
Abstract
Carnitine and its esters are physiologically present in all biological fluids, but carnitine is most abundant in tissues with high energy requirements, particularly skeletal and cardiac muscle. In 1973 the first two clinically relevant disorders affecting this pathway were described: primary carnitine deficiency and carnitine palmitoyltransferase type II deficiency (DiMauro and DiMauro 1973; Engel and Angelini 1973). To date, more than 20 different enzyme deficiency states affecting fatty acid transport and mitochondrial β-oxidation (FAO) are known and additional enzymes involved in this pathway are still being discovered (Bosch et al. 2011; Rinaldo et al. 2002).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Carnitine and its esters are physiologically present in all biological fluids, but carnitine is most abundant in tissues with high energy requirements, particularly skeletal and cardiac muscle. In 1973 the first two clinically relevant disorders affecting this pathway were described: primary carnitine deficiency and carnitine palmitoyltransferase type II deficiency (DiMauro and DiMauro 1973; Engel and Angelini 1973). To date, more than 20 different enzyme deficiency states affecting fatty acid transport and mitochondrial β-oxidation (FAO) are known and additional enzymes involved in this pathway are still being discovered (Bosch et al. 2011; Rinaldo et al. 2002).
Carnitine is involved as a detoxifying agent in branched-chain amino acid metabolism. Most of the classic organic acidurias are associated with secondary carnitine deficiency. In fact, the clinical utility of acylcarnitine analysis was first identified for several organoacidopathies, and urine was the preferred specimen. However, plasma became the specimen of choice because acylcarnitine profiles are less complex in plasma than in urine, and because the sensitivity of acylcarnitine analysis is higher when plasma is analyzed, especially for the diagnosis of long-chain FAO disorders (Millington et al. 1992). Today, urine acylcarnitine analysis is limited to specific analytes where diagnostic value is added to the metabolic workup of patients with organic acidemias but inconclusive or borderline abnormal urine organic acid and plasma acylcarnitine profiles (Ensenauer et al. 2004; Oglesbee et al. 2007; Tortorelli et al. 2005). Blood dried on filter paper is analyzed for newborn screening and, together with bile, in the postmortem evaluation of cases of sudden and unexpected death (Rinaldo et al. 2004). Cell-free supernatant of amniotic fluid is used for the prenatal diagnosis of selected inborn errors of metabolism (Rinaldo et al. 2001).
Cultured fibroblasts or amniocytes can be probed with FAO substrates and carnitine. Cell cultures deficient of an FAO enzyme will accumulate specific acylcarnitine species when incubated with substrates such as palmitate, allowing for the diagnosis of FAO disorders (Roe et al. 2001; Shen et al. 2000; Young et al. 2003). Modifications of this assay system have also been developed for the diagnosis of defects affecting the metabolism of branched-chain amino acids and for the study of peripheral blood mononuclear cells (Schulze-Bergkamen et al. 2005).
Acylcarnitine analysis is almost exclusively performed by tandem mass spectrometry (MS/MS) using stable isotope-labeled internal standards that allow quantitation of acylcarnitine species. However, to provide meaningful results to referring health care providers, it is critical to complement analytical proficiency with in-depth interpretation of results as is true for many other examples of complex metabolic profiles.
2 Carnitine and Acylcarnitines
Carnitine, l-3-hydroxy-4-(trimethylammonium)butyrate, is a water-soluble, trimethylammonium derivative of γ-amino-β-hydroxybutyric acid which is formed from trimethyllysine via γ-butyrobetaine (Vaz and Wanders 2002) (Fig. 51.1).

Structures of carnitine and acylcarnitine. The R represents the acylcarnitine species with up to 18 carbons which are typically the target of an acylcarnitine analysis
Carnitine originates to about 75 % from dietary intake of meat, fish, and dairy products containing proteins with trimethyllysine residues. Under normal conditions, endogenous synthesis from lysine and methionine plays a minor role. Carnitine is excreted in urine and bile as free carnitine or as conjugated carnitine esters. Adequate intracellular levels of carnitine depend on diet, endogenous synthesis, reabsorption, and cellular uptake.
Under physiologic conditions, carnitine is primarily required to shuttle long-chain fatty acids across the inner mitochondrial membrane for fatty acid β-oxidation and products of peroxisomal β-oxidation to the mitochondria for further metabolism in the citric acid cycle (Vaz and Wanders 2002). Acylcarnitines (the carnitine esters) are formed by conjugating acyl-CoA moieties to carnitine which for activated long-chain fatty acids is accomplished by carnitine palmitoyltransferase type I (CPT-I) (Fig. 51.1). The acyl group of the activated fatty acid (fatty acyl-CoA) is transferred by CPT-I from the sulfur atom of CoA to the hydroxyl group of carnitine. Carnitine-acylcarnitine translocase (CACT) then transfers the long-chain acylcarnitines across the inner mitochondrial membrane, where CPT-II reverses the action of CPT-I by formation of acyl-CoA and release of free carnitine.
In pathologic conditions, such as FAO disorders or organic acidemias due to acyl-CoA dehydrogenase deficiencies, the functions of carnitine as regulator of substrate flux and energy balance across cell membranes and as modulator of intracellular concentrations of free CoA become crucial. In such conditions acyl-CoAs accumulate inside the mitochondrial matrix, and carnitine is utilized to shuttle these compounds out of the mitochondria as acylcarnitines, thereby restoring free CoA.
Carnitine and its esters are present in all biological fluids albeit very low in the CSF and depending on the enzyme defect, a particular acylcarnitine pattern becomes apparent where those acylcarnitine species serving as direct substrates for the defective enzyme accumulate disproportionately to the down- and upstream metabolites (Table 51.1).
3 Indications for an Acylcarnitine Analysis
Acylcarnitine analysis has proven a useful tool in the evaluation of patients at risk for inborn errors of fatty acid oxidation and for organic acidemias that are primarily due to defects in branched-chain amino acid metabolism. Given the diverse clinical presentation of these conditions, acylcarnitine analysis has become an integral part of the biochemical genetic laboratory investigation of a large number of patients. Other laboratory studies that should be considered in such patient evaluations include urine organic acid and acylglycine, as well as plasma free fatty acid analyses. With the additional application to newborn screening, among esoteric tests acylcarnitine analysis has the highest sample throughput. Aside from clinical and newborn screening indications, a relevant family and/or prenatal history and sudden unexpected death also represent valid reasons to pursue an acylcarnitine analysis (Table 51.2). An unequivocal indication as a test to monitor treated patients has not yet been established, although it is frequently being performed for this purpose.
4 Methods
Several techniques have been described to differentiate and eventually quantify specific carnitine esters. These include gas chromatography-mass spectrometry (GC-MS), thin layer chromatography (TLC) and radioisotopic exchange/HPLC, liquid chromatography (LC) MS, and LC-MS/MS. However, the predominant method applied is flow injection analysis MS/MS using triple quadrupole analyzers combined with electrospray ionization (ESI). The advantage of this approach is its sensitivity that allows for simple but efficient preparation procedures of small sample volumes and fast analytical times, therefore providing for a rapid throughput of large numbers of samples. In a typical setup, acylcarnitines are extracted from the sample by mixing with methanol or an acidified acetonitrile solution containing isotopically labeled acylcarnitines of various chain lengths at defined concentrations as internal standards. Following centrifugation, the supernatant is evaporated and the residue derivatized with either n-butanol HCl or n-methanol HCl yielding the acylcarnitines for analysis by flow injection ESI-MS/MS. Following analysis, a graphical acylcarnitine profile is generated which can be interpreted qualitatively. (Semi-)quantitative calculation of the concentration of each individual acylcarnitine species is based on the abundance of the assigned internal standard (Smith and Matern 2010).
5 Specimen
A variety of body fluids can be used for acylcarnitine analysis, but testing of plasma or whole blood spotted on filter paper is most common. All sample types, except for dried blood and bile spots and fibroblast cultures which can be sent at room temperature, should be kept frozen until analysis. Reliable results, particularly for short-chain acylcarnitine species, for any sample, liquid or dried on filter paper, cannot be achieved following long-term storage at ambient temperatures (Matern et al. 1999).
6 Plasma and Serum
Heparinized plasma is the preferred specimen for acylcarnitine analysis, but EDTA plasma and serum are also acceptable. Hemolyzed or lipemic specimens can also be analyzed without negative impact on sensitivity or specificity. An amount of 100 μL is typically sufficient material to conduct the analysis and repeat it at least once if necessary. Most informative results are generally achieved when samples are obtained during acute illness. Because inborn errors of metabolism are traditionally not entered early into differential diagnostic considerations, sample collection should alternatively be timed before a meal, preferably after an overnight fast. A prolonged fasting challenge, however, should not routinely be undertaken as these require close surveillance typically not possible in an outpatient setting.
7 Dried Blood and Dried Bile Spots
Acylcarnitine analysis has been introduced into newborn screening laboratories in the late 1990s and is now part of almost all newborn screening programs. Because newborn screening tests make use of dried blood spots (DBS) collected after a heel prick on the second to fifth day of life, the DBS is the most common specimen used for acylcarnitine analysis. In addition, many biochemical genetics laboratories offer clinical testing of acylcarnitines in DBS for patients at any age. As is true for plasma samples, the most informative results are obtained when blood samples are collected during acute illness or at least prior to a meal. Blood should be obtained by capillary stick of well-perfused skin (heels in young infants or fingers) and free dripping of a few drops of blood directly on the filter paper card. For postmortem analysis, blood and bile are collected at the latest at the time of autopsy.
Following complete drying at room temperature for at least 3 h, the sample can be sent ambient. While diagnostic results can be obtained in most cases of medium- and long-chain FAO defects even after prolonged storage time at room temperature, samples should be stored frozen (with desiccant) because particularly short-chain acylcarnitine species are not reliably measurable several months after collection (Matern et al. 1999). At least one blood or bile spot with a diameter of 1 cm should be collected to allow for any necessary repeat testing which usually requires only a DBS punch of 3 mm in diameter.
8 Urine
While initially the favored specimen, urine acylcarnitine analysis is the least appropriate when a FAO disorder is under diagnostic consideration. Long-chain acylcarnitines are typically bound to plasma albumin and are not excreted by the kidney. Urine is collected from patients suspected to have an organic acidemia preferably during an acute metabolic decompensation. As this is often not possible, an early morning specimen should be collected. The minimum volume of urine is 1 mL which allows for acylcarnitine and creatinine analysis; the latter is essential to normalize quantitative acylcarnitine results. The sample should be sent frozen and without preservatives.
9 Fibroblast Culture Medium
Most FAO disorders present similarly, and their biochemical diagnosis can be difficult because common metabolite screens, such as urine organic acids, plasma acylcarnitines, and fatty acids, are influenced by dietary factors and the clinical status of the patient (Van Hove et al. 2000) leading to incomplete diagnostic information or even false-negative results (Browning et al. 2005). Enzyme assays are limited to one enzyme per assay, and molecular assays for common mutations are limited by the frequent occurrence of compound heterozygous patients with an uncommon, private mutation that must be distinguished from unaffected carriers. The in vitro probe assay offers screening for several defects of FAO and organic acid metabolism under controlled laboratory conditions using fibroblast cultures (Table 51.3). The principle of this assay relies on the assumption that skin fibroblasts of patients affected with relevant conditions will accumulate certain acylcarnitine species reflecting the metabolic defect when the cell medium is supplemented with a long-chain fatty acid, branched-chain amino acids, and l-carnitine. An acylcarnitine analysis can be performed in the post-incubation cell medium by tandem mass spectrometry as for the other sample types (Smith and Matern 2010).
Fibroblasts are typically grown from a small skin biopsy collected during an outpatient visit or as part of a planned surgical procedure following routine culturing techniques. Cell cultures may also derive from umbilical cord or, for prenatal diagnostic purposes, from amniocytes obtained by amniocentesis. Samples should be sent at ambient temperatures.
10 Interpretation and Reference Ranges
The laboratory director, typically a board certified clinical biochemical geneticist or equivalent, reviews all profiles and provides an interpretation based on pattern recognition and not on single abnormal values (Tables 51.1, 51.4, 51.5, 51.6, and 51.7). Simple reporting of numeric results is not appropriate because most physicians are not familiar with pattern recognition. A comprehensive interpretation takes into consideration any available clinical and dietary information and other laboratory results and provides possible differential diagnoses, recommendations for additional biochemical testing, and confirmatory studies if indicated, as well as contact information for the laboratory director in case the referring physician has additional questions.
Pitfalls
The ordering physician must be aware of the limitations of a laboratory test and should consider involving the biochemical genetics laboratory in discussions regarding the most appropriate diagnostic workup of patients.
Pitfalls in acylcarnitine analysis of plasma, serum, blood spots, and bile spots
Acylcarnitine profiles are dependent on the clinical status of the patient at the time of sample collection (Van Hove et al. 2000). Accordingly, all samples should be submitted to the biochemical genetics laboratory with information regarding the clinical context during which the sample was collected. The laboratory must be aware of the fact that carnitine deficiency states can cause seemingly normal acylcarnitine profiles due to the lack of carnitine as substrate for carnitine palmitoyltransferases. Accordingly, it is crucial to review the complete profile and to initiate follow-up when even borderline elevated acylcarnitines are noted in the presence of abnormally low free acetylcarnitine. If clinically indicated, a repeat sample should be collected as early as 24 h after l-carnitine supplementation.
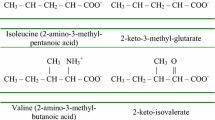
While MS/MS allows for unequivocal identification of most metabolites, there are a few exceptions (Table 51.1). In particular, the short-chain acylcarnitines of 4 and 5 carbons represent more than one analyte. C4-Acylcarnitine is known to be a mixture of butyrylcarnitine derived from fatty acid metabolism and isobutyrylcarnitine derived from the metabolism of valine (Oglesbee et al. 2007). C4-OH acylcarnitine can be a mixture of the d-3-OH-butyrylcarnitine, associated with ketosis; l-3-OH-butyrylcarnitine, associated with SCHAD deficiency; or 3-OH-isobutyrylcarnitine, characteristic of the valine degradation defect 3-OH-isobutyryl-CoA hydrolase deficiency. C5-Acylcarnitine is a mixture of isovalerylcarnitine and 2-methylbutyrylcarnitine derived from leucine and isoleucine degradation, respectively (Ensenauer et al. 2004; Matern et al. 2003). Samples of patients treated with antibiotics containing pivalic acid (e.g., pivampicillin) may contain pivaloylcarnitine, another C5 species. Several other metabolites are also nonspecific markers for several disorders. For example, C5-OH acylcarnitine, which represents 3-hydroxyisovalerylcarnitine and 2-methyl 3-hydroxybutyrylcarnitine, can be elevated in seven different organic acidemias. The interpretation of elevated C5-OH acylcarnitine in newborns or breast-fed infants is further complicated by the fact that it can indicate maternal 3-methylcrotonylglycinuria while the infant is only an unaffected carrier (Gibson et al. 1998). Differentiation is of clinical importance and most efficiently achieved by urine acylglycine and organic acid analyses.
The long-chain FAO disorders of CACT deficiency and CPT-II deficiency cannot be differentiated because both cause accumulation of the same long-chain acylcarnitine species which is explained by the fact that neither enzyme is involved in the chain-shortening action of FAO. Isolated LCHAD deficiency and complete mitochondrial TFP deficiency also cannot be differentiated by routine acylcarnitine analysis (Van Hove et al. 2000). When such profiles are encountered, delineation of the correct defect is only possible by either specific enzyme assay in cell cultures or molecular genetic analysis of the relevant genes. MCAD and MAD defects require the identification of medium-chain acylcarnitines. In this respect it is important to realize that patients on a medium-chain triglyceride-containing diet may accumulate C8 and C10 carnitine, potentially obscuring the diagnosis.
Isolated elevations of propionylcarnitine (C3) are not specific for propionic acidemia but also observed in methylmalonic acidemias of various etiologies. Because methylmalonylcarnitine (C4-DC) is not consistently elevated in methylmalonic acidemias, elucidation of the correct diagnosis requires at a minimum urine organic acid analysis.
Another antibiotic that may cause problems in the interpretation of butylated acylcarnitines is cefotaxime (Vianey-Saban et al. 2004). This antibiotic or metabolites thereof reveal itself by acylcarnitine analysis (following derivatization to butyl esters) at m/z 470 which otherwise is considered to represent the monounsaturated form of 3-hydoxyhexadecenoylcarnitine (C16:1-OH). In poorly resolved scans, this may be difficult to differentiate from m/z 472 which is a marker for LCHAD and TFP deficiencies. However, whereas m/z 472 (C16-OH) is more abundant than C16:1-OH in these FAO disorders, the profile of a patient treated with cefotaxime usually reveals an m/z 470 to m/z 472 ratio that is greater than 1. Furthermore and in contrast to cefotaxime treatment, both LCHAD and TFP deficiencies are usually accompanied by elevations of other long-chain species (Table 51.1) (Van Hove et al. 2000).
Formiminoglutamate (FIGLU), a marker for glutamate formiminotransferase deficiency, is revealed in acylcarnitine profiles by a peak with m/z 287 (Malvagia et al. 2006). In poorly resolved acylcarnitine profiles, this peak may be confused with iso-/butyrylcarnitine (m/z 288). To avoid the incorrect interpretation of acylcarnitine profiles, the analysis is best performed in product scan mode as opposed to multiple reaction monitoring (MRM) mode.
Pitfalls in acylcarnitine analysis of urine
Aside from the above mentioned potential problems such as the inability to discriminate isomers and interfering metabolites of antibiotics, urine also often contains a variety of nonidentified substances. This makes the interpretation of urine acylcarnitine profiles inherently more complex and overinterpretation must be avoided. Therefore, urine acylcarnitine analysis should not be included in the first line of screening investigations but be targeted to specific diagnostic considerations, in particular organic acidemias. A role of urine acylcarnitine analysis for the diagnosis of FAO disorders has not been established.
Pitfalls in acylcarnitine analysis of post-incubation fibroblast culture medium
The in vitro probe assay is performed under standardized conditions and is independent of the patient’s status at the time the skin biopsy is obtained. The accumulation of acylcarnitines in this assay system appears to be dependent on potentially present residual enzyme activity and therefore provides information regarding the severity of an enzyme deficiency state for some disorders such as VLCAD and SCAD deficiencies. Furthermore, as is true for acylcarnitine analysis of other sample types, TFP and isolated LCHAD deficiencies as well as CPT-II and CACT deficiencies cannot be differentiated. Finally, the analysis of fibroblasts does not allow for the diagnosis of enzyme deficiency states that are not expressed in this tissue (e.g., the liver-specific SCHAD deficiency).
References
Abdenur JE, Chamoles NA, Guinle AE, Schenone AB, Fuertes AN (1998) Diagnosis of isovaleric acidaemia by tandem mass spectrometry: false positive result due to pivaloylcarnitine in a newborn screening programme. J Inherit Metab Dis 21:624–630
Boemer F, Schoos R, de Halleux V, Kalenga M, Debray F-G (2014) Surprising causes of C5-carnitine false positive results in newborn screening. Mol Genet Metab (in press)
Bosch AM, Abeling NG, Ijlst L, Knoester H, van der Pol WL, Stroomer AE, Wanders RJ, Visser G, Wijburg FA, Duran M, Waterham HR (2011) Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J Inherit Metab Dis 34:159–164
Browning MF, Larson C, Strauss A, Marsden DL (2005) Normal acylcarnitine levels during confirmation of abnormal newborn screening in long-chain fatty acid oxidation Defects. J Inherit Metab Dis 28:545–550
Carrozzo R, Dionisi-Vici C, Steuerwald U, Lucioli S, Deodato F, Di Giandomenico S, Bertini E, Franke B, Kluijtmans LA, Meschini MC, Rizzo C, Piemonte F, Rodenburg R, Santer R, Santorelli FM, van Rooij A, Vermunt-de Koning D, Morava E, Wevers RA (2007) SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain 130:862–874
DiMauro S, DiMauro PM (1973) Muscle carnitine palmityltransferase deficiency and myoglobinuria. Science 182:929–931
Engel AG, Angelini C (1973) Carnitine deficiency of human skeletal muscle with associated lipid storage myopathy: a new syndrome. Science 179:899–902
Ensenauer R, Vockley J, Willard JM, Huey JC, Sass JO, Edland SD, Burton BK, Berry SA, Santer R, Grunert S, Koch HG, Marquardt I, Rinaldo P, Hahn S, Matern D (2004) A common mutation is associated with a mild, potentially asymptomatic phenotype in patients with isovaleric acidemia diagnosed by newborn screening. Am J Hum Genet 75:1136–1142
Gibson KM, Bennett MJ, Naylor EW, Morton DH (1998) 3-methylcrotonyl-coenzyme a carboxylase deficiency in amish/mennonite adults identified by detection of increased acylcarnitines in blood spots of their children. J Pediatr 132:519–523
Malvagia S, La Marca G, Casetta B, Gasperini S, Pasquini E, Donati MA, Zammarchi E (2006) Falsely elevated C4-carnitine as expression of glutamate formiminotransferase deficiency in tandem mass spectrometry newborn screening. J Mass Spectrom 41:263–265
Matern D, Strauss AW, Hillman SL, Mayatepek E, Millington DS, Trefz FK (1999) Diagnosis of mitochondrial trifunctional protein deficiency in a blood spot from the newborn screening card by tandem mass spectrometry and DNA analysis. Pediatr Res 46:45–49
Matern D, He M, Berry SA, Rinaldo P, Whitley CB, Madsen PP, Van Calcar SC, Lussky RC, Andresen BS, Wolff JA, Vockley J (2003) Prospective diagnosis of 2-methylbutyryl-CoA dehydrogenase deficiency in the Hmong population by newborn screening using tandem mass spectrometry. Pediatrics 112:74–78
Millington DS, Terada N, Chace DH, Chen YT, Ding JH, Kodo N, Roe CR (1992) The role of tandem mass spectrometry in the diagnosis of fatty acid oxidation disorders. Prog Clin Biol Res 375:339–354
Oglesbee D, He M, Majumder N, Vockley J, Ahmad A, Angle B, Burton B, Charrow J, Ensenauer R, Ficicioglu CH, Keppen LD, Marsden D, Tortorelli S, Hahn SH, Matern D (2007) Development of a newborn screening follow-up algorithm for the diagnosis of isobutyryl-CoA dehydrogenase deficiency. Genet Med 9:108–116
Rinaldo P, Studinski AL, Matern D (2001) Prenatal diagnosis of disorders of fatty acid transport and mitochondrial oxidation. Prenat Diagn 21:52–54
Rinaldo P, Matern D, Bennett MJ (2002) Fatty acid oxidation disorders. Annu Rev Physiol 64:477–502
Rinaldo P, Hahn S, Matern D (2004) Clinical biochemical genetics in the twenty-first century. Acta Paediatr 93:22–26
Roe DS, Vianey-Saban C, Sharma S, Zabot MT, Roe CR (2001) Oxidation of unsaturated fatty acids by human fibroblasts with very-long-chain acyl-CoA dehydrogenase deficiency: aspects of substrate specificity and correlation with clinical phenotype. Clin Chim Acta 312:55–67
Schulze-Bergkamen A, Okun JG, Spiekerkotter U, Linder M, Haas D, Kohlmuller D, Mayatepek E, Schulze-Bergkamen H, Greenberg CR, Zschocke J, Hoffmann GF, Kolker S (2005) Quantitative acylcarnitine profiling in peripheral blood mononuclear cells using in vitro loading with palmitic and 2-oxoadipic acids: biochemical confirmation of fatty acid oxidation and organic acid disorders. Pediatr Res 58:873–880
Shen JJ, Matern D, Millington DS, Hillman S, Feezor MD, Bennett MJ, Qumsiyeh M, Kahler SG, Chen YT, Van Hove JL (2000) Acylcarnitines in fibroblasts of patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency and other fatty acid oxidation disorders. J Inherit Metab Dis 23:27–44
Smith EH, Matern D (2010) Acylcarnitine analysis by tandem mass spectrometry. Curr Protoc Hum Genet (Chapter 17:Unit 17) 18:11–20
Tortorelli S, Hahn SH, Cowan TM, Brewster TG, Rinaldo P, Matern D (2005) The urinary excretion of glutarylcarnitine is an informative tool in the biochemical diagnosis of glutaric acidemia type I. Mol Genet Metab 84:137–143
Van Hove JL, Kahler SG, Feezor MD, Ramakrishna JP, Hart P, Treem WR, Shen JJ, Matern D, Millington DS (2000) Acylcarnitines in plasma and blood spots of patients with long-chain 3-hydroxyacyl-Coenzyme A dehydrogenase deficiency. J Inherit Metab Dis 23:571–582
Vaz FM, Wanders RJ (2002) Carnitine biosynthesis in mammals. Biochem J 361:417–429
Vianey-Saban C, Boyer S, Levrat V, Cheillan D, Piraud M, Guffon N, Maire I (2004) Interference of Cefotaxime in plasma acylcarnitine profile mimicking an increase of 3-hydroxypalmitoleylcarnitine (C16:1-OH) using butyl esters. J Inherit Metab Dis 27(Suppl 1):94
Young SP, Matern D, Gregersen N, Stevens RD, Bali D, Liu HM, Koeberl DD, Millington DS (2003) A comparison of in vitro acylcarnitine profiling methods for the diagnosis of classical and variant short chain acyl-CoA dehydrogenase deficiency. Clin Chim Acta 337:103–113
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Matern, D. (2014). Acylcarnitines. In: Blau, N., Duran, M., Gibson, K., Dionisi Vici, C. (eds) Physician's Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-40337-8_51
Download citation
DOI: https://doi.org/10.1007/978-3-642-40337-8_51
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-40336-1
Online ISBN: 978-3-642-40337-8
eBook Packages: MedicineMedicine (R0)