
Abstract
Human T-cell lymphotropic virus type 1 (HTLV-1) was originally discovered in the early 1980s. It is the first retrovirus to be unambiguously linked causally to a human cancer. HTLV-1 currently infects approximately 20 million people worldwide. In this chapter, we review progress made over the last 30 years in our understanding of HTLV-1 infection, replication, gene expression, and cellular transformation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Long Terminal Repeat
- Bovine Leukemia Virus
- Human Promyelocytic Leukemia HL60 Cell
- Tropical Spastic Paraparesis
- Hematopoietic Progenitor Stem Cell
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Human T-cell lymphotropic virus type 1 (HTLV-1) is the first identified human retrovirus. This virus belongs to the Deltaretrovirus genera of the Orthoretrovirinae subfamily which includes HTLV-2, HTLV-3, HTLV-4 (Mahieux and Gessain 2005; Mahieux and Gessain 2009), Bovine Leukemia Virus (BLV), and Simian T-cell lymphotropic virus (STLV). The virus was discovered in 1980–1981 by analyzing T cells from a patient suffering T-cell leukemias (ATL) (Poiesz et al. 1980; Hinuma et al. 1981; Miyoshi et al. 1981; Yoshida 1982; Watanabe et al. 1983; Gallo 2005). ATL is a rapidly fatal disease first described in Japan (Takatsuki 2005). Since then, a causal association between HTLV-1 and ATL has become firmly established (Gallo 2005). To date, HTLV-1 is the only known retrovirus that is directly linked to a human cancer. In addition to this cancer link, the virus can also cause inflammatory diseases such as HTLV-1-associated Myelopathy (HAM)/tropical spastic paraparesis (TSP), uveitis, infective dermatitis, and myositis (Gessain 2011; Goncalves et al. 2010).
2 HTLV-1 Infection
2.1 Epidemiology
Approximately 20 million people worldwide are infected with HTLV-1 (Proietti et al. 2005). However, HTLV-1 is not evenly distributed throughout the world. Indeed, the areas of highest prevalence of HTLV-1 are mainly southern Japan, the Caribbean islands, parts of South America and Central Africa, with foci in the Middle East, and Australia (Goncalves et al. 2010). This geographic distribution of HTLV-1 with some clustering of regions with high prevalence is still not understood (Proietti et al. 2005). Among HTLV-1-infected people, 2–5 % will develop ATL after a long latency period of 30–60-year post-infection; by comparison, approximately 0.25–5 % of the infected individuals will develop HAM/TSP. The development of ATL or TSP/HAM is not influenced by the subtype of HTLV-1 infection (Watanabe 2011; Ono et al. 1994). Indeed, while six subtypes of HTLV-1 (subtypes A-F) have been reported, the great majority of infections are caused by the cosmopolitan subtype A.
HTLV-1 has 3 modes of transmission: (1) mother to child, mainly through prolonged breastfeeding (>6 months); sexual, (2) mainly but not exclusively occurring from male to female; and (3) by blood products contaminated with infected lymphocytes (Goncalves et al. 2010; Matsuura et al. 2010). Male individuals and those infected in their early childhood are at the highest risk of developing ATL (Goncalves et al. 2010; Matsuura et al. 2010).
2.2 Tropism and Receptors
In vitro, HTLV-1 can infect many cell types including several non-lymphoid tumor cell lines such as human osteogenic sarcoma cells, lung cells, cervical carcinoma cells (HeLa), human gastric HGC-27 cells, human promyelocytic leukemia HL60 cells, as well as primary endothelial cells, monocytes, microglial cells, and mammary epithelial cells (Clapham et al. 1983; Hayami et al. 1984; Ho et al. 1984; Hiramatsu et al. 1986; Akagi et al. 1988; LeVasseur et al. 1998). However, in vivo, HTLV-1 is found primarily in CD4+ and CD8+ T lymphocytes (Nagai et al. 2001) and less frequently in other cell types such as monocytes, endothelial cells, myeloid, and plasmacytoid dendritic cells (Macatonia et al. 1992; Koyanagi et al. 1993; Jones et al. 2008), and CD34+ hematopoietic progenitor cells (Banerjee et al. 2008, 2010; Feuer et al. 1996; Grant et al. 2002; Tripp et al. 2003, 2005). Until the discovery of the glucose transporter GLUT1 as a receptor for HTLV-1 in 2003, little was known about the entry receptors for HTLV-1 (Manel et al. 2003). Currently, the published data from different laboratories support the idea of a multireceptor model for HTLV-1 entry (Fig. 1). Three cell surface proteins have been found to be involved in HTLV-1 entry: glucose transporter 1 (GLUT1), neuropilin-1 (NRP-1), and heparan sulfate proteoglycans (HSPG) (Jones et al. 2011). The following steps possibly explain HTLV-1 entry into cells. First, the surface subunit (SU) of the virally encoded envelope glycoprotein interacts with the heparan sulfate proteoglycans/neuropilin-1 complexes. Next, these interactions trigger conformational changes of the SU which are followed by the binding of the SU to GLUT1, and finally membrane fusion occur to allow the entry of the virus into the target cell (Jones et al. 2005, 2011; Pinon et al. 2003; Ghez et al. 2006; Lambert et al. 2009).

A multireceptor model for HTLV-1 entry. HSPG = heparan sulfate proteoglycans; SU = the subunit of HTLV-1 envelope glycoprotein; NRP-1 = neuropilin-1; GLUT-1 = glucose transporter 1; CTD = C-terminal domain; RBD = receptor-binding domain; PRR = proline-rich region. This drawing is modified after Jones et al. (2012) (Jones et al. 2011)
2.3 Viral Replication
At the cellular level, HTLV-1 is transmitted via two major routes: through cell-to-cell contact (horizontal transmission) and via clonal expansion of HTLV-1-infected cells (vertical transmission).
2.3.1 Cell-to-Cell Transmission
Naturally infected T lymphocytes produce little to no free viral particles, and the infectivity of these cell-free particles is very low. In vivo, HTLV-1 intercellular transmission, i.e., horizontal, reverse-transcription-based replication, requires close cell-to-cell contact. To date, three mechanisms have been reported in the literature (Fig. 2). First, in 2003, Igakura et al. showed the formation of a “virological synapse,” composed of viral and cellular molecules, at the point of contact between the HTLV-1-infected and recipient target cells (Igakura et al. 2003; Nejmeddine et al. 2005). Second, Pais-Correia et al. described that after viral budding, HTLV-1 virions are retained on the cell surface of infected cells in extracellular viral assemblies composed of collagen, agrin, and linker proteins such as tetherin and galectin-3 (Pais-Correia et al. 2010). When HTLV-1-infected cells attach to uninfected cells, the viral particles contained in these extracellular biofilm-like structures are rapidly transferred to the surface of the target cells, resulting in infection (Pais-Correia et al. 2010). Third, it was recently demonstrated by Franchini and colleagues that HTLV-1 encodes a protein, p12I, in its pX region. The processing of p12I generates p8I. This protein increases T-cell contact by clustering lymphocyte function-associated antigen-1 (LFA-1); it promotes T-cell conjugation through LFA-1 and intercellular adhesion molecule 1 (ICAM-1) interaction; and it enhances HTLV-1 cell-to-cell transmission by inducing the formation of cellular conduits (Van Prooyen et al. 2010; Fukumoto et al. 2009).

Mechanisms of cell-to-cell transmission of HTLV-1. This drawing is modified after Yasunaga and Matsuoka (2011)
2.3.2 Clonal Expansion
HTLV-1 infection is associated with an elevated proviral load, very low cell-to-cell transmission rate, and high viral genetic stability. This high genetic stability of HTLV-1 (and other deltaretroviruses) is due to its replication in vivo via “the clonal expansion of infected cells” (Wattel et al. 1995; Cavrois et al. 1996; Cavrois et al. 1996; Wattel et al. 1996; Zane et al. 2009). Indeed, instead of using the error-prone viral RT, the HTLV-1 genome is propagated as an integrated provirus that is replicated during cellular DNA synthesis. Since HTLV-1 mostly integrates randomly into the host genome, sequential analyses of integration sites have verified that the proliferation of HTLV-1-infected cells is clonal and persistent (Etoh et al. 1997; Cavrois et al. 1998). In some animal models [e.g., experimentally infected squirrel monkeys (Saimiri sciureus) and sheep with HTLV-1 and BLV, respectively], it has been shown that deltaretrovirus infection is a two-step process that includes an early (primo-infection) and transient phase of reverse transcription, before the establishment of an immune response, followed by the persistent multiplication of infected cells by clonal expansion (Mortreux et al. 2001; Pomier et al. 2008). The clonal cells survive over time, and it has been found that ATL originates from one of these clones present during the primo-infection (Moules et al. 2005).
2.4 Viral Expression
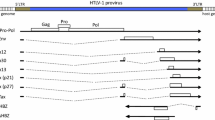
As shown in Fig. 3, the HTLV-1 proviral genome contains retroviral structural and non-structural genes. The viral gag, pro, pol, and env genes are flanked by the long terminal repeats (LTR) at both ends, and a region named pX is located between env and the 3′ LTR. The 5′ LTR serves as the viral promoter for transcription. The Pol open reading frame encodes reverse transcriptase, protease, and integrase. Gag provides the virion core proteins, and Env is used for viral infectivity. The pX region contains four partially overlapping open reading frames (ORFs); and through the use of alternative splicing and internal initiation codons, it encodes several regulatory proteins. Orf-I produces the p12I protein which can be proteolytically cleaved at the amino terminus to generate the p8I protein, while differential splicing of mRNAs from orf-II results in the production of the p13II and p30II proteins. Orf-III and orf-IV encode the Rex and Tax proteins, respectively; and an antisense mRNA transcribed from the 3′ LTR generates the HTLV-1 basic leucine zipper (HBZ) protein. Below, we will discuss in brief the roles of Tax and HBZ on the induction and the maintenance of leukemogenesis, respectively (Matsuoka and Jeang 2007).

The HTLV-1 proviral genome showing the expression of various spliced transcripts and the open reading frames (ORFs) that they encode. This drawing is modified from Matsuoka and Jeang (2007)
3 Tax Expression Dictates the Fate of HTLV-1-infected Cells
Expression of the viral Tax oncoprotein is sufficient to immortalize T cells (Grassmann et al. 1992), transform rodent cells (Tanaka et al. 1990), and induce tumorigenesis in mouse models (Hinrichs et al. 1987; Nerenberg et al. 1987; Green et al. 1989; Iwakura et al. 1991; Kwon et al. 2005; Hasegawa et al. 2006; Fu et al. 2011). Recently, Banerjee et al. have reported on the transformation of human cells into leukemic cells. Using immune-deficient NOD/SCID mice, they showed that CD4+ lymphomas can arise from mice that are injected with CD34+hematopoietic progenitor stem cells transduced to express Tax (Banerjee et al. 2010). These data raise the notion that a target of Tax transformation may be the CD34+ hematopoietic progenitor stem cells, instead of and perhaps in addition to the currently considered mature CD4+ or CD8+ T lymphocytes.
To become tumorigenic, cells have to grow more rapidly than non-transformed cells. The tumorigenic cells accumulate genetic changes (clastogenic damage or aneuploidy) and enforce the propagation of these aberrant changes by neutralizing the cell cycle checkpoints. To be effective, tumorigenic cells must also evade the host’s immune responses (Hanahan and Weinberg 2000, 2011).
Data from multiple laboratories over the past 25 years have begun to shed light on how Tax confers growth advantage to HTLV-1-infected cells, and how this viral oncoprotein triggers DNA damage accumulation and inhibits the cell cycle checkpoints during its transformation of a normal cell into a leukemic cell (Fig. 4).

Multistep processes that lead to the transformation of normal hematopoietic cells into ATL cells. The scheme incorporates the concept that ATL leukemogenesis is induced by Tax. This drawing is modified from Matsuoka and Jeang (2011)
3.1 Tax Promotes the Survival and the Proliferation of HTLV-1-infected Cells
3.1.1 Tax and Apoptosis and Senescence
Like other oncogenes, Tax confers pro-proliferative and pro-survival properties to cells (Schmitt et al. 1998; Xiao et al. 2001; Iwanaga et al. 2008). Curiously, its expression also has been reported to trigger apoptosis (Yamada et al. 1994; Chlichlia et al. 1995; Fujita and Shiku 1995; Chen et al. 1997; Hall et al. 1998; Kao et al. 2000; Nicot and Harrod 2000) and senescence (Kinjo et al. 2010; Yang et al. 2011; Zhi et al. 2011). These apparently contradictory findings are reconciled if one realizes that Tax performs a single signaling event that differentially elicits either a growth or death/senescence response depending on the context of the cell. Thus, the Tax signal for cells to grow capably stimulates cellular to proliferation under physiologically conditions favorable for growth conditions. On the other hand, under austere conditions that are non-permissive for cellular growth, the same Tax proliferative signal presumably attempts to initiate an increased metabolic program that cannot be consummated and instead the cells react by committing apoptosis or entering senescence (Kasai and Jeang 2004). Stated another way, Tax signaling is always intended to promote cell division. Cells, depending on context, can respond to that dictate to proliferate by growing or by executing apoptosis/senescence. Thus, Tax does not have two countervailing and contradictory functions; rather, it is the same function that elicits two different cellular outcomes (proliferation versus apoptosis/senescence) depending on the status of the infected cell (Jeang 2010; Boxus and Willems 2012). In vivo, because HTLV-1 infection ultimately leads to leukemogenesis and T-cell proliferation in some individuals, in these persons, it is clear that the prevailing effect of Tax is pro-proliferative and anti-apoptotic (Copeland et al. 1994; Kishi et al. 1997; Arai et al. 1998; Mulloy et al. 1998; Kawakami et al. 1999); in others who do not develop ATL, it is possible that apoptotic/senescent cellular responses predominate. For understanding the process of leukemogenesis, Tax’s activity on factors such as p53 (Mulloy et al. 1998; Haoudi and Semmes 2003; Jung et al. 2008) and NF-kB is consistent with the requirements in transformed cells of activating anti-apoptotic genes and suppressing pro-apoptotic genes (Kawakami et al. 1999; Tsukahara et al. 1999; Nicot et al. 2000; Mori et al. 2001; Pise-Masison et al. 2002; Krueger et al. 2006; Okamoto et al. 2006; Waldele et al. 2006).
3.1.2 Tax and NF-κB
NF-κB is a major survival factor engaged by HTLV-1. NF-κB is constitutively active in most tumor cells, and its suppression inhibits the growth of tumor (Chaturvedi et al. 2011; Perkins 2012). Although tightly controlled in normal cells, including T cells, NF-κB is constitutively activated in both transformed and untransformed HTLV-1-infected cells (Watanabe et al. 2005; Qu and Xiao 2011).
The NF-κB family of transcription factors has five closely related DNA-binding proteins (RelA (p65), RelB, c-Rel, NF-κB1/p50, and NF-κB2/p52) that can form various homodimers and heterodimers to regulate the transcription of genes containing κB motifs in their promoters. Latent or unstimulated cells sequester NF-κB dimers in the cytoplasm using inhibitors of kappa B (IκBs) proteins such as IκBα and p100. Upon activation, IκBs are degraded (canonical pathway) or p100 is processed to generate p52 (non-canonical pathway) leading to the translocation of active NF-κB proteins into the nucleus to activate transcription (Qu and Xiao 2011; Rauch and Ratner 2011). In the canonical pathway, IκBα degradation requires its phosphorylation by a specific IκB kinase (IKK) complex composed of two catalytic subunits IKKα (or IKK1) and IKKβ (or IKK2), and a regulatory subunit IKKγ (or NEMO). This phosphorylation results in rapid ubiquitination and proteasomal degradation of IκBα, allowing RelA (or p65), and other NF-κB members to localize to the nucleus in order to induce gene expression. In the non-canonical NF-κB pathway, IKKγ is specifically recruited into the p100 complex to phosphorylate p100, leading to p100 ubiquitination and processing to p52 which then associates with NF-κB-binding partners and translocates into the nucleus to induce or repress gene expression (Qu and Xiao 2011).
Work from many investigators has shown that Tax activates both canonical and non-canonical NF-κB signaling pathways in HTLV-1-infected cells (Xiao et al. 2001; Iha et al. 2003; Qu and Xiao 2011). Tax persistently activates IKK through binding to IKKγ, leading to the degradation of IκBα (canonical pathway) (Chu et al. 1999; Harhaj and Sun 1999; Jin et al. 1999; Xiao et al. 2000); and Tax promotes the formation of an IKKα-IKKγ-p100 complex followed by the processing of the NF-κB p100 precursor protein to its active p52 form (non-canonical pathway) (Xiao et al. 2001). Tax also binds to and increases the stability and activity of NF-κB (Hirai et al. 1992; Suzuki et al. 1993; Suzuki et al. 1994) and inactivates NF-κB inhibitors (Maggirwar et al. 1995; Suzuki et al. 1995; Good and Sun 1996; McKinsey et al. 1996; Petropoulos et al. 1996).
Recently, two independent studies using two different Tax transgenic mouse models have revealed that Tax-induced tumorigenesis is dependent on the NF-κB pathway and that both canonical and non-canonical NF-κB pathways are involved in this process (Kwon et al. 2005; Fu et al. 2011). The first study used mice expressing a wild-type Tax or a mutant form of Tax that is unable to activate the NF-κB pathway. A lethal cutaneous disease that shares several features in common with the skin disease that occurs during the preleukemic stage in HTLV-1-infected patients developed in the wild-type Tax–expressing mice (Kwon et al. 2005). In the second study, the investigators found that the genetic knockout of the NF-κB2 gene alone dramatically delayed tumor onset in Tax-expressing transgenic mice (Fu et al. 2011).
3.1.3 Tax and the Cell Cycle
Progression through the cell cycle is a tightly controlled process regulated by interactions between cyclins and cyclin-dependent kinases (CDKs). Tax deregulates the progression of infected cells through different phases of the cell cycle, especially the progression through G1.
Tax propels the cell through G1 by increasing the formation of complexes of cyclin D/CDK4, cyclin D/CDK6, and cyclin E/CDK6 via several mechanisms (Marriott and Semmes 2005). First, Tax can transcriptionally activate the expression of cyclins D2 (Akagi et al. 1996; Santiago et al. 1999; Iwanaga et al. 2001) and E (Iwanaga et al. 2001), CDK2 and 4 (Iwanaga et al. 2001), and transcriptionally repress CKIs such as p18INK4c, p19INK4D, and p27KIP1 (Suzuki et al. 1999; Iwanaga et al. 2001). Additionally, Tax can directly bind CDK4 (Haller et al. 2002; Fraedrich et al. 2005) and p16INK4a, thereby preventing the inhibitory p16INK4a molecule from binding to CDK4 and CDK6 (Low et al. 1997). Finally, Tax directly binds the retinoblastoma (RB) protein, which is a target substrate of cyclin D/CDK4/CDK6 and cyclin E/CDK2 complexes, and triggers proteosomal degradation of the RB protein; this then leads to the release of the E2F1 transcription factor from RB and the transcription of E2F1-responsive genes whose products are necessary for passage of the cells through G1 into S phase (Kehn et al. 2005). Moreover, it has also been reported that Tax expression activates the transcription of the E2F1 gene (Mori 1997; Lemasson et al. 1998; Ohtani et al. 2000).
Another fundamental property of Tax is that it can inhibit the G1/S checkpoint to allow cell cycle progression to happen even with the presence of DNA damage (Marriott and Semmes 2005). Accordingly, Tax can inhibit p53 activity which functions to monitor DNA structure integrity at the G1/S transition (Tabakin-Fix et al. 2006).
3.2 Tax-Expressing Cells Accumulate DNA Damage
Genetic instability of HTLV-1-infected cells generates the acquisition of eight biological changes predicted to be needed for the multistep development of ATL (Hanahan and Weinberg 2000, 2011). Two major types of genetic instability include the loss of DNA repair capabilities and the loss of euploidy. Indeed, Tax is able to disrupt normal cellular processes of DNA repair and chromosomal segregation (Majone et al. 1993; Saggioro et al. 1994, 1996; Lemoine and Marriott 2002).
3.2.1 Tax and Clastogenic Damage
The chromosomes in ATL cells show clastogenic damage (Marriott et al. 2002). Tax engenders direct DNA damage by increasing reactive oxygen species (Kinjo et al. 2010) and/or by inhibiting p53 checkpoint function (Tabakin-Fix et al. 2006). Two major mechanisms have been hypothesized to explain Tax-abrogation of p53 function. One model suggests that there is a competition between p53 and Tax for binding to the transcription coactivator CREB–binding protein (CBP)/p300 (Ariumi et al. 2000); a second model suggests that Tax activation of NF-κB is required for its inactivation of p53 (Miyazato et al. 2005). More recent data suggest that neither model satisfactorily explain Tax-p53 functional interaction, leaving incompletely answered the question of how Tax disables p53 function.
3.2.2 Tax and Aneuploidy
The majority of cancer cells including ATL cells are aneuploid. Aneuploidy has been proposed to be a cause of transformation. It has been shown that Tax can induce aneuploidy via several mechanisms. Tax can directly trigger chromosomal separation errors in two ways. First, Tax has been shown to cause multipolar mitosis (Peloponese et al. 2005; Ching et al. 2006; Nitta et al. 2006). Tax can also induce aberrant centrosomal multiplication by targeting the cellular TAX1BP2 protein, which normally blocks centriole over-duplication (Ching 2006). Second, during mitosis, Tax engages RANBP1 and fragments spindle poles, provoking multipolar segregation (Peloponese et al. 2005). Moreover, Tax has also been shown to promote premature mitotic exit by binding and activating the anaphase-promoting complex/cyclosome (APC/C). Finally, Tax-expressing cells are lost for the “aneuploidy” checkpoint in mitosis because of Tax-mediated inactivation of the critical spindle assembly checkpoint protein, Mad1 (Liu et al. 2005; Jin et al. 1998).
4 ATL
4.1 Absence of Tax Expression and Evasion of the Host’s Immune Surveillance
HTLV-1 chronic infection arises when an equilibrium is established between viral virulence and the host immunity. HTLV-1 requires Tax expression to transform cells, but Tax is also the main target of the host’s cytotoxic T Lymphocytes (CTLs) (Jacobson et al. 1990; Kannagi et al. 1991; Elovaara et al. 1993; Yamano et al. 2002). Thus, biologically, the virus has to evolve a process to control Tax expression to evade the host’s immune surveillance. Early after infection, the current view is that Tax is needed to initiate the cascade of events leading to transformation. On the other hand, Tax-expressing cells immediately become recognized as foreign entities and are targeted by the host’s immune system (cytotoxic T cells, CTL) for elimination. Accordingly, a balance has to be reached between growth advantage conferred by Tax to the cell and the susceptibility of the same cell to CTL killing. Early in virus infection when growth advantages conferred by Tax outweigh CTL killing, Tax expression is maintained in virus-infected cells; later in infection, the opposite may be the case which then explains why most HTLV-1 transformed cells become silenced for Tax expression. Thus, it is currently considered that although Tax is needed early to initiate transformation, when cells become transformed, Tax is no longer needed for maintenance of transformation. Given that situation and the need to evade CTL killing, it is not surprising that in ATLs cells late in the course of virus infection, more than 60 % of such cells show no detectable Tax transcripts (Takeda et al. 2004; Taniguchi et al. 2005; Miyazaki et al. 2007). While it is still not fully understood how Tax expression is silence, some of this likely occurs from genetic changes in the Tax gene (Furukawa et al. 2001; Takeda et al. 2004), epigenetic changes in the viral promoter in the 5′LTR (DNA hypermethylation and histone modifications) (Koiwa et al. 2002; Takeda et al. 2004; Taniguchi et al. 2005), and/or deletion of 5′LTR sequences (Tamiya et al. 1996).
4.2 HBZ Expression
The mechanism of how cells acquire Tax-independent proliferation is not completely understood. One explanation is that the genetic host chromosomal changes accumulated over time in HTLV-1-infected cells may have conferred sufficient virus-independent transformation/growth properties to those cells. An additional explanation may be the expression of the viral HBZ transcript/protein. Indeed, HBZ mRNA is highly expressed in ATL cells (Murata et al. 2006; Satou et al. 2006; Miyazaki et al. 2007). Using in vivo models, it has been shown that HBZ is expressed later than Tax in the infected cell, and its expression increases over time (Li et al. 2009). In contrast to Tax, HBZ sequence is not mutated in ATL cells (Fan et al. 2010), and the 3′LTR containing its promoter remains intact (Taniguchi et al. 2005; Fan et al. 2010). Moreover, although HBZ is an immunogenic protein, HBZ-specific CTLs seem unable to efficiently eliminate HTLV-1-infected cells (Suemori et al. 2009). HBZ further promotes virus-infected cell to proliferate late in infection (Satou et al. 2006), and its silencing of viral expression appears to enhance virus-infected cells to escape the host’s immune response (Gaudray et al. 2002). The complementary expression patterns of Tax and HBZ suggest that Tax and HBZ may act early and late, respectively, in virus infection with the former used to initiate transformation and the latter utilized to maintain the transformed phenotype of ATL cells.
5 Concluding Remarks
Despite robust progress, several questions regarding ATL leukemogenesis remain unresolved. First, what is the true cellular target of virus/Tax transformation? To date, only human CD34+ hematopoietic progenitor stem cells have been successfully transformed by Tax while other differentiated human primary cells have been refractory to Tax-mediated transformation. Thus, it is unclear what cellular factor differences between progenitor versus differentiated human cells govern Tax-induced transformation? Second, how does Tax fully inactivate p53 function? As mentioned above, current hypotheses on how Tax inactivates p53 appears to be unsatisfactory. Third, what factors are needed for the initiation of ATL versus those needed for maintenance of ATL? One anticipates that progress will be made on these and other questions in the coming years.
References
Akagi T, Ono H et al (1996) Expression of cell-cycle regulatory genes in HTLV-I infected T-cell lines: possible involvement of Tax1 in the altered expression of cyclin D2, p18Ink4 and p21Waf1/Cip1/Sdi1. Oncogene 12(8):1645–1652
Akagi T, Yoshino T et al (1988) Isolation of virus-producing transformants from human gastric cancer cell line, HGC-27, infected with human T-cell leukemia virus type I. Jpn J Cancer Res 79(7):836–842
Arai M, Kannagi M et al (1998) Expression of FAP-1 (Fas-associated phosphatase) and resistance to Fas-mediated apoptosis in T cell lines derived from human T cell leukemia virus type 1-associated myelopathy/tropical spastic paraparesis patients. AIDS Res Hum Retroviruses 14(3):261–267
Ariumi Y, Kaida A et al (2000) HTLV-1 tax oncoprotein represses the p53-mediated trans-activation function through coactivator CBP sequestration. Oncogene 19(12):1491–1499
Banerjee P, Sieburg M et al (2008) Human T-cell lymphotropic virus type 1 infection of CD34+ hematopoietic progenitor cells induces cell cycle arrest by modulation of p21(cip1/waf1) and survivin. Stem Cells 26(12):3047–3058
Banerjee P, Tripp A et al (2010) Adult T-cell leukemia/lymphoma development in HTLV-1-infected humanized SCID mice. Blood 115(13):2640–2648
Boxus M, Willems L (2012) How the DNA damage response determines the fate of HTLV-1 Tax-expressing cells. Retrovirology 9:2
Cavrois M, Gessain A et al (1996a) Proliferation of HTLV-1 infected circulating cells in vivo in all asymptomatic carriers and patients with TSP/HAM. Oncogene 12(11):2419–2423
Cavrois M, Leclercq I et al (1998) Persistent oligoclonal expansion of human T-cell leukemia virus type 1-infected circulating cells in patients with Tropical spastic paraparesis/HTLV-1 associated myelopathy. Oncogene 17(1):77–82
Cavrois M, Wain-Hobson S et al (1996b) Adult T-cell leukemia/lymphoma on a background of clonally expanding human T-cell leukemia virus type-1-positive cells. Blood 88(12):4646–4650
Chaturvedi MM, Sung B et al (2011) NF-kappaB addiction and its role in cancer: ‘one size does not fit all’. Oncogene 30(14):1615–1630
Chen X, Zachar V et al (1997) Role of the Fas/Fas ligand pathway in apoptotic cell death induced by the human T cell lymphotropic virus type I Tax transactivator. J Gen Virol 78(Pt 12):3277–3285
Ching YP, Chan SF et al (2006) The retroviral oncoprotein Tax targets the coiled-coil centrosomal protein TAX1BP2 to induce centrosome overduplication. Nat Cell Biol 8(7):717–724
Chlichlia K, Moldenhauer G et al (1995) Immediate effects of reversible HTLV-1 tax function: T-cell activation and apoptosis. Oncogene 10(2):269–277
Chu ZL, Shin YA et al (1999) IKKgamma mediates the interaction of cellular IkappaB kinases with the tax transforming protein of human T cell leukemia virus type 1. J Biol Chem 274(22):15297–15300
Clapham P, Nagy K et al (1983) Productive infection and cell-free transmission of human T-cell leukemia virus in a nonlymphoid cell line. Science 222(4628):1125–1127
Copeland KF, Haaksma AG et al (1994) Inhibition of apoptosis in T cells expressing human T cell leukemia virus type I Tax. AIDS Res Hum Retroviruses 10(10):1259–1268
Elovaara I, Koenig S et al (1993) High human T cell lymphotropic virus type 1 (HTLV-1)-specific precursor cytotoxic T lymphocyte frequencies in patients with HTLV-1-associated neurological disease. J Exp Med 177(6):1567–1573
Etoh K, Tamiya S et al (1997) Persistent clonal proliferation of human T-lymphotropic virus type I-infected cells in vivo. Cancer Res 57(21):4862–4867
Fan J, Ma G et al (2010) APOBEC3G generates nonsense mutations in human T-cell leukemia virus type 1 proviral genomes in vivo. J Virol 84(14):7278–7287
Feuer G, Fraser JK et al (1996) Human T-cell leukemia virus infection of human hematopoietic progenitor cells: maintenance of virus infection during differentiation in vitro and in vivo. J Virol 70(6):4038–4044
Fraedrich K, Muller B et al (2005) The HTLV-1 Tax protein binding domain of cyclin-dependent kinase 4 (CDK4) includes the regulatory PSTAIRE helix. Retrovirology 2:54
Fu J, Qu Z et al (2011) The tumor suppressor gene WWOX links the canonical and noncanonical NF-kappaB pathways in HTLV-I Tax-mediated tumorigenesis. Blood 117(5):1652–1661
Fujita M, Shiku H (1995) Differences in sensitivity to induction of apoptosis among rat fibroblast cells transformed by HTLV-I tax gene or cellular nuclear oncogenes. Oncogene 11(1):15–20
Fukumoto R, Andresen V et al (2009) In vivo genetic mutations define predominant functions of the human T-cell leukemia/lymphoma virus p12I protein. Blood 113(16):3726–3734
Furukawa Y, Kubota R et al (2001) Existence of escape mutant in HTLV-I tax during the development of adult T-cell leukemia. Blood 97(4):987–993
Gallo RC (2005) History of the discoveries of the first human retroviruses: HTLV-1 and HTLV-2. Oncogene 24(39):5926–5930
Gaudray G, Gachon F et al (2002) The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol 76(24):12813–12822
Gessain A (2011) Human retrovirus HTLV-1: descriptive and molecular epidemiology, origin, evolution, diagnosis and associated diseases. Bull Soc Pathol Exot 104(3):167–180
Ghez D, Lepelletier Y et al (2006) Neuropilin-1 is involved in human T-cell lymphotropic virus type 1 entry. J Virol 80(14):6844–6854
Goncalves DU, Proietti FA et al (2010) Epidemiology, treatment, and prevention of human T-cell leukemia virus type 1-associated diseases. Clin Microbiol Rev 23(3):577–589
Good L, Sun SC (1996) Persistent activation of NF-kappa B/Rel by human T-cell leukemia virus type 1 tax involves degradation of I kappa B beta. J Virol 70(5):2730–2735
Grant C, Barmak K et al (2002) Human T cell leukemia virus type I and neurologic disease: events in bone marrow, peripheral blood, and central nervous system during normal immune surveillance and neuroinflammation. J Cell Physiol 190(2):133–159
Grassmann R, Berchtold S et al (1992) Role of human T-cell leukemia virus type 1 X region proteins in immortalization of primary human lymphocytes in culture. J Virol 66(7):4570–4575
Green JE, Hinrichs SH et al (1989) Exocrinopathy resembling Sjogren’s syndrome in HTLV-1 tax transgenic mice. Nature 341(6237):72–74
Hall AP, Irvine J et al (1998) Tumours derived from HTLV-I tax transgenic mice are characterized by enhanced levels of apoptosis and oncogene expression. J Pathol 186(2):209–214
Haller K, Wu Y et al (2002) Physical interaction of human T-cell leukemia virus type 1 Tax with cyclin-dependent kinase 4 stimulates the phosphorylation of retinoblastoma protein. Mol Cell Biol 22(10):3327–3338
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Haoudi A, Semmes OJ (2003) The HTLV-1 tax oncoprotein attenuates DNA damage induced G1 arrest and enhances apoptosis in p53 null cells. Virology 305(2):229–239
Harhaj EW, Sun SC (1999) IKKgamma serves as a docking subunit of the IkappaB kinase (IKK) and mediates interaction of IKK with the human T-cell leukemia virus Tax protein. J Biol Chem 274(33):22911–22914
Hasegawa H, Sawa H et al (2006) Thymus-derived leukemia-lymphoma in mice transgenic for the Tax gene of human T-lymphotropic virus type I. Nat Med 12(4):466–472
Hayami M, Tsujimoto H et al (1984) Transmission of adult T-cell leukemia virus from lymphoid cells to non-lymphoid cells associated with cell membrane fusion. Gann 75(2):99–102
Hinrichs SH, Nerenberg M et al (1987) A transgenic mouse model for human neurofibromatosis. Science 237(4820):1340–1343
Hinuma Y, Nagata K et al (1981) Adult T-cell leukemia: antigen in an ATL cell line and detection of antibodies to the antigen in human sera. Proc Natl Acad Sci USA 78(10):6476–6480
Hirai H, Fujisawa J et al (1992) Transcriptional activator Tax of HTLV-1 binds to the NF-kappa B precursor p105. Oncogene 7(9):1737–1742
Hiramatsu K, Masuda M et al (1986) Mode of transmission of human T-cell leukemia virus type I (HTLV I) in a human promyelocytic leukemia HL60 cell. Int J Cancer 37(4):601–606
Ho DD, Rota TR et al (1984) Infection of human endothelial cells by human T-lymphotropic virus type I. Proc Natl Acad Sci USA 81(23):7588–7590
Igakura T, Stinchcombe JC et al (2003) Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299(5613):1713–1716
Iha H, Kibler KV et al (2003) Segregation of NF-kappaB activation through NEMO/IKKgamma by Tax and TNFalpha: implications for stimulus-specific interruption of oncogenic signaling. Oncogene 22(55):8912–8923
Iwakura Y, Tosu M et al (1991) Induction of inflammatory arthropathy resembling rheumatoid arthritis in mice transgenic for HTLV-I. Science 253(5023):1026–1028
Iwanaga R, Ohtani K et al (2001) Molecular mechanism of cell cycle progression induced by the oncogene product Tax of human T-cell leukemia virus type I. Oncogene 20(17):2055–2067
Iwanaga R, Ozono E et al (2008) Activation of the cyclin D2 and cdk6 genes through NF-kappaB is critical for cell-cycle progression induced by HTLV-I Tax. Oncogene 27(42):5635–5642
Jacobson S, Shida H et al (1990) Circulating CD8+ cytotoxic T lymphocytes specific for HTLV-I pX in patients with HTLV-I associated neurological disease. Nature 348(6298):245–248
Jeang KT (2010) HTLV-1 and adult T-cell leukemia: insights into viral transformation of cells 30 years after virus discovery. J Formos Med Assoc 109(10):688–693
Jin DY, Spencer F et al (1998) Human T cell Leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell 93(1):81–91
Jin DY, Giordano V et al (1999) Role of adapter function in oncoprotein-mediated activation of NF-kappaB. Human T-cell leukemia virus type I Tax interacts directly with IkappaB kinase gamma. J Biol Chem 274(25):17402–17405
Jones KS, Akel S et al (2005) Induction of human T cell leukemia virus type I receptors on quiescent naive T lymphocytes by TGF-beta. J Immunol 174(7):4262–4270
Jones KS, Lambert S et al (2011) Molecular aspects of HTLV-1 entry: functional domains of the HTLV-1 surface subunit (SU) and their relationships to the entry receptors. Viruses 3(6):794–810
Jones KS, Petrow-Sadowski C et al (2008) Cell-free HTLV-1 infects dendritic cells leading to transmission and transformation of CD4(+) T cells. Nat Med 14(4):429–436
Jung KJ, Dasgupta A et al (2008) Small-molecule inhibitor which reactivates p53 in human T-cell leukemia virus type 1-transformed cells. J Virol 82(17):8537–8547
Kannagi M, Harada S et al (1991) Predominant recognition of human T cell leukemia virus type I (HTLV-I) pX gene products by human CD8+ cytotoxic T cells directed against HTLV-I-infected cells. Int Immunol 3(8):761–767
Kao SY, Lemoine FJ et al (2000) HTLV-1 Tax protein sensitizes cells to apoptotic cell death induced by DNA damaging agents. Oncogene 19(18):2240–2248
Kasai T, Jeang KT (2004) Two discrete events, human T-cell leukemia virus type I Tax oncoprotein expression and a separate stress stimulus, are required for induction of apoptosis in T-cells. Retrovirology 1:7
Kawakami A, Nakashima T et al (1999) Inhibition of caspase cascade by HTLV-I tax through induction of NF-kappaB nuclear translocation. Blood 94(11):3847–3854
Kehn K, Fuente Cde L et al (2005) The HTLV-I Tax oncoprotein targets the retinoblastoma protein for proteasomal degradation. Oncogene 24(4):525–540
Kinjo T, Ham-Terhune J et al (2010) Induction of reactive oxygen species by human T-cell leukemia virus type 1 tax correlates with DNA damage and expression of cellular senescence marker. J Virol 84(10):5431–5437
Kishi S, Saijyo S et al (1997) Resistance to fas-mediated apoptosis of peripheral T cells in human T lymphocyte virus type I (HTLV-I) transgenic mice with autoimmune arthropathy. J Exp Med 186(1):57–64
Koiwa T, Hamano-Usami A et al (2002) 5′-long terminal repeat-selective CpG methylation of latent human T-cell leukemia virus type 1 provirus in vitro and in vivo. J Virol 76(18):9389–9397
Koyanagi Y, Itoyama Y et al (1993) In vivo infection of human T-cell leukemia virus type I in non-T cells. Virology 196(1):25–33
Krueger A, Fas SC et al (2006) HTLV-1 Tax protects against CD95-mediated apoptosis by induction of the cellular FLICE-inhibitory protein (c-FLIP). Blood 107(10):3933–3939
Kwon H, Ogle L et al (2005) Lethal cutaneous disease in transgenic mice conditionally expressing type I human T cell leukemia virus Tax. J Biol Chem 280(42):35713–35722
Lambert S, Bouttier M et al (2009) HTLV-1 uses HSPG and neuropilin-1 for entry by molecular mimicry of VEGF165. Blood 113(21):5176–5185
Lemasson I, Thebault S et al (1998) Activation of E2F-mediated transcription by human T-cell leukemia virus type I Tax protein in a p16(INK4A)-negative T-cell line. J Biol Chem 273(36):23598–23604
Lemoine FJ, Marriott SJ (2002) Genomic instability driven by the human T-cell leukemia virus type I (HTLV-I) oncoprotein, Tax. Oncogene 21(47):7230–7234
LeVasseur RJ, Southern SO et al (1998) Mammary epithelial cells support and transfer productive human T-cell lymphotropic virus infections. J Hum Virol 1(3):214–223
Li M, Kesic M et al (2009) Kinetic analysis of human T-cell leukemia virus type 1 gene expression in cell culture and infected animals. J Virol 83(8):3788–3797
Liu B, Hong S et al (2005) HTLV-I Tax directly binds the Cdc20-associated anaphase-promoting complex and activates it ahead of schedule. Proc Natl Acad Sci USA 102(1):63–68
Low KG, Dorner LF et al (1997) Human T-cell leukemia virus type 1 Tax releases cell cycle arrest induced by p16INK4a. J Virol 71(3):1956–1962
Macatonia SE, Cruickshank JK et al (1992) Dendritic cells from patients with tropical spastic paraparesis are infected with HTLV-1 and stimulate autologous lymphocyte proliferation. AIDS Res Hum Retroviruses 8(9):1699–1706
Maggirwar SB, Harhaj E et al (1995) Activation of NF-kappa B/Rel by Tax involves degradation of I kappa B alpha and is blocked by a proteasome inhibitor. Oncogene 11(5):993–998
Mahieux R, Gessain A (2005) New human retroviruses: HTLV-3 and HTLV-4. Med Trop (Mars) 65(6):525–528
Mahieux R, Gessain A (2009) The human HTLV-3 and HTLV-4 retroviruses: new members of the HTLV family. Pathol Biol (Paris) 57(2):161–166
Majone F, Semmes OJ et al (1993) Induction of micronuclei by HTLV-I Tax: a cellular assay for function. Virology 193(1):456–459
Manel N, Kim FJ et al (2003) The ubiquitous glucose transporter GLUT-1 is a receptor for HTLV. Cell 115(4):449–459
Marriott SJ, Lemoine FJ et al (2002) Damaged DNA and miscounted chromosomes: human T cell leukemia virus type I tax oncoprotein and genetic lesions in transformed cells. J Biomed Sci 9(4):292–298
Marriott SJ, Semmes OJ (2005) Impact of HTLV-I Tax on cell cycle progression and the cellular DNA damage repair response. Oncogene 24(39):5986–5995
Matsuoka M, Jeang KT (2007) Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat Rev Cancer 7(4):270–280
Matsuoka M, Jeang KT (2011) Human T-cell leukemia virus type 1 (HTLV-1) and leukemic transformation: viral infectivity, Tax, HBZ and therapy. Oncogene 30(12):1379–1389
Matsuura E, Yamano Y et al (2010) Neuroimmunity of HTLV-I Infection. J Neuroimmune Pharmacol 5(3):310–325
McKinsey TA, Brockman JA et al (1996) Inactivation of IkappaBbeta by the tax protein of human T-cell leukemia virus type 1: a potential mechanism for constitutive induction of NF-kappaB. Mol Cell Biol 16(5):2083–2090
Miyazaki M, Yasunaga J et al (2007) Preferential selection of human T-cell leukemia virus type 1 provirus lacking the 5′ long terminal repeat during oncogenesis. J Virol 81(11):5714–5723
Miyazato A, Sheleg S et al (2005) Evidence for NF-kappaB- and CBP-independent repression of p53’s transcriptional activity by human T-cell leukemia virus type 1 Tax in mouse embryo and primary human fibroblasts. J Virol 79(14):9346–9350
Miyoshi I, Kubonishi I et al (1981) Type C virus particles in a cord T-cell line derived by co-cultivating normal human cord leukocytes and human leukaemic T cells. Nature 294(5843):770–771
Mori N (1997) High levels of the DNA-binding activity of E2F in adult T-cell leukemia and human T-cell leukemia virus type I-infected cells: possible enhancement of DNA-binding of E2F by the human T-cell leukemia virus I transactivating protein, Tax. Eur J Haematol 58(2):114–120
Mori N, Fujii M et al (2001) Human T-cell leukemia virus type I tax protein induces the expression of anti-apoptotic gene Bcl-xL in human T-cells through nuclear factor-kappaB and c-AMP responsive element binding protein pathways. Virus Genes 22(3):279–287
Mortreux F, Kazanji M et al (2001) Two-step nature of human T-cell leukemia virus type 1 replication in experimentally infected squirrel monkeys (Saimiri sciureus). J Virol 75(2):1083–1089
Moules V, Pomier C et al (2005) Fate of premalignant clones during the asymptomatic phase preceding lymphoid malignancy. Cancer Res 65(4):1234–1243
Mulloy JC, Kislyakova T et al (1998) Human T-cell lymphotropic/leukemia virus type 1 Tax abrogates p53-induced cell cycle arrest and apoptosis through its CREB/ATF functional domain. J Virol 72(11):8852–8860
Murata K, Hayashibara T et al (2006) A novel alternative splicing isoform of human T-cell leukemia virus type 1 bZIP factor (HBZ-SI) targets distinct subnuclear localization. J Virol 80(5):2495–2505
Nagai M, Brennan MB et al (2001) CD8(+) T cells are an in vivo reservoir for human T-cell lymphotropic virus type I. Blood 98(6):1858–1861
Nejmeddine M, Barnard AL et al (2005) Human T-lymphotropic virus, type 1, tax protein triggers microtubule reorientation in the virological synapse. J Biol Chem 280(33):29653–29660
Nerenberg M, Hinrichs SH et al (1987) The tat gene of human T-lymphotropic virus type 1 induces mesenchymal tumors in transgenic mice. Science 237(4820):1324–1329
Nicot C, Harrod R (2000) Distinct p300-responsive mechanisms promote caspase-dependent apoptosis by human T-cell lymphotropic virus type 1 Tax protein. Mol Cell Biol 20(22):8580–8589
Nicot C, Mahieux R et al (2000) Bcl-X(L) is up-regulated by HTLV-I and HTLV-II in vitro and in ex vivo ATLL samples. Blood 96(1):275–281
Nitta T, Kanai M et al (2006) Centrosome amplification in adult T-cell leukemia and human T-cell leukemia virus type 1 Tax-induced human T cells. Cancer Sci 97(9):836–841
Ohtani K, Iwanaga R et al (2000) Cell type-specific E2F activation and cell cycle progression induced by the oncogene product Tax of human T-cell leukemia virus type I. J Biol Chem 275(15):11154–11163
Okamoto K, Fujisawa J et al (2006) Human T-cell leukemia virus type-I oncoprotein Tax inhibits Fas-mediated apoptosis by inducing cellular FLIP through activation of NF-kappaB. Genes Cells 11(2):177–191
Ono A, Miura T et al (1994) Subtype analysis of HTLV-1 in patients with HTLV-1 uveitis. Jpn J Cancer Res 85(8):767–770
Pais-Correia AM, Sachse M et al (2010) Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat Med 16(1):83–89
Peloponese JM Jr, Haller K et al (2005) Abnormal centrosome amplification in cells through the targeting of Ran-binding protein-1 by the human T cell leukemia virus type-1 Tax oncoprotein. Proc Natl Acad Sci USA 102(52):18974–18979
Perkins ND (2012) The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer 12(2):121–132
Petropoulos L, Lin R et al (1996) Human T cell leukemia virus type 1 tax protein increases NF-kappa B dimer formation and antagonizes the inhibitory activity of the I kappa B alpha regulatory protein. Virology 225(1):52–64
Pinon JD, Klasse PJ et al (2003) Human T-cell leukemia virus type 1 envelope glycoprotein gp46 interacts with cell surface heparan sulfate proteoglycans. J Virol 77(18):9922–9930
Pise-Masison CA, Radonovich M et al (2002) Transcription profile of cells infected with human T-cell leukemia virus type I compared with activated lymphocytes. Cancer Res 62(12):3562–3571
Poiesz BJ, Ruscetti FW et al (1980) Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci USA 77(12):7415–7419
Pomier C, Alcaraz MT et al (2008) Early and transient reverse transcription during primary deltaretroviral infection of sheep. Retrovirology 5:16
Proietti FA, Carneiro-Proietti AB et al (2005) Global epidemiology of HTLV-I infection and associated diseases. Oncogene 24(39):6058–6068
Qu Z, Xiao G (2011) Human T-cell lymphotropic virus: a model of NF-kappaB-associated tumorigenesis. Viruses 3(6):714–749
Rauch DA, Ratner L (2011) Targeting HTLV-1 activation of NFkappaB in mouse models and ATLL patients. Viruses 3(6):886–900
Saggioro D, Majone F et al (1994) Tax protein of human T-lymphotropic virus type I triggers DNA damage. Leuk Lymphoma 12(3–4):281–286
Santiago F, Clark E et al (1999) Transcriptional up-regulation of the cyclin D2 gene and acquisition of new cyclin-dependent kinase partners in human T-cell leukemia virus type 1-infected cells. J Virol 73(12):9917–9927
Satou Y, Yasunaga J et al (2006) HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci USA 103(3):720–725
Schmitt I, Rosin O et al (1998) Stimulation of cyclin-dependent kinase activity and G1- to S-phase transition in human lymphocytes by the human T-cell leukemia/lymphotropic virus type 1 Tax protein. J Virol 72(1):633–640
Semmes OJ, Majone F et al (1996) HTLV-I and HTLV-II Tax: differences in induction of micronuclei in cells and transcriptional activation of viral LTRs. Virology 217(1):373–379
Suemori K, Fujiwara H et al (2009) HBZ is an immunogenic protein, but not a target antigen for human T-cell leukemia virus type 1-specific cytotoxic T lymphocytes. J Gen Virol 90(Pt 8):1806–1811
Suzuki T, Hirai H et al (1993) A trans-activator Tax of human T-cell leukemia virus type 1 binds to NF-kappa B p50 and serum response factor (SRF) and associates with enhancer DNAs of the NF-kappa B site and CArG box. Oncogene 8(9):2391–2397
Suzuki T, Hirai H et al (1995) Tax protein of HTLV-1 destabilizes the complexes of NF-kappa B and I kappa B-alpha and induces nuclear translocation of NF-kappa B for transcriptional activation. Oncogene 10(6):1199–1207
Suzuki T, Hirai H et al (1994) Tax protein of HTLV-1 interacts with the Rel homology domain of NF-kappa B p65 and c-Rel proteins bound to the NF-kappa B binding site and activates transcription. Oncogene 9(11):3099–3105
Suzuki T, Narita T et al (1999) Down-regulation of the INK4 family of cyclin-dependent kinase inhibitors by tax protein of HTLV-1 through two distinct mechanisms. Virology 259(2):384–391
Tabakin-Fix Y, Azran I et al (2006) Functional inactivation of p53 by human T-cell leukemia virus type 1 Tax protein: mechanisms and clinical implications. Carcinogenesis 27(4):673–681
Takatsuki K (2005) Discovery of adult T-cell leukemia. Retrovirology 2:16
Takeda S, Maeda M et al (2004) Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int J Cancer 109(4):559–567
Tamiya S, Matsuoka M et al (1996) Two types of defective human T-lymphotropic virus type I provirus in adult T-cell leukemia. Blood 88(8):3065–3073
Tanaka A, Takahashi C et al (1990) Oncogenic transformation by the tax gene of human T-cell leukemia virus type I in vitro. Proc Natl Acad Sci USA 87(3):1071–1075
Taniguchi Y, Nosaka K et al (2005) Silencing of human T-cell leukemia virus type I gene transcription by epigenetic mechanisms. Retrovirology 2:64
Tripp A, Banerjee P et al (2005) Induction of cell cycle arrest by human T-cell lymphotropic virus type 1 Tax in hematopoietic progenitor (CD34+) cells: modulation of p21cip1/waf1 and p27kip1 expression. J Virol 79(22):14069–14078
Tripp A, Liu Y et al (2003) Human T-cell leukemia virus type 1 tax oncoprotein suppression of multilineage hematopoiesis of CD34+ cells in vitro. J Virol 77(22):12152–12164
Tsukahara T, Kannagi M et al (1999) Induction of Bcl-x(L) expression by human T-cell leukemia virus type 1 Tax through NF-kappaB in apoptosis-resistant T-cell transfectants with Tax. J Virol 73(10):7981–7987
Van Prooyen N, Gold H et al (2010) Human T-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc Natl Acad Sci USA 107(48):20738–20743
Waldele K, Silbermann K et al (2006) Requirement of the human T-cell leukemia virus (HTLV-1) tax-stimulated HIAP-1 gene for the survival of transformed lymphocytes. Blood 107(11):4491–4499
Watanabe M, Ohsugi T et al (2005) Dual targeting of transformed and untransformed HTLV-1-infected T cells by DHMEQ, a potent and selective inhibitor of NF-kappaB, as a strategy for chemoprevention and therapy of adult T-cell leukemia. Blood 106(7):2462–2471
Watanabe T (2011) Current status of HTLV-1 infection. Int J Hematol 94(5):430–434
Watanabe T, Seiki M et al (1983) Retrovirus terminology. Science 222(4629):1178
Wattel E, Cavrois M et al (1996) Clonal expansion of infected cells: a way of life for HTLV-I. J Acquir Immune Defic Syndr Hum Retrovirol 13(Suppl 1):S92–S99
Wattel E, Vartanian JP et al (1995) Clonal expansion of human T-cell leukemia virus type I-infected cells in asymptomatic and symptomatic carriers without malignancy. J Virol 69(5):2863–2868
Xiao G, Cvijic ME et al (2001) Retroviral oncoprotein Tax induces processing of NF-kappaB2/p100 in T cells: evidence for the involvement of IKKalpha. EMBO J 20(23):6805–6815
Xiao G, Harhaj EW et al (2000) Domain-specific interaction with the I kappa B kinase (IKK)regulatory subunit IKK gamma is an essential step in tax-mediated activation of IKK. J Biol Chem 275(44):34060–34067
Yamada T, Yamaoka S et al (1994) The human T-cell leukemia virus type I Tax protein induces apoptosis which is blocked by the Bcl-2 protein. J Virol 68(5):3374–3379
Yamano Y, Nagai M et al (2002) Correlation of human T-cell lymphotropic virus type 1 (HTLV-1) mRNA with proviral DNA load, virus-specific CD8(+) T cells, and disease severity in HTLV-1-associated myelopathy (HAM/TSP). Blood 99(1):88–94
Yang L, Kotomura N et al (2011) Complex cell cycle abnormalities caused by human T-lymphotropic virus type 1 Tax. J Virol 85(6):3001–3009
Yasunaga J, Matsuoka M (2011) Molecular mechanisms of HTLV-1 infection and pathogenesis. Int J Hematol 94(5):435–442
Yoshida M, Miyoshi I et al (1982) Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc Natl Acad Sci USA 79(6):2031–2035
Zane L, Sibon D et al (2009) Clonal expansion of HTLV-1 infected cells depends on the CD4 versus CD8 phenotype. Front Biosci 14:3935–3941
Zhi H, Yang L et al (2011) NF-kappaB hyper-activation by HTLV-1 tax induces cellular senescence, but can be alleviated by the viral anti-sense protein HBZ. PLoS Pathog 7(4):e1002025
Acknowledgments
Work in our laboratory is supported in part by intramural funding from the NIAID and by the IATAP program from the Office of the Director, NIH. We thank Lauren Lee for her assistance in the preparation of the figures.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Zane, L., Jeang, KT. (2014). HTLV-1 and Leukemogenesis: Virus–Cell Interactions in the Development of Adult T-Cell Leukemia. In: Chang, M., Jeang, KT. (eds) Viruses and Human Cancer. Recent Results in Cancer Research, vol 193. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-38965-8_11
Download citation
DOI: https://doi.org/10.1007/978-3-642-38965-8_11
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-38964-1
Online ISBN: 978-3-642-38965-8
eBook Packages: MedicineMedicine (R0)