
Abstract
Congenital nephrotic syndrome (CNS) is a group of rare conditions that present with high-grade urine protein (albumin) losses, hypoalbuminemia, and edema within the first 3 months of life. It is important to differentiate CNS from other presentations of nephrotic syndrome, mainly the infantile form (4–12 months of age) and childhood nephrotic syndrome (onset after the 1st year of life). Unless treatment is initiated, CNS is universally fatal.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Nephrotic Syndrome
- Glomerular Basement Membrane
- Slit Diaphragm
- Congenital Nephrotic Syndrome
- Unilateral Nephrectomy
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
FormalPara Core Messages-
Congenital nephrotic syndrome (CNS) is a rare condition diagnosed within the first 3 months of life by high-grade proteinuria, hypoalbuminemia, and edema.
-
The most common form of CNS is the Finnish type (CNF), but several other genetic and nongenetic forms do exist and need to be considered in the differential diagnosis.
-
Genetic testing is available and should be strongly considered.
-
At this point in time, kidney transplantation is the only curative therapy available.
-
The management of children with CNS is complex and dependent on the degree of protein losses.
A female newborn was admitted at 2.5 months of age with lethargy and edema. Her birth weight was 6 lbs 14 oz and her weight at the time of admission was close to 9 lbs. She was born at home as the 4th child of a Mennonite family. She had gained about 1 lbs since she was seen by her pediatrician for an early 2-month check-up. She was exclusively breastfeeding. She seemed to tire much more quickly in the recent past. She was diagnosed with congenital hypothyroidism and received daily Synthroid therapy. She became increasingly mottled, febrile, and floppy and was transferred to the Pediatric Intensive Care Unit at our institution. A blood culture grew Streptococcus pneumoniae. She was noted to have hyponatremia, hypokalemia, and hypoalbuminemia with a serum albumin of 1.2 g/dl. A 24-h urine showed a protein excretion of 650 mg/m2/h equivalent to about 4 g/day. During the hospitalization, she received several albumin infusions and was started on Lasix, captopril, and enoxaparin. Despite extensive counseling and several family meetings, the parents did agree to neither genetic testing nor possible interventions including nephrectomies, dialysis, or renal transplantation. The child passed away at the family’s home 6 weeks later.
Eighteen months later, the parents had another child which was admitted at 33 weeks of gestation to the NICU. The child was found to have nephrotic-range proteinuria with a serum albumin of 0.9 g/dl. Despite extensive counseling, the parents requested discharge to comfort care without additional interventions.
15.1 Introduction
Congenital nephrotic syndrome (CNS) is a group of rare conditions that present with high-grade urine protein (albumin) losses, hypoalbuminemia, and edema within the first 3 months of life. It is important to differentiate CNS from other presentations of nephrotic syndrome, mainly the infantile form (4–12 months of age) and childhood nephrotic syndrome (onset after the 1st year of life). Unless treatment is initiated, CNS is universally fatal.
Causes of CNS can be divided into primary and secondary. The most common primary form is CNS of the Finnish type which is inherited in an autosomal recessive fashion and due to mutations of the NPHS1 gene. The gene is located on chromosome 19, and two important mutations have been described (Fin-major and Fin-minor) in over 90 % of the Finnish patients [1]. Other mutations have been reported in non-Finnish patients. Pertinent to our introductory case presentation, a 1481delC mutation is common in Mennonites leading to a truncated form of the nephrin protein [2]. Other primary forms include mutations of the podocin gene (NPHS2) most often leading to focal and segmental glomerulosclerosis and WT1 gene mutations among others as will be discussed later on.
Secondary causes include several infectious etiologies like congenital TORCH infections, hepatitis B and C, HIV, as well as syndrome-associated CNS like Denys-Drash syndrome, Lowe syndrome, or nail-patella syndrome among others [3].
In the more recent past, significant scientific advances have led to a better understanding of the genetics and pathophysiologic factors involved in the disease mechanism (involving the glomerular filtration barrier) of the proteinuric syndromes [4]. Even though the management of children with CNS remains complex and complicated, one certainly hopes that our improved understanding will open new treatment modalities and ultimately translate into improved outcome.
15.2 Pathophysiology
Congenital nephrotic syndrome is distinguished from other forms of nephrotic syndrome by the fact that it compromises a heterogeneous group of renal diseases that results in significant postnatal glomerular permeability that ultimately leads to massive proteinuria and hematuria immediately or shortly after birth.
Each form of primary CNS is part of the large group of the hereditary proteinuric syndromes. Even though pathophysiologically similar, secondary forms of CNS do have identifiable nongenetic causes.
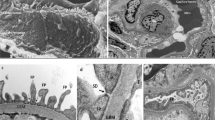
The glomerular filtration barrier is composed of three distinct layers: fenestrated endothelium, glomerular basement membrane, and the podocyte layer which is a composite of distal foot processes and interposed slit diaphragms. The glomerular basement membrane functions as the pre-filtration component while the slit diaphragm functions as the ultrafiltration unit of the filtration barrier. This filtration barrier functions to limit flux of solutes into the urine base and is charge and size selective. Figure 15.1 provides a diagram of the highly specialized filtration apparatus.

Overview of the renal filtration system (Reprinted from Khoshnoodi and Tryggvason [6], Copyright 2001, with permission from Elsevier)
The glomerular basement membrane is a precise network of proteins including but not limited to type IV collagen, laminin, nidogen, and proteoglycans which are the proteins responsible for giving the glomerular basement membrane its negative charge. The slit diaphragm – with the exact molecular ultrastructure still being unclear as mentioned by Jalanko – consists of podocyte proteins that form the backbone of the slit diaphragm that interacts with adapter proteins [5]. The adapter proteins are localized in the cytosolic component of the podocyte and connect the slit diaphragm with the actin cytoskeleton of the podocyte foot processes and play a pivotal role in signal transduction. Some of those adapter proteins are podocin, CD2AP, and ZO-1. The interaction of the actin cytoskeleton with other surrounding proteins, including α-actinin-4, plays a critical role in the maintenance of the podocyte structure. Figure 15.2 shows a cartoon of some of the most important components of the slit diaphragm and foot processes involved in CNS known today. The slit diaphragm plays a key role in restricting protein content into the ultrafiltrate reaching the Bowman space. Defects in either the slit diaphragms themselves or the structure/function of the podocyte foot processes are the primary reason for the high-grade proteinuria and lipiduria seen in CNS [5, 6]. It is the disruption of this highly intricate and specialized network of proteins that results in loss of the kidney filtration barrier located within the glomerular capillary wall which ultimately leads to CNS or other nephrotic disorders [5]. CNS is a result of excessive leakage of serum proteins into the urine. The exact mechanisms, even though still highly debated, are thought to be results of mutations in genes that encode for the structural and regulatory proteins that form the glomerular capillary wall which functions as a filtration barrier as mentioned above.

Important components of the glomerular filtration barrier altered by inherited forms of nephrotic syndrome (This figure was published in Weber [56], Copyright Elsevier)
15.3 Classification
CNS can be classified as primary when due to a known (or yet unknown) genetic mutation or secondary when it presents in the context of other, often infectious, disease. The primary forms can be divided in mutations involving the nephrin (NPHS1) and podocin gene (NPHS2) mutations as well as other genetic forms oftentimes associated with various syndromes. At least two thirds of newborns and infants diagnosed with CNS have a mutation in one of four genes: NPHS1, NHPS2, WT1, and LAMB2 [7]. The most common nongenetic forms are caused by certain infections or systemic disease:
-
1.
NPHS1: NPHS1 refers to nephrotic syndrome type 1 or congenital nephrotic syndrome of the Finnish type which was first described by Ahvenainen et al. in 1956 [8]. It is inherited in an autosomal recessive fashion occurring worldwide but most commonly found in the Finnish population. In 1994, the gene defect has been located on the long arm of chromosome 19, a year later to the exact location 19q13.1 [9, 10]. Affected neonates exhibit massive proteinuria in utero and develop nephrosis shortly after birth. It is due to a mutation in the nephrin gene which is a podocyte protein that is responsible for the slit diaphragm backbone [5]. Nephrin is a type 1 transmembrane protein that belongs in the immunoglobulin superfamily. It consists of eight immunoglobulin-like motifs followed by a fibronectin type III domain, a transmembrane region followed by a cytoplasmic carboxy-terminal part. Nephrin is expressed in multiple areas of the body. In the kidney, nephrin is expressed in the podocytes and exclusively localized in the slit diaphragm [6, 11–13]. NPHS1 is further divided into Fin-major and Fin-minor, with the most common form being Fin-major. Fin-major is due to a 2 base pair (2-bp) deletion in the stop codon on exon 2 (Fin-minor being a nonsense mutation in exon 26), which results in a complete loss of the slit diaphragms and ultimately massive nonselective proteinuria at birth [6, 14]. Recently, several new mutations of NPHS1 have been described including missense mutations and deletions [15]. Classic findings of NPHS1 include intrauterine growth restriction, muscular hypotonia, cardiac hypertrophy, microscopic hematuria with or without normal creatinine, and hyperlipidemia [5]. Most infants are born prematurely with the placenta being significantly enlarged accounting for more than 25 % of the neonate’s birth weight. Edema is present immediately at birth or within a few days. Massive proteinuria is accompanied by profound hypoalbuminemia with associated hypogammaglobulinemia. Renal biopsy shortly after birth presents with dilatations of the proximal tubules and microcyst formation with normal glomeruli. Radiographic features include increased cortical echogenicity, loss of corticomedullary differentiation, expansion of the glomerular mesangium, and dilatation of the proximal and distal tubules. Classic microscopic findings include interstitial fibrosis and inflammatory infiltrates, and effacement of the podocyte foot processes. Renal failure occurs most often between 3 and 8 years of age. Fin-minor is due to a missense mutation in exon 26 [14]. Figure 15.2 provides a diagram of the molecular structure of the glomerular filtration barrier and the role of nephrin.
-
2.
NPHS2: Mutations of the NPHS2 gene – which encodes a protein called podocin – are responsible for an autosomal recessive steroid-resistant nephrotic syndrome with often rapid progression to end-stage kidney disease. It is most common in children between 3 months and 5 years of age [6]. Several mutations have been described, the most common one due to a substitution of R138Q in the podocin gene that is a found on chromosome 1q25–q32. This mutation results in the retention of podocin in the endoplasmic reticulum and its inability to recruit nephrin in the lipid rafts [14, 16]. Podocin is a lipid raft-associated protein at the filtration slit that is exclusively expressed in the podocyte foot process. It interacts with nephrin at the level of the lipid rafts and is responsible for initiating nephrin signaling [14]. Podocin interacts with ion channels and the cytoskeleton [6]. Podocin also interacts with CD2AP, an adapter protein that stabilized contacts between podocytes, thus providing structural integrity and function to the slit diaphragm. This detailed interaction provides maintenance of the glomerular permselectivity [17]. There is wide variability in the degree of proteinuria that occurs with NPHS2. Classical microscopic findings include focal segmental glomerular sclerosis which may ultimately lead to end-stage renal disease. NPHS2 does not typically have any extrarenal manifestations primarily due to lack of expression of podocin in other areas [5]. Figure 15.2 provides a diagram of the molecular structure of the glomerular filtration barrier and the role of nephrin.
-
3.
Other Genetic Forms of CNS
-
Abnormalities in the Wilms tumor suppressor gene WT1 can lead to isolated nephrotic syndrome or nephrotic syndrome associated with developmental syndromes including Denys-Drash, Frasier, and WAGR syndromes. WT1 is a transcription factor that is responsible for the embryonic development of the genitourinary system. WT1 controls nephrin expression. Denys-Drash syndrome is defined by the combination of nephrotic syndrome, diffuse mesangial sclerosis, Wilms tumor, and male pseudohermaphroditism [18, 19]. The reason for associated gonadal anomalies is due to WT1 tumor gene is found on chromosome 11p13 and plays a vital role in the transcription of urogenital development. WT1 is highly expressed during embryonic development but in mature kidneys is only expressed in podocytes and epithelial cells of the Bowman’s capsule. The WT1 gene product results in the downregulatory effect on transcription factors such as PAX2, PAX8, and NOVH. Mutations ultimately result in absence or reduction in WT1 which leads to overexpression of PAX2 and possible glomerular lesions found in some patients with Denys-Drash syndrome [6]. Frasier syndrome has also been associated with mutations of the WT1 gene and is characterized by the association of male pseudohermaphroditism and progressive glomerulopathy usually due to FSGS. Proteinuria is usually detected between 2 and 6 years of age. Patients with Frasier syndrome typically have a slower progression to end-stage renal disease than those with Denys-Drash syndrome and progress to FSGS [14].
-
Pierson syndrome is a rare autosomal recessive condition caused by deficiency in laminin-β2 expressed in the glomerular basement membrane in which combined ocular and renal abnormalities are found in combination [20, 21]. Laminin-β2 (LAMB2) is responsible for anchoring the podocyte processes to the glomerular basement membrane. It was recently described as a cause of CNS [13].
-
Galloway-Mowat syndrome, first described in 1968, is an autosomal recessive disorder that results in CNS with associated central nervous system abnormalities and hiatus hernia [22]. The gene has not been identified. Central nervous system abnormalities result due to the fact that podocytes consist of similar structure proteins as neuronal cells [5].
-
The loss-of-function mutation causing the autosomal dominant disease hereditary onycho-osteodysplasia (nail-patella syndrome) was first described in 1998 [23]. LAMX1B is primary expressed by podocytes within the kidney and regulates several important proteins involved in the filtration barrier including nephrin, podocin, and the adapter protein CD2AP.
-
A new mutation involving the phospholipase C epsilon 1 (PLCE1, also referred to as NPHS3) gene more recently reported in 2006 [24].
-
Mitochondrial myopathies and mutations in LAMB3 have also been associated with CNS [25, 26].
-
-
4.
Nongenetic Forms: Congenital infections including HIV, CMV, toxoplasmosis, or syphilis can result in congenital proteinuric syndromes [27, 28]. Therefore, newborns with nephrotic-range proteinuria and evidence of CNS should undergo a thorough investigation for transmitted or acquired infections. Also, infantile systemic lupus erythematosus has been reported to be associated with CNS [29, 30]. To our knowledge, CNS has not been associated with neonatal lupus syndrome and it is important to differentiate the two entities [31].
15.4 Epidemiology
CNS is a rare disorder. The exact incidence and prevalence are unknown and most epidemiologic data available refer to congenital nephrotic syndrome of the Finnish type (CNF). With advances in prenatal screening, the prevalence of CNF has been estimated to be 4.2 per 10,000 live births. Due to termination of pregnancy, the incidence has decreased to 0.9 per 10,000 live births [32].
15.5 Clinical Presentation
The majority of patients born with CNS are born prematurely and are small for gestational age, with the placenta usually weighing more than 25 % of the newborn’s birth weight [6]. The patients typically have delayed ossification which attributes to widely enlarged sutures. Generalized edema presents within the first week of life for the majority of infants [5]. Typically urine analysis will reveal exclusively proteinuria without any evidence of hematuria which helps differentiate it from other possible etiologies for neonatal edema. The neonates also have profound hypoalbuminemia with associated severe hypogammaglobulinemia. Serum albumin is <10 g/l at presentation and urinary protein concentration is >20 g/l when serum albumin is corrected to >15 g/l [33]. These infants may present septic from lack of gamma globulin and possibly severe thromboembolic complications from excretion of coagulation factors. Infants may have symptoms typically found in hypothyroidism from urinary excretion of thyroxine-binding proteins. Renal sonography demonstrates enlarged hyperechogenic kidneys with lack of corticomedullary differentiation. Elevated creatinine is typically not common initially in the disease process but typically presents between 3 and 8 years of age [5].
15.6 Diagnosis
In CNF, high levels of α-fetoprotein (AFP) in the amniotic fluid and maternal serum can be useful for prenatal diagnosis in selected at-risk families [34, 35]. However, increased levels of α-fetoprotein (AFP) are unspecific and genetic analysis is the preferred method for making the diagnosis [5, 36]. Increased levels of amniotic fluid α-fetoprotein concentration may be used as a marker to distinguish congenital nephrotic syndrome Finnish type from diffuse mesangial sclerosis.
The classic clinical finding of CNS is generalized edema from reduced serum protein content from renal excretion. Clinical signs may not be initially present during the first weeks of life. Patients will present with urinary protein levels greater than 20 g per liter and serum albumin levels are less than 10 g per liter. Renal function is typically normal with all forms of CNS initially. Blood pressure readings are also extremely variable depending on the stage of CNS. Patient with renal failure will have associated hypertension and those with significant untreated proteinuria or hypoalbuminemia may potentially be hypotensive. Diagnostic imaging in the form of a renal ultrasound can be helpful. The kidneys are usually enlarged and echogenic with identifiable loss of corticomedullary differentiation as well as poor visualization of the renal pyramids [37]. Renal biopsy can be a useful tool to confirm the diagnosis in CNS but it does not identify the etiology of CNS due to significant overlap of different disease entities. Specific immunohistochemistry methods to identify expression of podocin or nephrin can be helpful and should be obtained if available. The purpose of a biopsy would be to identify any associated sclerosis or interstitial fibrosis which may guide therapy. Genetic analysis is the optimal method to identify CNS diagnosis. Analysis of NPHS1 and NPHS2 mutations is required for all CNS patients. In cases where NPHS1 and NPHS2 are not identified, WT1 or LAMB2 genetic analysis should be obtained. In some instances NPHS1 may be identified in utero in families with known risk for CNS by obtaining alpha-fetoprotein levels in the amniotic fluid prior to 20 weeks gestation [5].
15.7 Differential Diagnosis
Once CNS is suspected and nephrotic-range proteinuria is confirmed on 24-h collection (urine protein excretion >40 mg/m2/day), a thorough differential diagnosis needs to be built as outlined earlier in this chapter. The maternal and neonatal history as well as physical exam findings (presence or absence of associated syndromic features) will determine additional serologic and genetic testing of mother and child and aid in establishing a correct diagnosis which is important to determine the degree of necessary interventions as well as the expected course of the disease. For children who do have pathologic proteinuria but do not fit criteria for CNS, the differential diagnosis should include genetic or nongenetic tubular and/or glomerular disorders, and a comprehensive evaluation should be completed depending on associated findings as discussed elsewhere in this book.
15.8 General Aspects of Management
The management of children with CNS can be divided into a medical as well as a surgical component. Both of these have the goal to optimize the child’s cognitive and physical growth. The medical component includes treatment of edema by the use of albumin infusions and loop diuretics as well as medications. Surgical care primarily includes native nephrectomy and renal transplantation. Licht et al. have recommended a “stepwise treatment approach” to the disease that continues to be followed by most centers [38]. This stepwise approach has supportive therapies as base, followed by use of medications (ACE inhibitors and Indocin), unilateral nephrectomy, and ultimately bilateral nephrectomy with dialysis before ultimately renal transplantation.
15.8.1 Medical Management
15.8.1.1 Nutrition
Nutrition and growth deficits are particularly common in patients with congenital nephrotic syndrome and need to be minimized under all circumstances [5]. High-protein and high-calorie diets are usually necessary to provide patients with sufficient substrate for basic metabolic needs in addition to growth. Maintaining 120 kcal/kg/day with high-protein diet of 3–4 g/kg/day with a low-sodium diet has provided additional sources of therapy [33]. Breast milk and milk formulas are used first line and additional protein calories are provided using casein-based products [39]. Typical diets should consist of 10–14 % protein, 40–50 % fat, and 40–50 % carbohydrate energy. Monounsaturated and polyunsaturated fatty acids may be substituted with 10–15 ml of rape seed oil and 2 ml of fish oil daily. Initially patients are placed on breast milk and milk formulas that are fortified with additional protein [5]. Many patients may require nasogastric tube feeds to ensure that adequate nutrition is maintained to guarantee their basic metabolic needs. Children should receive additional supplements in magnesium and calcium secondary to deficits in parathyroid hormone which is secondary to excretion of albumin. Recommended magnesium supplement includes 50 mg/day in addition to 500–1,000 mg/day of calcium. Excretion of albumin and thus reduced levels of parathyroid hormone results requirement for vitamin D supplementation. All patients should receive 2,000 IU/day of vitamin D2 and 2.5–3.0 mg/day of rape seed oil in the form of vitamin E. Patients should also receive alphacalcidol pulse therapy to avoid hyperparathyroidism and a calcium carbonate as a phosphate binder. Other nutritional considerations should include low-salt diet and possible tube feeding or parenteral alimentation for provision of maximal growth for the ultimate goal of renal transplantation. Table 15.1 provides a list of nutritional recommendations for congenital nephrotic syndrome of the Finnish type during the first year of life.
15.8.1.2 Albumin and Diuretics
The basic problem in children with congenital nephrotic syndrome is the massive urinary loss of protein, 90 % of which is albumin. This loss leads to protein malnutrition, reduced growth, and secondary symptoms of hypoproteinemia [33]. Albumin infusions provide a necessary supplementation for the loss of necessary serum proteins that provide an osmotic gradient for hemodynamic stability, nutrition for growth, coagulation, and protection from infections in the form of immunoglobulin. A common method of substitution includes parenteral albumin infusions with a 20 % albumin solution via a central venous catheter followed by a furosemide bolus. Typical dosing includes 1–5 ml/kg albumin infusions divided into three infusions over 2 h. To guarantee normal growth, serum albumin concentration of 15–20 g/l should be maintained [33]. Maintenance therapy typically may consist of a single albumin infusion of up to 20 ml/kg over 6 h followed by a single Lasix bolus. This substitution only temporarily relieves hypoproteinemia but does not provide complete resolution of the generalized edema [5]. High-protein diets have also played a key role in the medical management of congenital nephrotic syndrome. Maintaining 120 kcal/kg/day with high-protein diet of 3–4 g/kg/day with a low-sodium diet has provided additional sources of therapy [33].
15.8.1.3 Antiproteinuric Therapies
In contrast to childhood nephrotic syndrome, immunosuppressive medications including corticosteroids play no role in the treatment. This certainly makes sense as the disease does not respond with the pathophysiologic process being not immune mediated [40]. The overall goal of therapy is to control generalized edema, maintain renal function, and avoid complications from the loss of anticoagulants and immunoglobulin [5]. Angiotensin-converting enzyme (ACE) inhibitors, used either as monotherapy or in combination with indomethacin, have been shown to be able to control proteinuria and maintain growth and health in some children with CNS [41–43]. Angiotensin-converting enzyme inhibitors lower intraglomerular pressure which ultimately results in a reduction in excretion of protein. Indomethacin exerts its antiproteinuric effects by inhibiting prostaglandin synthetase. With close monitoring of therapy response (as measured by degree of urine protein loss) and renal function, starting doses of 1 mg/kg/day of both captopril and Indocin have been recommended. This dose can be increased to a maximum of about 5 mg/kg/day [38]. In our center, mildly lower starting doses of 0.2–0.5 mg/kg/day with close monitoring of the serum creatinine have been used in the past.
15.8.1.4 Others
Patients with congenital nephrotic syndrome should have TSH and free T4 levels regularly obtained due to risk for hypothyroidism. This seems to be explained by high urinary excretion of thyroid-binding globulin as well as thyroxine and patients may benefit from thyroxine substitution. The hypothyroidism being a result of urinary protein losses is supported by the fact that hypothyroidism usually reverses after bilateral nephrectomies [44]. On laboratory evaluation, serum thyroxine concentrations are always low, TSH may be normal initially, but increase in most patients during the first months. Recommended dosing of thyroxine is typically 6.25 μg/day that should be started from birth and may be adjusted accordingly [33].
Immunoglobulin levels should be monitored due to urinary excretion of gamma globulin and complement factors B and D in patients with congenital nephrotic syndrome. Due to significant urinary excretion of gamma globulin and complement factors, these patients must have a high suspension for significant bacterial infections, specifically capsular organisms such as pneumococci. Parental antibiotics and immunoglobulin should be promptly initiated and admitted to the hospital for a thorough evaluation if at any time they presented with fevers or any other evidence of an infectious process.
Warfarin has been recommended by some centers in patients with NPHS1 [11]. This recommendation is in light of the fact that these patients have significant urinary losses of coagulation factors. Urinary excretion of plasminogen and AT III results in plasma deficiencies, and compensatory protein synthesis may result in increased production of macroglobulins, fibrinogen, thromboplastin, and factors II, V, VII, X, and XIII that result in a hypercoagulable state [33]. Some centers have recommended daily aspirin for all patients with congenital nephrotic syndrome due to this pathophysiology. Patients requiring any surgical procedures are recommended to discontinue warfarin and initiate antithrombin III 50 IU/kg to compensate for urinary losses prior to surgery.
15.8.2 Surgical Management
The role of uni- and bilateral nephrectomies has been investigated in the past [45]. Both methods therefore provide a bridge for the overall goal of renal transplantation [5]. Especially for children with WT1 gene mutations who are progressing quickly toward ESRD or have reached it, bilateral nephrectomy should be considered given the risk for development of Wilms tumor [46]. Some institutions routinely perform unilateral nephrectomies to reduce the amount of protein excretion. Unilateral nephrectomy does not eliminate the need for aggressive nutritional support and albumin infusions. Uremia also develops more often with unilateral nephrectomy [33]. In addition, other institutions perform bilateral nephrectomies and good outcomes have been reported [47]. Even though there are no guidelines, it is preferred to wait with nephrectomies until the patient reaches an acceptable weight and age. Following nephrectomy, the child will be maintained on peritoneal dialysis until renal transplantation becomes an option. In children under the age of 24 months, CNS is a common reason for initiation of peritoneal dialysis [48]. Typically following bilateral nephrectomy, hypoproteinemia dramatically will improve while the patients receive peritoneal dialysis. Most common complications that arise are peritonitis with staphylococci that are readily treated with intraperitoneal vancomycin. Bilateral nephrectomy at an age of 6–10 months and CCPD for 3–4 months provides correction of hypoproteinemia and further improves nutritional status. Subsequently, successful renal transplantation by 2 years of age provides long-term treatment for congenital nephrotic syndrome of any form [33].
Renal transplantation is the overall preferred treatment for congenital nephrotic syndrome. Typical age of renal transplantation is 1–2 years of age using an adult-size kidney if a graft is available [33]. Typical management of patients following transplantation should be maintained. Recurrence of nephrotic syndrome following graft is rare but is possible if anti-nephrin antibodies developed following transplantation. Typically this may be successfully managed with cyclophosphamide and plasmapheresis [5].
15.9 Prognosis
It is important to distinguish between the different types of nephrotic syndrome. Whereas children with CNF invariably progress toward dialysis- and transplant-dependent renal failure, the clinical course can be different in other forms even with reported spontaneous remission [49]. Several cases of recurrence of proteinuria, nephrotic syndrome, and high risk for graft loss after renal transplantation have been described in patients with CNSF [50, 51]. The pathophysiologic process is thought to be secondary to circulating anti-glomerular and anti-nephrin antibodies. Following transplantation, any patient can potentially develop an immune response to nephrin expressed in the graft which functions as a new antigen, and the anti-nephrin antibodies neutralize the action of nephrin. If proteinuria develops, it is typically observed 2–48 months after the initial transplantation. Several treatments for the recurrent proteinuria have been described, including cyclophosphamide, plasmapheresis, and most recently the anti-CD20 antibody rituximab [52–54]. FSGS has also been found on biopsy of patients with reoccurrence of proteinuria but has been more commonly found in patients who originally had heterozygous mutations in NPHS2. The overall prognosis of congenital nephrotic syndrome is significantly improved if successful renal transplantation is achieved. Reported patient survival in a study including a large number of children with CNF is greater than 90 % at 5 years posttransplant [55]. However, chronic allograft nephropathy is a long-term complication following transplantation that typically results in the need for a second renal transplant [5].
15.10 Conclusion
CNS is a rare but extraordinarily complex disease depending on etiology and degree of urine protein losses as well as its high potential for possible complications. Despite our markedly improved understanding of the genetics, pathophysiology, and pathology of the disease, the treatment options are limited and the prognosis remains overall guarded.
15.11 Take-Home Pearls
-
Congenital Nephrotic Syndrome (CNS) is defined as nephrotic syndrome presenting within the first 3 months of life. The most common form is the Finnish type (CNF) which usually progresses rapidly toward end-stage renal disease.
-
The management is focused on protein substitution and management of edema, optimal nutrition and provision of medications (anticoagulation, thyroxine, antiproteinurics), dialysis, and renal transplantation.
-
Recent advances in our understanding of the genetics and associated pathophysiology might at some point improve phenotype recognition, lead to new treatment options, and ultimately translate into improved outcome.
-
Careful guidance and education of the families during this process is of extreme importance.
References
Kestila M et al (1998) Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell 1(4):575–582
Bolk S et al (1999) Elevated frequency and allelic heterogeneity of congenital nephrotic syndrome, Finnish type, in the old order Mennonites. Am J Hum Genet 65(6):1785–1790
Papez KE, Smoyer WE (2004) Recent advances in congenital nephrotic syndrome. Curr Opin Pediatr 16(2):165–170
Tryggvason K, Patrakka J, Wartiovaara J (2006) Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med 354(13):1387–1401
Jalanko H (2009) Congenital nephrotic syndrome. Pediatr Nephrol 24(11):2121–2128
Khoshnoodi J, Tryggvason K (2001) Congenital nephrotic syndromes. Curr Opin Genet Dev 11(3):322–327
Hinkes BG et al (2007) Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics 119(4):e907–e919
Ahvenainen EK, Hallman N, Hjelt L (1956) Nephrotic syndrome in newborn and young infants. Ann Paediatr Fenn 2(3):227–241
Kestila M et al (1994) Congenital nephrotic syndrome of the Finnish type maps to the long arm of chromosome 19. Am J Hum Genet 54(5):757–764
Mannikko M et al (1995) Fine mapping and haplotype analysis of the locus for congenital nephrotic syndrome on chromosome 19q13.1. Am J Hum Genet 57(6):1377–1383
Savage JM et al (1999) Improved prognosis for congenital nephrotic syndrome of the Finnish type in Irish families. Arch Dis Child 80(5):466–469
Suh JH et al (2011) Forced expression of laminin beta1 in podocytes prevents nephrotic syndrome in mice lacking laminin beta2, a model for Pierson syndrome. Proc Natl Acad Sci USA 108(37):15348–15353
VanDeVoorde R et al (2006) Pierson syndrome: a novel cause of congenital nephrotic syndrome. Pediatrics 118(2):e501–e505
Niaudet P (2004) Genetic forms of nephrotic syndrome. Pediatr Nephrol 19(12):1313–1318
Heeringa SF et al (2008) Thirteen novel NPHS1 mutations in a large cohort of children with congenital nephrotic syndrome. Nephrol Dial Transplant 23(11):3527–3533
Huber TB et al (2003) Molecular basis of the functional podocin-nephrin complex: mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum Mol Genet 12(24):3397–3405
Schwarz K et al (2001) Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest 108(11):1621–1629
Denys P et al (1967) Association of an anatomo-pathological syndrome of male pseudohermaphroditism, Wilms’ tumor, parenchymatous nephropathy and XX/XY mosaicism. Arch Fr Pediatr 24(7):729–739
Drash A et al (1970) A syndrome of pseudohermaphroditism, Wilms’ tumor, hypertension, and degenerative renal disease. J Pediatr 76(4):585–593
Pierson M et al (1963) An unusual congenital and familial congenital malformative combination involving the eye and kidney. J Genet Hum 12:184–213
Zenker M et al (2004) Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet 13(21):2625–2632
Galloway WH, Mowat AP (1968) Congenital microcephaly with hiatus hernia and nephrotic syndrome in two sibs. J Med Genet 5(4):319–321
Dreyer SD et al (1998) Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet 19(1):47–50
Hinkes B et al (2006) Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 38(12):1397–1405
Goldenberg A et al (2005) Respiratory chain deficiency presenting as congenital nephrotic syndrome. Pediatr Nephrol 20(4):465–469
Hata D et al (2005) Nephrotic syndrome and aberrant expression of laminin isoforms in glomerular basement membranes for an infant with Herlitz junctional epidermolysis bullosa. Pediatrics 116(4):e601–e607
Besbas N et al (2006) Cytomegalovirus-related congenital nephrotic syndrome with diffuse mesangial sclerosis. Pediatr Nephrol 21(5):740–742
Shahin B, Papadopoulou ZL, Jenis EH (1974) Congenital nephrotic syndrome associated with congenital toxoplasmosis. J Pediatr 85(3):366–370
Dudley J et al (1996) Systemic lupus erythematosus presenting as congenital nephrotic syndrome. Pediatr Nephrol 10(6):752–755
Massengill SF, Richard GA, Donnelly WH (1994) Infantile systemic lupus erythematosus with onset simulating congenital nephrotic syndrome. J Pediatr 124(1):27–31
Westenend PJ (1995) Congenital nephrotic syndrome in neonatal lupus syndrome. J Pediatr 126(5 Pt 1):851
Levy M, Feingold J (2000) Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int 58(3):925–943
Holmberg C et al (1995) Management of congenital nephrotic syndrome of the Finnish type. Pediatr Nephrol 9(1):87–93
Seppala M et al (1976) Congenital nephrotic syndrome: prenatal diagnosis and genetic counselling by estimation of amniotic-fluid and maternal serum alpha-fetoprotein. Lancet 2(7977):123–125
Wiggelinkhuizen J et al (1976) Alpha fetoprotein in the antenatal diagnosis of the congenital nephrotic syndrome. J Pediatr 89(3):452–455
Mannikko M et al (1997) Improved prenatal diagnosis of the congenital nephrotic syndrome of the Finnish type based on DNA analysis. Kidney Int 51(3):868–872
Lanning P et al (1989) Ultrasonic features of the congenital nephrotic syndrome of the Finnish type. Acta Paediatr Scand 78(5):717–720
Licht C et al (2000) A stepwise approach to the treatment of early onset nephrotic syndrome. Pediatr Nephrol 14(12):1077–1082
Jalanko H, Holmberg C (2009) Congenital nephrotic syndrome. In: Avner ED, Niaudet P, Yoshikawa N (eds) Pediatric nephrology. Springer, Berlin, pp 601–619
Kovacevic L, Reid CJ, Rigden SP (2003) Management of congenital nephrotic syndrome. Pediatr Nephrol 18(5):426–430
Guez S et al (1998) Adequate clinical control of congenital nephrotic syndrome by enalapril. Pediatr Nephrol 12(2):130–132
Heaton PA, Smales O, Wong W (1999) Congenital nephrotic syndrome responsive to captopril and indometacin. Arch Dis Child 81(2):174–175
Pomeranz A et al (1995) Successful treatment of Finnish congenital nephrotic syndrome with captopril and indomethacin. J Pediatr 126(1):140–142
Chadha V, Alon US (1999) Bilateral nephrectomy reverses hypothyroidism in congenital nephrotic syndrome. Pediatr Nephrol 13(3):209–211
Kim MS, Primack W, Harmon WE (1992) Congenital nephrotic syndrome: preemptive bilateral nephrectomy and dialysis before renal transplantation. J Am Soc Nephrol 3(2):260–263
Hu M et al (2004) Prophylactic bilateral nephrectomies in two paediatric patients with missense mutations in the WT1 gene. Nephrol Dial Transplant 19(1):223–226
Slaughenhoupt BL et al (1998) Urologic management of congenital nephrotic syndrome of the Finnish type. Urology 51(3):492–494
Laakkonen H et al (2008) Peritoneal dialysis in children under two years of age. Nephrol Dial Transplant 23(5):1747–1753
Haws RM, Weinberg AG, Baum M (1992) Spontaneous remission of congenital nephrotic syndrome: a case report and review of the literature. Pediatr Nephrol 6(1):82–84
Laine J et al (1993) Post-transplantation nephrosis in congenital nephrotic syndrome of the Finnish type. Kidney Int 44(4):867–874
Patrakka J et al (2002) Recurrence of nephrotic syndrome in kidney grafts of patients with congenital nephrotic syndrome of the Finnish type: role of nephrin. Transplantation 73(3):394–403
Chaudhuri A et al (2011) Rituximab treatment for recurrence of nephrotic syndrome in a pediatric patient after renal transplantation for congenital nephrotic syndrome of Finnish type. Pediatr Transplant 16(5):E183–E187
Kuusniemi AM et al (2007) Plasma exchange and retransplantation in recurrent nephrosis of patients with congenital nephrotic syndrome of the Finnish type (NPHS1). Transplantation 83(10):1316–1323
Srivastava T et al (2006) Recurrence of proteinuria following renal transplantation in congenital nephrotic syndrome of the Finnish type. Pediatr Nephrol 21(5):711–718
Qvist E et al (1999) Graft function 5–7 years after renal transplantation in early childhood. Transplantation 67(7):1043–1049
Weber S (2008) Hereditary nephrotic syndrome. In: Geary DF, Schaefer F (eds) Comprehensive pediatric nephrology, 1st edn. Elsevier, Philadelphia
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Pleasant, L.D., Kiessling, S.G. (2014). Congenital Nephrotic Syndrome. In: Chishti, A., Alam, S., Kiessling, S. (eds) Kidney and Urinary Tract Diseases in the Newborn. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-39988-6_15
Download citation
DOI: https://doi.org/10.1007/978-3-642-39988-6_15
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-39987-9
Online ISBN: 978-3-642-39988-6
eBook Packages: MedicineMedicine (R0)