognitive impairment. Geriatrics 62:19–23
Smith EE, Egorova S, Blacker D et al (2008) Magnetic resonance imaging white matter hyperintensities and brain volume in the prediction of mild cognitive impairment and dementia. Arch Neurol 65:94–100
Snowden J, Neary D, Mann D (2007) Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol 114:31–38
Sojkova J, Resnick SM (2011) In vivo human amyloid imaging. Curr Alzheimer Res 8:366–372
Sojkova J, Driscoll I, Iacono D et al (2011a) In vivo fibrillar beta-amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Arch Neurol 68:232–240
Sojkova J, Zhou Y, An Y et al (2011b) Longitudinal patterns of beta-amyloid deposition in nondemented older adults. Arch Neurol 68:644–649
Song PJ, Bernard S, Sarradin P et al (2008) IMPY, a potential beta-amyloid imaging probe for detection of prion deposits in scrapie-infected mice. Nucl Med Biol 35:197–201
Sperling R, Johnson K (2010) Pro: can biomarkers be gold standards in Alzheimer’s disease? Alzheimers Res Ther 2:17
Sperling RA, Laviolette PS, O’Keefe K et al (2009) Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63:178–188
Sperling RA, Aisen PS, Beckett LA et al (2011a) Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:280–292
Sperling RA, Jack CR Jr, Aisen PS (2011b) Testing the right target and right drug at the right stage. Sci Transl Med 3:111cm133
Sperling RA, Jack CR Jr, Black SE et al (2011c) Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement 7:367–385
Sperling RA, Johnson KA, Doraiswamy PM et al (2012) Amyloid deposition detected with florbetapir F 18 ((18)F-AV-45) is related to lower episodic memory performance in clinically normal older individuals. Neurobiol Aging 34(3):822–831
Stern Y (2002) What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsychol Soc 8:448–460
Storandt M, Head D, Fagan AM et al (2012) Toward a multifactorial model of Alzheimer disease. Neurobiol Aging 33:2262–2271
Strozyk D, Blennow K, White LR et al (2003) CSF Abeta 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology 60:652–656
Sunderland T, Gur RE, Arnold SE (2005) The use of biomarkers in the elderly: current and future challenges. Biol Psychiatry 58:272–276
Sundgren-Andersson AK, Svensson SPS, Swahn BM et al (2009) AZD4694: fluorinated Positron Emission Tomography (PET) radioligand for detection of beta-amyloid deposits. Alzheimers Dement 5:P267–P268 [abstract]
Suo Z, Wu M, Citron BA et al (2004) Abnormality of G-protein-coupled receptor kinases at prodromal and early stages of Alzheimer’s disease: an association with early beta-amyloid accumulation. J Neurosci 24:3444–3452
Sur C, Zeng Z, Hostetler E et al (2010) In vitro characterization of MK-3328: a novel fluorinated positron emission tomography tracer for beta-amyloid plaques. Alzheimers Dement 6:S40
Taniguchi S, McDonagh AM, Pickering-Brown SM et al (2004) The neuropathology of frontotemporal lobar degeneration with respect to the cytological and biochemical characteristics of tau protein. Neuropathol Appl Neurobiol 30:1–18
Tenovuo O, Kemppainen N, Aalto S et al (2008) Posterior cortical atrophy: a rare form of dementia with in vivo evidence of amyloid-beta accumulation. J Alzheimers Dis 15:351–355
Thal DR, Del Tredici K, Braak H (2004) Neurodegeneration in normal brain aging and disease. Sci Aging Knowl Environ 2004:pe26
Thal LJ, Kantarci K, Reiman EM et al (2006) The role of biomarkers in clinical trials for Alzheimer disease. Alzheimer Dis Assoc Disord 20:6–15
Thompson PW, Ye L, Morgenstern JL et al (2009) Interaction of the amyloid imaging tracer FDDNP with hallmark Alzheimer’s disease pathologies. J Neurochem 109:623–630
Thurfjell L, Lotjonen J, Lundqvist R et al (2012) Combination of biomarkers: PET [F]Flutemetamol imaging and structural MRI in dementia and mild cognitive impairment. Neurodegener Dis 10:246–249
Tolboom N, van der Flier WM, Yaqub M et al (2009a) Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. J Nucl Med 50:1464–1470
Tolboom N, Yaqub M, van der Flier WM et al (2009b) Detection of Alzheimer pathology in vivo using both 11C-PIB and 18F-FDDNP PET. J Nucl Med 50:191–197
Tomiyama T, Nagata T, Shimada H et al (2008) A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann Neurol 63:377–387
Tosun D, Schuff N, Mathis CA et al (2011) Spatial patterns of brain amyloid-beta burden and atrophy rate associations in mild cognitive impairment. Brain 134:1077–1088
Trojanowski JQ (2002) Emerging Alzheimer’s disease therapies: focusing on the future. Neurobiol Aging 23:985–990
Tucker AM, Stern Y (2011) Cognitive reserve in aging. Curr Alzheimer Res 8:354–360
Vandenberghe R, Thurfjell L, Van Laere K et al (2010a) Comparison between cerebellum and pons as reference regions for quantification of the amyloid imaging agents [18F]flutemetamol and [11C]PIB. In: Proceedings of the human amyloid imaging Toronto, Canada, p. 42 [abstract]
Vandenberghe R, Van Laere K, Ivanoiu A, et al (2010b) 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Ann Neurol 68:319–329
Venneti S, Lopresti BJ, Wiley CA (2006) The peripheral benzodiazepine receptor (Translocator protein 18kDa) in microglia: from pathology to imaging. Prog Neurobiol 80:308–322
Verhoeff NP, Wilson AA, Takeshita S et al (2004) In-vivo imaging of Alzheimer disease beta-amyloid with [11C]SB-13 PET. Am J Geriatr Psychiatry 12:584–595
Vernooij MW, van der Lugt A, Ikram MA et al (2008) Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology 70:1208–1214
Villain N, Chetelat G, Grassiot B et al (2012) Regional dynamics of amyloid-beta deposition in healthy elderly, mild cognitive impairment and Alzheimer’s disease: a voxelwise PiB-PET longitudinal study. Brain 135:2126–2139
Villemagne VL, Rowe CC (2010) Amyloid ligands for dementia. PET Clin 5:33–53
Villemagne VL, Rowe CC, Macfarlane S et al (2005) Imaginem oblivionis: the prospects of neuroimaging for early detection of Alzheimer’s disease. J Clin Neurosci 12:221–230
Villemagne VL, Cappai R, Barnham KJ et al (2006) The Aβ centric pathway of Alzheimer’s disease. In: Barrow CJ, Small BJ (eds) Abeta peptide and Alzheimer’s disease. Springer, London, pp 5–32
Villemagne VL, Fodero-Tavoletti MT, Pike KE et al (2008a) The ART of loss: Aβ imaging in the evaluation of Alzheimer’s disease and other dementias. Mol Neurobiol 38:1–15
Villemagne VL, Pike KE, Darby D et al (2008b) Aβ deposits in older non-demented individuals with cognitive decline are indicative of preclinical Alzheimer’s disease. Neuropsychologia 46:1688–1697
Villemagne VL, Ataka S, Mizuno T et al (2009a) High striatal amyloid beta-peptide deposition across different autosomal Alzheimer disease mutation types. Arch Neurol 66:1537–1544
Villemagne VL, McLean CA, Reardon K et al (2009b) 11C-PiB PET studies in typical sporadic Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 80:998–1001
Villemagne VL, Pike K, Pejoska S et al (2010) 11C-PiB PET ABri imaging in Worster-Drought syndrome (familial British dementia): a case report. J Alzheimers Dis 19:423–428
Villemagne VL, Ong K, Mulligan RS et al (2011a) Amyloid imaging with 18F-florbetaben in Alzheimer disease and other dementias. J Nucl Med 52:1210–1217
Villemagne VL, Pike KE, Chetelat G et al (2011b) Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol 69:181–192
Villemagne VL, Furumoto S, Fodero-Tavoletti MT et al (2012a) In vivo tau imaging in Alzheimer’s disease and other dementias. Alzheimers Dement 8:P9
Villemagne VL, Klunk WE, Mathis CA et al (2012b) Abeta imaging: feasible, pertinent, and vital to progress in Alzheimer’s disease. Eur J Nucl Med Mol Imaging 39:209–219
Villemagne VL, Mulligan RS, Pejoska S et al (2012c) Comparison of (11)C-PiB and (18)F-florbetaben for Aβ imaging in ageing and Alzheimer’s disease. Eur J Nucl Med Mol Imaging 39:983–989
Villemagne VL, Okamura N, Pejoska S et al (2012d) Differential diagnosis in Alzheimer’s disease and dementia with Lewy bodies via VMAT2 and amyloid imaging. Neurodegener Dis 10:161–165
Viswanathan A, Rocca WA, Tzourio C (2009) Vascular risk factors and dementia: how to move forward? Neurology 72:368–374
Vlassenko AG, Mintun MA, Xiong C et al (2011) Amyloid-beta plaque growth in cognitively normal adults: longitudinal [11C] Pittsburgh Compound B data. Ann Neurol 70:857–861
Wahlund LO, Blennow K (2003) Cerebrospinal fluid biomarkers for disease stage and intensity in cognitively impaired patients. Neurosci Lett 339:99–102
Walker LC, Rosen RF, Levine H 3rd (2008) Diversity of Abeta deposits in the aged brain: a window on molecular heterogeneity? Rom J Morphol Embryol 49:5–11
Weller RO, Preston SD, Subash M et al (2009) Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimers Res Ther 1:6
Wermke M, Sorg C, Wohlschlager AM et al (2008) A new integrative model of cerebral activation, deactivation and default mode function in Alzheimer’s disease. Eur J Nucl Med Mol Imaging 35(Suppl 1):S12–S24
Wiley CA, Lopresti BJ, Venneti S et al (2009) Carbon 11-labeled Pittsburgh Compound B and carbon 11-labeled (R)-PK11195 positron emission tomographic imaging in Alzheimer disease. Arch Neurol 66:60–67
Winblad B, Palmer K, Kivipelto M et al (2004) Mild cognitive impairment–beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med 256:240–246
Wolk DA, Price JC, Saxton JA et al (2009) Amyloid imaging in mild cognitive impairment subtypes. Ann Neurol 65:557–568
Wolk DA, Grachev ID, Buckley C et al (2011) Association between in vivo fluorine 18-labeled flutemetamol amyloid positron emission tomography imaging and in vivo cerebral cortical histopathology. Arch Neurol 68:1398–1403
Wolk DA, Price JC, Madeira C et al (2012a) Amyloid imaging in dementias with atypical presentation. Alzheimers Dement 8:389–398
Wolk DA, Zhang Z, Boudhar S et al (2012b) Amyloid imaging in Alzheimer’s disease: comparison of florbetapir and Pittsburgh Compound-B positron emission tomography. J Neurol Neurosurg Psychiatry 83:923–926
Wong DF, Rosenberg PB, Zhou Y et al (2010) In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir [corrected] F 18). J Nucl Med 51:913–920
Wong DF, Moghekar AR, Rigamonti D et al (2013) An In vivo evaluation of cerebral cortical amyloid with [(18)F]flutemetamol using positron emission tomography compared with parietal biopsy samples in living normal pressure hydrocephalus patients. Mol Imaging Biol 15(2):230–237
Yates PA, Sirisriro R, Villemagne VL et al (2011) Cerebral microhemorrhage and brain {beta}-amyloid in aging and Alzheimer disease. Neurology 77:48–54
Ye L, Morgenstern JL, Gee AD et al (2005) Delineation of positron emission tomography imaging agent binding sites on beta-amyloid peptide fibrils. J Biol Chem 280:23599–23604
Ye L, Velasco A, Fraser G et al (2008) In vitro high affinity alpha-synuclein binding sites for the amyloid imaging agent PIB are not matched by binding to Lewy bodies in postmortem human brain. J Neurochem 105:1428–1437
Yee S, Mathis C, Klunk W et al (2007) Optimal time window for standardized uptake ratio as a simplified measure of PIB retention. J Nucl Med 48:404P
Yokokura M, Mori N, Yagi S et al (2011) In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer’s disease. Eur J Nucl Med Mol Imaging 38:343–351
Zhang W, Oya S, Kung MP et al (2005a) F-18 polyethyleneglycol stilbenes as PET imaging agents targeting Abeta aggregates in the brain. Nucl Med Biol 32:799–809
Zhang W, Oya S, Kung MP et al (2005b) F-18 stilbenes as PET imaging agents for detecting beta-amyloid plaques in the brain. J Med Chem 48:5980–5988
Acknowledgements
We thank Prof. Michael Woodward, Dr. John Merory, Dr. Peter Drysdale, Dr. Gordon Chan, Dr. Kenneth Young, Dr. Sylvia Gong, Dr. Greg Savage, Dr. Paul Maruff, Dr. David Darby, Ms. Fiona Lamb, Mr. Sean Thronton, Ms. Mary Kate Danaher, Ms. Joanne Robertson and the Brain Research Institute for their assistance with this study.
This work was supported in part by grant 1011689 of the National Health and Medical Research Council of Australia, the Science and Industry Endowment Fund, and the Austin Hospital Medical Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Villemagne, V.L., Fodero-Tavoletti, M., Yates, P., Masters, C.L., Rowe, C.C. (2014). Aβ Imaging in Aging, Alzheimer’s Disease and Other Neurodegenerative Conditions. In: Dierckx, R., Otte, A., de Vries, E., van Waarde, A., Leenders, K. (eds) PET and SPECT in Neurology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-54307-4_10
Download citation
DOI: https://doi.org/10.1007/978-3-642-54307-4_10
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-54306-7
Online ISBN: 978-3-642-54307-4
eBook Packages: MedicineMedicine (R0)