
Abstract
The potential for developing therapeutics for cancers driven by aberrant gene expression is becoming a reality in recent years, largely due to the identification and characterization of the enzymatic components regulating chromatin structure and function. One of the major classes of chromatin-modifying enzymes is the histone methyltransferases. These enzymes catalyze the methylation of lysine and arginine residues on the core nucleosomal histones. The methylation pattern present on the histones is associated with different chromatin states depending upon the particular site of methylation. The lysine and arginine methyltransferases (KMTs and RMTs) comprise enzyme families with promising therapeutic potential because they have been found to be altered in diseases with high unmet need (e.g., cancer) and are amenable to small molecule drug discovery efforts (Copeland et al., Nat Rev Drug Discov 8(9):724–732, 2009). The purpose of this chapter is to review the histone lysine and arginine methyltransferases with particular focus on their histone substrates and their association with cancer and status of the development of small molecule inhibitors.
Richard Chesworth and Tim J. Wigle contributed equally to the chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Mantle Cell Lymphoma
- Arginine Residue
- Arginine Methylation
- Arginine Methyltransferases
- H3K4 Methyltransferase
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
9.1 Introduction
Posttranslational modifications on the core nucleosomal histones play an important role in the regulation of chromatin structure and function. These modifications include methylation of lysine and arginine residues, acetylation and ubiquitinylation of lysine residues, and phosphorylation of serine residues. The histone modifications are referred to as “marks,” and the enzymes that catalyze the deposition of the marks are “writers,” while the enzymes that remove the marks are “erasers.” Proteins that recognize specific histone posttranslational marks or series of marks are referred to as “readers.” Thus, the methyltransferases “write” the methyl marks on lysine and arginine residues. The methyl marks on lysine residues can exist in a mono-, di-, or trimethylation state, while arginine residues are either mono- or dimethylated, with the dimethylation existing in an asymmetrical or symmetrical state (Fig. 9.1a). Mass spectrometry studies have identified specific sites of lysine and arginine methylation found on histone H3 and histone H4 (Fig. 9.1b). The pattern of modifications present on the histones is associated with specific functional states of the chromatin. For example, trimethylation of histone H3 lysine 4 (H3K4me3) is found in areas of chromatin that are either poised for transcription or actively transcribed (Bernstein et al. 2005, 2006). H3K4me3 is found in combination with trimethylation of histone H3 lysine 27 (H3K27me3) in areas that are poised for transcription. Histone H3K27me3 is generally associated with transcriptional silencing. Our understanding of the role of these posttranslational modifications of histones has increased tremendously in the past few years. The function and regulation of methyl marks on histone lysine and arginine residues are discussed in the sections in this review.

Methylation of lysine and arginine residues in proteins. (a) Lysine methyltransferases mono-, di- or trimethylate lysine residues while lysine demethylases remove these modifications. Arginine methyltransferases monomethylate and asymmetrically or symmetrically dimethylate arginine residues and there are no known enzymes that directly remove these modifications. (b) Sequences of human histone H3 and H4 N-terminal tails and known sites of methylation
Histone methyltransferases are divided into two major families based upon their amino acid substrate: the lysine methyltransferases and arginine methyltransferases (Copeland et al. 2009; Bedford and Richard 2005; Richon et al. 2011). The lysine methyltransferases (KMTs) consist of over 50 enzymes, and all of the enzymes in this class share a common catalytic domain referred to as the Su(var)3–9, Enhancer of Zeste, Trithorax, or SET domain except for one enzyme, DOT1L. The catalytic domain of DOT1L shares structural homology with the arginine methyltransferases (RMTs), based on the observation that the cofactor S-adenosyl methionine (SAM) displays a common extended conformation in DOT1L and the RMTs (Richon et al. 2011). This is in contrast to the U shape conformation that SAM adopts in the catalytic domain of the SET domain KMTs (Schubert et al. 2003). Nine enzymes that catalyze arginine methylation have been described and include PRMT1, PRMT2, PRMT3, CARM1, PRMT5, PRMT6, PRMT7, PRMT8, and PRMT10 (Bedford and Richard 2005; Di Lorenzo and Bedford 2011). The RMTs are divided into two major groups. The type 1 enzymes (PRMT1, PRMT2, PRMT3, CARM1, PRMT6) catalyze either monomethylation or asymmetric dimethylation, and the type II enzymes catalyze symmetric dimethylation (PRMT5 and PRMT7). PRMT7 has also been shown to catalyze monomethylation of specific substrates and is the sole member of the type III enzyme group (Miranda et al. 2004). Two additional, related putative RMTs have been described (PRMT9 and PRMT11), but have not been biochemically characterized (Di Lorenzo and Bedford 2011). A systematic survey of the human genome for RMT-related enzymes was performed, and an additional 33 putative enzymes were identified that contained related methyltransferase domains (Richon et al. 2011).
Alterations in histone lysine and arginine methylation and associated protein methyltransferases have been observed in a wide variety of cancers (Table 9.1). These alterations include translocations, mutations, amplification, and aberrant recruitment of methyltransferases and demethylases. One example is EZH2, a histone H3K27 methyltransferase that is altered in several cancer types through a variety of different mechanisms that leads to increased H3K27me3 levels. The increase in H3K27me3 is associated with repression of gene expression that is believed to result in the development of cancer.
The identification of genetic alterations in methyltransferases in cancer leads to a strategy for targeting patient populations with these alterations using small molecules designed to selectively inhibit the oncogenic methyltransferase. Drug discovery efforts have been initiated, and several small molecule inhibitors of this class of enzymes have been identified. The current state of the development of the methyltransferase inhibitors is summarized in Sect. 4.
9.2 Histone Methylation, Methyltransferases, and Cancer Association
9.2.1 Lysine Methylation
9.2.1.1 H3K4 Methylation
The regulation of methylation on lysine 4 on histone H3 (H3K4) is complex, with at least 20 enzymes reported to catalyze the addition or removal of methylation at this site. Methylation of H3K4 is almost exclusively associated with transcriptionally active chromatin (Sims and Reinberg 2006; Strahl et al. 1999), and high levels of H3K4 mono-, di-, and trimethylation can be found at or near transcription start sites (Barski et al. 2007). There is a strong relationship between the state of methylation and transcription; trimethylation is found at the 5′ end of active genes and correlates positively with active transcription, RNA polymerase II occupancy, and histone acetylation (Bernstein et al. 2005; Santos-Rosa et al. 2002; Schneider et al. 2004). Dimethylation of H3K4 often co-localizes with H3K4me3 in discrete zones 5–20 nucleosomes long proximal to actively transcribed genes. On its own, the dimethyl H3K4 mark has been found on genes that are maintained in a transcriptionally poised state (Bernstein et al. 2005; Schneider et al. 2004; Ruthenburg et al. 2007).
Unlike in yeast, where Set1 is the only H3K4 methyltransferase, humans have at least 13 confirmed or putative methyltransferases which regulate the addition of methyl groups at H3K4 (MLL1, MLL2, MLL3, MLL4, MLL5, Ash1, Set7/9, Set1A, Set1B, SMYD1, SMYD2, SMYD3, and PRDM9) (Ruthenburg et al. 2007; Abu-Farha et al. 2008; Hamamoto et al. 2004; Kouzarides 2007; Tan et al. 2006). Additionally, at least 7 H3K4 demethylases (LSD1, LSD2, JHMD1B, JARID1A, JARID1B, JARID1C, and JARID1D) have been reported to date (Kouzarides 2007; Lim et al. 2010a). The diverse array of human enzymes that have evolved to regulate the methylation status of the H3K4 indicates that exquisite control must be maintained over this mark in all cell types. In general, H3K4 methyltransferases are activators of gene expression, and H3K4 demethylases are repressors. The dynamic balance between these opposing classes of enzymes indicates the precision with which H3K4 methylation is regulated in diverse cell types.
While there is some overlap of function between H3K4 methyltransferases, the context-specific expression and activity of each enzyme is crucial to cell fate during differentiation and development (Eissenberg and Shilatifard 2010), cell functions, e.g., DNA repair, that maintain the status quo and in specialized processes, such as V(D)J recombination, which support physiological integrity (Shilatifard 2008). For example, the MLL family of H3K4 methyltransferases is known to exert control over the expression of HOX genes, and recent work from Shilatifard (2008) has described the phenomenon of redundancy versus specialization among this family in the activation of these gene clusters. Experiments performed using wild-type or Mll1 knockout MEF cells indicate that of 10,041 genes associating with H3K4me3, only 5 % show a reduction in H3K4me3 in the Mll1 knockout MEFs (Wang et al. 2009). Subsequent expression profiling indicated that the knockout of Mll1 resulted in the enhanced expression of 2,265 genes and reduced expression of 2,459 or a positive or negative regulation of 3 % of all genes (Wang et al. 2009). Overall this analysis supported the concept that a substantial overlap in H3K4 methyltransferase activity does exist, but precise control over the expression of a small subset of genes is dependent on MLL. Cell-type-specific expression of MLL family methyltransferases has also been observed. For example, MLL3 and MLL4 are observed to have minimal roles in HOX gene regulation in fibroblasts (Wang et al. 2009). The methylation product of the methyltransferases also adds to the level of regulation of H3K4me3 observed; during oogenesis, MLL2 controls levels of trimethylation but has no effect on monomethylation, indicating that a different methyltransferase produces H3K4me1, and MLL2 activity is coordinated to catalyze the trimethyl state (Andreu-Vieyra et al. 2010). Similar target and product specificity exists among the H3K4 demethylases. LSD1 and LSD2 are observed to primarily act upon mono- and dimethylated H3K4, and JARID family demethylases are specific for di- and trimethyl H3K4 (Lim et al. 2010a). Beyond cell- and temporal-specific expression of H3K4 methyltransferases, another level of regulation may involve accessory proteins. For example, enzymatic activity may be nonexistent or greatly reduced when the SET domain-containing subunit is present on its own; however, activity is stimulated greatly when present as part of a complex. The identity of the other partners in the complex differs for each member of the MLL family and presumably dictates the specificity and the context in which methylation occurs at the target H3K4 residue (Eissenberg and Shilatifard 2010).
H3K4 methylation is associated with an open chromatin configuration which enables access of the chromatin to transcription factors and machinery. It is becoming apparent that protein modules capable of “reading” these methyl marks are responsible for attracting effector proteins containing these modules to chromatin sites bearing H3K4, leading to a variety of outcomes (Ruthenburg et al. 2007). Methyl-lysine binding modules are generally part of the Royal Family of proteins and include MBT, Agenet, Tudor, Chromo, and PWWP domains. Additionally, methyl-lysine recognition motifs can be found in the PHD family and WD40 repeat protein WRD5 (Maurer-Stroh et al. 2003). The key lysine methylation recognition features of these domains are an aromatic electron-rich cage, interacting with the lysine cation, with additional charge neutralization and H-bonding by 0–2 acidic functionalities depending on the methylation state of lysine (Guo et al. 2009; Kaustov et al. 2011). The number of proteins that contain H3K4 methyl-lysine recognition modules far outnumbers the enzymes that modulate methylation of this residue. The ING-family of proteins contain SHD fingers that preferentially recognize H3K4me2/3, and members of this family usually bind to and affect the activity of histone acetyltransferases and deacetylases (Adams-Cioaba and Min 2009; Feng et al. 2002). The recruitment of effector proteins to H3K4me3 often stimulates gene expression, as is the case for the PHD finger of human bromodomain and PHD domain transcription factor (BPTF), the largest subunit of the NURF complex, which is responsible for remodeling chromatin at active loci (Li et al. 2006; Wysocka et al. 2006). However, there are examples of H3K4me3 effector modules that repress gene expression upon recruitment. In response to DNA damage, the PHD finger of ING2, a critical component of the mSin3a-HDAC1 deacetylase complex, binds H3K4me3 in proximity to the promoters of proliferation genes and shuts off transcription (Shi et al. 2006). The binding of effector proteins to H3K4me3 can also impact other neighboring marks. The demethylases PHF8 and JHDM1D harbor a PHD finger, allowing them to dock onto the target nucleosome. However, the length and rigidity of a linker region separating the Jumonji domain from the PHD finger dictates whether the enzyme is a H3K9 (PHF8) or H3K27 (JHDM1D) demethylase (Horton et al. 2010). Unlike ING-family and BPTF PHD fingers, there are even recognition modules that selectively bind to unmodified lysine. A component of the LSD1 complex, BHC80, binds unmethylated H3K4, prevents remethylation and aids in the propagation of the H3K4 demethylation (Lan et al. 2007). Similarly, DNMT3L is recruited to unmethylated H3K4 by a PHD-like domain to maintain a repressive effect on gene expression at nucleosomes not demarcated by H3K4 methylation (Ooi et al. 2007).
Histone H3K4 methylation is associated with oncogenesis at multiple levels. However, given the complexity of enzymes that add or remove methyl groups from H3K4, and the diverse array of effector proteins that bind to H3K4, selecting the appropriate targets for initiating drug discovery is challenging (Copeland et al. 2009). SMYD3 is an example of a H3K4 methyltransferase where there appears to be a link between overexpression and tumor progression (Hamamoto et al. 2004). SMYD3 contains a SET domain and a MYND-type zinc-finger domain that is common among developmental proteins (Hamamoto et al. 2004). SMYD3 forms a complex with RNA Pol II (Hamamoto et al. 2004), and its overexpression is associated with increased expression of oncogenes (e.g., N-Myc, CrkL, Wnt10b, RIZ, and hTERT), cell cycle control genes (e.g., cyclin G1 and CDK2), and regulators of signal transduction (e.g., STAT1, MAP3K11, and PIK3CB) (Luo et al. 2009; Liu et al. 2007). SMYD3 has been shown to be overexpressed in hepatic, colorectal, cervical, and breast cancers (Hamamoto et al. 2004, 2006; Luo et al. 2009; Wang et al. 2008a). In the case of breast cancers, recent evidence suggests that SMYD3 directly interacts with the ligand-binding domain of the estrogen receptor α (ERα), enhancing the levels of di- and trimethylation of H3K4 at ERα target genes (Kim et al. 2009), ultimately increasing their expression. RNAi-mediated knockdown of the SMYD3 has been shown to decrease invasiveness and proliferation and in some cases induce apoptosis.
MLL is another example of an H3K4 methyltransferase implicated in cancer. Rearrangements of the MLL gene have been identified in both pediatric and adult leukemias and are correlated with poor prognosis (Hess 2004; Krivtsov and Armstrong 2007; Krivtsov et al. 2008). The MLL rearrangements result in loss of the SET or catalytic domain while retaining the DNA-targeting domain. The translocation results in aberrant expression of a subset of the HOX genes, causing a block of hematopoietic differentiation. Targeting of the fusion proteins or mediators of the fusion partners could represent a novel and effective way to treat these leukemias. Several MLL translocation partners (AF4, AF9, AF10, and ENL) recruit another lysine methyltransferase, DOT1L (Okada et al. 2005). DOT1L catalyzes methylation of histone H3K79 and is described in a subsequent section. In addition to alterations in MLL, amplification of MLL4 is also observed in solid tumors (Hess 2004).
The histone H3K4 demethylases may also be important targets for therapeutic intervention using small molecules. LSD1 is overexpressed in prostate cancer, breast cancer, neuroblastoma and bladder cancer, and loss of H3K4me3 is postulated to result in the silencing of tumor suppressor gene expression (Lim et al. 2010a, b; Schulte et al. 2009; Shi and Whetstine 2007). Increasing local nonmethylated H3K4 not only impedes the recruitment of transcription factors but also enhances the activity of DNA methyltransferases proximal to the demethylated mark. Indeed, the use of LSD1 inhibitors in combination with a DNA methyltransferase inhibitor led to inhibition of the growth of human colon cancer xenografts (Huang et al. 2009). Similar to LSD1, JARID1B is found overexpressed in breast, testis, esophageal, and prostate cancers (Lim et al. 2010a; Barrett et al. 2007; Xiang et al. 2007). In addition to alterations in specific enzymes, alterations in H3K4 trimethylation have been described to be associated with cancer. For example, H3K4 trimethylation has been found to be associated with chromosome breakpoints in leukemia (Barski et al. 2007). While pursuing inhibitors of the reader domains that recognize H3K4 methylation is also a possibility (Kireev et al. 2010; Wigle et al. 2010), this chapter will focus on the opportunities presented by the histone methyltransferases.
9.2.1.2 H3K9 Methylation
Lysine methylation at H3K9 is generally associated with transcriptional silent chromatin and is an example of another mark where multiple enzymes regulate the methylation status. There are at least 7 methyltransferases (G9a, GLP, Suv39H1, Suv39H2, SETDB1, SETDB2, and PRDM2) and 7 demethylases (LSD1, JHDM2a, JHDM2b, JHDM3A, JMJD2B, JMJD2C, and JMJD2D) known to act upon this mark (Kouzarides 2007). Unlike H3K4, where there is no reported acetylation, the H3K9 mark is also prominently acetylated. Also, the serine (10) of histone H3 can become phosphorylated by the kinase Aurora B (Jeong et al. 2010).
Studies in organisms ranging from yeast to human show that H3K9 methylation is a hallmark of facultative and constitutive heterochromatin (Krishnan et al. 2011). High-resolution genomic mapping used to generate transcription start site (TSS) alignment has shown that both H3K9me2 and H3K9me3 were higher in chromatin of silent genes compared to transcriptionally active genes in a region of 10 kb surrounding the TSS. H3K9me1 localized to more active promoters surrounding the TSS (Barski et al. 2007). In addition, the di- and trimethylation states of H3K9 are implicated in the recruitment of DNA methyltransferases, which are also associated with areas of heterochromatin (Cheng and Blumenthal 2010).
Two of the most well-studied enzymes that catalyze methylation of H3K9 are G9a (also known as EHMT2) and G9a-like protein (GLP or EHMT1). G9a and GLP catalyze the mono- and dimethylation of H3K9 (Wu et al. 2010), but not trimethylation of H3K9. Unlike other methyltransferases, G9a and GLP do not rely on cross talk between neighboring marks to methylate H3K9, only the presence of a un- or monomethylated lysine (Rathert et al. 2008). DNA methyltransferases 3A and 3B are physically recruited to chromatin by G9a bound through its ankyrin repeats, facilitating the spread of heterochromatin (Xin et al. 2003). Functionally, G9a and GLP appear to be essential to early development, and knockouts of either in mouse embryos are lethal (Tachibana et al. 2002, 2005). In addition, G9a plays an essential role in embryonic stem cell differentiation through H3K9me2-mediated silencing of Oct3/4 (Feldman et al. 2006). There is substantial redundancy between G9a and GLP target genes, but specialization does exist. G9a knockdown leads to a global reduction of H3K9me2 at 67 % of promoters (Wagschal et al. 2008).
The Suv39 family of methyltransferases catalyze di- and trimethylation of H3K9, and like G9a and GLP, they interact with DNA methyltransferases to facilitate the formation of heterochromatin (Loyola et al. 2009). Suv39 members are essential in recruiting Heterochromatin Protein 1 (HP1), a chromodomain-containing protein selectively recognizing trimethyl lysine that is essential in the stability of telomeres and centromeres. In addition to the Suv39 enzymes, SETDB1 has also been shown to catalyze di- and trimethylation of H3K9 (Schultz et al. 2002; Wang et al. 2003). The accessory protein mAM stimulates the conversion of di- to trimethylation of H3K9 by SETDB1 (Wang et al. 2003). SETDB1 is localized at the 3′ ends of zinc-finger genes and appears to play an important role in gene silencing induced at these promoters (Frietze et al. 2010). Demethylation of the H3K9 mark is carried out mainly by two families: JMJD1 family proteins are specific for H3K9 mono- and dimethylation and members of JMJD2 family demethylate di- and trimethylated H3K9 (Shi and Whetstine 2007; Cloos et al. 2006; Klose et al. 2006).
Overexpression of G9a, Suv39H1 and SETDB1 has been linked to oncogenic transformation in a variety of cancers, including lung and prostate cancer (Watanabe et al. 2008). However, despite reports of the overexpression of G9a in cancer, there is a lack of definitive proof to this end. Indeed, small molecules that are nanomolar or sub-nanomolar inhibitors of G9a and GLP (Kubicek et al. 2007; Liu et al. 2009a, 2010) can reverse H3K9me2 in cells and regulate G9a target gene expression; however, these changes do not correlate with inhibition of proliferation. G9a may also contribute to the development of cancer by methylating and inactivating the p53 tumor suppressor protein (Huang et al. 2010). Recently, it has been shown that Suv39H1 overexpression may be oncogenic through its association and recruitment of DNMT1 (Kang et al. 2007).
Recurrent amplification and overexpression of SETDB1 has been described in melanoma (Ceol et al. 2011). Ceol et al. show that enforced expression of SETDB1 can accelerate melanoma onset in a zebrafish model, thus supporting the role of SETDB1 as an oncogene in melanoma.
There are also genetic alterations in the H3K9 demethylases that have been described in cancer. For example, recurrent amplifications and overexpression of JMJD2C/GASC1 have been found in squamous cell cancer cell lines and the basal-like subtype of breast cancer (Cloos et al. 2006; Yang et al. 2000; Liu et al. 2009b). Liu et al. show that GASC1 induces transformed phenotypes and regulates the expression of genes critical for stem cell self-renewal when ectopically expressed in immortalized breast cells.
9.2.1.3 H3K27 Methylation
Methylation of lysine 27 on histone H3, in particular trimethylation, is associated with gene silencing. The deposition of methyl groups onto H3K27 is catalyzed by the Polycomb-Repressive Complexes 2 (PRC2) containing either EZH1 or EZH2 as the catalytic subunit of the complex. EZH2-PRC2 is noted for being the prominent complex in actively dividing cells, while EZH1-PRC2 is ubiquitously expressed (Margueron et al. 2008). The core PRC2 complex is composed of EED, RbAp48, Suz12, and EZH2 (Cao and Zhang 2004a). Demethylation of H3K27 is principally carried out by UTX and JMJD3 and results in the activation of gene expression (Agger et al. 2007).
PRC2 catalyzes the mono- through trimethylation of H3K27 (Margueron et al. 2008; Cao et al. 2002). In vivo, H3K27me2 is the most abundant form, accounting for roughly 50 % of H3K27 methylation, with H3K27me1 and H3K27me3 representing 10 and 35 % of H3K27 methylation, respectively (Peters et al. 2003). Of these methylation states, the trimethylation of H3K27 (H3K27me3) is associated with increased DNA methylation in proximity to nucleosomes bearing this mark. This leads to chromatin condensation and ultimately the repression of genes that are located in these heterochromatic regions (Schlesinger et al. 2007). The establishment of the H3K27me3 is important during processes such as X-inactivation, germline development, and stem cell pluripotency, which require sustained gene suppression (Cao and Zhang 2004b). PRC2 has important roles in stem cell biology; there is strong overlap between binding of PRC2 to regions that are populated by genes controlled by the transcription factors Oct4, Sox2, and Nanog, which are critical to pluripotent stem cell generation and maintenance (Boyer et al. 2005, 2006). Loss of PRC2 derepresses embryonic stem cell differentiation genes (Boyer et al. 2006), and knockdown of Oct4 disrupts PRC2 association with target genes (Squazzo et al. 2006), indicating that PRC2 activity is important in inhibiting differentiation. Interestingly, H3K27me3 can sometimes co-localize with H3K4me3, an activating mark. In this chromatin state, termed “bivalent chromatin,” H3K27me3 is the dominant mark, and gene expression is repressed; however, the presence of the H3K4me3 is believed to maintain the genes in a poised state for activation of transcription upon demethylation by UTX or JMJD3 (Simon and Lange 2008).
The EED subunit of the PRC2 complex contains a WD40 domain that preferentially recognizes and binds to the higher methylation states of lysines in H3K9, H3K27, and H1K26, all of which contain a conserved ARKS sequence (Xu et al. 2010). Since H3K9me3 and H3K27me3 are repressive marks, their recognition by EED may facilitate the propagation of H3K27me3 across regions where gene expression is silenced. Additional proteins, such as PHF1, are not essential to EZH2 catalytic activity but may associate with PRC2 to enhance its activity. Loss of PHF1 correlates with a concomitant decrease in H3K27me3 at the HOXA6, HOXA9, and HOXA11 promoters. PHF1 contains a Tudor domain and two PHD fingers that most likely target PRC2 to discrete loci via recognition of adjacent methylated lysines (Cao et al. 2008a; Sarma et al. 2008). Also, it has been shown that large intervening noncoding RNAs (lincRNAs) interact with PRC2 and control the localization of H3K27 methylation via recognition of complementary DNA sequences (Gupta et al. 2010). The deposition of H3K27me3 is recognized by Polycomb-Repressive Complex 1 (PRC1), a complex with a core composition of Polycomb (PC), Polyhomeotic (PH), BMI-1, and RING1, which is thought to contribute to Polycomb silencing (Levine et al. 2002).
H3K27 trimethylation is strongly implicated in cancer, and several mechanisms leading to increased H3K27me3 (PRC2 overexpression, UTX loss of function mutations and PRC2 subunit overexpression) are associated with many human cancers (Simon and Lange 2008; Sarma et al. 2008; Bracken et al. 2003; Cao et al. 2008b; Kirmizis et al. 2003; Kleer et al. 2003; van Haaften et al. 2009; Varambally et al. 2002). As a result, PRC2, specifically its catalytic subunit EZH2, is one of the most pursued epigenetic targets. Overexpression and amplification of EZH2 (or other PRC2 subunits) is believed to silence genes that promote differentiation. One of the first reports of the involvement of PRC2 in cancer was a gene profiling study of metastatic and localized prostate cancer in which EZH2 scored as the most significant gene upregulated in metastatic cancer (Varambally et al. 2002). PRC2 correlates with poor outcome in prostate cancers and can be used as a prognostic indicator. Similarly, EZH2 overexpression in breast cancers is well documented and correlates with the aggressiveness of the tumor (Bracken et al. 2003; Kleer et al. 2003).
In addition to overexpression, somatic mutations in the catalytic domain of EZH2 have been described in diffuse large B-cell lymphoma (Morin et al. 2010). The mutations result in a single amino acid change in the catalytic SET domain of EZH2 at Y641 (Y641F, Y641N, Y641S, Y641H) in follicular lymphoma (FL) and germinal center B-cell-like (GCB) subtypes of diffuse large B-cell lymphoma (DLBCL). Initial biochemical experiments using recombinant PRC2 having these mutations demonstrated that mutant EZH2 complexes were unable to methylate a peptidic substrate corresponding to the H3K27 mark, while the wild-type enzyme methylated the same substrate. Morin et al. concluded, therefore, that the mutants appeared to be loss-of-function variants (Morin et al. 2010). However, subsequent studies using nucleosome, histone, and peptide substrates revealed that point mutations of Y641 to F, N, S, or H resulted in a change in the substrate specificity of EZH2 from unmethylated lysine to the dimethylated lysine. The Y641 mutants are much more proficient at catalyzing trimethylation than the wild-type enzyme (Sneeringer et al. 2010; Yap et al. 2011). Considering that these mutations were discovered as heterozygous, and both mutant and wild-type enzyme are expressed, it is believed that the coordinated activities of wild-type and mutant EZH2 drive H3K27 trimethylation and thus promote the development human lymphomas (Sneeringer et al. 2010; Yap et al. 2011). These findings suggest that lymphoma patients bearing the EZH2 mutation may be sensitive to small molecule inhibitors of EZH2. Ultimately, this may provide a framework for further genetic analysis of histone methyltransferase mutations, as catalytic site mutations that change the substrate specificity of the SET domain are not unprecedented (Wu et al. 2010; Del Rizzo et al. 2010).
9.2.1.4 H3K36 Methylation
H3K36 methylation is primarily an activating mark that is catalyzed by at least 6 methyltransferases (NSD1, NSD2, NSD3, SMYD2, SETD2, and SETMAR) and erased by at least 5 demethylases (JHDM1a, JHDM1b, JHDM3A, JMJD2B, and JMJD2C) (Kouzarides 2007; Lee et al. 2005; Li et al. 2009). Methylation at H3K36 competes with acetylation, as it is also recognized as a target for acetyltransferases. H3K36 trimethylation is generally observed downstream of transcriptional start sites, within the coding region of genes, and it peaks around the 3′ end (Barski et al. 2007). In yeast, H3K36 dimethylation is scarce or absent in upstream gene regulatory regions, telomeres, mating loci, and regions transcribed by RNA polymerase III and is predicted to be restricted to areas in contact with RNAP II. Localization of H3K36me2 is highly conserved from yeast to humans and is speculated to demarcate regulatory from coding regions in a similar fashion (Rao et al. 2005). It has been shown that SMYD2 associates with the Sin3A HDAC complex to generate H3K36me2 and restrain cell proliferation (Brown et al. 2006). WHSC1 (also known as NSD2) preferentially mono- and dimethylates H3K36 in the context of nucleosomes, but interestingly, in the absence of DNA, it can have its substrate preference changed to H4K44, a mark that has not been proven to exist in vivo (Li et al. 2009). While the NSD methyltransferases can only catalyze mono- or dimethylation, SETD2 can catalyze mono-, di-, and trimethylation (Yuan et al. 2009), and it is the key enzyme regulating trimethylation of H3K36.
Dysregulation of H3K36 methylation is a molecular determinant of several cancers. Overexpression of NSD3 (also known as WHSC1L1) resulting from amplification within chromosome 8p12 is observed in several tumor cell lines and breast carcinomas (Angrand et al. 2001). Chromosomal translocations that result in a fusion product of NSD methyltransferases to nucleoporin 98, NUP98-NSD1 (Jaju et al. 2001; Wang et al. 2007) or NUP98-NSD3 (Rosati et al. 2002), are defined genetic mutations associated with leukemogenesis. The NUP98-NSD1 fusion is believed to induce acute myeloid leukemia (AML) through the binding of genomic elements in proximity to Hox genes, maintaining H3K36 methylation and leading to overexpression of proto-oncogenes such as HoxA7, HoxA9, HoxA10, and Meis1 (Wang et al. 2007). Inactivating mutations in the NSD SET domain of the NUP98 fusions abolished the oncogenic potential of the chimera (Wang et al. 2007). In myeloma, t(4:14) translocations have been found to drive overexpression of NSD2 leading cancer cell proliferation and adhesion (Chesi et al. 1998; Lauring et al. 2008; Stec et al. 1998). The alterations in the NSD proteins cause methylation of H3K36 in genomic regions which do not normally contain this modification, resulting in aberrant gene expression and transformation. Therefore, one can envision small molecules that inhibit the catalytic activity of NSD family methyltransferases being useful in the treatment of several hematological malignancies.
9.2.1.5 H3K79 Methylation
H3K79 methylation is catalyzed by DOT1L. Unlike other histone modifications that are situated in the accessible N-terminal histone tails, H3K79 methylation occurs on the core of histone H3. DOT1L is the only known lysine methyltransferase that lacks a canonical SET domain (Dillon et al. 2005; Gao and Liu 2007), and structurally, the catalytic domain of DOT1L is more closely related to arginine methyltransferases than to lysine methyltransferases (Richon et al. 2011; Wu et al. 2010; Min et al. 2003). The mechanism of depletion of the H3K79 methylation mark appears to require replacement of histone H3 since no H3K79 demethylases have been described. DOT1L was originally identified for its role in telomere silencing (Singer et al. 1998), and it was later discovered that it possessed H3K79 methylating activity (Ng et al. 2002a, b; van Leeuwen et al. 2002). DOT1L-mediated methylation of H3K79 is considered to enhance gene expression (Steger et al. 2008). The catalytic activity of DOT1L is highly dependent on the presented of ubiquitinylated histone H2B, and it is only active in the context of nucleosomal substrates (Ng et al. 2002b; Shahbazian et al. 2005).
DOT1L is linked to oncogenic transformation in MLL-rearranged leukemias. DOT1L is recruited by MLL fusion partners, resulting in the mis-targeting of DOT1L and aberrant H3K79 methylation. This leads to increased transcription of MLL fusion target genes which then block differentiation and promote proliferation (Krivtsov et al. 2008; Okada et al. 2005; Bitoun et al. 2007). MLL translocations are found in approximately 5–10 % of acute leukemias (Slany 2009) and involve fusion of the N-terminal domain of MLL to partners such as AF10, AF4, AF9, ENL, ELL, and AF6 (Krivtsov and Armstrong 2007). DOT1L associates with the MLL fusions through the partner-binding domains. In these fusions, the DNA binding of MLL is retained; however, it lacks the SET domain. This concept of MLL fusion-mediated DOT1L recruitment was reinforced by an artificial DOT1L-MLL fusion which also was capable of inducing leukemic transformation (Okada et al. 2005). Therefore, patients with MLL-rearranged leukemias capable of recruiting DOT1L are an example of a genetically defined cancer population that may be candidates for treatment with small molecule inhibitors of DOT1L (Barry et al. 2010).
9.2.1.6 H4K20 Methylation
Methylation of H4K20 is catalyzed by at least 4 enzymes (SETD8, Suv420H1, Suv420H2, and NSD1), and there are currently no known demethylases for this mark (Kouzarides 2007). Top-down mass spectrometry analysis has revealed that H4K20me2 is the dominant modification, with H4K20me1 and H4K20me3 being less abundant (Pesavento et al. 2008; Yang et al. 2008). H4K20me2 is widely distributed and is observed to be bound by the Tudor domain of 53BP1 (Botuyan et al. 2006), which is involved in initiating the DNA damage response (Yang et al. 2008; Sanders et al. 2004). H4K20me1, catalyzed by SETD8, is localized to the chromatin of active genes (Oda et al. 2009). H4K20me1, however, is a known target of malignant brain tumor domain proteins such as L3MBTL1, which result in chromatin compaction (Kalakonda et al. 2008). Additionally, monomethylation is seen in X-inactivation (Kohlmaier et al. 2004). H4K20me1 appears to be either activating or repressive depending on the context. H4K20me3, catalyzed primarily by Suv420H1 and Suv420H2, is found in pericentromeric chromatin and is thought to be a silencing mark (Schotta et al. 2004).
Several cancers have decreased H4K20 trimethylation. In non-small cell lung cancer, loss of H4K20me3 increases as the disease progresses, and this correlates negatively with survival (Van Den Broeck et al. 2008). Additionally, a study comparing a panel of normal cell lines to cancer cell lines found cancer cell lines displayed a loss of H4K20me3 (Tryndyak et al. 2006). Additionally, a mouse model of skin cancer indicates that this loss of H4K20me3 occurs early in tumorigenesis (Fraga et al. 2005). Therefore loss of H4K20me3 may serve as a biomarker in early cancer detection.
9.2.2 Histone Arginine Methylation
Methylated histone arginine residues have also been linked to oncogenic signaling events and cancer phenotypes. The next section outlines the roles of specific arginine methylation sites and their association with cancer.
9.2.2.1 H3R8me2s and H4R3me2s
The two most studied histone arginine methyl modifications linked to human cancer are H3R8me2s and H4R3me2s and both are catalyzed by PRMT5 (Pal et al. 2004). Arginine methylation at these sites is associated with transcriptional repression. In fact, PRMT5 can be recruited to specific promoters by a variety of transcriptional repressors, including Snail and AJUBA proteins (Hou et al. 2008). Hypermethylation of H3R8 and H4R3 has been observed in several lymphoid cancer cell lines, such as mantle cell lymphoma (MCL) and chronic lymphocytic leukemia (B-CLL), and in tumor samples from MCL patients. Furthermore, these methyl marks have been associated with the repression of multiple tumor suppressor genes, including ST7, NME1 (Pal et al. 2004, 2007), and RB family members (Wang et al. 2008b). Importantly, RNAi-mediated knockdown of PRMT5 in these lymphoid cancer cell lines reduced proliferation, diminished hypermethylation of H3R8 and H4R3, and restored expression of tumor suppressor genes.
The mechanism governing increased methylation of H3R8 and H4R3 in lymphomas and leukemias appears to be increased PRMT5 protein translation. Surprisingly, PRMT5 mRNA is generally lower in transformed lymphoid cancer cell lines when compared to “normal” cells of the same lineage. However, PRMT5 protein translation efficiency is greatly enhanced in transformed B cells and MCL cell lines due to aberrant expression of certain subsets of PRMT5-targeting microRNAs (miRNAs) (Pal et al. 2007). In addition to enhanced translational efficiency, PRMT5 enzymatic activity is regulated by phosphorylation of its co-stimulatory protein, MEP50. Increased phosphorylation of MEP50 by cyclin D1/cdk4 complexes is associated with increased PRMT5 activity. These data suggest that PRMT5 may play an important role in cyclin D1-driven cancers (Aggarwal et al. 2010).
9.2.2.2 H4R3me2a
Asymmetric dimethylation of histone H4 on Arg-3 (H4R3me2a) is generally associated with gene promoters undergoing active transcription. H4R3me3a formation is catalyzed by at least three different type I arginine methyltransferases, PRMT1, PRMT6, and PRMT8 (Di Lorenzo and Bedford 2011). Of these RMTs, PRMT1 is viewed to be the predominant cellular asymmetric arginine methyltransferase, accounting for as much 85 % of physiological protein arginine methylation (Tang et al. 2000).
H4R3me2a has been linked to oncogenic signaling in several contexts. Notably, PRMT1 has been reported to be a key component of a novel MLL oncogenic transcriptional complex, which includes MLL-EEN, CBP, and the bridging protein Sam68 (Cheung et al. 2007). Using chromatin immunoprecipitation, Cheung et al. demonstrated that H4R3me2a promoter levels correlate with the expression of key MLL-EEN target genes, such as HOXA9. Additionally, these authors directly fused PRMT1 to the MLL protein and demonstrated that this fusion product increased self-renewal potential in primary hematopoietic cells. However, an enzymatically inactive point mutant version of PRMT1 was not able to bolster self-renewal capacity in this assay. Finally, this group demonstrated that shRNA-mediated knockdown of PRMT1 abrogated MLL-EEN-mediated transformation of primary hematopoietic cells. In summary, these data support the link between the H4R3me2a mark and MLL-EEN-mediated oncogenic signaling.
Recently, PRMT1 was reported to interact with a splice isoform of the oncogenic fusion protein AML-ETO (AE9a), the characteristic translocation product of t(8;21)-positive acute myeloid leukemia (Shia et al. 2012). PRMT1 directly methylates AE9a on Arg-142 and is recruited to AE9a target gene promoters, where it increases methylation on H4R3 (H4R3me2a). Genetic knockdown of PRMT1 decreased the levels of H4R3me2a at AE9a target gene promoters in AML-ETO translocation-positive cell lines. Most notably, knockdown of PRMT1 also decreased the self-renewal capacity of AE9a in colony growth assays, consistent with an important role for PRMT1 and H4R3me2a in t(8;21) leukemogenesis.
Interestingly, dimethylation of H4R3 can repress or promote gene expression at the same promoter depending on whether the modification is symmetric or asymmetric. Symmetric dimethylation of H4R3 by the type II protein arginine methyltransferase PRMT5 leads to recruitment of DNMT3A to the β-globin locus via direct binding of DNMT3A to H4R3me2s (Zhao et al. 2009). Subsequently, DNMT3A methylates CpG sites leading to transcriptional repression of the β-globin locus.
On the other hand, asymmetric methylation of H4R3 (H4R3me2a) by the type I protein arginine methyltransferase PRMT1 leads to repression of gene expression at the β-globin locus. The H4R3me2a mark, catalyzed by PRMT1, recruits the histone acetyltransferase KAT2B (PCAF) to the β-globin locus control region, and in turn, KAT2B acetylates H3K9 and K14 (Li et al. 2010). Acetylation of these sites is associated with increased transcription at the β-globin locus. Thus, the activities of PRMT5 and PRMT1 at the same histone H3 arginine residue have opposing functions on gene expression. Within the context of lymphomas, such as mantle cell and B-CLL, where the H4R3me2s mark is increased at key tumor suppressor promoters, it remains an open question as to whether H4R3me2a is concurrently downregulated.
9.2.2.3 H3R17me2a
Increased asymmetric dimethylation of H3R17, known to be catalyzed by CARM1, has been observed in breast (Frietze et al. 2008; El Messaoudi et al. 2006) and prostate (Majumder et al. 2006) cancers. CARM1 is recruited to nuclear hormone receptor complexes via interactions with members of the p160 steroid co-activator family (Chen et al. 1999). Upon hormone stimulation, CARM1 is recruited to p160-nuclear hormone receptor complexes and increases H3R17 dimethylation at androgen- and estrogen-responsive promoters. These signaling events lead to increased transcription of several proliferation genes, such as E2F1, CCNA1, CCNE1, CCNE2, and CDC25A. Disruption of CARM1 in both prostate and breast cancer cell lines decreases H3R17 methylation at hormone-responsive promoters and leads to decreased cancer cell proliferation. It should be noted that increased expression of nuclear CARM1 has been reported to correlate with the progression of prostate cancer malignancy (Majumder et al. 2006; Hong et al. 2004). However, these studies did not investigate whether H3R17me2a levels also correlate with disease progression.
9.2.2.4 H3R26me2a
Although not as well characterized as H3R17me2a, H3R26me2a is also generated by CARM1. This mark has been linked to oncogenic signaling by virtue of the observation that the CARM1/NCOA3 complex is recruited the promoters of a number of E2F-driven target genes, including CCNE1, DHFR, and CDC6 (El Messaoudi et al. 2006). Recruitment of the CARM1/NCOA3 complex to said promoters increases the levels of both H3R17me2a and H3R26me2a.
9.2.2.5 Nonhistone Substrates of Protein Arginine Methyltransferases in Cancer
The importance of nonhistone substrates in PRMT-mediated oncogenic signaling pathways is becoming increasingly clear. These substrates include a variety of signaling molecules such as transcription factors, transcriptional co-activators, chromatin modulating proteins, and other DNA-binding proteins. One of the most compelling examples of these nonhistone substrates is the tumor suppressor p53. Jansson and colleagues identified in the oligomerization domain of p53 three arginine residues which appear to be methylated by PRMT5 (Jansson et al. 2008). Disruption of PRMT5 reduces methylation of these residues and promotes a p53-mediated apoptotic response following DNA damage. Thus, PRMT5 may have a role in regulating p53-mediated cell fate (cell cycle arrest versus apoptosis) following a DNA damage stimulus.
As mentioned previously, the CARM1 has been shown to be recruited to nuclear hormone receptors via its interaction with the p160 steroid co-activator family of proteins. As a part of these transcriptional complexes, CARM1 can methylate not only nucleosomes (H3R17) but also its p160-binding partner NCOA3 (Feng et al. 2006; Naeem et al. 2007). Interestingly, this methylation event, which is increased by hormone stimulation, leads to disassembly of the NCOA3 transcriptional complex and downregulation of hormone-dependent transcription. Thus, the role of CARM1 in nuclear hormone receptor signaling is complex, as CARM1 can activate proliferative transcriptional pathways via methylation of nucleosomes (H3R17), while simultaneously attenuating hormone-dependent transcription by methylating NCOA3. The question as to how the balance of these stimulatory and inhibitory pathways becomes shifted in favor of proliferation in certain types of breast and prostate cancer is an area for future investigation.
9.2.2.6 DNA Damage Response Machinery and PRMT1
PRMT1 has been linked to asymmetric arginine methylation of number of DNA damage checkpoint machinery proteins. Recently, it was reported that PRMT1 methylates BRCA1 in a region that contains multiple lysine and arginine methylation sites and that arginine methylation of BRCA1 can be detected in multiple breast cancer lines and breast tumor samples (Guendel et al. 2010). Knockdown of PRMT1 in breast cancer cell lines caused a change in the binding pattern of BRCA1 to target promoters. It remains unclear exactly how BRCA1 arginine methylation correlates with cancer phenotypes. PRMT1 has also been reported to methylate MRE11, a member of the DNA double-strand break repair complex (Boisvert et al. 2005a). While methylation of MRE11 did not appear to affect formation of the MRE11-RAD40-NBS1 complex, mutation of arginine residues in the GAR (glycine-arginine rich) domain diminished its exonuclease activity. Like MRE11, 53BP1 has also been reported to contain a GAR motif and to be methylated by PRMT1 (Boisvert et al. 2005b). Mutation of arginine residues within the 53BP1 GAR motif abrogated its ability to localize to sites of DNA damage. Taken together, these observations suggest a role for PRMT1 in regulating DNA damage pathways which have critical importance to oncogenic signaling.
9.3 Mechanism of Methyltransfer
The KMT and RMT share a common mechanism to transfer the methyl group from a universal methyl donor, S-adenosyl-L-methionine (SAM) to the nitrogen atom of lysine and arginine side chains, respectively (Fig. 9.1). The transfer of the methyl group results in the production of S-adenosyl-L-homocysteine (SAH).
9.3.1 SET Domain Lysine Methyltransferases
As discussed above, the SET domain class of lysine methyltransferases includes enzymes known to methylate several sites on histone H3 (K4, K9, K27 & K36) and at least one site on histone H4 (K20). The KMTs can transfer 1, 2, or 3 methyl groups to lysine utilizing SAM as a methyl donor. The mechanism for methyl transfer has been extensively studied, and several crystal structures of KMTs bound to SAM/SAH, and a peptide substrate have been solved. The substrate specificity of the KMTs is not only specific to the lysine site on the histone but also depends on the methylation state of the lysine. Mutations and different binding proteins can change the substrate preference of some of the KMTs.
The SET domain family of methyltransferases is reported to contain 51 human proteins (Copeland et al. 2009; Richon et al. 2011). Generally the SET methyltransferases are thought to function as part of larger complexes such as the PRC2 complex. The accessory proteins act to target and regulate activity while the SET methyltransferase is the catalytic engine of the complex.
The SAM-binding motif contained in the SET domain of the KMTs has both highly conserved and divergent regions. In the highly conserved region, a NHS sequence is maintained for the majority of SET proteins. The histidine backbone carbonyl accepts a hydrogen bond from the N-6 of SAM, while the backbone amine of the same histidine donates a hydrogen bond to the N-7 of SAM (Dillon et al. 2005). The interaction with the conserved histidine has been seen in all of the co-crystal structures of SET methyltransferases with SAM or SAH. The remaining SAM-binding pocket can vary significantly between subfamilies, but all contain interactions with the amino acid portion of SAM. It is worth noting that while the interactions vary, the 3-dimensional configuration of SAM/SAH in the SET methyltransferase co-crystals are superimposable on each other and completely distinct from any of the other SAM-utilizing methyltransferases.
One of the most studied KMT is SET7/9 (SETD7). It has been the focus of several mechanistic papers regarding the process of transferring a methyl group from SAM to the lysine. There is consensus that the reacting lysine is initially protonated when binding to the KMT. While some disagreement exists on the nature of the base, the mechanism involves the deprotonation of lysine to give the neutral amine which then attacks the methyl of SAM in a SN2-like fashion. Guo et al. (Guo and Guo 2007) have suggested that the conserved tyrosine could be the base that deprotonates the lysine, while others have suggested water acts as the base. Xiao et al. (2005) pointed to bulk solvent to be the base, while Dirk et al. (2007) suggested a water molecule in the active site as the base. Zhang and Bruice (2007) favor the water being the base but point out that the water molecule in the active site is not basic enough to deprotonate the lysine and there is not access to bulk solvent in the active site. They indicate that a water channel forms upon binding the substrate, allowing the proton to be shuttled to bulk solvent. The water channel is proposed to form only when the lysine lacks a methyl group. Once the lysine is methylated, the methyl blocks the channel and prevents subsequent methylation, thus giving SET7/9 its specificity of catalyzing the 0–1 H3K4 methylation.
The lysine-binding portion of the active site of the SET domain has been shown to consist of an aromatic cage containing 1–3 tyrosine residues which can accept hydrogen bonds from the lysine. The aromatic cage stabilizes the charged species and lowers the pKa of the lysine, while the tyrosines play a role in proper alignment of lysine (Zhang and Bruice 2008). Mutational studies with GLP and G9a (Wu et al. 2010; Rathert et al. 2008) and with SET7/9 (Collins et al. 2005) have shown that mutation of one of the aromatic cage tyrosines to phenylalanine, and in one case to alanine, allows the enzyme to further methylate the lysine. Additionally for G9a, it was shown that mutating an aromatic cage phenylalanine to tyrosine restricted the number of methyl groups it could transfer, allowing only the unmethylated lysine to be an effective substrate. The two positions which control the ability of the SET to use methylated lysines as substrates have been termed the Phe/Tyr switch.
While initially these switches were identified by performing in vitro mutational studies, recently similar mutations have been described in follicular and germinal center B-derived diffuse large B-cell lymphomas (Morin et al. 2010). In these lymphomas, EZH2 was found to be mutated at Y641 (which corresponds to one of the residues identified previously as a Phe/Tyr switch) resulting in a change to several other amino acids (Morin et al. 2010). Enzymatic analysis of these mutant enzymes showed a change in substrate preference from H3K27me 0 > 1 > 2 for the wild-type enzyme to 2 > 1 > 0 for the Y641 mutants (Sneeringer et al. 2010; Yap et al. 2011). These change-of-function mutants led to elevated cellular levels of H3K27me3, which contributes to lymphomagenesis. Interestingly, all of the Y641 mutants displayed the same pattern of changes in substrate utilization, and all of the changes could be ascribed to transition state interactions with the enzyme (Sneeringer et al. 2010).
9.3.2 Arginine Methyltransferases
The arginine methyltransferases (RMTs) can be divided into two major subgroups, type I and II (Di Lorenzo and Bedford 2011). The type I RMTs consist of PRMT 1, 3, 4, 6, and 8 and type II of PRMT 5 and 7. Additionally, PRMT7 has been described to be a type III RMT on certain substrates (Fig. 9.2). The type I RMTs catalyze both the formation of monomethylated arginine (MMA) intermediates and asymmetrical dimethylated arginines. The type II RMTs catalyzes the formation of MMA and symmetrical dimethylated arginines. The type III RMT catalyzes only the formation of MMAs. In each case, SAM is utilized as the methyl donor as depicted in scheme A. The majority of methylated arginines found in vivo appear to be DMAs (dimethylated arginines) (Zhang and Cheng 2003).

Methylation of arginine residues by types I, II, and III RMTs
A number of RMTs including PRMT1 and 3 and CARM1 (PRMT4) have been found to exist in dimeric or oligomeric states (Zhang and Cheng 2003); this has led groups to speculate on whether the in vivo observation of preponderant DMA versus MMA is due to the dimeric/oligomeric structure leading to a processive mechanism. Kolbel et al. (2009), however, have demonstrated that PRMT1 and PRMT3 act via a distributive mechanism (Fig. 9.3). This was based on the analysis of multiple kinetic experiments, including the observation of a Poisson product distribution highly indicative of a distributive mechanism whereas a processive mechanism should result in a product profile biased towards dimethylated species. The authors suggest that release of the methylated arginine substrate is obligatory and rate determining.

Distributive mechanism of arginine methylation by type I RMTs
Additional work is necessary to understand why DMA products predominate in vivo as the distributive mechanism does not appear to account for this finding. Experiments with peptide substrates suggest little change in Km and kcat values for MMA residues over the corresponding unmethylated counterparts, i.e., the MMA arginine residues are not preferred substrates; however, further work with physiological substrates is required. Alternative explanations include that the RMTs are components of multi-protein complexes, where the other proteins may influence the substrate specificity of the RMT. CARM1, for example, is found in a complex of at least ten proteins called the nucleosomal methylation activator complex (NUMAC) (Xu et al. 2004). Interestingly, recombinant CARM1 in the absence of some, if not all, of the other proteins of the NUMAC complex preferentially methylates free histone H3 rather than the nucleosome. CARM1 activity has also been reported to be modulated by phosphorylation resulting in a decrease in RMT activity (Higashimoto et al. 2007). Neighboring residues on histone H3, such as H3K18, have been demonstrated to affect the rate of methylation on H3R17 by CARM1. For example, CARM1 activity on H3R17 is increased when H3K18 is acetylated (~5×). This is due to an increase in kcat rather than Km (Daujat et al. 2002). It is interesting to note that for CARM1 substrates, the +1 residue is neutral in known protein substrates except for histone H3. For example, the following CARM1 substrates contain a neutral +1 residue; PABP1 R455, R460; HuR R206, R217; HuD R225, R236; p300 (KIX domain) R580, R604; and CBP (post-KIX domain) R714, R742, R768 (Yue et al. 2007). This suggests a potential electrostatic sensing mechanism to explain the preference of a DMA product on H3R17. However, this does not provide an explanation for other substrates of CARM1. This example also highlights the inherent complexity of histone modifications, as posttranslational modification of specific residues can affect the methylation status of other residues. This may be important for drug discovery as changes in methylation status may be gene-specific due to the effect of other posttranslation modifications of chromatin. Therefore, it may be very difficult to detect changes from a methyltransferase inhibitor when measuring global methylation levels.
9.3.2.1 Structure and Catalytic Mechanism of RMTs
The X-ray crystal structure of PRMT1, PRMT3, and CARM1 in various states has been obtained (Zhang and Cheng 2003; Yue et al. 2007; Zhang et al. 2000). The CARM1 X-ray structure was obtained in the presence and absence of SAH, and it was found that upon binding to SAH, significant structural reorganization of the protein occurs resulting in encapsulation of SAH (and presumably SAM) and the formation of the substrate-binding pocket. The authors conclude that the structure is consistent with an ordered mechanism in which SAM binding occurs first and the intermediate MMA must be released from the active site prior to the replenishing of SAM, an observation which is consistent with a distributive mechanism (Yue et al. 2007).
Insight from the structures of PRMT1, PRMT3, and CARM1 has led to the proposal of a catalytic mechanism (Fig. 9.4). When the substrate binds to the RMT-SAM complex, a conserved carboxylate (for CARM1, this is Glu 267, E153 in PRMT1, E355 in PRMT3) interacts with one of the nitrogen atoms in the guanidine group of the arginine residue, resulting in increased localization of positive charge on the Nη1 nitrogen allowing the Nη2 nitrogen to react with SAM (Zhang and Cheng 2003; Yue et al. 2007; Zhang et al. 2000). This carboxylate residue has been shown to be critically important in catalysis, as the mutant Glu267Asn is catalytically inactive (Chen et al. 1999; Yue et al. 2007). It is likely, however, that the guanidine group is charged and needs to be deprotonated before reacting with SAM. The authors point out that the timing of the deprotonation step is unknown but is likely to occur via a His-Asp (CARM1-H415/D166, PRMT1-H476/D233) coupled proton relay similar to serine proteases (Zhang et al. 2000; Fersht and Sperling 1973). The His and Asp residues are also somewhat conserved throughout the RMTs (Zhang et al. 2000). It has been proposed that deprotonation is likely to be rate determining as in other SN2 enzymatic reactions (Yue et al. 2007). If the His-Asp couple is involved in the rate-determining step, then it is likely to be highly sensitive to the electronic state of neighboring residues (e.g., difference in kcat for H3K18 and acetylated H3K18).

Proposed catalytic mechanism of RMTs
The protein crystal structures have also provided a hypothesis for the selectivity differences between type I and II RMTs. The type I RMTs produce asymmetric dimethylated arginine residues. One possible explanation for this is due to the sulfur atom of M337 (PRMT3), which is only 3.6 A away from Nη1, essentially sterically blocking this nitrogen. The Nη2 has space to accommodate a monomethyl group which allows for the asymmetrical DMA product formation (Zhang et al. 2000).
9.4 Inhibitors of Histone Methyltransferases
Despite the discovery of the first HMT over 10 years ago (Rea et al. 2000) and the description of close to 100 related proteins (Richon et al. 2011), there has been a paucity of literature describing potent and selective inhibitors not based on SAM, SAH, or Sinefungin (Table 9.2). The majority of reports have focused on inhibitors of CARM1, G9a, and GLP.
9.4.1 Lysine Methyltransferase Inhibitors
9.4.1.1 G9a (EHMT2) and GLP (EHMT1)
The majority of published work on chemical inhibitors of SET domain-containing HMTs has focused on GLP and G9a. The first drug-like small molecule inhibitor that acted on these two enzymes was Compound (I) and is known as BIX-01294 (Kubicek et al. 2007). Compound (I) was identified via high-throughput screening and is closely related to the α-adrenoceptor antagonist bunazosin (Weidinger 1995) (Table 9.3). Compound (I) demonstrated an IC50 of 1.7 μM for G9a and 38 μM for GLP. Two independent groups have replicated the IC50 for G9a but found the compound to be significantly more active at GLP with an IC50 ~ 0.7 μM (Liu et al. 2009a; Chang et al. 2010). The authors attribute the discrepancy to the fact that the original data was not performed under linear assay conditions. Compound (I) appears to be selective against other HMTs as only minimal activity was observed at PRMT1, ESET, SET7/9, Suv420H1, and SUV39H1.
Compound (I) is uncompetitive with SAM, suggesting that it only binds to the SAM-G9a complex and not the free enzyme. These data are consistent with the X-ray structure of Compound (I) bound to GLP (~80 % sequence similarity to G9a) where the small molecule is bound in the substrate-binding pocket and SAH is also bound (Chang et al. 2009).
In mouse embryonic stem cells treated with 4.1 μM of (I) the H3K9me2 mark was reduced by 20 % with a concomitant rise in unmethylated H3K9 and no change in H3K9me3 or H3K9me1 levels. No changes on the global methylation status of H3K27, H3K36, or H4K20 were observed indicating that the inhibitor selectively inhibits G9a/GLP in cells. The inhibitor (I) did reduce H3K9me2 in G9a null ES cells, and the authors ascribe this finding to the activity on GLP which also affects H3K9 methylation. This explanation is even more likely as the work of other groups has shown that Compound (I) is essentially equipotent against GLP. Therefore, the current evidence suggests that Compound (I) is a dual GLP/G9a inhibitor.
ChIP analysis demonstrated that Compound (I) affected H3K9me2 levels at target genes such as Mage-a2. Approximately 60 % loss of the H3K9me2 mark at the Mage-a2 promoter was observed in mouse ES cells that were treated with 4.1 μM Compound (I). This inhibition was reversible in that the mark returned to original levels within 48 h once the inhibitor was removed.
Two independent groups (Liu et al. 2009a; Chang et al. 2010) expanded upon the crystal structure findings of the GLP-Compound (I)-SAH complex, hypothesizing that an appropriately placed lysine mimic added to Compound (I) would result in improved activity at G9a and or GLP. The hypothesis was derived from the observation that when the X-ray structures of GLP-(I) and GLP-peptide substrate were compared, it was noted that Compound (I) resembled the bound conformation of histone H3 Lys4 to Arg8 but leaving the lysine-binding channel unoccupied. Using this observation, both Chang et al. and Liu et al. replaced the 7-OMe with an aminoalkoxy groups. The first result of the Lui et al. effort was UNC-0224 (III) which demonstrated improved potency compared to (I) (see Table 9.3). It was also found to be inactive in inhibiting other HMTs including SETD7, SET8/PreSET7, PRMT3, and the H3K9 demethylase JMJD2E.
A high-resolution (1.7Å) X-ray crystal structure of the G9a-UNC0224 (III) complex was obtained. On the basis of this structural information, the authors indicate the improved potency of UNC-0224/Compound (III) over Compound (I) is explained by interactions gained from the additional lysine mimetic moiety. In particular, the electrostatic interaction of the protonated dimethyl amine and Leu1086 and the π-cation interaction of the protonated dimethyl amine with Tyr 1154 add to the affinity.
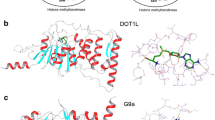
Key structural insights were identified from this analysis, and these are illustrated in Fig. 9.5. Two features of particular note are that (1) the bulk of UNC-0224 (III) occupies the histone peptide-binding site as in the GLP-(I) structure and (2) the lysine-binding channel is not fully occupied by the amino-propoxy lysine mimic. This last observation suggested the potential for further improvement in potency with a longer lysine mimetic. This hypothesis was confirmed by the synthesis of Compound (IV) which shows improved potency against G9a (Table 9.3).

Key interactions of compound (III)/UNC0224 with G9a
The inhibitor (IV) demonstrated a high Hill slope suggesting that the potency limit of the assays used had been reached (Liu et al. 2009a). Using microfluidic capillary electrophoresis, the Ki values of 2.6 nM and 63 pM were established for Compound (III)/UNC0224 and Compound (IV), respectively, compared to 16 nM for Compound (I). This analysis shows that Compound (IV) has greatly improved potency over both Compound (I)/BIX-01924 and Compound (III)/UNC0224.
Vedadi et al. describe the chemical probe UNC0638 (Vedadi et al. 2011) (Table 9.3). UNC0638 shares common features with (III)/UNC0224 and (IV) but interestingly the basic and polar 1-methyl-1,4-diazepane ring system has been replaced with the neutral and lipophilic cyclohexyl ring. This modification was presumably made in order to improve cell permeability. Indeed, Compound (V) showed reduction of H3K9me2 in MDA-MB-231 cells with an IC50 of 81 nM and with a better separation of methyl mark effects and cell toxicity compared to Compound (I).
Chang et al. (2010) also found that a longer chain at the seven position resulted in improved potency. This change combined with replacement of the diazepane ring and benzyl groups of Compound (I) with a 3-diemethylaminopropyl and a 5-aminopentyl chain, respectively, led to Compound (VI) (Table 9.3). The cumulative effect of these changes is 7-fold improvement in affinity for GLP relative to Compound (I) under identical assay conditions.
It was noted from the (VI)-GLP-SAH X-ray structure that the sulfur atom of SAH was only 4.7Å from the nitrogen of the lysine mimetic, very close to the distance seen in the GLP-SAH-H3K9me1peptide substrate structure of 4.2Å (Chang et al. 2010). Superposition of the (VI)/GLP complex with the GLP/H3K9me1 peptide side-chain complex suggested that the 5-aminopentoxy side chain was appropriately positioned to be a substrate for methylation. Mono-, di-, and trimethylated derivatives of (VI) were detected by mass spectrometry following overnight incubations of nearly equimolar ratio of (VI) and GLP. Despite the nonphysiological conditions of this experiment, it does provide evidence that the inhibitors may also be substrates.
9.4.1.2 DOT1L
EPZ004777 has been reported as a potent and selective inhibitor of DOT1L (Daigle et al. 2011). This molecule is an aminonucleoside analog that was shown to be SAM competitive with an IC50 of 0.4 nM for DOT1L biochemical activity and greater than 1,000-fold selectivity against other KMTs and RMTs (Table 9.2). EPZ004777 causes dose-dependent reduction of histone H3K79me2 in cultured cells and inhibits proliferation of leukemia cells bearing the MLL translocation. It is noteworthy that cells lacking the MLL translocation were insensitive with respect to antiproliferative effects despite undergoing a reduction in the H3K79me2 mark. EPZ004777 also demonstrated in vivo efficacy in a mouse model of disseminated MLL-rearranged leukemia, at doses that were well tolerated. These data demonstrate the high specificity of targeting DOT1L for MLL-rearranged leukemias.
9.4.2 Arginine Methyltransferase Inhibitors
9.4.2.1 CARM1
One of the first non-SAM like small molecule inhibitors of CARM1 was described by Purandare et al. (2008). High-throughput screening efforts identified Compound (VII) with a CARM1 IC50 of 1.8 μM (Table 9.4). Progression of this hit led to the identification of Compound (VIII) with a 10-fold improvement in CARM1 IC50, 0.08 μM (Purandare et al. 2008). Compound (VIII) was also tested for activity against PRMT1 and PRMT3, and IC50 values of >25 μM were obtained for each enzyme. No cellular data was reported, presumably because the series was hampered by poor permeability. Compound (VIII) scored poorly in a PAMPA assay with a permeability of less than 0.015 mm/s. An improvement in the permeability was observed when the secondary amide was replaced with the bio-isosteric 1,3,4-oxadiazole ring. This change led to an increase of approximately 10-fold in permeability with no loss of CARM1 inhibitory activity.
Further optimization of the oxadiazole (IX) led to the discovery of (X) which improved CARM1 potency (IC50 0.04 μM) and permeability to 0.267 μm/s (Huynh et al. 2009) (Table 9.4). The compound was further characterized for ADMET properties and found to not inhibit P-450 enzymes (human hepatocyte assay-HHA) or to activate the human pregnane-X receptor (hPXR).
Allan et al. have also developed a related series of thienyl pyrazoles of which Compound (XI) is representative (Allan et al. 2009). They found that the series was of similar potency as earlier compounds described above. Compound (XI) was also selective over the RMT PRMT1 and the KMT SETD7 (IC50 >100 μM for each enzyme). Compound (XI), however, was reported to lack cellular activity in two different cell types after 48 h of exposure to the inhibitor at a concentration of 5 μM. A number of reasons for the lack of cell activity are possible, such as cell permeability (the authors report that the thienyl-amide series also possessed poor permeability similar to the phenyl-amide series (e.g., Compound (VIII)) (Huynh et al. 2009). It is also possible that the compounds lack sufficient biochemical potency to elicit cellular effects. Alternatively, the lack of changes in global levels of H3R26 methylation may be due to redundancy due to other RMTs. In light of this, Compounds (XI) and (XII) were evaluated in a series of functional assays. The assays targeted both estrogen dependent transcription, estrogen growth and androgen dependent transcription. Neither Compound (XI) nor (XIII) demonstrated any effect in these assays consistent with the lack of methylation changes observed previously.
Therrien et al. (2009) also noted the poor PK profile of the phenyl-amide and thienyl-amide series, low oral bioavailability in rats and low exposure in mice when dosed by intraperitoneal injection. Compound (XII) which is representative of the phenyl-amide series was characterized with extremely high clearance (1,433 ml/min/kg) and a short t 1/2 (<0.25 h) when dosed by intravenous injection in rats. The high clearance precluded the accurate measurement of oral bioavailability, volume of distribution and half-life. Therrien et al. hypothesized that the high clearance was due to the presence of the (S)-alanine benzylamide present in both chemical series.
After SAR exploration into the replacement of this moiety, it was found that a N1-benzyl-N2methylethane-1,2 diamine unit could adequately replace the alanine amide group with minimal loss in CARM1 potency, as exemplified by Compound (XIII). The PK profile of Compound (XIII) was evaluated in rats and was found to have a much reduced clearance (20 ml/min/kg) which resulted in an improved t 1/2 of 2.1 h. The volume of distribution and oral bioavailability, however, remained low: 1.4 l/kg and 4 %, respectively.
The low oral bioavailability may be due to the amide functionality, as both Therrien et al. (2009) and Huynh et al. (2009) indicate that this group confers low permeability in both the phenyl and thienyl series. To address these concerns, Therrien et al. (2009) synthesized Compound (XIV) with the 1,3,4-oxadiazole ring which had been shown to improve permeability (Huynh et al. 2009). As can be seen from Compound (XIV), bio-steric replacement did not improve oral bioavailability or increase the volume of distribution; however, no PAMPA data was reported for this analog, so it is unclear if this analog had improved permeability.
Troffer-Charlier et al. also proposed a binding mode based on docking (XIII) into a published X-ray structure of CARM1-SAH (Troffer-Charlier et al. 2007). A graphical representation of the critical interaction of the N1-benzyl-N2methylethane-1,2 diamine group is shown schematically in Fig. 9.6. The diamine group is proposed to bind to a water molecule as well as the carboxylate group of Asp191 and the backbone carbonyl of Gly193. A significant SAR effort around the diamine side chain was undertaken, with small changes producing large changes in activity. It was proposed that the linker needs to be flexible in order to adopt the required conformation to interact with the water molecule, Asp 191 and Gly193, while avoiding a potentially energetically costly interaction with the positively charged Arg 169 (which interacts with the carboxylate of SAH). No kinetic or crystallographic data has been reported on this diamine series to date to support the idea that these compounds bind in the SAM-binding site.

Proposed binding of compound (XIII) in the SAM binding pocket of CARM1
Wan et al. also found a benzo[d]imidazole hit (XV) via high-throughput screening which inhibited CARM1 with an IC50 of 0.84 μM (Wan et al. 2009). This series also contains a diamine moiety similar to that in the Compounds (XIII) and (XIV), though in this series a tertiary amine is incorporated into the diamine, and no such corresponding analog was reported (Allan et al. 2009; Therrien et al. 2009). Therefore, it is unclear if the two series are related in regard to how they interact with CARM1.
Wan et al. performed a hit to lead effort on (XV) and found most changes to the molecule were detrimental (Wan et al. 2009). The C-2 substituent on the benzo[d]imidazole, however, proved to be a fruitful area to analog resulting in Compound (XVI) with over 10-fold improvement in CARM1 IC50. Further studies from Wan et al. led to the discovery of the indole Compound (XVII) (presented at ACS Symposium, Boston, 2010). Compound (XVI) is a potent CARM1 inhibitor and inhibits histone H3 methylation (IC50 0.081 μM). It was reported to inhibit the methylation of nonhistone CARM1 substrates (PABP and HuR) with similar potency. This group also performed additional pharmacology on the indole compound (XVII) and found that the compound is metabolically stable in mouse, rat, and human microsomes and displays no inhibition of P-450 enzymes but does, however, possess poor permeability as measured in Caco-2 and PAMPA experiments. The compound also is an hERG inhibitor which is likely due to the basic side chain coupled with high lipophilicity (clogP ~4.9). A co-crystal of Compound (XVII) with Sinefungin and CARM1 was also presented. Unlike the model proposed by Therrien et al. (2009), it was found that the inhibitor was bound in the substrate-binding pocket rather than the SAM-binding pocket which was occupied by Sinefungin.
No cellular data was reported for any of the new analogs. This remains an outstanding issue for the CARM1 inhibitors described to date, as poor permeability is observed in multiple series and until robust cellular effects are observed, it is unclear whether these compounds are appropriate tools for examining the role of CARM1 mediated biology.
9.5 Summary and Future Perspectives
The HMTs are a promising new class of targets for cancer. Members of the enzyme family have demonstrated strong cancer relevance, and recent genome studies have linked genetic alterations in members of this family with specific cancer indications. Until recently, very few small molecule inhibitors have been described, and currently the first inhibitors are entering evaluation in clinical trials. The field has begun identifying inhibitors of these novel enzymes, initially with nucleoside or natural product-derived inhibitors. The development of a rich set of selective inhibitors with diverse chemical properties will enable the field to evaluate the potential of these targets in both preclinical and ultimately clinical studies. Many question remain, but with the increasing biological understanding of these enzymes and their role in cancer and other diseases, it is likely that this field will rapidly evolve over the coming years to provide important new therapeutics for cancer treatment.
References
Abu-Farha M et al (2008) The tale of two domains: proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol Cell Proteomics 7(3):560–572
Adams-Cioaba MA, Min J (2009) Structure and function of histone methylation binding proteins. Biochem Cell Biol 87(1):93–105
Aggarwal P et al (2010) Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell 18(4):329–340
Agger K et al (2007) UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449(7163):731–734
Allan M et al (2009) N-Benzyl-1-heteroaryl-3-(trifluoromethyl)-1H-pyrazole-5-carboxamides as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1). Bioorg Med Chem Lett 19(4):1218–1223
Andreu-Vieyra CV et al (2010) MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol 8(8):e1000453
Angrand PO et al (2001) NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics 74(1):79–88
Barrett A et al (2007) Breast cancer associated transcriptional repressor PLU-1/JARID1B interacts directly with histone deacetylases. Int J Cancer 121(2):265–275
Barry ER, Corry GN, Rasmussen TP (2010) Targeting DOT1L action and interactions in leukemia: the role of DOT1L in transformation and development. Expert Opin Ther Targets 14(4):405–418
Barski A et al (2007) High-resolution profiling of histone methylations in the human genome. Cell 129(4):823–837
Bedford MT, Richard S (2005) Arginine methylation: an emerging regulator of protein function. Mol Cell 18:263–272
Bernstein BE et al (2005) Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120(2):169–181
Bernstein BE et al (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125(2):315–326
Bitoun E, Oliver PL, Davies KE (2007) The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet 16(1):92–106
Boisvert FM et al (2005a) Arginine methylation of MRE11 by PRMT1 is required for DNA damage checkpoint control. Genes Dev 19(6):671–676
Boisvert FM et al (2005b) The GAR motif of 53BP1 is arginine methylated by PRMT1 and is necessary for 53BP1 DNA binding activity. Cell Cycle 4(12):1834–1841
Botuyan MV et al (2006) Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127(7):1361–1373
Boyer LA et al (2005) Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122(6):947–956
Boyer LA et al (2006) Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441(7091):349–353
Bracken AP et al (2003) EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 22(20):5323–5335
Brown MA et al (2006) Identification and characterization of Smyd2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol Cancer 5:26
Cao R, Zhang Y (2004a) SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell 15(1):57–67
Cao R, Zhang Y (2004b) The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev 14(2):155–164
Cao R et al (2002) Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298(5595):1039–1043
Cao R et al (2008a) Role of hPHF1 in H3K27 methylation and Hox gene silencing. Mol Cell Biol 28(5):1862–1872
Cao Q et al (2008b) Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 27(58):7274–7284
Ceol CJ et al (2011) The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 471(7339):513–517
Chang Y et al (2009) Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat Struct Mol Biol 16(3):312–317
Chang Y et al (2010) Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J Mol Biol 400(1):1–7
Chen D et al (1999) Regulation of transcription by a protein methyltransferase. Science 284(5423):2174–2177
Cheng X, Blumenthal RM (2010) Coordinated chromatin control: structural and functional linkage of DNA and histone methylation. Biochemistry 49(14):2999–3008
Chesi M et al (1998) The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 92(9):3025–3034
Cheung N et al (2007) Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol 9(10):1208–1215
Cloos PA et al (2006) The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 442(7100):307–311
Collins RE et al (2005) In vitro and in vivo analyses of a Phe/Tyr switch controlling product specificity of histone lysine methyltransferases. J Biol Chem 280(7):5563–5570
Copeland RA, Solomon ME, Richon VM (2009) Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov 8(9):724–732
Daigle SR et al (2011) Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20(1):53–65
Daujat S et al (2002) Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr Biol 12(24):2090–2097
Del Rizzo PA et al (2010) SET7/9 catalytic mutants reveal the role of active site water molecules in lysine multiple methylation. J Biol Chem 285(41):31849–31858
Di Lorenzo A, Bedford MT (2011) Histone arginine methylation. FEBS Lett 585(13):2024–2031
Dillon SC et al (2005) The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol 6(8):227
Dirk LM et al (2007) Kinetic manifestation of processivity during multiple methylations catalyzed by SET domain protein methyltransferases. Biochemistry 46(12):3905–3915
Eissenberg JC, Shilatifard A (2010) Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol 339(2):240–249
El Messaoudi S et al (2006) Coactivator-associated arginine methyltransferase 1 (CARM1) is a positive regulator of the Cyclin E1 gene. Proc Natl Acad Sci USA 103(36):13351–13356
Feldman N et al (2006) G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol 8(2):188–194
Feng X, Hara Y, Riabowol K (2002) Different HATS of the ING1 gene family. Trends Cell Biol 12(11):532–538
Feng Q et al (2006) Signaling within a coactivator complex: methylation of SRC-3/AIB1 is a molecular switch for complex disassembly. Mol Cell Biol 26(21):7846–7857
Fersht AR, Sperling J (1973) The charge relay system in chymotrypsin and chymotrypsinogen. J Mol Biol 74(2):137–149
Fraga MF et al (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37(4):391–400
Frietze S et al (2008) CARM1 regulates estrogen-stimulated breast cancer growth through up-regulation of E2F1. Cancer Res 68(1):301–306
Frietze S et al (2010) ZNF274 recruits the histone methyltransferase SETDB1 to the 3′ ends of ZNF genes. PLoS One 5(12):e15082
Gao WL, Liu HL (2007) DOT1: a distinct class of histone lysine methyltransferase. Yi Chuan 29(12):1449–1454
Guendel I et al (2010) Methylation of the tumor suppressor protein, BRCA1, influences its transcriptional cofactor function. PLoS One 5(6):e11379
Guo HB, Guo H (2007) Mechanism of histone methylation catalyzed by protein lysine methyltransferase SET7/9 and origin of product specificity. Proc Natl Acad Sci USA 104(21):8797–8802
Guo Y et al (2009) Methylation-state-specific recognition of histones by the MBT repeat protein L3MBTL2. Nucleic Acids Res 37(7):2204–2210
Gupta RA et al (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464(7291):1071–1076
Hamamoto R et al (2004) SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol 6(8):731–740
Hamamoto R et al (2006) Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci 97(2):113–118
Hess JL (2004) Mechanisms of transformation by MLL. Crit Rev Eukaryot Gene Expr 14(4):235–254
Higashimoto K et al (2007) Phosphorylation-mediated inactivation of coactivator-associated arginine methyltransferase 1. Proc Natl Acad Sci USA 104(30):12318–12323
Hong H et al (2004) Aberrant expression of CARM1, a transcriptional coactivator of androgen receptor, in the development of prostate carcinoma and androgen-independent status. Cancer 101(1):83–89
Horton JR et al (2010) Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat Struct Mol Biol 17(1):38–43
Hou Z et al (2008) The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol Cell Biol 28(10):3198–3207
Huang Y et al (2009) Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin Cancer Res 15(23):7217–7228
Huang J et al (2010) G9a and Glp methylate lysine 373 in the tumor suppressor p53. J Biol Chem 285(13):9636–9641
Huynh T et al (2009) Optimization of pyrazole inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1). Bioorg Med Chem Lett 19(11):2924–2927
Jaju RJ et al (2001) A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood 98(4):1264–1267
Jansson M et al (2008) Arginine methylation regulates the p53 response. Nat Cell Biol 10(12):1431–1439
Jeong YS et al (2010) Phosphorylation of serine-10 of histone H3 shields modified lysine-9 selectively during mitosis. Genes Cells 15(3):181–192
Kalakonda N et al (2008) Histone H4 lysine 20 monomethylation promotes transcriptional repression by L3MBTL1. Oncogene 27(31):4293–4304
Kang MY et al (2007) Association of the SUV39H1 histone methyltransferase with the DNA methyltransferase 1 at mRNA expression level in primary colorectal cancer. Int J Cancer 121(10):2192–2197
Kaustov L et al (2011) Recognition and specificity determinants of the human cbx chromodomains. J Biol Chem 286(1):521–529
Kim H et al (2009) Requirement of histone methyltransferase SMYD3 for estrogen receptor-mediated transcription. J Biol Chem 284(30):19867–19877
Kireev D et al (2010) Identification of non-peptide malignant brain tumor (MBT) repeat antagonists by virtual screening of commercially available compounds. J Med Chem 53(21):7625–7631
Kirmizis A, Bartley SM, Farnham PJ (2003) Identification of the polycomb group protein SU(Z)12 as a potential molecular target for human cancer therapy. Mol Cancer Ther 2(1):113–121
Kleer CG et al (2003) EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA 100(20):11606–11611
Klose RJ et al (2006) The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature 442(7100):312–316
Kohlmaier A et al (2004) A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biol 2(7):E171
Kolbel K et al (2009) Type I arginine methyltransferases PRMT1 and PRMT-3 Act distributively. J Biol Chem 284(13):8274–8282
Kouzarides T (2007) Chromatin modifications and their function. Cell 128(4):693–705
Krishnan S, Horowitz S, Trievel RC (2011) Structure and function of histone H3 lysine 9 methyltransferases and demethylases. Chembiochem 12(2):254–263
Krivtsov AV, Armstrong SA (2007) MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 7(11):823–833
Krivtsov AV et al (2008) H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 14(5):355–368
Kubicek S et al (2007) Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell 25(3):473–481
Lan F et al (2007) Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature 448(7154):718–722
Lauring J et al (2008) The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood 111(2):856–864
Lee SH et al (2005) The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc Natl Acad Sci USA 102(50): 18075–18080
Levine SS et al (2002) The core of the polycomb repressive complex is compositionally and functionally conserved in flies and humans. Mol Cell Biol 22(17):6070–6078
Li H et al (2006) Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 442(7098):91–95
Li Y et al (2009) The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J Biol Chem 284(49):34283–34295
Li X et al (2010) H4R3 methylation facilitates beta-globin transcription by regulating histone acetyltransferase binding and H3 acetylation. Blood 115(10):2028–2037
Lim S et al (2010a) Epigenetic regulation of cancer growth by histone demethylases. Int J Cancer 127(9):1991–1998
Lim S et al (2010b) Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 31(3):512–520
Liu C et al (2007) The telomerase reverse transcriptase (hTERT) gene is a direct target of the histone methyltransferase SMYD3. Cancer Res 67(6):2626–2631
Liu F et al (2009a) Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J Med Chem 52(24):7950–7953
Liu G et al (2009b) Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene 28(50):4491–4500
Liu F et al (2010) Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J Med Chem 53(15):5844–5857
Loyola A et al (2009) The HP1alpha-CAF1-SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep 10(7):769–775
Luo XG et al (2009) Effects of SMYD3 overexpression on transformation, serum dependence, and apoptosis sensitivity in NIH3T3 cells. IUBMB Life 61(6):679–684
Majumder S et al (2006) Involvement of arginine methyltransferase CARM1 in androgen receptor function and prostate cancer cell viability. Prostate 66(12):1292–1301
Margueron R et al (2008) Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 32(4):503–518
Maurer-Stroh S et al (2003) The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem Sci 28(2):69–74
Min J et al (2003) Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 112(5):711–723
Miranda TB et al (2004) PRMT7 is a member of the protein arginine methyltransferase family with a distinct substrate specificity. J Biol Chem 279(22):22902–22907
Morin RD et al (2010) Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 42(2):181–185
Naeem H et al (2007) The activity and stability of the transcriptional coactivator p/CIP/SRC-3 are regulated by CARM1-dependent methylation. Mol Cell Biol 27(1):120–134
Ng HH et al (2002a) Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev 16(12):1518–1527
Ng HH et al (2002b) Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J Biol Chem 277(38):34655–34657
Oda H et al (2009) Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol Cell Biol 29(8):2278–2295
Okada Y et al (2005) hDOT1L links histone methylation to leukemogenesis. Cell 121(2):167–178
Ooi SK et al (2007) DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448(7154):714–717
Pal S et al (2004) Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol 24(21):9630–9645
Pal S et al (2007) Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J 26(15):3558–3569
Pesavento JJ et al (2008) Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol Cell Biol 28(1):468–486
Peters AH et al (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 12(6):1577–1589
Purandare AV et al (2008) Pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1). Bioorg Med Chem Lett 18(15):4438–4441
Rao B et al (2005) Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Mol Cell Biol 25(21):9447–9459
Rathert P et al (2008) Protein lysine methyltransferase G9a acts on non-histone targets. Nat Chem Biol 4(6):344–346
Rea S et al (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406(6796):593–599
Richon VM et al (2011) Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des 78(2):199–210
Rosati R et al (2002) NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood 99(10):3857–3860
Ruthenburg AJ, Allis CD, Wysocka J (2007) Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell 25(1):15–30
Sanders SL et al (2004) Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119(5):603–614
Santos-Rosa H et al (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419(6905):407–411
Sarma K et al (2008) Ezh2 requires PHF1 to efficiently catalyze H3 lysine 27 trimethylation in vivo. Mol Cell Biol 28(8):2718–2731
Schlesinger Y et al (2007) Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 39(2):232–236
Schneider R et al (2004) Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol 6(1):73–77
Schotta G et al (2004) A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev 18(11):1251–1262
Schubert HL, Blumenthal RM, Cheng X (2003) Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci 28(6):329–335
Schulte JH et al (2009) Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res 69(5):2065–2071
Schultz DC et al (2002) SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 16(8):919–932
Shahbazian MD, Zhang K, Grunstein M (2005) Histone H2B ubiquitylation controls processive methylation but not monomethylation by Dot1 and Set1. Mol Cell 19(2):271–277
Shi Y, Whetstine JR (2007) Dynamic regulation of histone lysine methylation by demethylases. Mol Cell 25(1):1–14
Shi X et al (2006) ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442(7098):96–99
Shia WJ et al (2012) PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood 119(21):4953–4962
Shilatifard A (2008) Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol 20(3):341–348
Simon JA, Lange CA (2008) Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res 647(1–2):21–29
Sims RJ 3rd, Reinberg D (2006) Histone H3 Lys 4 methylation: caught in a bind? Genes Dev 20(20):2779–2786
Singer MS et al (1998) Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics 150(2):613–632
Slany RK (2009) The molecular biology of mixed lineage leukemia. Haematologica 94(7):984–993
Sneeringer CJ et al (2010) Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci USA 107(49):20980–20985
Squazzo SL et al (2006) Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res 16(7):890–900
Stec I et al (1998) WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum Mol Genet 7(7):1071–1082
Steger DJ et al (2008) DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol 28(8):2825–2839
Strahl BD et al (1999) Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc Natl Acad Sci USA 96(26):14967–14972
Tachibana M et al (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev 16(14):1779–1791
Tachibana M et al (2005) Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev 19(7):815–826
Tan X et al (2006) SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc Natl Acad Sci USA 103(8):2713–2718
Tang J, Kao PN, Herschman HR (2000) Protein-arginine methyltransferase I, the predominant protein-arginine methyltransferase in cells, interacts with and is regulated by interleukin enhancer-binding factor 3. J Biol Chem 275(26):19866–19876
Therrien E et al (2009) 1,2-Diamines as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1). Bioorg Med Chem Lett 19(23):6725–6732
Troffer-Charlier N et al (2007) Functional insights from structures of coactivator-associated arginine methyltransferase 1 domains. EMBO J 26(20):4391–4401
Tryndyak VP, Kovalchuk O, Pogribny IP (2006) Loss of DNA methylation and histone H4 lysine 20 trimethylation in human breast cancer cells is associated with aberrant expression of DNA methyltransferase 1, Suv4-20h2 histone methyltransferase and methyl-binding proteins. Cancer Biol Ther 5(1):65–70
Van Den Broeck A et al (2008) Loss of histone H4K20 trimethylation occurs in preneoplasia and influences prognosis of non-small cell lung cancer. Clin Cancer Res 14(22):7237–7245
van Haaften G et al (2009) Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet 41(5):521–523
van Leeuwen F, Gafken PR, Gottschling DE (2002) Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109(6):745–756
Varambally S et al (2002) The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419(6907):624–629
Vedadi M et al (2011) A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol 7(8):566–574
Wagschal A et al (2008) G9a histone methyltransferase contributes to imprinting in the mouse placenta. Mol Cell Biol 28(3):1104–1113
Wan H et al (2009) Benzo[d]imidazole inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1)–Hit to Lead studies. Bioorg Med Chem Lett 19(17):5063–5066
Wang H et al (2003) mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol Cell 12(2):475–487
Wang GG et al (2007) NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol 9(7):804–812
Wang SZ et al (2008a) Knockdown of SMYD3 by RNA interference inhibits cervical carcinoma cell growth and invasion in vitro. BMB Rep 41(4):294–299
Wang L, Pal S, Sif S (2008b) Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol Cell Biol 28(20):6262–6277
Wang P et al (2009) Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol 29(22):6074–6085
Watanabe H et al (2008) Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int 8:15
Weidinger G (1995) Pharmacokinetic and pharmacodynamic properties and therapeutic use of bunazosin in hypertension. A review. Arzneimittelforschung 45(11):1166–1171
Wigle TJ et al (2010) Screening for inhibitors of low-affinity epigenetic peptide-protein interactions: an AlphaScreen-based assay for antagonists of methyl-lysine binding proteins. J Biomol Screen 15(1):62–71
Wu H et al (2010) Structural biology of human H3K9 methyltransferases. PLoS One 5(1):e8570
Wysocka J et al (2006) A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442(7098):86–90
Xiang Y et al (2007) JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci USA 104(49):19226–19231
Xiao B et al (2005) Specificity and mechanism of the histone methyltransferase Pr-Set7. Genes Dev 19(12):1444–1454
Xin Z et al (2003) Role of histone methyltransferase G9a in CpG methylation of the Prader-Willi syndrome imprinting center. J Biol Chem 278(17):14996–15000
Xu W et al (2004) A methylation-mediator complex in hormone signaling. Genes Dev 18(2):144–156
Xu C et al (2010) Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2). Proc Natl Acad Sci USA 107(45):19266–19271
Yang ZQ et al (2000) Identification of a novel gene, GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer Res 60(17):4735–4739
Yang H et al (2008) Preferential dimethylation of histone H4 lysine 20 by Suv4-20. J Biol Chem 283(18):12085–12092
Yap DB et al (2011) Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117(8):2451–2459
Yuan W et al (2009) Heterogeneous nuclear ribonucleoprotein L Is a subunit of human KMT3a/Set2 complex required for H3 Lys-36 trimethylation activity in vivo. J Biol Chem 284(23):15701–15707
Yue WW et al (2007) Insights into histone code syntax from structural and biochemical studies of CARM1 methyltransferase. EMBO J 26(20):4402–4412
Zhang X, Bruice TC (2007) Histone lysine methyltransferase SET7/9: formation of a water channel precedes each methyl transfer. Biochemistry 46(51):14838–14844
Zhang X, Bruice TC (2008) Enzymatic mechanism and product specificity of SET-domain protein lysine methyltransferases. Proc Natl Acad Sci USA 105(15):5728–5732
Zhang X, Cheng X (2003) Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure 11(5):509–520
Zhang X, Zhou L, Cheng X (2000) Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J 19(14):3509–3519
Zhao Q et al (2009) PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol 16(3):304–311
Acknowledgement
We thank Dr. Robert A. Copeland for helpful discussions and careful reading of the Chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Chesworth, R., Wigle, T.J., Kuntz, K.W., Smith, J.J., Richon, V.M. (2014). Histone Methyltransferases: Opportunities in Cancer Drug Discovery. In: Lübbert, M., Jones, P. (eds) Epigenetic Therapy of Cancer. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-38404-2_9
Download citation
DOI: https://doi.org/10.1007/978-3-642-38404-2_9
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-38403-5
Online ISBN: 978-3-642-38404-2
eBook Packages: MedicineMedicine (R0)