
Abstract
The chapter initially discusses normal spinal development and variants. Pathology is addressed first in terms of general structural vertebral anomalies. These are then applied to congenital scoliosis, kyphosis, and lordosis, as well as anomalies involving the craniovertebral junction. Discussion of spinal dysraphism follows, including tethered filum terminale, hydrosyringomyelia, and diastematomyelia. Congenital intraspinal tumors are briefly reviewed. Klippel-Feil syndrome and caudal regression are presented, as well as spine manifestations of neurofibromatosis and several other syndromes. Idiopathic scoliosis is discussed in detail, including curve measurement, imaging techniques, and treatment options. Neuromuscular scoliosis is briefly reviewed. The chapter concludes with juvenile kyphosis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Congenital abnormalities of the spine are relatively common and occur as an isolated finding or as part of a syndrome or association. Anomalies can be severe and lead to debilitating physical deformities with significant neurological consequences. Others have relatively mild clinical manifestations and may even be initially overlooked. Correct categorization depends in part on an adequate understanding of vertebral development, as well as the ability to recognize variant anatomy. Congenital scoliosis may result from segmentation abnormalities, including hemivertebrae and vertebral fusion. In addition, depending on severity, spinal dysraphism may be associated with extensive anomalous development of the spine.
1 Normal Development and Variant Ossification
1.1 Normal Development
Vertebral body development begins during embryogenesis and continues until the third decade [1]. Development occurs in four overlapping stages: the mesenchymal stage, chondrification, primary ossification, and secondary ossification. During the mesenchymal stage, the notochord forms; cells migrate from the bilaminar embryonic disc toward the primitive knot where the notochord forms. This occurs between the endodermal yolk sac and the ectodermal neural plate; the latter then folds, forming the neural tube. The notochord then induces differentiation of the ectoderm and mesoderm. By day 20 of embryonic life, the paraxial mesoderm is ready to separate into lateral plates, from which the genitourinary system later forms (thus explaining the close relationship between vertebral and renal anomalies), and medial columns, from which the myotomes and sclerotomes develop. By the end of the fifth gestational week, the paraxial mesoderm has divided into between 42 and 44 somites (Fig. 3.1). Each consists of three cell groups: the sclerotome, or future vertebra; the myotome, or future skeletal muscle; and the dermatome, or future dermis [2].


Sclerotome division and reconvergence. Sclerotome division allows the spinal nerves to emerge from the neural tube and extend to the periphery (a). The sclerotome then reconverges to form the final vertebrae (b) (Reprinted with permission from Schoenwolf et al. [127])
The sclerotome grows to surround the notochord and neural tube and then forms the annulus fibrosus. The enveloped notochord goes on to form the infantile and childhood nucleus pulposus. Once adjacent to the notochord and neural tube, the sclerotome divides, allowing the spinal nerves to emerge from the neural tube. When division of the sclerotome is complete, the caudal half of the sclerotome above merges with the cranial half of the sclerotome below to form the vertebra precursor. Fibrocartilagenous cells gradually replace the original notochord cells of the nucleus pulposus.
Chondrification of the membranous vertebrae occurs between fetal weeks 4 and 7. Two cartilaginous centers form in the centrum (either dorsal-ventral or side-by-side). Every neural arch and every costal/transverse process develop one center as well. Ossification begins in the neural arches at fetal weeks 8–9, starting at the upper cervical region. Ossification of the vertebral bodies starts in the lower thoracic and upper lumbar region and moves caudally and cranially; this process is complete by about week 20. All vertebrae below C2 and above the level of the sacrum have three ossification sites: one for the vertebral body and for each neural arch (Fig. 3.2). The single vertebral body ossification center results from coalescence of the two chondral centers that are transiently present (dorsal-ventral or side-by-side), which helps explain the occurrence of posterior and lateral hemivertebrae.

Formation of a lower cervical vertebra. Axial (a) and sagittal (b) views. The body (1) appears by the fifth fetal month. Neural arches (2) appear by the seventh to ninth fetal week and are separated from the body by the neurocentral synchondroses (3), which close between 3 and 7 years of age. The posterior synchondrosis (4) between the spinous processes usually closes by age 2–4 years. Superior and inferior epiphyseal rings (5) appear at puberty, contribute to the vertical growth of the vertebral body, and unite with it at about age 25 years (Reprinted from Ghanem et al. [4] with kind permission from Springer Science and Business Media)
The strip of cartilage that separates the two neural arch centers from the single vertebral centrum, the neurocentral synchondrosis, is slightly anterior to the base of the pedicles (Fig. 3.3). The age at closure of the neurocentral synchondroses varies by position—cervical (3–7 years), lumbar (4–10 years), and thoracic (5 to >10 years) [3, 4]. The ossified portions of each neural arch fuse with each other dorsally by 2–4 years of age; the radiolucent lines seen on the frontal projection of the spine before this age should therefore be recognized as normal (Fig. 3.3). Between 3 and 6 years of age, this synchondrosis disappears as bone bridges the segment, although more distal levels may fuse later [5].

Normal spine synchondroses. (a) Frontal view shows unfused neural arches (arrow) in a 3-month-old infant. (b) Normal neurocentral synchondrosis (arrow) in a neonate, shown on a lateral radiograph. (c) Computed tomography (CT) shows normal unfused neural arch (asterisk) and bilateral unfused neurocentral synchondroses (arrowhead)
Around puberty, secondary ossification centers appear at the tips of the spinous, transverse, and mammillary processes, as well as in the cartilaginous ring apophysis surrounding the vertebral end plate (note that this ossification can appear as early as 2–6 years of age) [6]. Complete rings are rarely visible—on lateral radiographs, they appear as small ossification centers in notches adjacent to the anterior and posterior borders of the vertebral end plates. These secondary ossification centers fuse with the vertebral body in the third decade, usually by age 25.
Ossification of the atlas and axis is somewhat different. Most of the cranial half of the first cervical sclerotome normally merges with the occipital condyles, with a small portion forming the tip of the dens. The caudal half of the first cervical sclerotome forms the anterior, lateral, and posterior masses as well as the posterior arch of the atlas (C1). In the atlas, three separate ossification centers are present in the lateral masses and anterior arch (Fig. 3.4). The anterior arch’s center is ossified at birth in only 20 % of infants and may not be radiographically evident until 2 years of age [7]. Incomplete incorporation or failure of segmentation of the first cervical sclerotome can cause a broad spectrum of fusion anomalies involving the craniocervical junction and accessory structures.

Development of the atlas (C1). Axial (a), coronal (b), and sagittal (c) views. The neurocentral synchondrosis (1) unites the neural arch (2) to the anterior arch (3) of the atlas. The ossification center of the anterior arch (3) appears between age 6 months and 2 years. The posterior synchondrosis at the spinous process (4) unites by 4–6 years of age, followed by the neurocentral synchondrosis (1), which fuses by about age 7 (Reprinted from Ghanem et al. [4] with kind permission from Springer Science and Business Media)
The usual pattern of ossification occurs in the centrum and neural arches of the axis. However, two additional centers form in the base of the dens and fuse into a single odontoid center, either by birth or a few months later (Fig. 3.5) [8, 9]. The ossification centers of the body and dens are separated by a well-demarcated synchondrosis, the homologue of the disc between C1 and C2 (Fig. 3.6). This synchondrosis closes between 3 and 11 years [5] and the neurocentral synchondroses by 3–7 years. A small ossification center appears at the tip of the odontoid between 3 and 10 years; it fuses between 10 and 12 years [5].

Development of the axis (C2). Coronal (a) and sagittal (b) views. The dens (odontoid process) (1) develops from two primary ossification centers that usually coalesce within the first 3 months of life. The C2 centrum (2) appears by the fifth fetal month and is separated from the dens (1) by the dentocentral synchondrosis (3). The dentocentral synchondrosis fuses between ages 3 and 11, though it usually closes by age 6–7 years. The bilateral neural arches (4) appear by the seventh fetal month and fuse posteriorly by age 3 years. The neurocentral synchondroses (7), located on each side between the dens/vertebral body and neural arch, close between 3 and 7 years. The ossiculum terminale (5) at the tip of the dens appears between 3 and 10 years of age and fuses to the dens between ages 10 and 12. The inferior epiphyseal ring (6) appears at puberty and fuses at about age 25

Dentocentral synchondrosis at the base of the odontoid (curved arrow) in a 3-year-old boy
At the sacrum, in addition to the usual three centers for the body and neural arches of each segment, there are two epiphyseal plates that provide secondary ossification centers to form the superior and inferior portions of the sacral ala of the first through third segments.
Most people have 33 vertebrae: 7 cervical, 12 thoracic, 5 lumbar, 5 sacral, and 4 coccygeal. About 5 % have variant distribution (e.g., 6 lumbar and 11 thoracic vertebrae) but still have a total of 33 vertebral bodies. Three percent have one or two extra vertebrae, and 2 % have only 32 vertebrae [10, 11].
The lumbosacral junction is unstable during embryogenesis, and variations in formation and ossification are common in this area. Transitional vertebrae occur in approximately 20 % and often involve the sacrococcygeal and lumbosacral junctions. These vertebrae retain some features of the adjacent segments and often do not alter the overall number of vertebrae. Common variants include “sacralized” L5 and “lumbarized” S1 vertebrae (Fig. 3.7). Hemi-sacralization or lumbarization can be seen in up to 6.5 % and can cause back pain due to nonuniform articulation. Symptomatic abnormal articulation, known as Bertolotti syndrome, may be characterized on magnetic resonance imaging (MRI) by adjacent edema (Fig. 3.8). Confusion is common when it comes to labeling levels of transitional vertebrae—complete evaluation of the spinal axis before surgical intervention will allow correct assignment of vertebral levels.

Transitional S1 vertebra in a 14-year-old girl with scoliosis. Note pseudoarticulation of S1 and S2 on the left (arrow) (Courtesy of Thomas Ray Sanchez)

Bertolloti syndrome in a 15-year-old girl with back pain. (a) Transitional vertebral segment with pseudoarticulation on the left (black arrow). (b) Coronal inversion recovery (IR) image shows hyperintensity due to marrow edema on both sides of the pseudoarticulation (white arrow) (Courtesy of Chirag V. Patel)
The individual vertebral bodies enlarge circumferentially by the process of periosteal apposition and grow vertically by endochondral ossification; the cartilage of the vertebral end plates functions like other growth plates. The ring apophyses do not contribute to vertical growth. Following a relatively rapid length increase during the first 2 years of life, the spine grows linearly until age 16 [12]. Normal weight bearing helps balance vertical and horizontal growth and maintains an acceptable ratio between axial diameter and vertebral height. Without normal upright or walking postures, as in children with spinal muscular atrophy or cerebral palsy, vertebral bodies grow disproportionately tall and thin, developing biconvex end plates and decreased disc space height (Fig. 3.9).

Increased height of vertebral bodies (caninization) in an 11-year-old boy with congenital myopathy
1.2 Variant Ossification
Common developmental features as well as unusual normal variants can be easily mistaken for pathologic processes. The variants described below are common but not exhaustive. Ultimately, consultation with atlases of normal variants when bony defects are uniform, disproportionate to physical exam findings, or asymptomatic should prevent mischaracterization of congenital variants as disease.
The multiple primary and secondary ossification centers and their intervening synchondroses are often mistaken for fractures or avulsion fragments. Ossification in the superior and inferior ring apophysis may begin in children as young as 6 years of age and may persist well into adolescence.
During adolescence, the nucleus pulposus may herniate through the vertebral end plate, resulting in Schmorl nodes (Fig. 3.10). Additionally, notochord remnants may appear as impressions on consecutive inferior end plates, mimicking erosive or lytic lesions. Similarly, anterior intraosseous herniation of the intervertebral disc, also referred to as a limbus vertebra (Fig. 3.11), may be mistaken for a teardrop fracture. These normal variants may be differentiated from fractures by examining the edge of the lesion: physeal plates should be smooth and regular, with subchondral sclerotic lines, whereas acute fractures are irregular and non-sclerotic; fractures can occur at any location.

Multiple Schmorl nodes in a 17-year-old male. Arrows delineate L3–4 on radiograph (a) and sagitally reformatted CT (b) (Courtesy of John Hunter)

Limbus vertebra at L2 (arrow) in a 16-year-old girl
During the first two decades, the appearance and physiology of the cervical spine varies tremendously. Until age 15, a normal atlantoaxial interval can be as large as 5 mm and occasionally even 6 mm. More accurate assessment evaluates for change in distance between passive flexion and extension, which should not exceed 2 mm.
Physiologic displacement of C2 anteriorly on C3 is common in normal children due to ligamentous laxity (Box 3.1). Known as pseudosubluxation, this occurs because until age 10, flexion and extension are centered at the C2–3 level (Figs. 3.12 and 3.13). (In adolescence and throughout adulthood, maximum mobility is at C4–6.) In order to determine whether subluxation results from this normal variant or from a C2 arch fracture and pathologic subluxation, one can draw a curved “posterior cervical” line, connecting the anterior cortices of the C1 and C3 spinous processes (Fig. 3.12). With physiologic subluxation, this line either passes through the C2 posterior arch or passes less than 2 mm anterior to it. In the presence of an arch fracture, the distance will be greater, since the atlas and the C2 centrum slip forward at the fracture site while the spinous process remains posterior [13, 14].

Normal pseudosubluxation of C2 on C3. A discontinuous line connects the posterior C2 and C3 vertebral bodies, but the posterior cervical line along C1–3 is continuous and normal (Reprinted from Cakmakci [14] with kind permission from Springer Science and Business Media)

Normal pseudosubluxation of C2 on C3. Radiograph (a) and sagitally reformatted CT (b) show forward displacement of C2 on C3. However, the posterior cervical line is normal
Box 3.1: Pseudosubluxation of C2–3
Before about age 10 years |
Due to normal ligamentous laxity |
Posterior cervical line connects the anterior cortex of spinous processes of C1 and C3 |
Passes through the posterior arch of C2 or <2 mm anterior to it |
Developmental pseudospread of the atlas is a normal phenomenon that occurs because the growth of the atlas is linked to the relatively rapid growth of the skull base, whereas the growth of the axis is influenced by the slower growth of the spine. With pseudospread, the lateral margins of C1 lie symmetrically lateral to the margins of C2, mimicking the bilateral lateral displacement of a Jefferson fracture of the arch of the atlas. This disparity generally resolves after age 2. The overlap is usually less than 2 mm, whereas in the case of fracture, it generally exceeds 3 mm [15].
In up to 5 % of newborns, a coronal cleft is apparent on a lateral radiograph of the thoracic or lumbar spine. This appears as a vertical lucency at the junction of the anterior two thirds and posterior third of the vertebral body [16]. The cleft is filled with hyaline cartilage, possibly a remnant of the division between dorsal and ventral ossification centers. The coronal cleft ossifies during infancy and does not persist or lead to other anomalies.
A horizontal cleft in the midportion of the vertebral body, also referred to as a Hahn cleft, may also be visible on a lateral view. It marks both the boundary of fusion between sclerotomes and the nutrient foramen (Fig. 3.14) and may persist as a sclerotic line throughout adolescence.

Hahn clefts (arrow), the boundary of fusion of the reformed sclerotomes and also the level of arterial entry into the vertebral bodies (sagittally reformatted CT)
Bone-in-bone is another common finding during infancy (Fig. 3.15). This results from rapid growth during the first postnatal months, before remodeling of the fetal spine begins [14]. The outline of a smaller replica of the vertebral body is seen within the centrum.

Bone-in-bone (arrow) in a newborn with malrotation (curved arrow) (Lateral view from a fluoroscopic study)
Incomplete bony fusion at the neural arch of L5 or S1 is very common in children, but the incidence decreases during childhood. Cartilage usually fills this apparent gap, so it is not functionally open [17]. The S1 neural arch is incompletely fused in up to 58 % at age 8, 48 % at age 12, and 10–26 % of adults. A separation of 3 mm or less is normal and will often ossify during childhood. Occasionally, a similar narrow cleft may be encountered as an isolated finding at the T11 or T12 level. The term spina bifida occulta has been applied to this appearance (at any isolated level), but this term may be misleading as it implies pathology. Misreporting of this finding may result in confusion and unnecessary imaging.
At the T12 and L1 vertebrae, localized moderate to marked pedicle thinning occurs in 7 % of normal children and adults. The developmentally aberrant pedicle may be differentiated from a pedicle that is being deformed by mass effect because the lateral border of the aberrant pedicle is flat or concave, rather than rounded.
2 Structural Vertebral Abnormalities
Spinal defects may be restricted to the vertebrae or may involve neural and non-neural structures [18]. Sixty-one percent of those with congenital spinal defects have other abnormalities as well. For example, a prenatal neural crest and sensory nerve injury may lead to a field defect that causes the constellation of upper limb, vertebral, and rib anomalies. The mnemonic VACTERL describes the clinical confluence of vertebral, anorectal, cardiac, tracheoesophageal, renal, and limb anomalies [19]. It is important to recognize that VACTERL is an association not a syndrome. Less common associated abnormalities are found in the ear, choanae, lips, palate, intestinal tract, abdominal wall (hernias), and genital system.
Scoliosis may develop if vertebrae fail to form, as with wedge vertebrae (unilateral hypoplasia) or hemivertebrae (unilateral aplasia). Less often, a hemivertebra may be supernumerary, resulting from partial duplication. Failure of segmentation may be unilateral or bilateral, resulting in an osseous bar (or bars) connecting the posterior elements, vertebral bodies, or both (Fig. 3.16). If anomalies are multiple and balanced, alignment may be normal.

Diagrammatic representation of the classification system of congenital scoliosis. (a) Failures of formation: various types of hemivertebrae (semisegmented, fully segmented, wedge vertebra). (b) Failures of segmentation: block vertebra, unsegmented bar. (c) Mixed: combination of hemivertebra and unsegmented bar (Reprinted with permission from Erol et al. [128])
2.1 Vertebral Anomalies
Wedge vertebrae (Fig. 3.16) form asymmetrically such that one side is hypoplastic and the other develops normally. Both pedicles are present, but one is larger than the other. A single wedge vertebra results in scoliosis, with the apex at the abnormal vertebra. However, bilateral wedge vertebrae may balance each other and result in a straight spine [20].
Hemivertebrae (Figs. 3.16, 3.17, and 3.18) are completely aplastic on one side, resulting in a single unpaired pedicle attached to a triangular vertebral half-body [21]. Hemivertebrae usually replace a normal vertebra but if supernumerary tend to be positioned slightly more laterally than are the other vertebrae. The hemivertebra may fuse with an adjacent vertebral body (“nonsegmented”), creating a tripediculate block vertebra with two pedicles on one side but only one on the other. If the hemivertebra is not fused to adjacent vertebrae (“fully segmented”), it is more likely to cause continued asymmetrical growth [21], since it has two functioning—and tilted—growth plates. However, curve progression is less likely with a tripediculate block vertebra. The hemivertebra may also be semisegmented, in which case one growth plate is fused to the adjacent vertebra and the other is not. Computed tomography (CT) or magnetic resonance imaging (MRI) helps assess the extent to which a hemivertebra is fused to the adjacent vertebrae [22].

Congenital scoliosis. Radiograph (a) and 3-dimensional (3D)-CT reconstruction (b) show three hemivertebrae on the left. The most cephalad (arrowhead) is fully segmented, whereas the more caudad are semisegmented (arrows), fused to the vertebra below

Congenital rotatory scoliosis. Radiograph (a) and 3-dimensional (3D)-CT reconstruction (b) demonstrate a right-sided hemivertebra at T9–T10 interspace (arrow). Even though the ribs end at same level, the right side has 12 ribs and the left has 11, because of the extra rib induced by the right hemivertebra. Also note fusion of the left 9th and 10th ribs (arrowhead)
The pedicle of the hemivertebra may induce rib development. Additionally, ribs at the most concave portion of the scoliosis may fuse. Counting all the ribs on both sides is an essential step in assessing the configuration and significance of wedge vertebrae and hemivertebrae. Since hemivertebrae may be supernumerary, accurate assessment of vertebral level requires evaluation of the entire spine, from C1 through the coccyx.
Butterfly vertebrae (Fig. 3.19) have a partial sagittal cleft resulting in two approximately symmetric segments that can be otherwise normal or appear as bilateral wedge vertebrae or—if completely divided—hemivertebrae. If balanced, butterfly vertebrae do not cause significant scoliosis, but they may suggest the presence of a syndrome or other anomalies.

Butterfly vertebra (arrow) in an 11-year-old boy with Alagille syndrome
Isolated absence of the pedicle (Fig. 3.20) is most common in the cervical and lumbar region and is uncommon in the thoracic spine. It is important to distinguish this congenital anomaly from pathologic destruction of the pedicle. With congenital absence of the pedicle, the pedicle outline is completely absent, and the associated intervertebral foramen is enlarged. In older children, the opposite pedicle and lamina may be hypertrophied.

Transitional lumbosacral vertebra in a 6-year-old girl. The pedicle on the right (arrow) is absent
Congenital vertebral bars (Fig. 3.21) develop when the laminae or pediculate bases of adjacent vertebrae fuse. Fusion that is limited to the laminae and facets dorsal to the intervertebral foramina leads to lordo-scoliosis. If the vertebral bodies at the base of the pedicles are also fused, the intervertebral foramina will be small. If there are no ribs in this region, a large conglomerate foramen develops, through which multiple nerve roots pass. If these anomalies are bilateral and symmetric, growth will be restricted but alignment may be normal. If unilateral, more problematic angular deformity occurs.

Laminopediculate bar. The lamina of the hemivertebra has fused to the lamina of the pedicle above (arrow)
If cleavage of somites fails, normal segmentation does not occur, and block vertebrae form. Vertebral bodies do not separate, and there is no (or very little) intervening disc space. The vertebral block appears constricted at the level of the disc, and the height of the structure is slightly less than that of two normal vertebral bodies (excluding the height of a disc). A thin sclerotic linear density may replace the disc, or a rudimentary disc may be present.
Fusion in young children is often cartilaginous and thus may be radiographically occult. In older children, a frontal radiograph demonstrates a laminopediculate bar as a discrete sclerotic curved line connecting pedicles at two or more levels (see Fig. 3.21). The ipsilateral vertebral body and disc space are often hypoplastic, and the vertebral bodies may be fused as well. If the fusion is coronal rather than sagittal, the anterior aspect of the vertebral body is almost invariably wedge shaped or entirely absent, resulting in kyphosis or lordosis (further discussed below). In young children, radiographs may fail to demonstrate fusion, but MRI readily depicts block vertebra formation by demonstrating absence of intervertebral disc signal (Fig. 3.22).

Lack of normal hyperintense intervertebral disc signal in the upper thoracic and lower cervical region (arrows) denotes areas that will develop into block vertebrae when ossification is complete (sagittal T2-weighted (T2-W) fat-suppressed (FS) magnetic resonance image (MRI)) (Incidental flow artifact dorsal to the spinal cord)
CT is valuable in showing associated spinous process obliquity, transverse process hypoplasia, abnormally shaped spinal canal, anomalous facet and rib articulations, and hypoplasia of pedicles at adjacent levels. It also delineates hypoplastic or malpositioned pedicles that are not apparent on routine radiography. MRI best images nerve root compression, myelomalacia, and contralateral laminar stress fracture, all of which can cause back pain.
2.2 Congenital Scoliosis (Table 3.1)
Spinal curvature in the coronal plane is caused by unbalanced anatomy and growth potential of the two sides of the spine. Unlike idiopathic scoliosis, congenital scoliosis progresses in up to 75 % of patients and most often affects the thoracic spine (thoracolumbar scoliosis is the second most common). Once curvature starts to increase, it will continue to do so, often accelerating during the preadolescent growth spurt. Since ribs and vertebrae form from the same mesenchymal somites, associated developmental and fusion anomalies are common. However, it is difficult to predict exactly what effect a single anomaly will have.
Multiple lesions are sometimes balanced, canceling each other out and causing little overall effect on spinal alignment. Ribs that fuse centrally near the spine may contribute to scoliosis, but peripheral rib fusion generally does not affect spinal curvature [23].
Hemivertebrae function both as the apex of the curve and as an unbalanced growth center that contributes to increased curvature. Congenital bars have the opposite effect of hemivertebrae, restricting growth on one side of the spine while the other side continues to grow. Congenital bars are present in 40 % of patients with multiple hemivertebrae. A hemivertebra at the same level as a contralateral bar has the worst prognosis, especially if thoracolumbar. However, in young children it can be difficult to predict exactly where and how a segment will fuse to adjacent bodies.
Imaging
Radiographic evaluation includes frontal and lateral radiographs. Curves can be measured according to a variety of techniques; the Cobb technique is commonly employed and is presented later in this chapter. Cross-sectional imaging is indicated for exact delineation of anomalies [24]. CT and MRI may demonstrate occult hemivertebrae as well as vertebral fusion. Additionally, MRI of the entire spine is typically performed before surgery in order to assess asymptomatic associated anomalies such as tethered cord, syringomyelia, and diastematomyelia, which are cumulatively described in up to 40 % of patients with congenital scoliosis [25].
Up to 18 % of patients with congenital scoliosis have associated genitourinary tract abnormalities, with renal agenesis being the most common [26]. Urologic evaluation is therefore indicated in all patients with congenital anomalies of the spine. Screening ultrasound (US) to assure the normal site and structure of both kidneys usually suffices, although occasionally additional imaging is necessary. Patients may also have congenital heart disease (7 % incidence), undescended scapula (6 %), thumb anomalies (4 %), and other malformations.
2.3 Congenital Kyphosis and Lordosis
Congenital kyphosis is another manifestation of unbalanced longitudinal development of vertebrae, with the curvature occurring along the sagittal axis. As with coronal scoliosis, these developmental anomalies generally manifest as failure of formation, failure of segmentation, or both.
Kyphosis in the thoracic spine is defined as curvature greater than the 40° [27–29]. It can be caused by posterior overgrowth, restricted anterior growth, or anomalies of segmentation [30]. The most common form of congenital kyphosis is caused by a dorsal hemivertebra (although infrequently the entire vertebral body is absent but the neural arch elements are formed) (Fig. 3.23). Both the anterior and the lateral portions of the vertebra may be deficient, causing a combination of scoliosis and kyphosis [31]. If kyphosis is severe, an anterior pseudoarticulation may develop above or below the defect. Anterior block vertebra formation rarely causes kyphosis, usually without scoliosis, since such fusion tends to be symmetric [32].

Congenital thoracolumbar kyphosis and posterior spinal dysraphism. (a-frontal, b-lateral) Multiple anterior vertebral body segments are hypoplastic, and posterior elements have not fused. The patient is in a brace, and there is a ventriculoperitoneal shunt catheter
Congenital nonsegmentation and fusion must be distinguished from progressive anterior vertebral body fusion, in which erosion of the anterior end plates is followed by anterior fusion and later disc fusion. Muscle imbalance in severe myelomeningocele may also cause kyphosis.
Congenital lordosis, a lumbar angle over 40°, is usually severe but fortunately uncommon. In this condition, posterior elements fuse while the anterior vertebral bodies continue to grow. Extreme thoracolumbar lordosis may also result from intrauterine positioning. Posterior rib synostosis contributes to growth inhibition.
Kyphosis and lordosis are usually assessed with standing radiographs, with flexibility determined by placing the patient on a bolster. As with congenital scoliosis, CT clearly delineates osseous causes of kyphosis or lordosis. MRI best evaluates for spinal cord compression and other cord abnormalities. A complete investigation includes cardiac and urologic screening due to the high associated incidence of these anomalies.
2.4 Craniovertebral Junction
Like the lumbosacral area, the craniovertebral junction is developmentally unstable. Congenital anomalies and developmental variants in this region are therefore common. Anomalous development here often results in multiple abnormalities, and therefore recognition of a single variant should trigger evaluation for additional problems. Many patients with abnormal development at the craniocervical junction can further be classified as syndromic [33]. While many abnormalities at the craniocervical junction are minor and asymptomatic, clinically significant spinal cord compression can result from somatic growth or recurrent motion.
2.4.1 Occipitalization of the Atlas
Occipitalization of the atlas (Fig. 3.24) occurs in up to 0.75 % of the population and ranges from complete osseous fusion between the atlas and occiput to more focal osseous bridging. Most commonly, the anterior arch of the atlas fuses to the skull base. However, fusion may involve the posterior arch or the lateral masses, and multiple sites of fusion are not uncommon [33]. Atlantoaxial instability is common in those with occipitalization of the atlas, as is encroachment on the spinal cord, sometimes resulting in myelopathy. Since development of the neural elements is closely intertwined with that of bony elements, basilar invagination, Chiari 1 malformation, and tonsillar ectopia may be present as well. Fifty-seven percent of patients with occipitalization of the atlas also have a syndrome [33].

Occipitalization of atlas. There is continuity of the cortex between the skull base and C1 (Same patient as Fig. 3.27)
Occipitalization of the atlas must be distinguished from an unusually high atlas, which can occur as a normal variant due to relatively flat occipital condyles. In some cases, occipitalization is associated with a short clivus, the latter due to primary growth failure (as in achondroplasia) or premature fusion of the spheno-occipital synchondrosis [4].
The posterior arch at C1 may be open in some patients with craniocervical fusion [33], but this finding can be seen until age 4 years in normal children and indeed persists as a normal variant in up to 4 % of adults. Complete absence of the posterior arch of the atlas is rare (Fig. 3.25). Defects in the anterior arch of C1 are less common than defects in the posterior arch. The anterior arch of the atlas rarely fuses to the dens. The occipital condyles may be asymmetrical, and similar asymmetry may be seen at the atlantal and axial articulations. One third of patients have associated fusion of C2–3 vertebrae.

Near complete absence of the posterior arch of C1 in a 12-year-old girl
Patients with craniocervical junction abnormalities may complain of neck pain and stiffness, with neurological signs and symptoms usually beginning insidiously in the third decade and progressing slowly. However, earlier onset is being reported with increasing frequency. Sometimes symptoms occur acutely, and sudden death may result from relatively mild trauma at the craniocervical junction.
Imaging
Fusion may be apparent on a lateral radiograph, showing a fused block of bone or continuous cortex. Lack of mobility on flexion and extension radiographs can indicate fusion as well. This finding is especially valuable in skeletally immature children. However, if there is reasonable suspicion for fusion, CT with sagittal and coronal reformats as well as three-dimensional (3D) reconstructions is most useful to delineate bony anatomy, and MRI may be useful to evaluate cord impingement or myelopathy.
2.4.2 C2 Developmental Anomalies
Multiple anomalies can affect the dens. Some have significant clinical implications, whereas others may contribute to diagnostic uncertainty in the setting of trauma. The dens may be hypoplastic or hypertrophied, or it may be composed of ossicles that fail to fuse to each other or to C2.
The normal odontoid terminates just below the foramen magnum, with a normal basion to (top of) dens measurement of 8.3–10 mm [34]. If the odontoid extends above or below this level, neurologic symptoms may result, especially if there is also transverse ligament laxity [35]. In odontoid hypoplasia, also known as stubby dens, the odontoid process is underdeveloped and does not reach its normal height at the superior surface of the atlas. In the severe form, odontoid aplasia, the dens does not extend above the body of C2. These congenital anomalies of formation may be isolated or associated with syndromes such as mucopolysaccharidoses, spondyloepiphyseal dysplasia, and metatropic dwarfism.
Anomalous development of the odontoid can take several forms. Os odontoideum describes a small fragment of bone that does not fuse with the subjacent portion of the odontoid. The ossific fragment usually lies directly behind the anterior arch of the atlas, but it is sometimes displaced (Fig. 3.26). Occasionally, there is a transverse cleft between the odontoid and the adjacent C2 vertebral body. A history of neck trauma is common in patients with this finding, and the phenomenon may sometimes result from an incomplete or partially healed fracture rather than aberrant development. The unfused ossicle usually moves with the atlas and occasionally fuses to the anterior lip of the foramen magnum.

Os odontoideum in an 11-year-old girl with Down syndrome. Sagittally reformatted CT shows the os (arrow) is displaced, positioned just behind and superior to the anterior arch of C1, with the remainder of the dens posterior to the os. There is marked narrowing of the spinal canal (double arrow)
Differentiating os odontoideum from acute fracture can be difficult [36]. Features that suggest a chronic process such as os odontoideum include a relatively thick anterior arch of C1 with a convex posterior border. MRI can also help differentiate by demonstrating normal signal intensity in the bone and surrounding soft tissues.
2.4.3 Atlantoaxial Subluxation
In atlantoaxial subluxation, the transverse, alar, and other ligaments binding the odontoid to the atlas are lax or absent whereas bones are normal. Laxity of the transverse and other stabilizing ligaments allows the anterior arch of the atlas to subluxate anteriorly [37] (Box 3.2). Atlantoaxial subluxation may be associated with structural abnormalities. Alternatively, it may be a component of various syndromes, such as Ehlers-Danlos syndrome and other connective tissue disorders or trisomy 21. The condition can also follow trauma or be associated with local osteomyelitis, pharyngitis and cervical adenitis, and juvenile idiopathic arthritis. Rotational and atlanto-occipital instability as well as skeletal anomalies may be present, and scoliosis occurs in up to 50 % of cases.
Imaging
In normal older children and adults, the gap between the posterior surface of the anterior arch of the atlas and the anterior surface of the odontoid is 2–3 mm, whereas in younger children the presence of cartilage may cause this distance to be as large as 5 mm or occasionally even 6 or 7 mm. Although slight laxity and instability can be present in normal children, the atlanto-dens interval usually does not change with flexion or extension. In atlantoaxial subluxation, the distance is usually normal on standard neutral lateral radiographs but increases 2 mm or more with flexion (Fig. 3.27). However, in some patients with trisomy 21, the atlanto-dens interval is abnormal on neutral images yet normal with flexion [37].

Abnormal motion between the anterior arch of C1 and the dens (double arrows) in patient with occipitalization of the atlas (Same patient as Fig. 3.24)
Box 3.2: True Atlantoaxial Subluxation
Associated with structural abnormalities, trisomy 21 and other syndromes, arthritis, trauma, and pharyngitis |
Increased distance between the anterior arch of C1 and dens |
Normal <5 mm in young children |
Normal <2–3 mm in older children |
Increases with flexion |
2.4.4 Atlantoaxial Rotary Subluxation
Atlantoaxial rotary subluxation may follow trauma or occur with Grisel syndrome (local inflammation in the pharynx) [38]. In the latter condition, inflammation around the dens and pharyngo-vertebral venous plexus causes the ligaments to weaken [39]. Relatively minor trauma to this area that would not affect an adult may lead to rotary subluxation in children, perhaps due to more steeply angled facets.
Imaging
The atlantoaxial facets and occiput-C1-C2 articulation are best investigated with CT as the patient passively rotates their head. Sagittal and coronal reformatted images or 3D-CT reconstructions demonstrate the abnormal relationships well (Fig. 3.28). On fluid-sensitive MRI sequences, abnormal signal in the transverse and alar ligaments is sometimes evident; this may be reactive or causative.

Atlantoaxial rotary fixation in a preteen. (a) Frontal radiograph shows asymmetry between the lateral masses of C1 and the dens, with the head tilted to the right. (b) Coronally reformatted CT in neutral confirms this finding. Axial CT images with the head turned to the right (c) and left (d) show asymmetrical widening on the right persists. (e) Sagittal T1-weighted (T1-W) MRI shows subacute hemorrhage at the alar ligament (arrow)
3 Spinal Dysraphism Complexes
Dysraphism describes the failure of midline vertebral structures to fuse. The term encompasses not only structural vertebral defects but also associated anomalies of neurologic, gastrointestinal, genitourinary, and other systems. The sequence of neural tube-notochord-yolk sac differentiation is disrupted, and thus structures that develop from the primordial layers may be affected. There may be abnormal induction of structures that develop later as well. Dysraphism is categorized as open (uncovered) or closed (covered) and the later further subdivided according to configuration (Fig. 3.29) [40]. Most open defects (meningocele (Fig. 3.30), meningomyelocele (Fig. 3.31), and myelocele) are clinically obvious, but dermal sinuses may be difficult to detect. Closed defects are completely covered with skin, which may be dysplastic and have an associated nevus, hairy patch, hemangioma, or lipoma. The spinal cord may be duplicated or split (diplomyelia and diastematomyelia), or there may be overgrowth of normal tissues (lipoma and dermoid) or herniation of one germ layer through another (neurenteric cyst).

Diagnostic algorithm of closed spinal dysraphism presenting with (a) and without (b) a subcutaneous mass (Reprinted from Badve et al. [129] with kind permission from Springer Science and Business Media)

Anterior pelvic meningocele. Sagittally reformatted (a) and axial CT (b) images show an anterior pelvic meningocele at the mid-sacral level. Dorsal elements are dysmorphic, and there is lower lumbar and sacral dysraphism with partial sacral agenesis

Meningomyelocele. Axial (a) and sagittal (b) T2-W images demonstrate a myelomeningocele at the L5–S1 level with posterior spinal dysraphism and extension of the neural placode into the dorsal aspect of the cyst (arrow). Note the trabeculated neurogenic bladder
Imaging
In both open and closed lesions, radiographs reveal structural vertebral defects in most patients. Neural arches appear widely spread, and laminar/pediculate fusion or intersegmental fusion is common, often affecting more than one level. The spinal canal may appear diffusely enlarged. Vertebral bodies are often hypoplastic, deformed, or scalloped, and there may be hemivertebrae (see Fig. 3.23). These findings may be present at the level of the neurologic defect or elsewhere in the spine. Associated mediastinal or pelvic masses may result from ectopic neurenteric rests.
Cross-sectional imaging provides essential information for skin-covered lesions. If cutaneous stigmata are present, US can help evaluate for tethered cord until about age 3 months (Figs. 3.32 and 3.33), with subsequent MRI if needed. CT is rarely performed to better delineate the bony abnormalities, while MRI exquisitely depicts the extent of neural and soft tissue abnormalities (see Figs. 3.30 and 3.31). MRI may occasionally be performed before surgery for open lesions, as it allows more precise diagnosis and surgical planning.

Tethered cord in a newborn. Longitudinal ultrasound (US) shows the spinal cord extends to the sacral-level dysraphism (arrow). Additionally there is a lipoma of the filum. Note the fused vertebrae compatible with VACTERL association (arrowhead)

Tethered cord. Longitudinal US shows conus at the S1 level, with increased fibro-fatty tissue in the canal below this level
3.1 Tethered Filum Terminale
Low conus or tethered cord generally occurs because either the terminal cord fails to involute or the filum terminale fails to lengthen. Low conus (Fig. 3.33) is characterized by abnormal termination of the cord, whereas abnormal thickening of the filum, fibrolipoma, and/or a filar cyst characterize an abnormal filum terminale (Fig. 3.34). If the conus is abnormally low, nerve roots emerge more horizontally than usual, and stretching of the vascular supply may lead to myelopathy. Tethering occasionally presents late with such symptoms as lower extremity spasticity, bladder incontinence, and toe walking. Hairy tufts, midline vascular malformations, and other cutaneous findings should prompt evaluation.

Tethered cord and multiple segmentation anomalies in a 3-year-old. Sagittal T2-W image shows the distal cord is dorsally positioned and kinked over dysmorphic posterior elements at the thoracolumbar junction. Multiple dorsal and ventral hemivertebrae
A conus terminating at the superior half of the L3 vertebral body can be considered normal in newborns, provided that normal ascent follows [41]. The inferior tip of the conus should lie at the adult level, T12 to L2/3, by 2 months of age. Absence of normal motion with cerebrospinal fluid (CSF) pulsation is another feature of tethered cord [42]. Even if surgery reveals that the conus is not tethered, symptoms sometimes occur. Tethering can also occur or recur after surgical repair of myelomeningocele, diastematomyelia, and other lesions.
Imaging
Ultrasound (US), performed between birth and 3 months of age, readily evaluates the level of the conus. Additionally, evaluation of cerebrospinal fluid pulsations can identify abnormal cord motility. If US identifies tethered cord, MRI of the entire neural axis should be performed to evaluate for occult concomitant issues. The filum is abnormally thick if it measures more than 2 mm in diameter at L5–S1 or if it is infiltrated by fibrous or fatty tissue. In addition, the filum may adhere to other structures, which limits its normal compliance and may lead to signs of traction, even if the conus terminates at a normal level. Very thin sagittal T2-weighted (T2-W) or balanced steady-state free precession images may be necessary to image the filum.
3.2 Syringohydromyelia
Hydromyelia and syringomyelia are both characterized by the presence of fluid within the central cord. Hydromyelia involves the central canal, whereas syringomyelia does not. However, the pathogenesis of these abnormalities is similar, and they are clinically indistinguishable and frequently lumped together as “syrinx”, or syringohydromyelia. A congenital syrinx may develop from an obstructed fourth ventricle in the fetus [43], but a syrinx can also develop from posttraumatic/ischemic myelomalacia or obstruction due to adhesions, compression, or tumor. Syrinxes occur in 63 % of patients with myelomeningocele and scoliosis, and therefore, preoperative MRI is recommended prior to scoliosis instrumentation [44]. A syrinx often accompanies cord tethering, congenital hydrocephalus, and other congenital abnormalities. Hydrosyringomyelia is a common component of Chiari I malformations, probably due to obstruction. Occasionally, isolated primary hydromyelia becomes evident only in late childhood [45].
Imaging
If the syrinx is large, radiographs rarely may reveal fusiform enlargement of the spinal canal, with widening of the interpediculate distance and of the sagittal diameter of the canal. This may be limited to a portion of the spine (often cervical) but in severe cases extends inferiorly into the thoracic and even lumbar spine. MRI is necessary to evaluate the spinal cord, allowing differentiation of syrinx from, for example, a long-standing neoplasm. It can also demonstrate tonsillar herniation, indicating Chiari malformation.
3.3 Diastomatomyelia
This refers to a longitudinal split in the spinal cord that develops when the notochord divides during cell migration [11]. The two parts are usually asymmetric in size and are separated by a bony (Fig. 3.35), cartilaginous, or fibrous septum; the cord usually unites distally [46]. Diastematomyelia is most common in the upper lumbar region (Fig. 3.36) but can occur anywhere from the lower thoracic spine to the sacrum. Cervical and basicranial diastematomyelia are rare and usually associated with Klippel-Feil (see Fig. 3.40 below) and Chiari malformations [47].

Diastematomyelia with bony septum (arrow)

Diastematomyelia. (a) Coronal T1-W image shows the two hemicords unite below a bony spur. (b) Axial fluid-sensitive sequence shows two hemicords
The conus is almost always low, and neurologic signs are likely related to tethering or stretching of the spinal cord. In up to 70 % of cases, each hemicord is encased in its own dural sheath. Skin changes are present in up to 80 % of patients and are similar to those found in other occult spinal dysraphisms. The location of the skin abnormality does not necessarily correlate with the septum level. Diastematomyelia should be distinguished from cord duplication or diplomyelia.
Because the notochord gives rise to vertebral bodies, diastematomyelia can be accompanied by neurenteric cysts, dorsal enteric fistulas, and abnormal vertebral bodies such as hemi- and/or butterfly vertebrae.
Imaging
Radiographs may demonstrate associated increased interpediculate distance and narrow intervertebral discs with possible vertebral body fusion. A subtle linear density may be apparent at the midline if the septum is osseous (see Fig. 3.35). CT depicts a bony septum but will identify the hemicords and a fibrous or cartilaginous septum only if CT myelography is performed. MRI depicts the cord anomaly as well as the dural sac(s) and demonstrates a fibrous, cartilaginous, or bony septum (see Fig. 3.36). The septum often extends anteriorly from the neural arch and thus may not be visibly fused to the vertebral body.
3.4 Congenital Intraspinal Tumors
Lipomas represent 1 % of all spinal cord tumors and may be intramedullary, extradural, or mixed; these lesions are often associated with meningocele, in which case they are termed lipomeningocele. Extravertebral lipomas may enter the spinal canal and cord or remain totally subcutaneous; associated anomalous bony structures may be present. Intraspinal lipomas are strongly associated with large sacral sacs, a low conus, and vertebral abnormalities. Occasionally, neural elements may enter these extraspinal tumors.
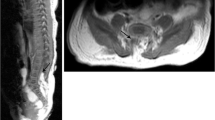
Sacrococcygeal teratomas (SCT) (Figs. 3.37 and 3.38) are the most common tumor of the fetus and newborn and are more common in girls [48]. They are closely associated with the coccyx and may involve the spinal canal. By definition they contain three germ cell layers (ectoderm, mesoderm, and endoderm). SCT may be mature or immature and may develop malignant transformation especially if internal (see staging below), in males, or if diagnosed after 2 months of age (Box 3.3). Fetal SCT may present with maternal increased fundal height and polyhydramnios, prompting imaging evaluation and diagnosis of the mass by US and subsequently MRI (Fig. 3.39) [48]. Prenatal hydrops due to a highly vascular tumor portends a poor outcome.

Huge exophytic type I sacrococcygeal teratoma in a newborn boy. There are ill-defined calcifications within the mass (arrows) (Umbilical catheters are malpositioned)

Type IV sacrococcygeal teratoma. Sagittally reformatted contrast-enhanced CT (a) and sagittal T2-W MRI (b) show a large, inhomogeneous, predominantly solid presacral mass, which elevates and obstructs the bladder

Large exophytic predominantly cystic sacrococcygeal teratoma in a 30-week fetus (single-shot fast-spin echo MRI). Arrows delineate teratoma. H fetal head, S fetal spine
Newborns usually present with a mass. Associated genitourinary anomalies include hydronephrosis and renal dysplasia. Rectal atresia/stenosis or Currarino triad (presacral mass, scimitar sacrum, and anal malformation) may be present. Treatment is complete surgical resection, including resection of the coccyx.
Box 3.3: Sacrococcyngeal Teratoma (SCT)
Most common tumor of fetus/newborn |
Contains all three germ cell layers |
May be malignant, especially if detected after 2 m of age, in male, or completely internal |
Associated genitourinary abnormalities |
Heterogeneous mixed solid/cystic mass |
Sacrococcygeal teratomas may be classified [49] as type I, external (these account for half of the cases [50]) (see Fig. 3.37); type II, predominantly external with presacral internal extension; type III, external and internal with extension into the abdominal cavity; and type IV, entirely internal (see Fig. 3.38).
Imaging
Radiographs may show segmentation abnormalities in the case of lipomas and lipomyelomeningoceles or show a presacral or external SCT mass (see Fig. 3.37). Pre or postnatal US may identify presacral masses and characterize SCT as solid, cystic, or—most often—heterogeneous (15 % are purely cystic) [48]. The vascular/shunting nature of the solid portion of the tumor may be ascertained by Doppler interrogation. MRI provides greater anatomic detail for lipomas, lipomyelomeningoceles, and associated spinal cord abnormalities and may better delineate the extent and characteristics of internal SCT (see Fig. 3.38).
4 Syndromes with Spinal Dysplasia
4.1 Klippel-Feil Syndrome
In Klippel-Feil syndrome, the cervical and upper thoracic spine fuses partially or completely, forming a bony mass (Box 3.4). In its most extreme form, the neck is essentially absent [51]. However, the term can describe any degree of congenital cervical spine fusion if the clinical triad of short neck, low posterior hairline, and limited range of motion at the neck is evident. External and middle ear atresia and malformation may be present as well. Many patients have brain stem or high vertebral anomalies, making them vulnerable to cord impingement if manipulated.
Box 3.4: Klippel-Feil Syndrome
Clinical triad of short neck, low posterior hairline, and limited range of motion at neck |
Cervical and upper thoracic spine fusion and/or segmentation anomalies |
C2–3 and C5–6, the most commonly affected |
May also have rib fusion anomalies |
Sprengel anomaly in up to 50 % |
Renal anomalies in about 50 % |
Neurologic dysfunction is associated with increased mobility at unfused levels and may progress slowly or decompensate acutely after trauma. Spinal cord compression occurs either anteriorly in flexion or posteriorly due to an hypertrophied ligamentum flavum. Additionally, excess motion above or below fused levels may result in vertebral artery occlusion [52].
Twenty-five percent of patients with congenital scoliosis or kyphosis demonstrate structural malformations of the cervical spine, and 60 % of children with Klippel-Feil have scoliosis that exceeds 15° [53]. Undescended scapula, or Sprengel anomaly (see Chap. 5), occurs in up to 42 % of patients with Klippel-Feil syndrome. In up to half of children with Sprengel deformity, there is an omovertebral bone which extends from the medial border of the scapula to the posterior elements of C5–C7 [54].
Primitive renal tissue is closely linked with the developing cervicothoracic junction, and therefore renal anomalies accompany Klippel-Feil syndrome in more than 50 % of patients. Unilateral renal agenesis is most common, but ectopia and horseshoe kidney may be seen. US should therefore be performed on all patients with congenital cervical spine abnormalities.
Imaging
Radiographs demonstrate total or partial fusion of cervical spinous processes, laminae, pedicles, and vertebral bodies (Fig. 3.40). C2–3 and C5–6 are most often affected. Upper thoracic segmentation anomalies, scoliosis, rib fusion abnormalities, and Sprengel deformity may also be seen. Progressive fusion incorporating additional vertebral bodies has been observed as these patients mature. The complex anatomy in this area lends itself to CT evaluation, and sagittal or coronal reformatted images as well as 3D reconstructions are often helpful. MRI may delineate associated pathology, such as diastematomyelia (Fig. 3.40).

Klippel-Feil syndrome with associated diastematomyelia in a teen. (a) Multilevel fusion of vertebral bodies and posterior elements. (b) Axial T1-W MRI shows split cord (arrow). Also note asymmetric musculature (Courtesy of Rebecca Stein-Wexler)
4.2 Lumbosacral Agenesis (Caudal Regression)
As with many abnormalities of the spine, caudal regression varies from mild to severe. At least one sacral segment is usually present, but the entire sacrum and lumbar spine can be absent, and in especially severe cases, even the distal thoracic spine may fail to form. Unilateral sacral maldevelopment/agenesis results in a sickle-shaped sacrum and sometimes a protruding anterior meningocele. The constellation of scimitar sacrum, presacral mass, and anorectal malformation is termed Currarino triad. Currarino syndrome is the familial autosomal dominant form of the triad related to mutations of a homeobox gene, HLXB9, on chromosome 7 [55].
The spinal cord segments distal to the aplastic vertebrae are usually absent, and bowel/bladder dysfunction is common (indeed, the astute radiologist may diagnose partial sacral agenesis when performing fluoroscopy to assess vesicoureteral reflux, neurogenic bladder, or constipation). Flexion contractures may be seen at the hips and knees. Hip dysplasia occurs in 20 % of cases (Fig. 3.41) and foot deformities in up to 50 %. More than 50 % of patients have scoliosis [56]. Up to 20 % of patients with this abnormality are children of diabetic mothers.

Sacral agenesis and left hip dysplasia in a 1-year-old girl. The iliac wings articulate with each other. The left hip is dislocated superolaterally, and the left acetabulum is dysplastic and shallow (Image copyright Shriners Hospital for Children Northern California)
Imaging
The radiographic appearance varies with the level of agenesis. If the sacrum is completely absent, the iliac bones may fuse at the midline or articulate with each other below the lowest intact vertebral segment—lumbar or thoracic. If any portion of the sacrum is present, its position is normal, between the iliac wings.
US evaluation of the urinary tract is essential, since all patients with more than one absent sacral segment have neurogenic bladder and many also have abnormalities of the upper urinary tract. Lower tract disorders such as megalourethra and congenital urethral valves as well as strictures and duplications are common [57]. Gastrointestinal anomalies may be encountered as well.
MRI of the abdomen and spine should be performed early. Spine MRI (Fig. 3.42) may demonstrate dural sac stenosis, intradural adhesions, and cord tethering, whereas abdomen MRI may demonstrate visceral and musculoskeletal abnormalities important for staged surgical repair.

Sacral agenesis in a 6-month-old. (a) The iliac wings are positioned too closely together, and most of the sacrum is absent. (b) There are no vertebral segments below S1 (arrow). (c) Sagittal T2-W image shows no vertebral segments below S1 (arrow), along with tethering of the spinal cord into a large lipoma that involves the canal, retrorectal space, and subcutaneous tissues
4.3 Neurofibromatosis Type 1
Neurofibromatosis type 1 (NF1) is inherited as an autosomal dominant trait that involves chromosome 17. Mesodermal dysplasia results in a wide variety of skeletal and soft tissue findings (see Chap. 23 for more extensive discussion of NF1). More than 50 % of patients have skeletal abnormalities, including scoliosis and kyphosis. Mesodermal dysplasia may result in vertebral wedging, concave scalloping of the anterior and lateral borders of vertebrae, and irregularities of the vertebral margins (Fig. 3.43). Posterior vertebral scalloping may result from lack of a strong, intact dura (dural ectasia) or from intraspinal neurofibromas (Fig. 3.44). This may also be seen with other mesenchymal abnormalities and collagen vascular diseases, such as Marfan, Ehlers-Danlos, and Déjerine-Sottas syndromes [58].

Neurofibromatosis. (a-frontal, b-lateral) Radiographs in brace show a sharp-angle lower thoracic dystrophic scoliosis and anterior vertebral scalloping (black arrows). There is also penciling of the posterior left tenth and 11th ribs (white arrows) (Images copyright Shriners Hospital for Children Northern California)

Neurofibromatosis. There are massive plexiform neurofibromas (arrows) as well as posterior vertebral scalloping (arrowheads)
Spinal deformity—most often kyphoscoliosis—is seen in 10–69 % of patients with NF1 and is most common in the thoracic region [59]. Curves may be dystrophic or nondystrophic. Nondystrophic scoliosis is more common and resembles the idiopathic form, although it can modulate to the dystrophic form. As many as 80 % of curves in children younger than 7 years of age will indeed eventually become dystrophic [60].
Dystrophic scoliosis is characterized by a sharply angulated, short-segment curve of four to six vertebral levels (see Fig. 3.43). Neurofibromas may be present, and vertebral bodies at the apex are dysplastic and may appear wedged, scalloped, and rotated. Foramina may be enlarged (usually due to meningeal ectasia and CSF pulsations), and transverse processes can be quite narrow; this configuration contributes to spine instability. The ribs may appear thin (“penciled”), twisted, and irregular. If very thin, they may penetrate the spinal canal at the apex of the curve and cause neural compression [59]. Dystrophic kyphoscoliosis progresses rapidly and requires aggressive treatment.
Severe kyphosis of the cervical spine can suggest the diagnosis of NF1. Cervical spine abnormalities resemble those elsewhere in the spine and occur in as many as 40 % of those with scoliosis [61]. Instability, including atlantoaxial instability, and neurologic deficits may be present in addition to kyphosis [62].
Imaging
Radiographs depict the bony findings described above. Preoperative MRI is essential for patients with NF1, and many advocate routine spine MRI. This allows characterization of dural ectasia, anterior and lateral meningoceles, myelopathy, thecal volume (often increased), and potential intraoperative cord compression. Neurofibromas are readily identified because of their markedly increased T2 signal, characteristic dumbbell or plexiform appearance, and avid enhancement.
4.4 Additional Syndromes
Abnormal formation and growth of the spine are common in many bone dysplasias and heritable syndromes. Kyphosis occurs in 70 % of some dysplasias, especially achondroplasia (Fig. 3.45), mucopolysaccharidoses, spondyloepiphyseal dysplasia congenita, diastrophic dysplasia, and metatropic dwarfism. Scoliosis occurs in about one fourth of patients with spondyloepiphyseal dysplasia (Fig. 3.46), diastrophic dysplasia, and metatropic dwarfism. Clinical and skeletal factors can assist with diagnosis, but genetic mapping is necessary in order to differentiate between similar syndromes.

Achondroplasia. (a) There is progressive decrease in the interpediculate distance in the lumbar spine. The pelvis also shows typical features of achondroplasia. (b) The pedicles are short, and there is focal thoracolumbar kyphosis. The discs are nearly as tall as the vertebral bodies (Images copyright Shriners Hospital for Children Northern California)

Spondyloepiphyseal dysplasia. There is diffuse platyspondyly and biphasic scoliosis. Fragmentation of the femoral heads
Some of the more common syndromes and dysplasias are discussed in Chap. 23 (such as achondroplasia, osteogenesis imperfecta, and trisomy 21). A few unusual syndromes are discussed briefly below, but thorough presentation is beyond the scope of this text. Texts on skeletal dysplasias offer a more complete review [63].
Spondylocostal dysostosis (Jarcho-Levin syndrome) is an autosomal recessive disorder, characterized by congenital abnormalities of the spine and associated dysplasia of the ribs, leading to severe lordosis and variable thoracic insufficiency (Fig. 3.47). Spondylothoracic dysostosis (Lavy-Moseley syndrome) has less severe lumbar and rib anomalies, but there is a crab-like thoracic deformity due to posterior element fusion, and the clinical prognosis is worse (Fig. 3.48). It is seen predominantly in children of Puerto Rican descent [64].

Spondylocostal dysostosis with multiple hemivertebrae and butterfly vertebrae

Spondylothoracic dysostosis with crab-like deformity of the thorax due to posterior rib fusion
Marfan syndrome is characterized by reduced amounts of stable collagen and such abnormalities as ectopia lentis, thoracic kyphoscoliosis, dural ectasia, arachnodactyly, pectus excavatum deformity, and cardiovascular lesions. Any of these traits may be partially expressed. Scoliosis is present in 40–60 % of patients. Vertebral scalloping and intrasacral meningoceles may also occur. With autosomal dominant inheritance, Marfan syndrome should be considered in tall, myopic children with pectus excavatum deformity and a family history of scoliosis.
The most important feature of trisomy 21 is the malformation, aplasia, or laxity of the transverse ligament that holds the odontoid to the axis, resulting in C1–2 subluxation in 20 % of patients (discussed in the section above on atlantoaxial subluxation). In addition, cervicothoracic kyphosis may develop in young adults. It resembles juvenile kyphosis radiographically, with decreased disc space height and with end plate irregularities.
Significant abnormalities of the spine occur in many other dysplasias. For example, kyphoscoliosis is a prominent feature of diastrophic dwarfism, and extreme platyspondyly is seen with metatropic dwarfism. Patients with osteogenesis imperfecta (Fig. 3.49) may have severe scoliosis, or their spine may be normal. Butterfly vertebrae may be seen in patients with Alagille syndrome (see Fig. 3.19), also known as arteriohepatic dysplasia [65]; these patients also have a paucity of bile ducts (resulting in chronic cholestasis) and congenital heart abnormalities such as tetralogy of Fallot and pulmonary artery stenosis.

Osteogenesis imperfecta, characterized by severe scoliosis, biconcave vertebral bodies, osteoporosis, and acetabular protrusion (a-frontal, b-lateral)
5 Idiopathic Scoliosis
The word scoliosis, derived from the Greek word for curvature or crookedness, refers to curvature of the spine in the coronal plane. About 80 % of cases of scoliosis [66] are of seemingly spontaneous origin and thus termed idiopathic. Non-idiopathic causes of scoliosis include congenital spinal anomalies, neuromuscular disease, and irritative, traumatic, metabolic, or extraspinal lesions (Table 3.2). Non-idiopathic scoliosis may also be associated with neuroectodermal or mesodermal diseases or syndromic dysplasias.
Many theories have been advanced to explain idiopathic scoliosis, etiology, including connective tissue abnormalities, unusual growth patterns, neuromuscular and central nervous system (CNS) aberrations, and hormonal variations, but none seems to explain all the features that may be encountered [67]. There is evidence for multifactorial inheritance [67–69], as the rate of scoliosis occurring in first-degree (10–11 %) or first- through third-degree (26 %) relatives is up to 20 times that expected in the normal population. Geographic variation in the incidence of scoliosis is also well recognized. Estimates of the prevalence of adolescent idiopathic scoliosis in the normal population (in patients 10–16 years old) varies from 2 % to 4 %, depending on how scoliosis is defined [68].
It is important to remember that spinal curvature is three dimensional [66]. Biomechanical initiation of scoliosis appears to begin with axial rotation of the involved thoracic vertebral bodies, resulting in increased pressure on the more ventrally placed vertebrae and decreased pressure on the more dorsally placed vertebrae [70]. In the immature skeleton, bone growth is inhibited by growth plate compression and accelerated by growth plate tension; this results in the more ventral part of the spine becoming the concave side of the curve and the dorsally located vertebrae the convex side. Mechanical imbalance accelerates during skeletal growth, with resultant progression of scoliosis. Supporting this, the pedicles and laminae are short and thick on the concave side of the curve, and the cartilage of the ipsilateral end plates demonstrates degeneration and decreased chondrogenesis along with osteogenesis, consistent with growth inhibition.
Scoliosis may progress during growth but ceases with skeletal maturity unless the curve is severe (more than 30°) [66].
5.1 Clinical Variants
Idiopathic scoliosis has been divided into three temporal classes, defined by age of onset/recognition/main period of progression: infantile (0–3 years), juvenile (4–10 years), and adolescent (11–18 years) (Box 3.5). It can alternatively be grouped into early onset (<5 years of age) and late onset (>5 years of age), based upon the risk of developing cardiopulmonary complications; progressive early-onset scoliosis can lead to cardiopulmonary compromise in later life, whereas late onset does not [71]. The following discussion employs the classic division into three different age groups.
Box 3.5: Idiopathic Scoliosis
Infantile | |
Onset <3 years old, 75 % resolve by age 2 years | |
0.5–4 % of cases of scoliosis | |
Usually thoracic levoconvex | |
More likely to progress if | onset after age 1 year |
Double major curve | |
>35° | |
Altered articulation between ribs and vertebrae | |
Juvenile | |
Onset 4–10 years old, almost all will progress | |
12 % of cases of scoliosis | |
Usually dextroconvex | |
Adolescent | |
Onset 11 years old to skeletal maturity | |
Most common | |
Usually thoracic dextroconvex, compensatory lumbar levoconvex | |
Curve increases most quickly during growth spurt (Risser 0–1) | |
Infantile scoliosis is diagnosed before the age of 3 years and accounts for 0.5–4 % of all idiopathic scoliosis. The clinical picture is typical: it is more common in boys than girls, is usually not present at birth but develops during the first 6 months of life, and is associated with hip dysplasia in 25 % [72]. Plagiocephaly is also commonly associated [73]. Almost all (85–90 %) curves are thoracic and convex to the left (levoconvex) (Fig. 3.50). The curves resolve without treatment during the first 2 years of life in three quarters of these patients. Factors that suggest a curve will progress include: (1) double major curves, (2) curvature of more than 35°, (3) onset after the age of 1 year, and (4) changes in the normal relation of the rib articulation with the vertebra [74, 75]. The rib-vertebral angle difference (RVAD) also predicts progression (see discussion on measurement, below).

Infantile scoliosis. There is levoconvex thoracolumbar scoliosis, but no segmentation anomalies. The RVAs are 74° and 30°, and the RVAD is 74°–30° = 34°, indicating increased risk of curve progression (see Fig. 3.58 and text) (Image copyright Shriners Hospital for Children Northern California)
Juvenile idiopathic scoliosis is first diagnosed between the ages of 4 and 10 years and represents about 12 % of all idiopathic scoliosis. There is a male predominance in younger patients (4–6 years), but after that age, the curve is more common in girls. Most curves in patients in the United States are convex to the right (dextroconvex). Scoliosis first recognized at this age almost invariably progresses.
Adolescent scoliosis is the most common type of idiopathic scoliosis and is diagnosed in patients after age 11 years and before skeletal maturity has been attained. On average, girls present at 13 years of age and boys at 14. There is a female predominance of 4:1–8:1. The majority of these curvatures are dextroconvex in the thoracic region with a levoconvex lumbar compensatory curve (Fig. 3.51), but other patterns are common. Curvature increases most rapidly during the normal growth spurt, coincident with development of pubic hair and breast buds; by the time of menarche and male axillary hair development, about 18 months of growth (and potential curvature increase) remain. Curves are thus most likely to progress in patients between age 9 and 13 years, in those with progression skeletal maturity between 9 and 14 years, if the Risser stage is 0 or 1 (see following discussion), or if annual vertical growth exceeds 2 cm [66]. The younger the patient and the greater the curve, the faster the curve will progress [76].

Adolescent idiopathic scoliosis in a 15-year-old girl. Primary dextroconvex thoracic and secondary levoconvex curves. Incidental ununited spinous process at T12 (Same patient as Fig. 3.69) (Image copyright Shriners Hospital for Children Northern California)
5.2 Curve Patterns and Terminology
The Scoliosis Research Society has attempted to standardize terminology [77]. The curve is identified by the level of the apical and end vertebrae, as well as the side of convexity. There are five possible patterns: (1) cervicothoracic, (2) thoracic (most common), (3) thoracolumbar, (4) lumbar, and (5) double structural (double thoracic, combined thoracic and lumbar, combined thoracic and thoracolumbar) [77]. Although the length and severity of the curve may change during growth, the pattern usually does not.
The direction of curvature is labeled according to its convexity. Thus, a curve whose convex side faces the left is termed “levoconvex,” whereas a curve whose convex side faces the right is termed “dextroconvex.”
All curves can be identified as structural (also called major) or nonstructural (minor). Nonstructural curves are flexible with lateral bending to both sides, straightening when the patient bends toward the convexity (Fig. 3.52). Structural curves are less flexible and retain some deformity with lateral bending. With severe or long-standing curves, vertebral bodies become wedged and thoracic kyphosis is reduced, leading to straightening of the thoracic spine. A compensatory curve that returns spinal alignment to the midline is usually present above or below a structural curve of any significant magnitude. This compensatory scoliosis is initially flexible but may become structural with time. The most characteristic idiopathic pattern is a structural dextroconvex thoracic curve with a compensatory levoconvex lumbar curve.

Scoliosis with lateral bending views. Neutral view shows primary dextroconvex thoracic and secondary levoconvex lumbar scoliosis. With rightward bending, the rightward thoracic curve does not significantly change. With leftward bending, there is complete correction of the leftward compensatory curve (Images copyright Shriners Hospital for Children Northern California)
Finally, the severity of the curvature can be graded into the following groups: I, less than 20°; II, 20 to 30°; III, 31 to 50°; IV, 51 to 75°; V, 76 to 100°; VI, 101 to 125°; and VII, more than 125°.
5.3 Imaging Techniques
Radiology plays an essential role in identifying and following any type of scoliosis. To analyze alignment from C5 to the level of the hips, a frontal (coronal) standing projection of the spine is obtained (without shoes), from the lower edge of the mandible to the level of the top of the greater trochanters. Although some centers obtain the initial frontal image in the anteroposterior projection in order to achieve greater bone detail at the spine, the posteroanterior projection is generally preferred, since it delivers a lower radiation dose to the breasts and thyroid. If the patient is unable to stand, a sitting or supine view may be performed.
Structures outside the field of view are excluded by collimation and using a gonadal shield. To reduce breast and thyroid exposure, a combination of posteroanterior positioning, breast shields, high tube voltage, contoured beam filtration, high-speed films and screens, replacement of anti-scatter grids with air gap techniques, and segmented field radiography with beam collimation can be employed [78, 79]. Using computed radiography or digital imaging systems also reduces dosage. For example, the EOS 2D/3D low-dose digital imaging device (EOS Imaging SA, Paris, France) simultaneously captures frontal and lateral standing images that can then undergo 3D reconstruction to create 3D surface images (Fig. 3.53). These can be manipulated further to allow detailed analysis of curve structure [80]. All of these measures should be employed in any facility performing a significant number of spine examinations, as radiographic monitoring of scoliosis has been shown to increase the risk of breast cancer, thyroid cancer, and infertility [76, 81–84].

Spine image obtained with low-dose digital EOS 2D/3D device. Frontal (a) and lateral (b) images of the spine can be processed into a variety of 3D images, including coronal (c), sagittal (d), and axial (e). In this case, the apical and end vertebrae have been highlighted with yellow and blue. The axial 3D image shows the degree of rotation and displacement (Courtesy of Leslie E. Grissom)
Since scoliosis is a three-dimensional process and the sagittal component of the thoracic curve must also be assessed, initial evaluation includes a lateral projection of the spine. Further lateral projections are usually only performed if there is evidence of abnormal kyphosis or lordosis. The sagittal image is obtained with the same field limits, shielding, and technique described above. To remove the effect of upper extremity positioning on forward or backward lean, the arms should be extended parallel to the floor with the wrists resting on a support (usually an intravenous [IV] pole) so they do not bear weight.
Other imaging techniques may be necessary for surgical planning or to determine the need for bracing. In these situations, the degree of flexibility and correctability of curvatures is evaluated with supine maximal bending images. If the patient cannot cooperate with bending studies, a stretch or traction supine view may be obtained (Fig. 3.54). Since dose-reduction techniques do not provide optimal visualization of osseous anatomy, detailed anteroposterior (AP) images are indicated to further evaluate suspicious areas. In addition, complex osseous abnormalities may be assessed on noncontrast CT with three-dimensional (3D) reformation. All of these examinations should be ordered judiciously to minimize radiation exposure.

Neurogenic scoliosis in a 7-year-old girl. (a) Standing view shows severe dextroconvex thoracolumbar C-shaped scoliosis. Note pelvic tilt, gastrostomy button, and gracile long bones. (b) Stretch supine view shows improvement in the rightward scoliosis. (c) Sitting view in brace after posterior instrumentation and fusion shows significantly improved curvature (compared to preoperative study) (Images copyright Shriners Hospital for Children Northern California)
Additional imaging may be helpful. For example, skeletal maturity can affect treatment decisions; the hand, elbow, and iliac crest apophyses provide information about skeletal maturity at different ages (see Chap. 2). If leg length discrepancy (Fig. 3.55) is suggested by clinical evaluation or significantly unequal iliac crest height, leg length measurement may be performed by a variety of techniques (see Chap. 13). In order to eliminate the effect of leg length disparity on spinal curvature, erect spine radiographs can be obtained with the patient sitting (Fig. 3.56), or a standing view can be obtained with blocks elevating the short extremity. Nuclear scintigraphy is reserved for demonstrating whether an irritative focus (e.g., osteoid osteoma) is causing scoliosis and for evaluating postoperative pseudarthrosis.

Scoliosis resulting from leg length inequality in an 8-year-old boy. (a) The right hemi-pelvis is higher than the left, and there is levoconvex thoracolumbar scoliosis. (b) Left femoral (and to a lesser degree tibial and fibular) hypoplasia (Images copyright Shriners Hospital for Children Northern California)

Flexible positional spinal curvature due to leg length inequality. (a) Standing view shows dextroconvex spinal curvature with pelvic tilt (left side higher due to short right lower extremity). (b) Curvature flips when the patient sits (Images copyright Shriners Hospital for Children Northern California)
In cases of congenital scoliosis, the urologic system should be evaluated by screening US, whereas the presence of symptoms dictates the need for renal US in cases of idiopathic scoliosis. MRI is useful in assessing underlying or occult spinal pathology in scoliosis. MRI should be performed when intraspinal tumors are suspected based on clinical or radiologic examination; indeed, 27 % of patients with intraspinal tumors also have scoliosis [85] (Fig. 3.57). Other clinical indications for spine MRI include spinal dysraphism, early age of onset of scoliosis, painful scoliosis, abnormal neurologic exam, rapidly progressive curve (more than 1° per month), atypical curve (such as levoconvex thoracic), developmental abnormalities such as neurofibromatosis or Klippel-Feil syndrome, and for surgical clearing or to assess for postoperative complications [79, 86].

Scoliosis in a 13-year-old boy with hyperactive reflexes and and positive Babinski test. (a) Levoconvex thoracic and dextroconvex lumbar spinal curve. Sagittal T1-W gadolinium-enhanced (b) and T2-W (c) images show an enhancing, partially cystic ependymoma in the cervical and upper thoracic cord, bulging upward into the posterior fossa. There is also a thoracic syrinx (arrow)
Two to four percent of neurologically normal patients with idiopathic adolescent scoliosis have CNS abnormalities [87–89], the most common being Arnold-Chiari I malformation, subtle syrinx, tethered cord, and filar lipoma [86]. In the absence of a neurologic deficit, the first two would usually not be treated [90]. However, treating a CNS lesion may prevent neurological deterioration and progression of scoliosis, and the presence of an untreated lesion may compromise successful surgical correction [91].
5.4 Radiologic Analysis and Imaging Techniques (Boxes 3.6, 3.7)
Box 3.6: Techniques for Measuring Scoliosis
Cobb: assesses angle of curvature in coronal or sagittal plane |
Angle between lines parallel to end plates or pedicles of upper and lower vertebrae |
Difference of 6° indicates progression |
Rib-vertebral angle difference (RVAD): indicates likelihood of progression |
Evaluate apical vertebra on frontal projection |
Based on comparing bilateral rib-vertebral angles (RVA) |
RVA: angle between line connecting/extending from the head and neck of each rib (where neck widens into shaft) |
Vertical line perpendicular to vertebral end plate |
RVAD = concave side RVA minus convex side RVA |
RVAD >20° indicates progression is likely |
Box 3.7: Nash and Moe Technique to Assess Vertebral Body Rotation
Evaluates position of pedicle on convex side of curve on frontal projection | |
Divides convex side of vertebral body into thirds | |
1+ | Pedicle in lateral third |
2+ | Pedicle in middle third |
3+ | Pedicle in medial third or at midline |
4+ | Pedicle projects over other side of the vertebral body |
Several key components of every curve must be assessed, including identifying the apical and transitional vertebrae. The apical vertebra is the one that is most rotated and most laterally displaced; its end plates are the least tilted (Fig. 3.58). Rotation and lateral displacement of the vertebrae above and below this apical vertebra will diminish until either no curve remains or an opposite curve begins. The vertebrae at the termination of the curvature are called either “end” or “transitional,” respectively. These vertebrae may be identified by their lack of rotation and by the fact that lines drawn along their superior and inferior end plates point maximally toward the concavity of the curve. Additionally, at the transitional or end vertebrae, the intervertebral disc space will be maximally wedged, with the greatest width on the convex side of the curve.

Triphasic idiopathic scoliosis in a 16-year-old girl. The primary dextroconvex thoracic curve Cobb angle measures 54° (A apical and E end vertebrae of this curve). There are compensatory levoconvex lumbar and upper thoracic curves. The thorax is centered to the right of the pelvis
Once the curve extent has been determined, all subsequent measurements should use the same vertebral end points, unless the curve becomes longer or shorter. In that case, the new examination should be compared with the most recent, assessing the alteration in curve extent.
The Scoliosis Research Society recommends the standardized Cobb method of calculation. A line is drawn along the superior end plate of the upper end (or transitional) vertebra, and another is drawn along the inferior end plate of the lower end (or transitional) vertebra (Fig. 3.58). If the end plates are indistinct, the tops or bottoms of the pedicles may be used. The angle between these two lines (or between lines drawn perpendicular to them, if the angle is not sufficiently acute to allow measurement) is the degree of curvature. Severe vertebral wedging may exaggerate the true curve measurement.
When two oppositely convex curves are present but contiguous, the lower end vertebra of the upper curve represents the upper end vertebra of the lower curvature and is therefore included in both measurements. This technique has an interobserver error of 7–9° [92, 93]. The 95 % confidence level for any observed change between serial measurements is 3.3–10° for scoliosis and 11° for kyphosis [93–95]. The variation is less with computer-generated measurements [79]. Generally, a difference of 6° between serial radiographs indicates curve progression [79].
The rib-vertebral angle difference (RVAD) predicts curves that are more likely to progress. The rib-vertebral angle (RVA), which is determined on a frontal radiograph at the apical vertebra and its corresponding ribs, is formed by the intersection between a line drawn along the midpoint of the head and neck of each rib and a vertical line drawn perpendicular to the superior or inferior end plate of the vertebra (Fig. 3.59). The RVA is usually smaller on the convex side of the curve. The RVAD is obtained by subtracting the RVA on the convex side from the RVA on the concave side. If the RVAD is greater than 20° and if the head of the rib projects over the vertebra, the curve will likely progress. In addition, when both the RVA is less on the concave side than the convex side of the curve and the eleventh and twelfth ribs are very oblique to the spine, a double structural progressive curve is likely.

Rib-vertebral angle (RVA) and rib-vertebral angle difference (RVAD) calculation. The RVA is the angle formed between a perpendicular line drawn to either the upper or lower end plate of the apical vertebra with a second line drawn from the midpoint in the rib head to the midpoint in the rib neck just medial to the region where the neck widens into the shaft of the rib (angle A for concave side, B for convex side). The RVAD is the difference between the RVA on the concave and convex sides of the curve (A-B)
The Cobb method is also used to determine the degree of lordosis or kyphosis on the lateral projection (Fig. 3.60). Thoracic kyphosis is usually measured between the superior end plate of T3 and the lower end plate of T12; lumbar lordosis is measured between the top of Ll and the lower edge of L5. Normal thoracic kyphosis ranges from 21 to 42°, and normal lumbar lordosis ranges from 31 to 50° [27]. These values do not change with patient age, sex, height, or weight [96]. Severe scoliosis is often associated with thoracic lordosis and straightening of normal lumbar lordosis.

Mildly increased thoracic kyphosis in an 8-year-old girl. Cobb measurement technique delineates 45° of kyphosis
Rotation is maximal in the apical vertebra of the structural curve. One or more of the vertebrae above or below the maximally tilted end vertebra may also be rotated. Generally, the vertebral body rotates anteriorly toward the convexity, although CT studies have shown both opposite rotation with scoliosis and the presence of some rotation in patients without scoliosis [97].
Rotation is common but not obligatory; occasionally, fairly severe curves have no rotation. Nash and Moe describe a method for estimating rotation, mild to severe, and this should be sufficient for clinical purposes [66, 98] (Fig. 3.61). The pedicles of a non-rotated vertebra are symmetrically positioned, tangent to the edges of the vertebral body. With rotation, the body usually turns toward the convex side of the curve, and the pedicle on this side appears to move away from the edge of the vertebral body and toward the midline. The pedicle on the concave side becomes progressively less apparent.

Grading of vertebral rotation by the Nash-Moe method. (a) No rotation. (b) Grade 1. (c) Grade 2. (d) Grade 3. (e) Grade 4. On a frontal spine radiograph, each involved vertebra is bisected by an imaginary line, and the half vertebra on the convex side is then divided into outer, middle, and inner thirds. Rotation is quantified on the basis of the location of the convex-side pedicle in one of these segments and the visibility of the concave-side pedicle, which gradually disappears as rotation progresses. The pedicles are shown as vertically oriented ovals (Reprinted with permission from Kim et al. [66])
Rotation can be estimated by assessing the pedicle position relative to the vertebral body (evaluating the pedicle on the convex side of the curve). The distance between the vertebral edge and its midline is divided into thirds. If the pedicle lies in the lateral one third, rotation is termed 1+; in the middle one third, 2+; and in the medial one third or in the midline, 3+. Rotation greater than this is denoted 4+. Roughly speaking, the percentage of migration of the pedicle equals the degree of rotation of the body; for example, 50 % pedicle displacement (pedicle in midline, or 3+) equals 50° rotation. CT may be employed if this determination affects critical surgical decisions.
How a curve changes with bending determines whether or not it is structural (see Fig. 3.52). While a nonstructural curve corrects scoliosis and vertebral rotation (returning to normal, straight alignment), morphologic changes prevent structural curves from correcting. Nonstructural curves are usually either compensatory to structural scoliosis or secondary to irritative or extraspinal causes, most often leg length discrepancy. With leg length discrepancy, the convexity of the curve is ipsilateral to the short limb (see Fig. 3.55). A nonstructural curve usually does not progress; however, it may become structural due to growth retardation on the concave side of the curve and subsequent ligament shortening [66]. Additionally, wedging of the vertebral body indicates a permanent structural change. The degree of wedging can be graded from 1+ to 4+ [99].
Coronal and sagittal balance is measured using a plumb line from the center of the C7 vertebral body (Fig. 3.62). On the frontal view, this line is compared with the central sacral vertebral line (CSVL). The CSVL bisects the sacrum and is drawn perpendicular to a tangent connecting the iliac crests [66]. The normal CSVL is within 2 cm of the plumb line. On the lateral view, the plumb line should be within 2 cm of the sacral promontory (defined as the most cephalad part of the ventral sacrum) [100].

Idiopathic scoliosis in a 14-year-old girl.(a-frontal, b-lateral) The C7 plumb line is the solid white line on the frontal and lateral views dropped from the center of the C7 vertebral body. The central sacral vertebral line (CSVL) (dashed white line), which bisects the sacrum, is drawn perpendicular to a line connecting the tops of the iliac crests (dashed black line)
On the lateral view, the gravity line and pelvic incidence assess gravitational alignment between the trunk and feet. The center of balance, or gravity line, should be located anterior to the spine on the lateral view [101]. Since anteroposterior translation and/or rotation of the pelvis attempts to maintain sagittal balance, evaluating pelvic alignment is essential in patients with scoliosis [101]. Pelvic incidence, which is the sum of pelvic tilt and sacral slope, is utilized to describe pelvic alignment and guide corrective surgery (Fig. 3.63).

Determining pelvic incidence. The sacral slope is the angle between the horizontal and the sacral plate. Pelvic tilt is the angle between the vertical and a line drawn between the midpoint of the femoral head and the midpoint of the sacral plate. Pelvic incidence is the sum of these two angles (Reprinted with permission from Schwab et al. [101])
Evaluation of maturity is crucial when following idiopathic scoliosis, as curvature may increase until the spine is mature. Radiologic observation of the iliac apophyses can gauge maturity [102]. The Risser stages of maturity, which correlate with standard bone age determinations, progress as follows: the iliac apophysis first ossifies laterally (stage 1), and the maturation process extends medially until there is complete capping of the crest (stage 4); complete fusion of the apophysis (stage 5) occurs some years later, often after age 20 (Table 3.3) (Fig. 3.64). However, average age of menarche—and thus maximal spinal growth—occurs 8 months before stage 1. During the period of accelerational growth, before ossification of the iliac crest apophysis, skeletal maturation may be assessed by evaluating the olecranon apophysis (Sauvegrain method) or the configuration of the hand (Greulich and Pyle, among others) (see Chap. 2 for additional discussion of techniques for assessing skeletal maturation). The vertebral ring apophyses tend to fuse at the same time as the iliac crest apophyses. Thus the spine may continue to grow and scoliosis to progress, until Risser stage 5 and after extremity growth plate fusion [103].

Risser staging. The iliac crest apophysis ossifies from anterior and lateral to posterior and medial. At stage 0, the apophysis has not ossified. For stages I to IV, divide the iliac crest into four equal parts and assign the Risser stage according to the most advanced point of ossification of the apophysis (this patient is Risser stage IV). At stage V, the apophysis has fused (Image copyright Shriners Hospital for Children Northern California)
Sequential monitoring is performed at intervals of 4–12 months, depending on the nature of the curve and age of the child [71]. At each visit, the same end vertebrae should be measured and compared to several previous examinations. After growth stops, curves greater than 30° should be monitored for progression about every 5 years [66].
5.5 Treatment
Treatment depends on the etiology and severity of the scoliosis, associated symptoms, and the patient’s perception of the deformity. Options include observation, bracing, and surgery.
5.5.1 Orthotic Management
Nonoperative treatment of scoliosis generally involves orthotic (brace) control of curvature and is usually indicated when curvature approaches the 25–30° range and when children have significant growth remaining (Fig. 3.65); it is also indicated at lesser degrees of scoliosis in younger children with rapid progression. For most curvatures, however, a plastic, molded, underarm thoracolumbosacral orthosis (TLSO) is adequate. The brace can be expected to somewhat decrease the curvature (e.g., from 30 to 20°) and maintain the curvature at this value. In general, the Cobb angle should decrease about 30 % when the brace is in place [104]. After bracing therapy concludes, the curvature may remain improved or gradually return to its original value by the time spinal growth is complete. In that case, the orthosis has served to halt progression.

Idiopathic scoliosis. At age 11 years, this girl had a 43° dextroconvex scoliosis (a) that decreased to 33° after bracing (b). At age 12, after completion of brace therapy, her thoracic curve had worsened to 56° (c). Bending views show that the dextroconvex thoracic curve decreases to 28° with rightward bending (d) and the 25° levoconvex secondary curve completely corrects with leftward bending (e). After posterior spinal fusion (f), the thoracic curve measures 7°, and the secondary levoconvex lumbar curve has resolved (Images copyright Shriners Hospital for Children Northern California)
5.5.2 Surgical Management
Spinal fusion is indicated when scoliosis approaches or exceeds 50°. The primary goal is to prevent curve progression by fusing the involved segments; secondary goals include restoration of truncal balance, improvement in spinal curvature, and improved cosmesis (such as diminishing a rib hump) [71]. Occasionally, surgery corrects cardiopulmonary impairment caused by thoracic deformity. As each case is different, the decision for surgical treatment varies, based on likely outcomes with or without surgery [71]. Fusion is most commonly performed posterior to the laminae; fusion of the anterior vertebral column is generally combined with posterior fusion and performed for cases of severe neuromuscular, congenital, and dysplastic scoliosis. Hardware, technique, and nomenclature are continually being modified. While detailed explanation is beyond the scope of this text, some typical procedures are illustrated in Chap. 27.
5.5.2.1 Posterior Spinal Fusion (see Fig. 3.65)
Permanent growth arrest and halt in curve progression are obtained by bone grafting and facet obliteration, with instrumentation holding the spine in place during the period of graft maturation. The original stabilization apparatus was the Harrington rod, but many methods of segmental fusion offering greater correction and stability have since been developed. These include custom rods, pedicle screws, laminar hooks, sublaminar wires, and crossbars (see Fig. 3.65). Growing implants may be employed in young patients with severe early-onset congenital scoliosis, syndromic scoliosis, or scoliosis due to cerebral palsy, spinal muscular atrophy, or chromosomal abnormalities. When growing rods are used, the spine is not fused; this allows for continued spinal growth while stabilizing or improving curvature (Fig. 3.66).

VEPTR instrumentation in an 8-year-old boy. Radiographs before (a) and after (b) lengthening show distraction (arrow vs. arrowhead) at the tandem connector (Image copyright Shriners Hospital for Children Northern California)
5.5.2.2 Anterior Spinal Fusion
This may be performed alone or in combination with posterior fusion; the latter is useful when severe neuromuscular forces would defeat isolated posterior fusion or when bone is dystrophic or the configuration of posterior elements is suboptimal (e.g., with spinal dysraphism). In this procedure, the intervertebral discs are excised and the gap is filled with bone. Instrumentation is applied to the vertebral bodies along the convex side of the structural curve, compressing the vertebrae and correcting the curve (Fig. 3.67). If needed, bone struts are applied to the anterior aspect of the vertebral bodies. With anterior fusion, fewer levels may need to be fused.

Focal lumbar rotoscoliosis in a teenage girl. (a) Asymmetric pedicles indicate rotation. (b, c) Postoperative images show scoliosis has improved after anterior spinal instrumentation
5.6 Postoperative Evaluation
In the immediate postoperative period, a chest radiograph is often obtained to evaluate lung expansion and possible pneumothorax (especially if a transthoracic approach was used for anterior fusion). Very rarely, acute neurological postoperative symptoms may necessitate CT to evaluate for hardware malplacement/displacement or MRI to evaluate for cord or nerve injury. Postoperative infection or abscess is best defined by contrast-enhanced CT. Before hospital discharge, it is necessary to confirm that instrumentation is intact and has not displaced.
Occasionally, gastroduodenal obstruction develops during treatment, attributed to compression of the third part of the duodenum by the superior mesenteric artery (SMA). Although the exact mechanism is uncertain, the duodenum may become trapped in the angle formed by the SMA and the aorta due to elongation of the spine and stretching of the mesentery. Weight loss may contribute to compression of the duodenum between the SMA and the aorta. Abdominal radiographs show gastric dilatation, and the diagnosis can be confirmed by administration of a small amount of barium after gastric decompression. Extrinsic compression of the SMA is characteristic, appearing as a sharp, oblique termination of the barium column in the transverse duodenum (Fig. 3.68). This obstruction may partially correct after the patient is placed in the left lateral decubitus or prone position. Surgical intervention is rarely necessary; instead, nasojejunal feeding with continued gastric drainage usually suffice until symptoms resolve.

Superior mesenteric artery (SMA) syndrome in a patient with cerebral palsy and scoliosis. (a) Abdomen radiograph shows lumbar levoscoliosis and gaseous distension of the stomach, along with bilateral dislocated hips. (b) Image from the upper gastrointestinal tract shows a dilated proximal duodenum with abrupt vertical cutoff at the level where the SMA crosses the transverse duodenum (Images copyright Shriners Hospital for Children Northern California)
After spinal instrumentation, the patient must be monitored at least for skeletal maturity to ensure security of fixation, confirm integrity of hardware, exclude pseudarthrosis development, and evaluate for recurrent or new deformity [71]. Successful surgical spinal fusion prevents further increase in the length of the fused segment, although some anterior growth may occur and result in narrowing of the anterior disc space. This may then lead to lateral bulging of intervertebral discs [71, 105].
The fusion mass is best seen along the concavity of the curvature. Oblique radiographs may be necessary to evaluate all portions. Eventually, the facets and intervertebral discs are obliterated by bone ingrowth. Surgical complications may include rod and wire breakage (Fig. 3.69), hook slippage, spondylolysis at levels of increased stress below the fusion, and dislocation of facets above the site of fusion after severe trauma.

Fractured spinal instrumentation rods (arrows)
At each postoperative evaluation, the density and maturity of bone fusion should be evaluated. Pseudarthroses are recognized as clearly demarcated gaps in the continuity of bone fusion, with sclerosis at the edges of the discontinuity; motion shown on bending films proves true functional pseudarthrosis is present. However, only 68 % of surgically proven pseudarthroses are visible on radiographs [106]. Furthermore, areas of decreased bone density on radiographs as well as irregular uptake on nuclear scintigraphy, without sharp edges or sclerosis, are very common during the healing process and do not indicate nonunion unless they persist unchanged for longer than 6 months. Although oblique images can be obtained, CT is the standard to confirm pseudarthrosis (Fig. 3.70) [107]. Hardware failure late after fusion suggests but is not always associated with pseudarthrosis.

Pseudarthrosis after posterior spinal fusion. Axial CT images at the (a) T12–L1 and (b) L1–2 levels show a pseudarthrosis on the right (arrow) at L1–2 while the fusion mass at T12–L1 is intact. Pseudarthrosis (arrow) is also seen on the 3D-CT reformatted image (c) (Same patient as Fig. 3.51)
Gallstones have been reported in almost 20 % of patients on average of 4.5 years postoperatively, attributed to blood loss and hemolysis [108].
5.7 Differential Diagnosis (Box 3.8)
Box 3.8: Clues to Non-idiopathic Scoliosis
Findings | Diagnosis |
|---|---|
Dysmorphic vertebrae | Congenital scoliosis |
Levoconvex thoracic curve | Syrinx or spinal cord tumor |
Short segment severe dystrophic curve with kyphosis | Neurofibromatosis |
Rib dysplasia, vertebral deformities | |
Lumbar curve convex to short leg | Compensatory (leg length discrepancy) |
Flexible, corrects with lateral bending | |
Long, C-shaped, without compensatory curves | Neuromuscular |
Marked pelvic obliquity | |
Hip may be dislocated on concave side | |
Levoconvex or double thoracic curve | Congenital cardiac disease |
Early onset, fails bracing | |
Pedicle or lamina at curve apex is sclerotic or overgrown | Vertebral osteoid osteoma |
Tumor, fracture, infection, spondylolysis/spondylolisthesis | Pain |
Although the usual pattern of idiopathic scoliosis is a dextroconvex thoracic structural curve with levoconvex nonstructural curve(s) above and/or below, almost any pattern can be seen. There are, however, some intrinsic or associated features that may suggest the scoliosis is not completely idiopathic.
Intraspinal pathology. Levoconvex thoracic curves are reported to be more often associated with hydrosyringomyelia and spinal cord tumors (see Fig. 3.57). If clinical findings suggest this, MRI is indicated.
Congenital scoliosis (see Figs. 3.17, 3.18, 3.21, and 3.23). Any congenital vertebral anomaly, when not sufficiently severe to cause the patient’s scoliosis, should prompt a search for an occult bar or fusion.
Neurofibromatosis (see Figs. 3.43 and 3.44). Dystrophic curves associated with neurofibromatosis are frequently short, severe, and accompanied with kyphosis. The presence of rib dysplasia and vertebral deformities, along with other findings of neurofibromatosis, may confirm this etiology (see Chap. 23).
Compensatory scoliosis due to underlying leg length discrepancy (see Figs. 3.55 and 3.56). Lumbar curvature is convex on the side of the short leg; it is flexible and corrects with lateral bending. There should be significant disparity in iliac crest level on standing radiographs. Scoliosis improves with a lift under the short leg.
Neuromuscular scoliosis (see Fig. 3.54 and further discussion below). These curves are frequently long and C shaped, without compensatory curves above or below. Pelvic obliquity is usually marked, often with hip dislocation on the concave side of the curve. The femoral necks have valgus deformity. The combination of scoliosis and pelvic obliquity is almost pathognomonic for a neuromuscular imbalance.
Vertebral osteoid osteoma. The pedicle or lamina at the curve apex is sclerotic or overgrown. Back pain is the presenting complaint (see Chap. 4).
Painful scoliosis. Idiopathic scoliosis is not painful. If spine pain is significant, consideration should be given to the presence of osteoid osteoma or other vertebral tumor, fracture, infection, spondylolisthesis, or intraspinal tumor.
Scoliosis in association with other diseases. The incidence of scoliosis is increased in patients with a great variety of disorders, often without an underlying structural osseous anomaly. Scoliosis occurs in 5–16 % of patients with congenital heart disease [109]. These patients show an increased incidence of levoconvex and double thoracic curves, and early onset and brace failure are characteristic [110]. As many as 48 % of patients with ulnar longitudinal deficiency, radial longitudinal deficiency, or phocomelia have scoliosis, and up to 50 % of those with unilateral and 100 % of those with bilateral amelia have scoliosis [111]. Scoliosis has been reported in 13–23 % of those with spondylolysis and in 25–48 % of those with spondylolisthesis (see Chap. 4). About 20 % of patients with pectus excavatum have scoliosis [112]. Scoliosis is common in diseases characterized by abnormal collagen and osteopenia, such as osteogenesis imperfecta (see Fig. 3.49) and idiopathic juvenile osteoporosis. Patients with posterior rib resection and prior thoracotomy may develop scoliosis whose curve is concave to the abnormal side, whereas those with a fractured transverse process may develop a curve that is convex to the injured side.
6 Neuromuscular Scoliosis
Scoliosis is a common and frequently disabling concomitant of neuromuscular disease (see Chap. 24). Its incidence varies with the type of disease and patient age; progression of scoliosis correlates with the severity and symmetry of muscle involvement. Gravity and growth affect the unstable spinal column and significantly influence otherwise nonprogressive conditions, such as myelomeningocele and poliomyelitis.
A discussion of all of the types of neuromuscular disease associated with scoliosis is beyond the scope of this chapter. A few of the more common are presented below.
Scoliosis is common in patients with cerebral palsy and tends to be worse in those who are more severely neurodevelopmentally impaired (Fig. 3.71). Most of these patients eventually develop associated pelvic obliquity, and this is thus an important radiologic sign. Furthermore, severe pelvic tilt causes the pelvis to move away from the femoral head on the concave side of the spinal curvature, contributing to hip dysplasia/dislocation.

Biphasic scoliosis in a 13-year-old boy with severe cerebral palsy. There is also a small pelvis compared to the size of the chest, bilateral coxa valga, and superolateral subluxation of the left femoral head (supine view)
Muscular dystrophies are characterized by a variety of spinal curvatures. In Duchenne muscular dystrophy, early muscular changes are usually symmetric, so scoliosis typically does not progress until the child is wheelchair bound (Fig. 3.72) (see Chap. 24). At that point, pelvic obliquity, combined with gravitational and growth effects, induces progressive and frequently severe scoliosis. Chest radiography is important during adolescence, since the cardiomyopathy associated with this disease will eventually cause cardiac failure and death. Chest radiographs characteristically show soft tissue wasting, humeral shaft thinning, and obliquity of the clavicles and ribs due to weak intercostal muscles. The different subtypes of spinal muscular atrophy (Fig. 3.73) manifest scoliosis at different ages. Scoliosis occurs in 60–70 % of patients with spinal muscular atrophy, and the average age at onset is between 7 and 8 years.

Duchenne muscular dystrophy. Sitting radiographs show neurogenic (a) levoconvex C-shaped thoracolumbar curve and (b) accentuated thoracic kyphosis and straightening of lumbar lordosis (Images copyright Shriners Hospital for Children Northern California)

Neuromuscular scoliosis in an 8-year-old girl with spinal muscular atrophy. 53° long C-shaped scoliosis (sitting, braced). Incidental hiatal hernia (Image copyright Shriners Hospital for Children Northern California)
Imaging
The typical neuromuscular curve is single, C shaped, dextroconvex, and long thoracolumbar. Radiographs may show the patient is sitting (often supported) or supine. Traction radiographs assess flexibility of the curvature, as these patients cannot participate in bending exams. Other imaging clues can include decreased bone density, caninization of vertebral bodies (see Fig. 3.9), coxa valga with or without hip dislocation, prominent fecal burden, and/or indwelling gastrostomy button.
7 Juvenile Kyphosis
Thoracic kyphosis and lumbar lordosis develop as the child adopts an erect posture. Interference with this normal process results in persistence of the infantile continuous smooth upper thoracic-to-lower-lumbar kyphotic curve. Recognizing persistence of the infantile pattern allows diagnosis of developmental delay or generalized muscle weakness.
Norms for thoracic kyphosis depend on patient age, measurement limits, and placement of the subject’s arms. To standardize radiologic evaluation, the patient should stand with arms extended forward and parallel to the floor, supported to reduce any tendency to lean forward. Measurements are made from T3, or the highest visible superior end plate below this, to the inferior end plate of T12. In children aged 2–9, kyphosis averages 21–24°, with a maximum of 40° [28]. In those aged 10–19, kyphosis averages 25–34°, with a maximum of 45° [28, 29]. In general, the normal upper limit in adolescents is 40°. For repeated measurements, the 95 % confidence limit is 11° [94].
Abnormal kyphosis either results from a primary structural cause or is associated with another condition. Structural causes include congenital anomalies, fracture, neoplasm, dysplasias, syndromes, and post-laminectomy and postradiation states. Conditions that are associated with kyphosis include neuromuscular disorders, myelomeningocele, vertebral and spinal cord inflammation, and collagen, metabolic, and skeletal diseases. There is of course some overlap among these categories. The two main types of idiopathic juvenile kyphosis are postural kyphosis and Scheuermann kyphosis (or disease). Postural kyphosis exceeds 40° and lacks additional abnormalities including those described below.
Scheuermann disease (Box 3.9) is characterized by gradual development of abnormal kyphosis during adolescence (Fig. 3.74) [113]. Kyphosis exceeds 40°, and at least three adjacent vertebrae are wedged 5 or more degrees [114]. Clinical vertebral deformity and back pain are present in slightly more than half of these patients. Males and females are affected equally, and symptoms are rarely described in those younger than 11 years of age. Statistics obtained from young male military recruits indicate that 5–6 % of the population may be affected, although most have no symptoms [115]. Scheuermann kyphosis appears to be inherited, with probable autosomal dominant transmission and variable penetrance and expression [113, 116]. There is an increased incidence of spondylolyses and spondylolisthesis in patients with Scheuermann disease; this may cause low back pain.

Scheuermann disease in a 15-year-old girl. (a) Standing radiograph shows disc space narrowing and anterior vertebral wedging at T8–10, with 82° of kyphosis. (b) Lateral radiograph with patient lying on a bolster placed at the apex of the kyphosis shows marked improvement. (c) Standing radiograph after anterior and posterior fusion shows correction of abnormal kyphosis
Box 3.9: Scheuermann Kyphosis
Etiology unknown |
5–6 % of population, M = F |
Asymptomatic deformity or back pain |
Thoracic kyphosis >42° with anterior wedging of at least three adjacent vertebrae |
Decreased disc space height (worse anteriorly), Schmorl nodes |
Increased vertebral anterior-posterior length |
Irregular ossification of ring apophyses |
Hyperlordosis of lumbar spine |
Many cases are incorrectly attributed to poor posture, delaying correct diagnosis and treatment. Clinically, Scheuermann disease is differentiated from postural kyphosis by the Adam test, in which the patient is observed from the side while forward bending. Those with Scheuermann disease have acute angular deformity, while those with postural kyphosis have uniform rounding of the spine [114].
Scheuermann disease is considered benign, as it causes negligible deformity and limited symptomatology after skeletal maturity [113]. Treatment depends on patient age, the severity and position of the deformity, and the degree of pain; surgery is infrequently performed [114]. If the thoracic kyphosis is between 55 and 80°, the patient should participate in an exercise regimen to increase flexibility and also wear a brace. Brace treatment of Scheuermann kyphosis may not only halt progression but also decrease the severity of kyphosis. If the thoracic kyphosis is more than 80°, posterior instrumentation and fusion with posterior column shortening may be performed [113]. Severe or rigid kyphotic curves may require additional anterior release. It is important to consider preoperative MRI to evaluate for cord compression from thoracic disc herniation or from accompanying spinal epidural lipomatosis [114, 117, 118].
The etiology of this condition is incompletely understood; extensive pathologic investigation has shown no evidence of inflammation, osteochondritis, or avascular necrosis. Histopathological evaluation shows disorganized enchondral ossification similar to Blount disease, thinning or absence of vertebral end plates and ring apophyses, as well as a decrease in collagen fibrilism, altered proteoglycan content, and increased muccopolysaccharides in the end plates [113]. However, it is unclear whether these changes are primary or secondary. Biomechanical abnormalities have been suggested, including lack of anterior pelvic tilt with forward bending resulting from hamstring tightness commonly found in these patients [113]. Since Scheuermann disease has been reported in chimpanzees and gorillas, upright posture may not be essential in the development of this deformity [119]. Trabecular bone structure is normal [120].
Classic work by Schmorl has found fissures in the cartilaginous end plate adjacent to the nucleus pulposus, with nuclear material herniating through these areas [121]. It is uncertain why these fissures develop, however—perhaps because cartilage in this area is relatively thin or because unrecognized variability in cartilage structure or cartilage-osseous boundary predisposes to fissuring. In any case, once trauma or the usual stresses of childhood open these fissures, herniation of disc material occurs, followed by fibrosis and reactive sclerosis. The so-called Schmorl nodes are formed by sclerosis outlining the cystic areas where nucleus pulposus has herniated through fissures (see Fig. 3.10). However, Schmorl nodes alone do not indicate the presence of Scheurmann disease, since isolated central end plate disc herniation without vertebral wedging is seen in almost the same proportion of normal teenagers.
Loss of disc material causes decreased disc space height, most notable anteriorly, since the posterior height of the intervertebral space is maintained by facet articulations. The loss of elasticity and increased pressure anteriorly retard growth in this area, eventually resulting in wedging of the vertebral body. It is important to recognize that the wedging is an anterior growth dysfunction and not analogous to the compressive wedging seen with osteoporosis. Abnormal pressure dynamics presumably cause irregular ossification of the ring apophysis.
Imaging
Radiologic examination should include erect frontal and lateral projections of the thoracic and lumbar spine. The vertebral bodies are anteriorly wedged, slightly flattened, and often elongated from front to back. The cortical outlines may be irregularly marginated or indistinct. Only one or two vertebrae may be affected, but in most cases three or more are abnormal; half of the patients have five or more wedged vertebrae. Vertebrae between T3 and T12 are characteristically involved, with T7 most frequently abnormal. Occasionally, the upper lumbar vertebrae are affected—many of these cases are associated with severe stress, such as weightlifting. Persistent vascular channels in the anterior face have been reported in 24 % of patients [115]. Ring apophyses may ossify irregularly. Instead of the usual homogeneous continuous pattern, a fragmented pattern of irregular thickness is noted, and the steplike corners into which the ossification centers fit may be enlarged. In 10 % of cases, at least one ring will remain unfused into adulthood. There is usually hyperlordosis of the lumbar spine [114].
Scoliosis is present in one third of patients but is rarely severe, usually around 15°. In about two thirds of patients, the scoliotic curve is above or (more often) below the level of the kyphosis, in the region of compensatory lordosis. In one third of patients, the level of scoliosis is the same as the level of kyphosis [122]. Secondary hypertrophic changes are common later in life. Spastic paraparesis may develop, associated with a posteriorly herniated thoracic disc.
The angle of kyphosis should be measured according to the Cobb technique, with lines drawn along the superior and inferior vertebral end plates at each end of the curvature (Fig. 3.74). The degree of flexibility is estimated by comparing the curvature when the patient lies supine on the radiographic table versus supine over a bolster placed at the apex of the kyphosis (Fig. 3.74).
Radiographic findings are consistent with pathologic findings. The intervertebral disc spaces are decreased in height, with greater narrowing anteriorly. Herniation of nuclear material into the anterior or central portions of the vertebral body may appear as typical Schmorl nodes in up to 75 % of patients, visible on frontal or lateral projections as well as reformatted CT images.
Nuclear scintigraphy is normal [123]. MRI demonstrates the bony and disc findings described above as well as abnormal disc signal (decreased signal of the nucleus pulposus on T2-W images). Discs with the most decreased height demonstrate the most striking signal abnormalities (Fig. 3.75) [124]. MRI may also define disc herniation, usually at the apex of the kyphosis (often affecting T7–T9). Spinal epidural lipomatosis and extradural cysts have been reported [116, 118].

Scheuermann disease in a 15-year-old boy. Sagittal T2-W image shows mild anterior wedging of T7–9, with disc space narrowing and irregularity as well as decreased signal of the nucleus pulposus at T7–8, T8–9, and T9–10. Schmorl nodes at T10–11, T11–12, and T12–L1 (For reference, the conus medullaris is at L1)
Differential diagnosis of juvenile kyphosis is occasionally problematic. Simple postural round back has none of the disc changes or vertebral wedging described above and will be fairly supple in hyperextension. Tumor and infection rarely affect a series of vertebrae so uniformly, without extensive deformity or destructive changes. Isolated severe trauma may cause anterior disc herniation and vertebral wedging.
References
Bess S, Varma V. Embryology and anatomy: spine/spinal cord. In: Akbarnia B, Yazici M, Thompson G, editors. The growing spine. Berlin/Heidelberg: Springer; 2010. p. 3–12.
Diel J, Ortiz O, Losada RA, Price DB, Hayt MW, Katz DS. The sacrum: pathologic spectrum, multimodality imaging, and subspecialty approach. Radiographics. 2001;21(1):83–104.
Zhang H, Sucato DJ, Nurenberg P, McClung A. Morphometric analysis of neurocentral synchondrosis using magnetic resonance imaging in the normal skeletally immature spine. Spine. 2010;35(1):76–82. Epub 2010/01/01.
Ghanem I, El Hage S, Rachkidi R, Kharrat K, Dagher F, Kreichati G. Pediatric cervical spine instability. J Child Orthop. 2008;2(2):71–84.
Lustrin ES, Karakas SP, Ortiz AO, Cinnamon J, Castillo M, Vaheesan K, et al. Pediatric cervical spine: normal anatomy, variants, and trauma. Radiogr Rev Publ Radiol Soc N Am Inc. 2003;23(3):539–60. Epub 2003/05/13.
Hindman BW, Poole CA. Early appearance of the secondary vertebral ossification centers. Radiology. 1970;95(2):359–61. Epub 1970/05/01.
Ogden JA. Radiology of postnatal skeletal development. XI. The first cervical vertebra. Skeletal Radiol. 1984;12(1):12–20. Epub 1984/01/01.
Ogden JA. Radiology of postnatal skeletal development. XII. The second cervical vertebra. Skeletal Radiol. 1984;12(3):169–77. Epub 1984/01/01.
Ogden JA, Murphy MJ, Southwick WO, Ogden DA. Radiology of postnatal skeletal development. XIII. C1-C2 interrelationships. Skeletal Radiol. 1986;15(6):433–8.
Moore K. The developing human: clinically oriented embryology. 5th ed. Philadelphia: Saunders; 1997.
Barkovich AJ. Pediatric neuroimaging. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2005.
Currarino G, Williams B, Reisch JS. Linear growth of the thoracic spine in chest roentgenograms from birth to 16 years. Skeletal Radiol. 1986;15(8):628–30. Epub 1986/01/01.
Swischuk L. Normal variations. Imaging of the cervical spine in children. New York: Springer; 2002. p. 13–38.
Cakmakci H. Essentials of trauma: head and spine. Pediatr Radiol. 2009;39(3):391–405.
de Beer JD, Hoffman EB, Kieck CF. Traumatic atlantoaxial subluxation in children. J Pediatr Orthop. 1990;10(3):397–400.
Wollin DG, Elliott GB. Coronal cleft vertebrae and persistent notochordal derivatives of infancy. J Can Assoc Radiol. 1961;12:78–81. Epub 1961/09/01.
Boone D, Parsons D, Lachmann SM, Sherwood T. Spina bifida occulta: lesion or anomaly? Clin Radiol. 1985;36(2):159–61. Epub 1985/03/01.
Osborne J, Masel J, McCredie J. A spectrum of skeletal anomalies associated with pulmonary agenesis: possible neural crest injuries. Pediatr Radiol. 1989;19(6–7):425–32.
Beals RK, Rolfe B. VATER association. A unifying concept of multiple anomalies. J Bone Joint Surg Am. 1989;71(6):948–50. Epub 1989/07/01.
Hensinger RNMD. Congenital scoliosis: etiology and associations. Spine. 2009;34(17):1745–50.
McMaster MJ, David CV. Hemivertebra as a cause of scoliosis. A study of 104 patients. J Bone Jt Surg Br. 1986;68(4):588–95.
Kawakami NMD, Tsuji TMD, Imagama SMD, Lenke LGMD, Puno RMMD, Kuklo TRMD, et al. Classification of congenital scoliosis and kyphosis: a new approach to the three-dimensional classification for progressive vertebral anomalies requiring operative treatment. Spine. 2009;34(17):1756–65.
Marks DSFF, Qaimkhani SAF. The natural history of congenital scoliosis and kyphosis. Spine. 2009;34(17):1751–5.
Chan GMD, Dormans JPMD. Update on congenital spinal deformities: preoperative evaluation. Spine. 2009;34(17):1766–74.
McMaster MJ. Occult intraspinal anomalies and congenital scoliosis. J Bone Joint Surg Am. 1984;66(4):588–601. Epub 1984/04/01.
MacEwen GD, Winter RB, Hardy JH. Evaluation of kidney anomalies in congenital scoliosis. J Bone Joint Surg Am. 1972;54(7):1451–4. Epub 1972/10/01.
Lee CS, Noh H, Lee DH, Hwang CJ, Kim H, Cho SK. Analysis of sagittal spinal alignment in 181 asymptomatic children. J Spinal Disord Tech. 2012;25(8):E259–63. Epub 2012/06/13.
Fon GT, Pitt MJ, Thies Jr AC. Thoracic kyphosis: range in normal subjects. AJR Am J Roentgenol. 1980;134(5):979–83. Epub 1980/05/01.
Singer KP, Jones TJ, Breidahl PD. A comparison of radiographic and computer-assisted measurements of thoracic and thoracolumbar sagittal curvature. Skeletal Radiol. 1990;19(1):21–6. Epub 1990/01/01.
Mayfield JK, Winter RB, Bradford DS, Moe JH. Congenital kyphosis due to defects of anterior segmentation. J Bone Joint Surg Am. 1980;62(8):1291–301. Epub 1980/12/01.
Tsou PM. Embryology of congenital kyphosis. Clin Orthop Relat Res. 1977;128:18–25. Epub 1977/10/01.
Winter RB. Congenital kyphosis. Clin Orthop Relat Res. 1977;128:26–32.
Gholve PA, Hosalkar HS, Ricchetti ET, Pollock AN, Dormans JP, Drummond DS. Occipitalization of the atlas in children morphologic classification, associations, and clinical relevance. J Bone Jt Surg. 2007;89(3):571–8.
Bulas DI, Fitz CR, Johnson DL. Traumatic atlanto-occipital dislocation in children. Radiology. 1993;188(1):155–8. Epub 1993/07/01.
Hosalkar HS, Sankar WN, Wills BPD, Goebel J, Dormans JP, Drummond DS. Congenital osseous anomalies of the upper cervical spine. J Bone Jt Surg. 2008;90(2):337–48.
Schuler TC, Kurz L, Thompson DE, Zemenick G, Hensinger RN, Herkowitz HN. Natural history of os odontoideum. J Pediatr Orthop. 1991;11(2):222–5. Epub 1991/03/01.
Pueschel SM, Scola FH. Atlantoaxial instability in individuals with Down syndrome: epidemiologic, radiographic, and clinical studies. Pediatrics. 1987;80(4):555–60. Epub 1987/10/01.
Kawabe N, Hirotani H, Tanaka O. Pathomechanism of atlantoaxial rotatory fixation in children. J Pediatr Orthop. 1989;9(5):569–74.
Parke WW, Rothman RH, Brown MD. The pharyngovertebral veins: an anatomical rationale for Grisel’s syndrome. J Bone Joint Surg Am. 1984;66(4):568–74. Epub 1984/04/01.
Tortori-Donati P, Rossi A, Cama A. Spinal dysraphism: a review of neuroradiological features with embryological correlations and proposal for a new classification. Neuroradiology. 2000;42(7):471–91. Epub 2000/08/22.
Thakur N, Lowe L. Borderline low conus medullaris on infant lumbar sonography: what is the clinical outcome and the role of neuroimaging follow-up? Pediatr Radiol. 2011;41(4):483–7.
Unsinn KM, Geley T, Freund MC, Gassner I. US of the spinal cord in newborns: spectrum of normal findings, variants, congenital anomalies, and acquired diseases. Radiographics. 2000;20(4):923–38.
Porensky P, Muro K, Ganju A. Nontraumatic cervicothoracic syrinx as a cause of progressive neurologic dysfunction. J Spinal Cord Med. 2007;30(3):276–81. Epub 2007/08/10.
Ozerdemoglu RA, Denis F, Transfeldt EE. Scoliosis associated with syringomyelia: clinical and radiologic correlation. Spine (Phila Pa 1976). 2003;28(13):1410–7. Epub 2003/07/03.
Nakamura M, Ishii K, Watanabe K, Tsuji T, Matsumoto M, Toyama Y, et al. Clinical significance and prognosis of idiopathic syringomyelia. J Spinal Disord Tech. 2009;22(5):372–5. Epub 2009/06/16.
Rufener SL, Ibrahim M, Raybaud CA, Parmar HA. Congenital spine and spinal cord malformations–self-assessment module. AJR Am J Roentgenol. 2010;194(3 Suppl):S38–40. Epub 2010/03/05.
Huisman TAGM, Rossi A, Tortori-Donati P. MR imaging of neonatal spinal dysraphia: what to consider? Magn Reson Imaging Clin N Am. 2012;20(1):45–61.
Woodward PJ, Sohaey R, Kennedy A, Koeller KK. From the archives of the AFIP: a comprehensive review of fetal tumors with pathologic correlation. Radiogr Rev Publ Radiol Soc N Am Inc. 2005;25(1):215–42. Epub 2005/01/18.
Altman RP, Randolph JG, Lilly JR. Sacrococcygeal teratoma: American Academy of Pediatrics Surgical Section Survey-1973. J Pediatr Surg. 1974;9(3):389–98. Epub 1974/06/01.
Kocaoglu M, Frush DP. Pediatric presacral masses. Radiogr Rev Publ Radiol Soc N Am Inc. 2006;26(3):833–57. Epub 2006/05/17.
Hensinger RN, Lang JE, MacEwen GD. Klippel-Feil syndrome; a constellation of associated anomalies. J Bone Joint Surg Am. 1974;56(6):1246–53. Epub 1974/09/01.
Winter RB, Moe JH, Lonstein JE. The incidence of Klippel-Feil syndrome in patients with congenital scoliosis and kyphosis. Spine (Phila Pa 1976). 1984;9(4):363–6. Epub 1984/05/01.
Theiss SM, Smith MD, Winter RB. The long-term follow-up of patients with Klippel-Feil syndrome and congenital scoliosis. Spine (Phila Pa 1976). 1997;22(11):1219–22.
Azouz EM. CT demonstration of omovertebral bone. Pediatr Radiol. 2007;37(4):404. Epub 2007/02/03.
Lynch SA, Wang Y, Strachan T, Burn J, Lindsay S. Autosomal dominant sacral agenesis: Currarino syndrome. J Med Genet. 2000;37(8):561–6. Epub 2000/08/03.
Pang D. Sacral agenesis and caudal spinal cord malformations. Neurosurgery. 1993;32(5):755–78. Discussion 78–9; Epub 1993/05/01.
Torre M, Buffa P, Jasonni V, Cama A. Long-term urologic outcome in patients with caudal regression syndrome, compared with meningomyelocele and spinal cord lipoma. J Pediatr Surg. 2008;43(3):530–3. Epub 2008/03/25.
Mitchell GE, Lourie H, Berne AS. The various causes of scalloped vertebrae with notes on their pathogenesis. Radiology. 1967;89(1):67–74. Epub 1967/07/01.
Ton J, Stein-Wexler R, Yen P, Gupta M. Rib head protrusion into the central canal in type 1 neurofibromatosis. Pediatr Radiol. 2010;40(12):1902–9.
Tsirikos AI, Saifuddin A, Noordeen MH. Spinal deformity in neurofibromatosis type-1: diagnosis and treatment. Eur Spine J Off Publ Eur Spine Soc Eur Spinal Deformity Soc Eur Sect Cervical Spine Res Soc. 2005;14(5):427–39.
Calvert PT, Edgar MA, Webb PJ. Scoliosis in neurofibromatosis. The natural history with and without operation. J Bone Jt Surg Br. 1989;71(2):246–51. Epub 1989/03/01.
Vitale MG, Guha A, Skaggs DL. Orthopaedic manifestations of neurofibromatosis in children: an update. Clin Orthop Relat Res. 2002;401:107–18. Epub 2002/08/02.
Taybi HLR. Radiology of syndromes, metabolic disorders, and skeletal dysplasias. 5th ed. Chicago: Year Book Medical Publishers; 2006.
Berdon W, Lampl B, Cornier A, Ramirez N, Turnpenny P, Vitale M, et al. Clinical and radiological distinction between spondylothoracic dysostosis (Lavy-Moseley syndrome) and spondylocostal dysostosis (Jarcho-Levin syndrome). Pediatr Radiol. 2011;41(3):384–8.
Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet Eur J Hum Genet. 2012;20(3):251–7. Epub 2011/09/22.
Kim H, Kim HS, Moon ES, Yoon CS, Chung TS, Song HT, et al. Scoliosis imaging: what radiologists should know. Radiogr Rev Publ Radiol Soc N Am Inc. 2010;30(7):1823–42. Epub 2010/11/09.
Miller NH. Cause and natural history of adolescent idiopathic scoliosis. Orthop Clin North Am. 1999;30(3):343–52, vii. Epub 1999/07/07.
Roach JW. Adolescent idiopathic scoliosis. Orthop Clin North Am. 1999;30(3):353–65, vii–viii. Epub 1999/07/07.
Yang T, Jia Q, Guo H, Xu J, Bai Y, Yang K, et al. Epidemiological survey of idiopathic scoliosis and sequence alignment analysis of multiple candidate genes. Int Orthop. 2012;36(6):1307–14. Epub 2011/12/21.
Stokes IA. Mechanical effects on skeletal growth. J Musculoskelet Neuronal Interact. 2002;2(3):277–80. Epub 2005/03/11.
Cassar-Pullicino VN, Eisenstein SM. Imaging in scoliosis: what, why and how? Clin Radiol. 2002;57(7):543–62. Epub 2002/07/05.
Ceballos T, Ferrer-Torrelles M, Castillo F, Fernandez-Paredes E. Prognosis in infantile idiopathic scoliosis. J Bone Joint Surg Am. 1980;62(6):863–75. Epub 1980/09/01.
Wynne-Davies R. Infantile idiopathic scoliosis. Causative factors, particularly in the first six months of life. J Bone Joint Surg. 1975;57(2):138–41. Epub 1975/05/01.
James JI. The management of infants with scoliosis. J Bone Joint Surg. 1975;57(4):422–9. Epub 1975/11/01.
Thompson SK, Bentley G. Prognosis in infantile idiopathic scoliosis. J Bone Joint Surg. 1980;62-B(2):151–4. Epub 1980/05/01.
Jaramillo D, Poussaint TY, Grottkau BE. Scoliosis: evidence-based diagnostic evaluation. Neuroimaging Clin N Am. 2003;13(2):335–41, xii. Epub 2003/09/19.
Goldstein LA, Waugh TR. Classification and terminology of scoliosis. Clin Orthop Relat Res. 1973;93:10–22. Epub 1973/06/01.
Fearon T, Vucich J, Butler P, McSweeney WJ, Taylor GA, Markle BM, et al. Scoliosis examinations: organ dose and image quality with rare-earth screen-film systems. AJR Am J Roentgenol. 1988;150(2):359–62. Epub 1988/02/01.
Khanna G. Role of imaging in scoliosis. Pediatr Radiol. 2009;39 Suppl 2:S247–51. Epub 2009/05/19.
Wybier M, Bossard P. Musculoskeletal imaging in progress: the EOS imaging system. Jt Bone Spine revue du rhumatisme. 2013;80(3):238–43.
Levy AR, Goldberg MS, Hanley JA, Mayo NE, Poitras B. Projecting the lifetime risk of cancer from exposure to diagnostic ionizing radiation for adolescent idiopathic scoliosis. Health Phys. 1994;66(6):621–33. Epub 1994/06/01.
Levy AR, Goldberg MS, Mayo NE, Hanley JA, Poitras B. Reducing the lifetime risk of cancer from spinal radiographs among people with adolescent idiopathic scoliosis. Spine (Phila Pa 1976). 1996;21(13):1540–7. Discussion 8. Epub 1996/07/01.
Doody MM, Lonstein JE, Stovall M, Hacker DG, Luckyanov N, Land CE. Breast cancer mortality after diagnostic radiography: findings from the U.S. Scoliosis Cohort Study. Spine. 2000;25(16):2052–63.
Goldberg C, Thompson F, Dowling F, Regan BF, Blake NS. Pilot study for a scoliosis screening project in South Dublin. Ir Med J. 1980;73(7):265–8. Epub 1980/07/01.
Tachdjian MO, Matson DD. Orthopaedic aspects of intraspinal tumors in infants and children. J Bone Joint Surg Am. 1965;47:223–48. Epub 1965/03/01.
Musson RE, Warren DJ, Bickle I, Connolly DJ, Griffiths PD. Imaging in childhood scoliosis: a pictorial review. Postgrad Med J. 2010;86(1017):419–27. Epub 2010/07/17.
Do T, Fras C, Burke S, Widmann RF, Rawlins B, Boachie-Adjei O. Clinical value of routine preoperative magnetic resonance imaging in adolescent idiopathic scoliosis. A prospective study of three hundred and twenty-seven patients. J Bone Joint Surg Am. 2001;83-A(4):577–9. Epub 2001/04/24.
Maiocco B, Deeney VF, Coulon R, Parks Jr PF. Adolescent idiopathic scoliosis and the presence of spinal cord abnormalities. Preoperative magnetic resonance imaging analysis. Spine. 1997;22(21):2537–41. Epub 1997/12/31 23:39.
Davids JR, Chamberlin E, Blackhurst DW. Indications for magnetic resonance imaging in presumed adolescent idiopathic scoliosis. J Bone Joint Surg Am. 2004;86-A(10):2187–95. Epub 2004/10/07.
Malfair D, Flemming AK, Dvorak MF, Munk PL, Vertinsky AT, Heran MK, et al. Radiographic evaluation of scoliosis: review. AJR Am J Roentgenol. 2010;194(3 Suppl):S8–22. Epub 2010/03/05.
Inoue M, Minami S, Nakata Y, Otsuka Y, Takaso M, Kitahara H, et al. Preoperative MRI analysis of patients with idiopathic scoliosis: a prospective study. Spine. 2005;30(1):108–14. Epub 2005/01/01.
Oda M, Rauh S, Gregory PB, Silverman FN, Bleck EE. The significance of roentgenographic measurement in scoliosis. J Pediatr Orthop. 1982;2(4):378–82. Epub 1982/10/01.
Morrissy RT, Goldsmith GS, Hall EC, Kehl D, Cowie GH. Measurement of the Cobb angle on radiographs of patients who have scoliosis. Evaluation of intrinsic error. J Bone Joint Surg Am. 1990;72(3):320–7. Epub 1990/03/01.
Carman DL, Browne RH, Birch JG. Measurement of scoliosis and kyphosis radiographs. Intraobserver and interobserver variation. J Bone Joint Surg Am. 1990;72(3):328–33. Epub 1990/03/01.
Shea KG, Stevens PM, Nelson M, Smith JT, Masters KS, Yandow S. A comparison of manual versus computer-assisted radiographic measurement. Intraobserver measurement variability for Cobb angles. Spine. 1998;23(5):551–5. Epub 1998/04/08.
Propst-Proctor SL, Bleck EE. Radiographic determination of lordosis and kyphosis in normal and scoliotic children. J Pediatr Orthop. 1983;3(3):344–6. Epub 1983/07/01.
Armstrong GW, Livermore 3rd NB, Suzuki N, Armstrong JG. Nonstandard vertebral rotation in scoliosis screening patients. Its prevalence and relation to the clinical deformity. Spine. 1982;7(1):50–4. Epub 1982/01/01.
Nash Jr CL, Moe JH. A study of vertebral rotation. J Bone Joint Surg Am. 1969;51(2):223–9. Epub 1969/03/01.
McAlister WH, Shackelford GD. Measurement of spinal curvatures. Radiol Clin North Am. 1975;13(1):113–21. Epub 1975/04/01.
Van Goethem J, Van Campenhout A, van den Hauwe L, Parizel PM. Scoliosis. Neuroimaging Clin N Am. 2007;17(1):105–15. Epub 2007/05/12.
Schwab F, Lafage V, Boyce R, Skalli W, Farcy JP. Gravity line analysis in adult volunteers: age-related correlation with spinal parameters, pelvic parameters, and foot position. Spine. 2006;31(25):E959–67. Epub 2006/12/02.
Risser JC. The iliac apophysis; an invaluable sign in the management of scoliosis. Clin Orthop. 1958;11:111–9. Epub 1958/01/01.
Dickson RA, Deacon P. Spinal growth. J Bone Joint Surg. 1987;69(5):690–2. Epub 1987/11/01.
Thomsen M, Abel R. Imaging in scoliosis from the orthopaedic surgeon’s point of view. Eur J Radiol. 2006;58(1):41–7. Epub 2006/01/28.
Hefti FL, McMaster MJ. The effect of the adolescent growth spurt on early posterior spinal fusion in infantile and juvenile idiopathic scoliosis. J Bone Joint Surg. 1983;65(3):247–54. Epub 1983/05/01.
Lauerman WC, Bradford DS, Transfeldt EE, Ogilvie JW. Management of pseudarthrosis after arthrodesis of the spine for idiopathic scoliosis. J Bone Joint Surg Am. 1991;73(2):222–36. Epub 1991/02/01.
Selby MD, Clark SR, Hall DJ, Freeman BJ. Radiologic assessment of spinal fusion. J Am Acad Orthop Surg. 2012;20(11):694–703. Epub 2012/11/03.
Teele RL, Nussbaum AR, Wyly JB, Allred EN, Emans J. Cholelithiasis after spinal fusion for scoliosis in children. J Pediatr. 1987;111(6 Pt 1):857–60. Epub 1987/12/01.
Roth A, Rosenthal A, Hall JE, Mizel M. Scoliosis and congenital heart disease. Clin Orthop Relat Res. 1973;93:95–102. Epub 1973/06/01.
Farley FA, Phillips WA, Herzenberg JE, Rosenthal A, Hensinger RN. Natural history of scoliosis in congenital heart disease. J Pediatr Orthop. 1991;11(1):42–7. Epub 1991/01/01.
Powers TA, Haher TR, Devlin VJ, Spencer D, Millar EA. Abnormalities of the spine in relation to congenital upper limb deficiencies. J Pediatr Orthop. 1983;3(4):471–4. Epub 1983/09/01.
Hong JY, Suh SW, Park HJ, Kim YH, Park JH, Park SY. Correlations of adolescent idiopathic scoliosis and pectus excavatum. J Pediatr Orthop. 2011;31(8):870–4. Epub 2011/11/22.
Tsirikos AI. Scheuermann’s kyphosis: an update. J Surg Orthop Adv. 2009;18(3):122–8. Epub 2009/10/22.
Lowe TG. Scheuermann’s kyphosis. Neurosurg Clin N Am. 2007;18(2):305–15. Epub 2007/06/09.
Williams HJ, Pugh DG. Vertebral epiphysitis: a comparison of the clinical and roentgenologic findings. Am J Roentgenol Radium Ther Nucl Med. 1963;90:1236–47. Epub 1963/12/01.
Graat HC, van Rhijn LW, Schrander-Stumpel CT, van Ooij A. Classical Scheuermann disease in male monozygotic twins: further support for the genetic etiology hypothesis. Spine. 2002;27(22):E485–7. Epub 2002/11/19.
Kapetanos GA, Hantzidis PT, Anagnostidis KS, Kirkos JM. Thoracic cord compression caused by disk herniation in Scheuermann’s disease: a case report and review of the literature. Eur Spine J Off Publ Eur Spine Soc Eur Spinal Deformity Soc Eur Cervical Spine Res Soc. 2006;15 Suppl 5:553–8. Epub 2006/01/20.
Abul-Kasim K, Schlenzka D, Selariu E, Ohlin A. Spinal epidural lipomatosis: a common imaging feature in Scheuermann disease. J Spinal Disord Tech. 2012;25(7):356–61. Epub 2011/06/28.
Farrell BM, Kuo CC, Tang JA, Phan S, Buckley JM, Kondrashov DG. Scheuermann kyphosis in nonhuman primates. Spine. 2012;37(23):E1432–7. Epub 2012/08/28.
Gilsanz V, Gibbens DT, Carlson M, King J. Vertebral bone density in Scheuermann disease. J Bone Joint Surg Am. 1989;71(6):894–7. Epub 1989/07/01.
Schmorl G. Uber Knorpelknotchen an der Wirbelbandscheiben. Fortschr Roentgenstr. 1928;38:265–79.
Deacon P, Berkin CR, Dickson RA. Combined idiopathic kyphosis and scoliosis. An analysis of the lateral spinal curvatures associated with Scheuermann’s disease. J Bone Joint Surg. 1985;67(2):189–92. Epub 1985/03/01.
Winter WA, Veraart BE, Verdegaal WP. Bone scintigraphy in patients with juvenile kyphosis (M. Scheuermann). Diagn Imaging. 1981;50(4):186–90.
Swischuk LE, John SD, Allbery S. Disk degenerative disease in childhood: Scheuermann’s disease, Schmorl’s nodes, and the limbus vertebra: MRI findings in 12 patients. Pediatr Radiol. 1998;28(5):334–8. Epub 1998/05/07.
Winter RB. Congenital scoliosis. Clin Orthop Relat Res. 1973;93:75–94. Epub 1973/06/01.
Scoles PV, Salvagno R, Villalba K, Riew D. Relationship of iliac crest maturation to skeletal and chronologic age. J Pediatr Orthop. 1988;8(6):639–44. Epub 1988/11/01.
Schoenwolf GC, Bleyl SB, Brauer PR, Francis-West PH. Development of the musculoskeletal system. In: Schoenwolf GC, Bleyl SB, Brauer PR, Francis-West PH, editors. Larsen’s human embryology. 4th ed. New York: Churchill Livingstone; 2009.
Erol B, Kusumi K, Lou J, et al. Etiology of congenital scoliosis. Univ Pennsylvania Orthop J. 2002;15:37–42.
Badve CA, Khanna PC, Phillips GS, Thapa MM, Ishak GE. MRI of closed spinal dysraphisms. Pediatr Radiol. 2011;41(10):1308–20.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Taragin, B.H., Wootton-Gorges, S.L. (2015). The Spine: Congenital and Developmental Conditions. In: Stein-Wexler, R., Wootton-Gorges, S., Ozonoff, M. (eds) Pediatric Orthopedic Imaging. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-45381-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-642-45381-6_3
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-45380-9
Online ISBN: 978-3-642-45381-6
eBook Packages: MedicineMedicine (R0)