
Abstract
Organ morphogenesis requires coordinated communications between and within different populations of progenitors. Development of the mammalian asymmetric aortic arch artery tree is an excellent model system to study genes and mechanisms orchestrating complex inter-tissue interactions during organ morphogenesis. In this system, the initially symmetric vascular tree connecting the heart to the embryonic circulation undergoes asymmetric remodeling in a highly stereotyped manner. This morphogenetic process is essential for the separation of arterial and venous circulations and requires coordinated communication between cells of mesoderm, endoderm, surface ectoderm, and the neural crest. While a number of key signals have been identified, the means by which these signals are integrated to drive this morphogenetic process is not well understood. One possible means by which signaling by various growth factors can be integrated into precise developmental programs is via the extracellular matrix. This review will examine roles of extracellular matrix proteins in mediating growth factor signaling between neural crest cells and the surrounding tissues during development of the cardiac outflow tract and aortic arch arteries.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
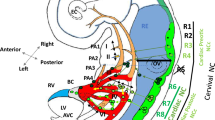
Neural crest (NC) derivatives play a major role in cardiovascular development by modulating morphogenesis of the cardiac outflow tract (OFT) and aortic arch arteries (AAAs) (Fig. 5.1). Proper development of this vascular system is essential for delivering deoxygenated blood to the lungs and oxygenated blood to the rest of the body. Congenital malformations of this system lead to the most common and severe forms of human birth defects, which are often lethal if not corrected by surgery in the first few days of life (Roger et al. 2011). A high proportion of AAA remodeling defects are observed in patients with DiGeorge syndrome, the most common chromosome microdeletion syndrome in humans, in 50 % of patients with Down syndrome and in up to 90 % of patients with heterotaxy (defective left–right patterning) (Moon 2006). Therefore, understanding genes and mechanisms orchestrating morphogenetic processes leading to formation and remodeling of the cardiac OFT and AAAs is essential for gaining insights into etiology, diagnosis, prevention, and treatment of a large proportion of human congenital heart defects.

Cardiac neural crest and morphogenesis of cardiac outflow tract and aortic arch arteries. (a) Left-side view of E9.5-10 mouse embryo. NC-derived cells are shown as blue dots. Bracket marks the position of the CNC in the neural tube. Pharyngeal arch arteries are numbered. dAo dorsal aorta. (b) Ventral view of the symmetric arch arteries at E10.5. (c–d) Ventral views of the remodeled arch arteries in normal configuration and (e–f) in examples of disease configurations. L left; R right; RCA, LCA right and left carotid arteries; RSA, LSA right, left subclavian arteries; PT pulmonary trunk; Ao aorta; da ductus arteriosus. (d) da closes after birth and degenerates, leading to separate aortic and pulmonary circulations. (e) Premature degeneration or defective formation of the left fourth arch artery (orange) leads to interrupted aortic arch type B (IAAB). (f) Degeneration or aberrant formation of the right fourth pharyngeal arch results in the absent RSA; instead, the persistent right segment of the dorsal aorta (grey) forms vascular ring around the trachea, termed RERSA, retroesophageal right subclavian artery
NC is a population of progenitor cells located in the dorsal neural tube of vertebrate embryos. Cardiac neural crest (CNC) resides within a particular region of the neural tube located between the otic pit and the fourth somite (Chan et al. 2004; Leatherbury et al. 1990). Descendants of these cells play pivotal roles in OFT septation resulting in formation of two separate vessels, the aorta and the pulmonary trunk, development of cardiac conduction system, OFT valves, as well as in asymmetric morphogenesis of the AAAs. Mutations in genes compromising CNC development (directly or indirectly) give rise to lethal cardiovascular defects in model vertebrate organisms and in humans (Wurdak et al. 2006) (Fig. 5.1a, b). Development of the asymmetric cardiac outflow vasculature requires coordinated communication between and within different populations of progenitor cells. This review will examine the role of cell–extracellular matrix (ECM) interactions in mediating inter-tissue communications between NC-derived cells and the surrounding tissues during development of the cardiac OFT and AAAs.
Similar to other NC-derived populations, CNC-derived cells encounter changing extracellular microenvironment as they transit from the dorsal neural tube to their final destinations, and their interactions with various tissues are fundamental, not only to the development of the CNC-derived lineages but also to the development of tissues in close proximity to the CNC (Graham et al. 2005; Wurdak et al. 2006). For example, signaling between CNC-derived cells and precardiac mesoderm is essential for addition of the cardiac precursors to the OFT of the heart. Defects in these interactions result in shortened OFT, leading to malrotation and malalignment of major arteries with respect to the left and right ventricles of the heart (Waldo et al. 2005). Similarly, interactions between blood vessel endothelial cells (derived from the mesoderm) within the pharyngeal arches 3, 4, and 6 and the CNC-derived cells are essential for recruitment of the CNC, their differentiation toward arterial vascular smooth muscle cell (VSMC) fate, and remodeling of the initially symmetric pharyngeal arch artery blood vessels into their final asymmetric arrangement (High et al. 2008) (Fig. 5.1b). Signaling between the pharyngeal endoderm, mesoderm, surface ectoderm, and the CNC is essential for navigation, proliferation, survival, and differentiation of the CNC-derived cells, as well as for the downstream morphogenetic events such as OFT rotation, septation, and asymmetric remodeling of the pharyngeal arch arteries (Fig. 5.3).
2 Extracellular Microenvironment Regulates Migration and Navigation of CNC-Derived Cells
How interactions between transiting NC-derived cells and surrounding tissue microenvironment mediate cardiovascular morphogenesis is not well understood. There is evidence that short- and long-range signaling interactions between all embryonic germ layers are important for NC migration, navigation, cell fate specification, and cardiovascular morphogenesis (Calmont et al. 2009; Goddeeris et al. 2007; Graham et al. 2005; Kirby and Hutson 2010; Liu et al. 2004; Macatee et al. 2003; Park et al. 2008; Rentschler et al. 2010; Stuhlmiller and Garcia-Castro 2012; Urness et al. 2011) (Fig. 5.2). The extracellular milieu, which includes large ECM glycoproteins, proteoglycans, and their cellular receptors, plays essential roles in modulating interactions between the CNC and components of the pharyngeal microenvironment.
2.1 Migration of NC Cells Toward Their Target Destinations
Genes and mechanisms regulating routing of NC-derived cells toward their eventual destinations are not well understood. A process termed contact inhibition of locomotion (CIL) has been hypothesized to facilitate directional migration of cell cohorts during embryogenesis (Rovasio et al. 1983). In the case of the NC, CIL involves transient homotypic interactions between NC-derived cells, preventing cells at the front from moving back, while heterotypic interactions between NC-derived cells and cells of other tissues do not have the same effect and allow NC cell invasion. Noncanonical Wnt signaling and activation of Rho GTPases play essential roles in this process by limiting polarized lamellipodial protrusions to the leading edge of NC cells traveling in front of the group (Carmona-Fontaine et al. 2008). In addition to CIL, NC cell co-attraction mediates migration of NC cells in cohorts. In fish and frog, this process is mediated by the complement fragment C3a and the C3a receptor, both of which are produced by NC-derived cells and are required for cohesive migration of NC streams (Carmona-Fontaine et al. 2011). Thus, NC cell homotypic CIL and co-attraction cooperate to promote directional movement of swarms of NC-derived cells toward their target destinations.
Recent experiments in chick and genetic manipulations in mouse indicate the existence of cues playing fine-tuning roles in migration and navigation of NC-derived cells (Erickson et al. 1980; Gammill and Roffers-Agarwal 2010; Kulesa and Gammill 2010). In vitro studies suggest that locomotion of NC cells depends on composition of the ECM and the complement of ECM receptors expressed by NC-derived cells. For example, compared with CNC, cephalic NC cells isolated from the neural tube regions anterior to the otic pit exhibit different migratory properties when presented with increasing levels of FN. Migration speed and persistence of cephalic NC cells do not change when these cells are plated on a wide range of FN concentrations. However, increasing concentrations of FN lead to decreased speed and increased migrational persistence of CNC cells (Xu et al. 2006). Migrational persistence is defined as a length of time traveled in a straight line.
One reason for different migratory responses of NC cells originating at different rostrocaudal levels of the neural tube could be due to a varied complement of integrins, a major class of cell surface ECM receptors (Delannet et al. 1994; Haack and Hynes 2001; Hynes 2002; Testaz et al. 1999). Cranial NC cells migrate faster on laminin1 [LM-111 according to new nomenclature (Aumailley et al. 2005)] relative to trunk NC cells. This has been attributed to a fast recycling rate of integrin α6 in these cells (Strachan and Condic 2004). Trunk NC cells migrate 2–3 times faster on FN than on LM-111 or collagen I, on which they exhibit a more persistent migration (Rovasio et al. 1983). Trunk NC cells preferentially adhere and migrate on FN when given a choice between FN, collagen I, LM-111, or glass. Although when these cells are cultured on LM-111 for more than 24 h, their adhesion to LM-111 improves, suggesting that NC-derived cells can adjust their properties depending on the type of ECM substrate they encounter (Rovasio et al. 1983).
Navigation of NC-derived cells in the embryo may be modulated by a combination of attractive and repulsive cues. For example, attractive properties of fibronectin (FN) can be modulated by versican and type 6 semaphorins, Sema6A and Sema6B, all of which act as NC cell repellents (Dutt et al. 2006; Testaz et al. 2001; Toyofuku et al. 2008). Different classes of semaphorins expressed along the dorsoventral axis further modulate the routing of NC cells toward their destinations (Gammill and Roffers-Agarwal 2010; Kulesa and Gammill 2010; Toyofuku et al. 2008). For example, NC cells transiting through somites migrate through the anterior of somite’s sclerotome. This pattern is determined by neuropilin (Nrp) 2 expressed both by NC cells in the anterior part of each somite and by semaphorin (Sema) 3F expressed in somites’ posterior, repelling NC-derived cells from this region. Global genetic ablation of either Nrp2 or Sema3F allows NC cells to traverse through the entire width of a somite, without disturbing somite’s anterior–posterior polarity itself. Interestingly, wild-type NC cells avoid Sema3F+ territories, while Nrp2-deficient NC cells are not repelled by Sema3F (Gammill et al. 2006).
2.2 Extracellular Microenvironment and Differentiation of NC-Derived Cells
NC progenitor cell fate can be changed by surgical transplantation from one position to another along the neural tube (Trainor et al. 2002). Such transplantation experiments indicated that NC progenitor cell fate is plastic and can be determined by short- and long-range signals emanating from tissue microenvironment (Le Douarin and Kalcheim 1999). In addition to modifying NC migration, various components of ECM play distinct roles in modulating differentiation of NC-derived cells. For example, plating NC cells from either cephalic or trunk regions on FN promotes their differentiation toward smooth muscle cell lineage, while plating on LM-111 or collagen IV does not (Costa-Silva et al. 2009). These experiments suggest that varying levels and distributions of ECM molecules in vivo modulate migration and differentiation of NC-derived cells.
3 Examples of ECM Macromolecules Modulating Neural Crest Development In Vivo
ECM proteins modulate a multitude of signaling pathways involved in cell survival, proliferation, migration, and differentiation and are prominently expressed within embryonic tissues (Giancotti and Tarone 2003). Intriguingly, there is a large body of evidence showing that ECM proteins are synthesized and distributed in nonuniform patterns within embryos (e.g., Fig. 5.3). For example, immunofluorescence staining experiments showed that vitronectin, collagen, laminin, fibulin1, perlecan, and FN proteins are highly abundant in some and greatly underrepresented in other sites of the embryo and genetic ablation of fibulin1, α5 chain-containing laminins, perlecan, or FN indicated that ECM plays essential and distinct roles in NC (and CNC) development in vivo (Coles et al. 2006; Cooley et al. 2008; Copp et al. 2011; Costell et al. 2002; Delannet et al. 1994; Duband and Thiery 1987; Gammill et al. 2007; Mittal et al. 2010; Peters and Hynes 1996). Intriguingly, expression studies of genes encoding ECM macromolecules indicate that ECM mRNAs and proteins are dynamically regulated during embryogenesis and are expressed and/or accumulate as “hot spots” at specific embryonic sites implying that ECM proteins play tissue-specific roles during embryonic development (Coles et al. 2006; Cooley et al. 2008; Copp et al. 2011; Mittal et al. 2010) (Fig. 5.3).

Extracellular matrix proteins are expressed in nonuniform patterns during embryogenesis. (a, c, d, h) E9.5 right side views. (a) Laminin alpha 5 (LAMA5) mRNA. (b) E8.5. Transverse section. LAMA5 protein (green) (Coles et al. 2006). (c) Fibulin1 mRNA. (d) Fibulin1 protein. Sagittal section (Cooley et al. 2008). (e–j) Expression of FN mRNA and protein. (e) FN1 mRNA is expressed at the edges of the dorsal neural tube in 8–10 somite embryos (black arrows). Dotted line is an approximate plane of section shown in (f). (f) FN mRNA is expressed by the dorsal neural ectoderm (long arrow in the inset) and the surface, nonneural ectoderm (arrowhead in the inset). Asterisk marks FN1 mRNA in pharyngeal pocket endoderm and short arrow points at pharyngeal ectoderm. (g) Transverse section through a region containing CNC cells in a 10-somite wild-type embryo. FN protein (green) is present between TFAP2α+ neural (arrow, inset) and surface (arrowhead, inset) ectoderm. (h) FN1 mRNA expression is maintained at E9.5 in the dorsal neural tube (black arrow) and within the region of branchial arches 3, 4, and 6 (bracket). FN1 mRNA is also expressed by microvascular endothelial cells in the head (open arrow). (i) Sagittal section through E9.5 embryo showing expression of FN protein. (j) FN protein is enriched in branchial arches 3 and 4, compared with arches 1 and 2 (Mittal et al. 2010). (Printed with permission from Developmental Biology)
3.1 Laminin α5
Laminins are extracellular protein heterotrimers composed of α, β, and γ chains. The mRNA and protein of laminin α5 chain, a component of laminins 511, 521, and 523 (Aumailley et al. 2005) are distributed nonuniformly in the mouse embryo (Copp et al. 2011). At the early times of NC development, at embryonic day (E) E8.5, LAMA5 mRNA is expressed by cells in the upper third of the dorsal neural tube, in endoderm, surface ectoderm including that of pharyngeal arches, and, weakly, in the pharyngeal arch mesenchyme (Coles et al. 2006). By E9.5, LAMA5 mRNA becomes downregulated in the neural tube anterior to the forelimb bud, while it remains expressed in the posterior neural tube, pharyngeal arches, and the endoderm (Copp et al. 2011). However, laminin α5 protein is enriched at the basal sides of foregut endoderm, surface ectoderm, and the neural tube. This pattern is distinct from the pattern of LAMA5 mRNA (Coles et al. 2006; Copp et al. 2011) (Fig. 5.3a, b). The presence of laminin α5 protein in the basement membrane surrounding the neural tube suggests that laminin α5 protein diffuses away from the sites of its synthesis and that its enrichment at certain embryonic sites could be determined by expression patterns of cognate laminin receptors. Genetic ablation of LAMA5 leads to widening and fusion of NC cell cohorts, reduction in the size, and a delay in the formation of initial ganglionic condensations during early neurogenesis between E8.5 and E10.5, but remarkably gangliogenesis largely recovers by E11. These observations (supported by in vitro data) suggest that α5 chain-containing laminins play restrictive role(s) in NC migration by facilitating proper routing of NC-derived cells to their destinations (Coles et al. 2006). The authors of this paper also suggest an intriguing idea that the initial delay followed by recovery of gangliogenesis at a later developmental time in LAMA5 mutants implies a role for LAMA5 in cessation of NC cell migration upon arrival at their destinations (Coles et al. 2006).
3.2 Fibulin1
The mRNA of the ECM protein fibulin1 is highly expressed at the dorsal neural tube, somitic and pre-somitic mesoderm, and in mesoderm containing presumptive cardiac progenitors at E8.5 (Cooley et al. 2008). At E9.5, fibulin1 mRNA is downregulated from the trunk neural tube anterior to the forelimb bud, while it remains expressed in all pharyngeal arches. Interestingly, the localization of fibulin1 protein does not match that of its mRNA at this time: fibulin1 protein is highly enriched in the embryo mesenchyme lateral to the neural tube and dorsal to the foregut, while it is virtually excluded from the pharyngeal arch mesenchyme, containing Pax3+ cells (NC cells); in addition, fibulin1 is highly enriched within the cardiac OFT (Cooley et al. 2008) (Fig. 5.3c, d). Global deletion of fibulin1 leads to defects in cranial NC-derived structures such as facial skeleton, CNC-related defects in thymus, and aortic arch artery development and in the development of cranial nerves. Elevated apoptosis in NC-rich areas suggests that fibulin1 is important for NC cell survival. Fusion of cranial NC streams in fibulin1 mutants suggests that fibulin1 is important for the establishment and/or maintenance of proper routes or boundaries of NC paths and implies that fibulin1 acts as a repellent for NC cells contributing to cranial nerves IX and X (Cooley et al. 2008).
3.3 Fibronectin
FN gene, multipotent neural crest, and a closed, endothelial-lined vasculature are vertebrate innovations (Hynes 2012). Investigation into the fate of the CNC in global FN-null mouse mutants demonstrated that FN is not required for specification of the CNC precursors or for their exit from the dorsal neural tube. In global FN-null mutants, CNC cells reach their distal destinations in the pharyngeal arches and the cardiac OFT at appropriate developmental times and in grossly normal patterns, indicating that FN is not required for CNC migration. Instead, FN is required to maintain normal CNC cell numbers, by promoting CNC survival and proliferation (Mittal et al. 2010).
FN is alternatively spliced at EIIIA, EIIIB, and V regions, and there is evidence that alternative splice variants of FN containing EIIIA and/or EIIIB exons are also involved in modulating CNC development. (Astrof et al. 2007; Astrof and Hynes 2009; Peters and Hynes 1996). The sequences of EIIIA and EIIIB are virtually 100 % conserved among vertebrates, while the sequence similarity between EIIIA and EIIIB is low. Alternative splicing of these exons is dynamically regulated such that they are nearly always included into FN protein around newly developing blood vessels during embryogenesis or in pathologies involving new blood vessel growth, while their expression is undetectable around mature, quiescent vasculature in an adult (Astrof and Hynes 2009; Ffrench-Constant and Hynes 1989; Ffrench-Constant et al. 1989; Peters et al. 1996). Concurrent deletion of EIIIA and EIIIB exons from the FN gene leads to embryonic lethality depending on the genetic background (Astrof et al. 2007). 80 % of EIIIA/EIIIB-double nulls die by E10.5 and exhibit severe cardiovascular defects on mixed 129/C57BL/6J genetic background. Of note are the small size of pharyngeal arches, dilated arch arteries, and paucity of mesenchymal cells in the cardiac OFT, suggesting that EIIIA and/or EIIIB isoforms of FN play important role(s) in the development of the CNC (Astrof et al. 2007). Further studies are needed to explore potential roles of these splice variants in CNC development and in morphogenesis of the cardiac OFT and AAAs.
3.4 FN mRNA Is Dynamically Regulated During Embryogenesis
Detailed knowledge of cellular source(s) of ECM proteins and the identity of their target cell populations is important for understanding the role of ECM during organ morphogenesis. Examination of FN mRNA and protein expression at different stages of embryogenesis indicated that the expression of this gene is dynamically regulated in time and space (Mittal et al. 2010; Pulina et al. 2011). For example, by the late headfold stage, FN protein surrounds the ventral node, and absence of FN protein in global FN-null embryos leads to defective intercalation of cells within the node leading to piling of nodal cells on top of each other and defective morphogenesis of the node (Pulina et al. 2011). This leads to aberrant specification of the left–right asymmetric body plan, which in turn is required for the proper asymmetric development of the cardiovascular system (Ramsdell 2005). At E8.0, FN mRNA is highly expressed by the lateral plate mesoderm, by anterior definitive endoderm, and by endothelial cells of the nascent heart tube. At a slightly later time point, in embryos containing 8–10 somites, FN mRNA is upregulated in the NC as well as in the pharyngeal pocket endoderm, in pharyngeal surface ectoderm, and in splanchnic mesoderm, containing presumptive myocardial progenitors (Mittal et al. 2010).
At E9.5, FN mRNA is maintained in the dorsal neural tube and is present in the newly emigrating NC cells (Mittal et al. 2010). At this time, FN is also expressed at distinct locations within pharyngeal epithelia: pharyngeal pocket endoderm and surface ectoderm (Figs. 5.2f and 5.3a) and in the pharyngeal mesoderm. Accordingly, we found that FN protein is enriched in specific embryonic locations, which correlate with the “hot spots” of FN mRNA (Figs. 5.3g, i, j, and 5.2a). Pharyngeal endoderm, ectoderm, and mesoderm comprise pharyngeal microenvironment regulating CNC cell proliferation, survival, differentiation, and remodeling of the AAAs and the cardiac OFT. The presence of FN in this area suggests that this protein could play an important role in intercellular and inter-tissue interactions regulating development of organs containing NC-derived cells.

Signaling within pharyngeal microenvironment. (a) Tissue-specific enrichment of FN protein in the pharyngeal region. A schematic cross section through E9.5 embryo. NT neural tube, CV cardinal vein, dA dorsal aorta, PAA pharyngeal arch artery, AS aortic sac, OFT outflow tract. Green marks areas enriched in FN protein. Neural crest-derived cells are red ovals. Dorsal is at the top; ventral is at the bottom. (b) Enlarged pharyngeal region shown in (a) depicting examples of signaling pathways known to orchestrate signaling between endoderm, mesoderm, surface ectoderm, and the CNC during cardiovascular morphogenesis. FN protein is enriched at strategic positions and is predicted to modulate some of the depicted intercellular signaling pathways
Our mRNA expression studies indicated that FN is expressed by populations of progenitors known to give rise to all components of the heart and the cardiac OFT vasculature: CNC, precardiac mesoderm, endothelium, and epicardium. Being highly conserved among vertebrates, the distinct spatiotemporal distribution of FN mRNA and protein implies that tissue-specific and timed synthesis of FN is biologically important. What is the significance of tissue-specific expression of FN (or other ECM macromolecules)? And is there a difference in intracellular signaling responses to paracrine and autocrine sources of FN? Recent studies indicated that tissue-specific synthesis of FN by breast epithelial cells or by retinal astrocytes plays important roles in morphogenesis of these organs. FN produced by breast epithelial cells is important for their proliferation and lobuloalveolar development (Liu et al. 2010). FN produced by retinal astrocytes is required for directional, VEGF-dependent migration of blood vessels during retinal angiogenesis (Stenzel et al. 2011b). We also have evidence that tissue-specific synthesis of FN by pharyngeal endoderm, mesoderm, or neural ectoderm in the embryo plays essential roles in morphogenesis of the cardiovascular system (S. Astrof, unpublished). Because FN is essential for cardiovascular development and because cell adhesion to FN is known to modulate a multitude of intracellular signaling pathways in vitro, investigation of tissue-specific autocrine and paracrine functions of FN would provide invaluable insights into mechanisms of intercellular communications essential for cardiovascular morphogenesis.
In summary, nonuniform expression patterns of ECM proteins suggest that NC-derived cells traversing the embryo encounter nonuniform and dynamic tissue microenvironment, containing different gradients and assortments of ECM proteins in time and space. Cell adhesion to different ECM proteins elicits diverse intracellular signaling responses based on the presence of other signaling molecules and on mechanical properties of tissues (Engler et al. 2006; Giancotti and Tarone 2003; Hynes 2009). Studies cited above support the notion that interactions between the NC-derived cells and the ECM modulate NC cell fate and morphogenesis of tissues containing, derived from, or influenced by the transiting NC cells.
4 Integrins Are Important for the Development of the Cardiac Neural Crest
Integrins are a major class of cell surface receptors that bind ECM and activate ECM-dependent intracellular signaling cascades regulating cell adhesion, migration, survival, proliferation, self-renewal, and differentiation in a context-dependent manner. There are 18 alpha and 8 beta subunits in mammals; they are known to comprise 24 distinct integrin heterodimers. Integrin beta 1 chain can combine with 12 distinct alpha chains forming 12 different integrin heterodimers with diverse ligand specificities (Hynes 2002). Thus genetic deletion of integrin beta 1 leads to the ablation of 12 distinct integrins, depleting cells from receptors with binding specificities for a wide range of ECM proteins, including laminins, collagens, FN, and other RGD-containing constituents of ECM.
Genetic experiments in mice indicate that integrins play important roles in NC development (Breau et al. 2006) and the CNC in particular. Ablation of integrin beta 1 using Wnt1-Cre transgenic mice leads to embryonic lethality by E12.5 (Turlo et al. 2012). Unexpectedly, CNC migration does not depend on β1-containing integrins. Instead, beta 1-deficient CNC cells cannot support proper development of the pharyngeal arch arteries, and these mutants die due to severe dilation and hemorrhage of these vessels (Turlo et al. 2012). Consistent with this finding, prior experiments in the mouse and chick showed that integrin heterodimers containing alpha 5 or integrin alpha 4 subunits (forming α5β1, α4β1, and α4β7 integrins) were also not required for NC migration (Haack and Hynes 2001). Global ablation of integrin alpha 5 indicated that this integrin was important for proliferation and survival of the CNC (Mittal et al. 2010). Taken together these studies indicate that individual integrin heterodimers or a combination of 12 β1-containing integrins are not required to mediate NC migration.
Cell–ECM interactions mediated by integrins engage a number of downstream signaling cascades, and NC-specific mutations in genes encoding downstream effectors of integrin activation, such as Rac1, Cdc42, and FAK, give rise to defective development of the cardiac OFT and reduced differentiation of CNC-derived cells around pharyngeal arch arteries toward VSMC cell fate leading to aberrant remodeling of the pharyngeal vascular tree (Fuchs et al. 2009; Thomas et al. 2010; Vallejo-Illarramendi et al. 2009).
However, mechanisms by which integrins and their downstream effectors mediate CNC-dependent cardiovascular development are not well understood.
ECM proteins are thought to function as platforms for integrating signaling by diverse components of extracellular milieu and orchestrating intercellular interactions during tissue morphogenesis (Hynes 2009). Analogously, we propose that integrin-mediated cell–ECM interactions in the pharynx integrate combinatorial inputs from a variety of growth factor signaling pathways to orchestrate the complex process of AAA morphogenesis (Fig. 5.2). In the section below, we provide examples of how this integration could take place.
5 Roles of Cell–ECM Interactions in Modulating Signaling Pathways During CNC Development and Cardiovascular Morphogenesis
5.1 Fibroblast Growth Factor Family
Signaling by fibroblast growth factors (Fgf) Fgf3, Fgf8, Fgf10, and Fgf15 individually and/or in combination is intimately involved in morphogenesis of the cardiovascular outflow system. In vitro studies demonstrated that cell adhesion to FN facilitates Fgf signaling, and it is possible that cell–ECM interactions modulate tissue-specific responses to Fgf in vivo as well (Miyamoto et al. 1996). Fgfs including Fgf8 and other growth factors bind FN (and probably other ECM components), and Fgf8 was shown to function as a morphogen in vivo (Martino and Hubbell 2010; Toyoda et al. 2010). Therefore, FN and probably other ECM macromolecules could facilitate cellular responses to Fgf8 and other growth factors by modulating their spatial distribution and bioavailability. Understanding how and which components of the ECM are important for Fgf8 signaling in vivo is important because Fgf8 plays a requisite (albeit an indirect) role in CNC development and regulates morphogenesis of the cardiac outflow system. Deregulated signaling by Fgf8 is implicated in the pathogenesis of human DiGeorge syndrome, the most common chromosomal microdeletion syndrome in humans (Moon 2006).
While Fgfs signal to CNC cells, Fgf signaling to the CNC is not required for OFT septation and pharyngeal arch artery remodeling (Park et al. 2006, 2008; Zhang et al. 2008). Instead, Fgf signaling to the pharyngeal surface ectoderm, to pharyngeal endoderm, and to cardiac progenitors in the splanchnic mesoderm is required indirectly, in part by regulating BMP signaling, to modulate CNC cell fate, number, proliferation, survival, and activation of MAPK signaling pathway. Consequently, tissue-specific autocrine Fgf8 signaling to surface ectoderm is required for the formation of the fourth pharyngeal arch arteries, and autocrine signaling to precardiac mesoderm and pharyngeal endoderm mediates rotation and septation of the cardiac OFT. Interestingly, in addition to regulating gene expression of transforming growth factor family members, Fgf8 signaling regulates expression of semaphorins and plexins. Thus defective septation of the cardiac OFT in the conditional Fgf8;Isl1Cre/+ mutants may be due in part to defective navigation of the CNC cells (Cai et al. 2008; Macatee et al. 2003; Moon 2006; Park et al. 2006, 2008; Urness et al. 2011; Vincentz et al. 2005).
The precise complement of intracellular components mediating Fgf signaling is not fully understood. Similar to other RTKs, cell adhesion to FN leads to Fgf receptor (FGFR) phosphorylation in a ligand-independent manner (Zou et al. 2012). Downstream signaling mediated by FN via integrins and by Fgfs via FGFRs may involve common intracellular mediators activated by both pathways, including FAK, src family kinases, src substrate, p130Cas, and Crkl (Li et al. 2003; Zou et al. 2012). In vivo signaling by Fgf8 is mediated in part by phosphorylation of FRS2α and Crkl (Moon et al. 2006; Zhang et al. 2008). Fgf-mediated phosphorylation of Crkl and Erk1/2 in vivo is dependent on FAK, a well-known downstream effector of integrin signaling (Vallejo-Illarramendi et al. 2009). Global deletion of Crkl leads to similar (albeit milder) phenotypes seen in hypomorphic Fgf8 mutants (Moon et al. 2006). Upon integrin-mediated cell–ECM adhesion, Crkl associates with p130Cas and becomes recruited to focal adhesions in integrin-, src-, and p130Cas-dependent manner (Li et al. 2003). In this light, it would be interesting to investigate the detailed cardiovascular abnormalities in global p130Cas mutants, which are reported to die at about E11.5–E12.5 from severe cardiac and vascular defects (Honda et al. 1998). Crkl can also facilitate Fgf8 signaling in adhesion-independent manner, by binding directly to the phosphorylated Tyr463 within the YELP motif of FGFR1 upon cell stimulation with Fgf8. This binding leads to activation of Rac1, Cdc42, and PAK in DOCK180-dependent manner and allows full activation of Erk1/2, Raf1, and MEK1 (Seo et al. 2009). The importance of these studies is underscored by the finding that Crkl is required for NC survival in vivo (Moon et al. 2006). Significantly, Crkl is located within the 22q11 region commonly deleted in human DiGeorge patients (Moon et al. 2006). Therefore, mutations in Fgf8, Crkl, and p130Cas and in other mediators of Fgf8 signaling may contribute to the severity and variability of symptoms observed in these patients. As mentioned above, ECM may facilitate Fgf signaling by binding to and localizing Fgf ligands at specific embryonic locations. In addition, syndecans serve as common co-receptors for FN-mediated integrin signaling and Fgf-mediated FGFR signaling (Arrington and Yost 2009; Horowitz et al. 2002; Saoncella et al. 1999), suggesting the existence of common FN–integrin–syndecan–Fgf–FgfR signaling complexes in cells. Taken together, there exists compelling evidence for the crosstalk and possible synergy between FN-, integrin-, and Fgf-mediated signaling pathways. Future studies investigating details of these interactions would provide important insights into mechanisms underlying defects in cardiovascular morphogenesis in human patients with congenital heart defects.
5.2 Platelet-Derived Growth Factor Family
Signaling by platelet-derived growth factor (PDGF) ligands is mediated by homo- or heterodimers of PDGF receptors α and β. Concurrent deletion of both PDGFRs from the NC is early neonatal lethal due to cardiovascular insufficiency and results in 100 % penetrant defects in thymic development, ventricular septation, cardiac OFT septation, and premature regression of the right fourth pharyngeal arch artery, leading to the presence of vascular rings around trachea (Richarte et al. 2007). These abnormalities are classically attributed to defective development of the CNC. Formation, delamination, migration, proliferation, and survival of CNC cells were not significantly affected in NC-specific PDGFRα/β-double null mutants. Interestingly, there were fewer CNC cells present within the cardiac OFT implying a role for CNC-specific PDGF signaling in the navigation of CNC cells into the heart (Richarte et al. 2007). Usually, defective persistence and remodeling of pharyngeal arch arteries is associated with low numbers or defective differentiation of CNC cells into αSMA+ VSMCs. However, normal numbers of αSMA+ cells were present around the AAAs between E11.5–E13.5, the window of time during which the symmetric pharyngeal arch artery vascular tree attains its mature asymmetric configuration (Fig. 5.1b). Expression of PDGFR ligands by the pharyngeal epithelia (PDGF-AA) and endothelium (PDGF-BB) suggests that signaling interactions between pharyngeal microenvironment and CNC play requisite roles in asymmetric remodeling of pharyngeal arch arteries even in the presence of normal numbers and differentiation of CNC-derived VSMCs (Richarte et al. 2007).
Seminal studies in the 1990s demonstrated that cell adhesion, and in particular, cell adhesion to FN mediated by integrin α5β1, is required for RTK activation and signaling downstream of EGF, PDGF, and FGF receptors (Miyamoto et al. 1996). Recent studies indicate that cell adhesion leads to pre-activation of RTKs in a ligand-independent manner and potentiates growth factor signaling (Giancotti and Tarone 2003). In case of PDGF signaling, cell adhesion to FN (but not to collagen I, IV, or LM-111) leads to a ligand-independent activation of PDGFRs, mediated by α5β1 and/or by αv-containing integrins (Veevers-Lowe et al. 2011). Extracellular domains of PDGFRs and integrin α5β1 interact by virtue of their binding to heparin sulfate (FN is heparinated) and co-localize in lamellipodia of migrating cells. FN and PDGF-BB additively activate PDGFRβ signaling, and PDGF-BB can directly bind to the extracellular domain of integrin α5β1 (Veevers-Lowe et al. 2011). Cell adhesion to FN enhances PDGF-BB-mediated cell migration in integrin α5β1-dependent manner. FAK is required for FN- and integrin α5β1-mediated phosphorylation of PDGFRβ and for the subsequent phosphorylation and activation of Akt. Taken together, these experiments suggest that cell–FN adhesion regulates PDGF signaling and directional migration by a combination of at least two modes: (1) by facilitating assembly of integrin–FN–growth factor–RTK signaling complexes and (2) by activating integrin signaling (Veevers-Lowe et al. 2011).
5.3 Vascular Endothelial Growth Factor Family
Vascular endothelial growth factor (VEGF) family consists of VEGF-A, VEGF-B, VEGF-C, and VEGF-D members, which have different roles in vascular and lymphatic development in normal and pathological conditions (Lohela et al. 2009). VEGF-A growth factors exist in several isoforms arising from alternative splicing of a single gene. VEGF-A isoforms of increasing lengths have increased capacity for binding ECM macromolecules, including FN, and these interactions potentiate VEGF-A signaling to cells (Wijelath et al. 2002). The shortest VEGF-A isoform, VEGF-A120, does not bind ECM. Individual VEGFs play integral roles in vascular development, and global VEGF-A haploinsufficiency in VEGF+/− mutants leads to early embryonic lethality due to severe vascular insufficiency (Carmeliet et al. 1996). Soluble and matrix-bound VEGF-A isoforms play distinct roles in cardiovascular development (Stalmans et al. 2003; van den Akker et al. 2007). VEGF-A164 isoform has intermediate ECM-binding capacity compared with VEGF-A120 and VEGF-A188 and is the only VEGF-A isoform that can bind Nrp1 (Lohela et al. 2009). Mice lacking VEGF-A164 isoform (e.g., mutants expressing either VEGF-A120 or VEGF-A188 isoforms) develop a range of facial, thymic, and cardiovascular abnormalities similar to those observed in patients with DiGeorge syndrome, and mutations in the promoter of human VEGF-A are associated with increased severity in cardiovascular defects of DiGeorge patients (Stalmans et al. 2003). Cardiovascular abnormalities in mice lacking VEGF-A164 include defective septation of the cardiac OFT and aberrant/random remodeling of AAAs. Interestingly, VEGF-A mRNA is highly expressed in the pharyngeal pocket endoderm, while its co-receptor Nrp1 is expressed in the pharyngeal endoderm, in arch core mesoderm, and in NC-derived cells; Nrp1 mutants also exhibit defective septation of the cardiac OFT and aberrant remodeling of the AAAs (Kawasaki et al. 1999). These observations suggest that signaling by VEGF-A within the pharyngeal microenvironment and/or to the CNC is important for CNC development and vascular morphogenesis. Signaling by VEGF-A164 regulates transcription of Tbx1, a gene located within human 22q11 interval and often mutated in DiGeorge patients; VEGF-A and Tbx1 genetically interact to mediate the proper development of pharyngeal arch arteries (Stalmans et al. 2003). Taken together, these studies support the notion that VEGF-A164-Nrp1-Tbx1 axis indirectly regulates CNC development, vascular patterning, and penetrance of cardiovascular congenital phenotypes in human patients (Calmont et al. 2009; Guris et al. 2006).
Recent studies indicate that cell adhesion to ECM via β1-containing integrins is required for differential signaling activities elicited by soluble and matrix-bound VEGF-A164 isoforms. Matrix-bound but not soluble VEGF promotes integrin β1–VEGFR2 association and trafficking to focal adhesions. This association is required for the prolonged (~30 min) phosphorylation of specific VEGFR2 residues induced by the matrix-bound (but not soluble) VEGF and activation of P38 downstream of VEGFR2. In contrast, VEGFR2 phosphorylation by soluble VEGF is downregulated within 5 min of stimulation and does not lead to activation of P38 (Chen et al. 2010). Taken together, these studies suggest that cooperation between matrix-bound VEGF and signaling pathways engaged by cell–ECM interactions facilitate VEGF-mediated septation of the cardiac OFT and remodeling of the pharyngeal arch arteries.
5.4 Sonic Hedgehog
Sonic hedgehog (Shh) signaling to the CNC is tightly regulated. Either increased or reduced Shh signaling to the CNC-derived cells leads to defective cardiovascular development (Goddeeris et al. 2007). Shh produced by the pharyngeal endoderm is required for CNC survival and consequently for the aorticopulmonary septation and arch artery remodeling (Goddeeris et al. 2007). Autocrine Shh signaling to the endoderm is also important for these processes, while paracrine Shh signaling to mesodermal precursors of the cardiac OFT is important for the OFT septation and arch artery remodeling. These studies indicate that autocrine signaling by Shh to the pharyngeal endoderm and paracrine signaling to early cardiac precursors lead to production of a secreted factor(s), modulating CNC cell fate and vascular remodeling; however, mechanisms of this regulation are not well understood (Goddeeris et al. 2007).
How cell–ECM interactions regulate Shh-mediated signaling and cardiovascular development within the pharyngeal microenvironment is not well understood; however, they are probably important since cell–ECM communications are known to modulate Shh activity (and vice versa) in other biological contexts. For example, conditional ablation of β1-containing integrins in intestinal epithelial cells leads to downregulation of Shh, Ihh, and FoxA2. Conversely, ectopic expression of integrin β1 leads to increased expression of Shh and FoxA2 in a manner dependent on activation of Akt, one of the downstream integrin effectors (Jones et al. 2006).
Extracellular ligands, mechanical forces, and intracellular factors can activate integrin signaling by modulating integrin’s three-dimensional conformation (Astrof et al. 2006; Hynes 2002). Shh negatively modulates NC cell adhesion and migration by inhibiting conformational changes that lead to integrin β1 activation but not its cell surface expression (Fournier-Thibault et al. 2009; Testaz et al. 2001). This inhibition is mediated by solid phase rather than soluble Shh, and remarkably, it is independent of Shh-mediated gene transcription, translation, cell fate specification, or signaling involving the Patched-Smoothened-Gli pathway (Fournier-Thibault et al. 2009; Jarov et al. 2003). These experiments show that in addition to and independent of its role in patterning the neural tube (NT), Shh modulates the balance between cell–cell and cell–ECM adhesion by modulating integrin activation in NC progenitors in vivo (Fournier-Thibault et al. 2009). Ectopic expression of Shh in the NT abrogates activation of integrin β1, attenuating cell–ECM interactions necessary for the detachment of the NC cells from the NT, and promotes cell–cell adhesion by activating Cad6B, Daam1, and RhoB. These changes in cell–cell and cell–ECM interactions occur much earlier than the expected alterations in dorsal–ventral NT cell fates induced by ectopic Shh. The inhibitory role of Shh in integrin activation and NC cell adhesion and migration can be counteracted by agents that artificially activate integrins, such as Mn2+ (Fournier-Thibault et al. 2009) but not by cyclopamine or forskolin, known inhibitors of canonic Shh signaling. Reducing the dose of Shh in vivo by sequestration of Shh leads to excessive delamination of NT cells (Testaz et al. 2001). Interestingly, the inability of CNC to regenerate following ablation of the CNC progenitor territory in the dorsal NT in the chick has been linked to excessive Shh signaling (Hutson et al. 2009). Therefore, it would be interesting to determine whether CNC regeneration in this model could be induced by integrin-activating agents such as Mn2+. The role of a potential cross talk between Shh and cell–ECM signaling pathways within the pharyngeal microenvironment in CNC development, OFT septation, and arch artery remodeling remains to be investigated.
5.5 Wnt
Wnt signaling to the NC is important for the maintenance/abundance of NC progenitors as well as specification/balance of NC progenitor cell fates (Hari et al. 2002, 2012; Ikeya et al. 1997). Wnt signaling regulates and is regulated by cell–ECM interactions (De Langhe et al. 2005; Rallis et al. 2010); however, the role(s) of cell–ECM interactions and Wnt signaling during cardiovascular morphogenesis are not well understood.
5.6 Notch
Notch signaling to the CNC and to cardiac progenitors of the OFT myocardium is important for regulating OFT septation and vascular remodeling in the pharynx and has been the subject of several recent reviews (Jain et al. 2010; Rentschler et al. 2010). Recent in vivo studies indicate that cell–ECM interactions modulate Notch signaling during embryogenesis. For example, α4 chain-containing laminins facilitate blood vessel morphogenesis by regulating Notch signaling through integrin β1-mediated upregulation of VEGFR2 and Dll4 mRNA (Stenzel et al. 2011a). Signaling by β1-containing integrins during somitogenesis is important for activation of Notch signaling in a cell-autonomous manner by activation of integrin-linked kinase (ILK) (Rallis et al. 2010). It will be interesting to investigate whether cell–ECM interactions modulate Notch signaling during OFT septation and arch artery remodeling.
5.7 Transforming Growth Factor Beta Family
Signaling by transforming growth factor beta (TGFβ) family members to the CNC is required for the OFT septation and pharyngeal arch artery remodeling, in part by regulating differentiation of CNC cells into VSMCs and expression of αSMA (Kaartinen et al. 2004; Stottmann et al. 2004; Stottmann and Klingensmith 2011; Wurdak et al. 2006). Numerous BMP and TGFβ ligands and their antagonists orchestrate development of the CNC and CNC-dependent cardiovascular morphogenesis (Kirby and Hutson 2010). TGFβ ligands regulate synthesis of the ECM proteins (Massague 1998); conversely cell–ECM interactions play an integral role in regulating signaling by TGFβ family members (Shi et al. 2011). Most members of this superfamily are synthesized in the inactive, latent form. At least two modes of regulation are known to activate TGFβ ligands: (1) by proteolysis, to release TGFβ homodimers from their pro-domains; this could be facilitated by association between αv-containing integrins with cellular or secreted metalloproteinases or (2) by mechanical forces, via integrin αv-mediated pulling and stretching of the inactive, TGFβ-containing latent complexes (Wipff and Hinz 2008).
Members of TGFβ family of proteins are synthesized as pro-peptides; their pro-domains are cleaved off during exocytosis but remain non-covalently associated with the mature TGFβ homodimers. These complexes (termed small latent complexes, SLCs) render TGFβ inactive. TGFβ–SLCs are covalently bound to latent TGFβ-binding proteins (LTBPs 1, 3, or 4), forming large SLC–LTBP latent complexes via disulfide bonding of the pro-domain to the LTBPs (Todorovic et al. 2005). In the extracellular milieu, LTBP1 is covalently cross-linked to ECM proteins including FN, VN, and/or fibrillin via transglutamination (Wipff and Hinz 2008). TGFβ-containing SLC–LTBP1 complexes are inactive but can be activated in vivo by proteolytic cleavage severing the covalent association of the pro-domain with LTBP and releasing active TGFβ homodimers free of their pro-domains or by a pulling force exerted by cells on SLC–LTBP1 complexes (Munger et al. 1999). The latter has been best illustrated for the activation of TGFβ1–SLC–LTBP1 complexes. The pro-domains of TGFβ(1–3) have an RGD motif that can bind to and activate αv-containing integrin heterodimers. The tensile force generated by the interactions between integrin cytoplasmic tails and the actin cytoskeleton exerts a pulling force on the SLC and is counteracted by the covalently bound LTBP1 anchored to the ECM, leading to stretching of the SLC–LTBP1 complex (McMahon et al. 1996; Sivakumar et al. 2006). Upon stretching of the SLC–LTBP1, the embedded TGFβ homodimers pop out, and free from their pro-domains, they are competent to bind TGFβ receptors and activate downstream signaling; mutations that interfere with stretching of the SLC–LTBP1 complex inhibit release of the active TGFβ (Shi et al. 2011).
Consistent with these studies, TGFβ signaling is downregulated in global LTBP1 mutant mouse embryos, which develop lethal cardiovascular defects characterized by aberrant septation of the cardiac OFT and AAA remodeling defects (Todorovic et al. 2007). In addition, elegant recent studies demonstrated the role of αvβ8-mediated activation of TGFβ signaling during remodeling of retinal vasculature (Hirota et al. 2011). In these studies, αvβ8 integrin expressed by retinal astrocytes is required to activate TGFβ signaling in the closely apposed endothelial cells of retinal blood vessels, and the absence of αv or β8 on the astrocytes leads to reduced TGFβ response in the endothelial cells, blunted endothelial tip cell morphology, and aberrant development of the retinal vascular plexus (Hirota et al. 2011). FN expressed by retinal astrocytes is also required for retinal vascularization by modulating directionality, alignment, and numbers of endothelial filopodia, in part by binding to and facilitating signaling by VEGF-A (Stenzel et al. 2011a).
The importance of ECM proteins in modulating TGFβ signaling and human pathology is underscored by the discovery that integral ECM proteins fibrillins modulate activity of TGFβ and BMPs (Neptune et al. 2003; Nistala et al. 2010). Fibrillin-1 binds and modulates activity of TGFβs, and deficiency of fibrillin-1 causes human Marfan syndrome characterized in part by dilation and rupture of the ascending aorta (Neptune et al. 2003). Decrease or absence of fibrillins causes increased availability of active TGFβ, leading to increased canonical and noncanonical TGFβ signaling (Holm et al. 2011). Interestingly, the increase in noncanonical TGFβ signaling pathway mediated by activation of Erk1/2 appears to be the main cause of abnormal aortic pathology in the fibrillin-1 mutant mouse model of Marfan syndrome (Habashi et al. 2011; Holm et al. 2011). In conjunction with integrins, abundance, composition, and mechanical compliance of the ECM modulate activity of TGFβ family members by regulating mechanical properties of the microenvironment (Wipff and Hinz 2008). For example, while integrin α5 does not appear to bind TGFβ-containing SLC complexes, FN fibrillogenesis mediated by integrin α5 is important for αvβ6-mediated TGFβ signaling, and activation of latent TGFβ is compromised in FN-null or integrin α5-null cells (Fontana et al. 2005). Because FN binds LTBP1 and facilitates activation of latent TGFβ1-containing complexes (Fontana et al. 2005) and because both LTBP1 and FN are synthesized by the endoderm within the pharyngeal microenvironment, it would be interesting to examine whether FN regulates TGFβ signaling and AAA remodeling.
5.8 Semaphorins, Plexins, and Neuropilins
Semaphorin signaling is essential for navigation of NC-derived cells through the embryo, for the proper development of the CNC and, subsequently, for septation of the cardiac OFT and AAA remodeling (Feiner et al. 2001; Schwarz et al. 2008; Toyofuku et al. 2008). CNC cells traversing the embryo encounter repellent and attractant types of semaphorins. Recent experiments in the chick and mouse suggest that cues provided by the action of several diffusible types of semaphorins expressed at the time of CNC migration and their receptors expressed by the CNC cells could facilitate (a) exit of the CNC from the dorsal neural tube, (b) navigation of the CNC to their destinations, and (c) CNC entry into the cardiac OFT (Feiner et al. 2001; Schwarz et al. 2008).
Semaphorin and neuropilin signaling regulate (and could be probably regulated by) cell–ECM interactions during vascular remodeling. Class 3 semaphorins, Sema3A and 3F, provide repulsive cues for NC migration by signaling through NC-expressed Nrp1 and Nrp2, to pattern cranial neurons and ganglia (Schwarz et al. 2008). This mode of action is independent of the requirement for Nrp signaling to the endothelium, which plays an important role in vascular remodeling (Kawasaki et al. 1999; Schwarz et al. 2008). Signaling by Sema3A and 3F inhibits integrin activation in vitro and in vivo, negatively regulates directional persistence of migrating vascular endothelial cells, and is required for remodeling of the primitive vascular plexus in chick and mouse embryos into the stereotypic, yet nonuniform plexus of large and small blood vessels (Serini et al. 2003). Nrp1 regulates integrin-mediated endothelial cell adhesion in a manner that is separable from its roles in VEGF-mediated activation or Sema3-mediated inhibition of cell adhesion (Valdembri et al. 2009). Interestingly, Nrp1 potentiates cell adhesion to FN but not to other large ECM glycoproteins vitronectin, collagen I, or LM-111, suggesting that composition of extracellular microenvironment imparts specificity to Nrp1-mediated vascular remodeling. Nrp1 potentiates integrin α5β1-dependent cell spreading and FN matrix assembly on cell surfaces by facilitating integrin endocytosis and recycling. The extracellular portion of Nrp1 binds activated integrin α5β1, while the intracellular SEA motif present within the short, 50-amino acid cytoplasmic tail of Nrp1 facilitates binding of Nrp1 to endocytic adaptor protein GIPC1 and a minus-directed motor protein Myo6 and targets activated α5β1 integrins into Rab5/Rab21-containing endocytic vesicles, which rapidly recycle back to the plasma membrane. These experiments demonstrated that endothelial Nrp1, GIPC1, and Myo6 are required to stimulate α5β1-mediated cell adhesion to FN (Valdembri et al. 2009).
Cardiovascular defects in Nrp1-null embryos are similar to those observed in Sema3C mutants, suggesting that Sema3C signaling mediated by Nrp1 participates in AAA remodeling and OFT septation (Feiner et al. 2001; Kawasaki et al. 1999). Interestingly, Sema3C signaling to endothelial cells mediated by endothelial PlexinD1 and its Nrp co-receptors is required for cardiac OFT septation and AAA remodeling (Gitler et al. 2004; Zhang et al. 2009). Thus a possible mechanism underlying aberrant remodeling of pharyngeal arch arteries in Nrp1, PlexinD1, and Sema3C mutants could be due to defects in integrin-mediated cell–ECM interactions and signaling. In this regard, NC-specific mutations in genes encoding downstream effectors of integrin activation, such as Rac1, Cdc42, and FAK, give rise to defective development of the cardiac OFT and reduced differentiation of CNC-derived cells around pharyngeal arch arteries toward smooth muscle cell fate and aberrant remodeling of the pharyngeal arch arteries (Fuchs et al. 2009; Thomas et al. 2010; Vallejo-Illarramendi et al. 2009). Intriguingly, FAK-deficient CNC cells within the OFT do not express Sema3C (Vallejo-Illarramendi et al. 2009), implicating integrin-mediated activation of FAK in regulation of semaphorin expression and signaling. Future studies will determine whether cell–ECM interactions modulate attractive and/or repulsive properties imparted by semaphorin signaling to endothelial, NC, or other cell types during cardiovascular development.
Mutations leading to decreased expression of Sema3C and PlexinA2 have been observed in human patients with defective septation of the cardiac OFT (Kodo et al. 2009). These mutations disrupted the function of the transcription factor Gata6, which directly binds to and activates transcription from Sema3C and PlexinA2 promoters (Kodo et al. 2009; Lepore et al. 2006). Function of Gata6 in the CNC cells is required for the septation of the cardiac OFT and remodeling of AAAs (Lepore et al. 2006). Therefore, understanding genes, pathways, and interactions regulating expression and signaling by semaphorins and plexins would provide further insight into human congenital cardiovascular disorders.
6 Conclusions
ECM components such as FN, laminins, collagens, and others have long been known to modulate growth factor signaling in vitro; however, signaling pathways engaged by the ECM macromolecules in vivo and especially during embryogenesis are not well understood. Examples given above illustrate how cell–ECM interactions could facilitate signaling by a variety of growth factors important for the proper morphogenesis of the cardiac outflow vasculature. We hypothesize that the dynamic and nonuniform distributions of ECM macromolecules within embryos impact combinatorial signaling outcomes and orchestrate inter-tissue communications, which affect cellular positions, shapes, fates, and functions during OFT septation and AAA morphogenesis.
References
Arrington CB, Yost HJ (2009) Extra-embryonic syndecan 2 regulates organ primordia migration and fibrillogenesis throughout the zebrafish embryo. Development 136:3143–3152
Astrof S, Hynes RO (2009) Fibronectins in vascular morphogenesis. Angiogenesis 12:165–175
Astrof NS, Salas A, Shimaoka M, Chen J, Springer TA (2006) Importance of force linkage in mechanochemistry of adhesion receptors. Biochemistry 45:15020–15028
Astrof S, Crowley D, Hynes RO (2007) Multiple cardiovascular defects caused by the absence of alternatively spliced segments of fibronectin. Dev Biol 311:11–24
Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, Engel J, Engvall E, Hohenester E, Jones JC et al (2005) A simplified laminin nomenclature. Matrix Biol 24:326–332
Breau MA, Pietri T, Eder O, Blanche M, Brakebusch C, Fassler R, Thiery JP, Dufour S (2006) Lack of beta1 integrins in enteric neural crest cells leads to a Hirschsprung-like phenotype. Development 133:1725–1734
Cai CL, Martin JC, Sun Y, Cui L, Wang L, Ouyang K, Yang L, Bu L, Liang X, Zhang X et al (2008) A myocardial lineage derives from Tbx18 epicardial cells. Nature 454:104–108
Calmont A, Ivins S, Van Bueren KL, Papangeli I, Kyriakopoulou V, Andrews WD, Martin JF, Moon AM, Illingworth EA, Basson MA et al (2009) Tbx1 controls cardiac neural crest cell migration during arch artery development by regulating Gbx2 expression in the pharyngeal ectoderm. Development 136:3173–3183
Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C et al (1996) Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380:435–439
Carmona-Fontaine C, Matthews HK, Kuriyama S, Moreno M, Dunn GA, Parsons M, Stern CD, Mayor R (2008) Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature 456:957–961
Carmona-Fontaine C, Theveneau E, Tzekou A, Tada M, Woods M, Page KM, Parsons M, Lambris JD, Mayor R (2011) Complement fragment C3a controls mutual cell attraction during collective cell migration. Dev Cell 21:1026–1037
Chan WY, Cheung CS, Yung KM, Copp AJ (2004) Cardiac neural crest of the mouse embryo: axial level of origin, migratory pathway and cell autonomy of the splotch (Sp2H) mutant effect. Development 131:3367–3379
Chen TT, Luque A, Lee S, Anderson SM, Segura T, Iruela-Arispe ML (2010) Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J Cell Biol 188:595–609
Coles EG, Gammill LS, Miner JH, Bronner-Fraser M (2006) Abnormalities in neural crest cell migration in laminin alpha5 mutant mice. Dev Biol 289:218–228
Cooley MA, Kern CB, Fresco VM, Wessels A, Thompson RP, McQuinn TC, Twal WO, Mjaatvedt CH, Drake CJ, Argraves WS (2008) Fibulin-1 is required for morphogenesis of neural crest-derived structures. Dev Biol 319:336–345
Copp AJ, Carvalho R, Wallace A, Sorokin L, Sasaki T, Greene ND, Ybot-Gonzalez P (2011) Regional differences in the expression of laminin isoforms during mouse neural tube development. Matrix Biol 30:301–309
Costa-Silva B, da Costa MC, Melo FR, Neves CM, Alvarez-Silva M, Calloni GW, Trentin AG (2009) Fibronectin promotes differentiation of neural crest progenitors endowed with smooth muscle cell potential. Exp Cell Res 315:955–967
Costell M, Carmona R, Gustafsson E, Gonzalez-Iriarte M, Fassler R, Munoz-Chapuli R (2002) Hyperplastic conotruncal endocardial cushions and transposition of great arteries in perlecan-null mice. Circ Res 91:158–164
De Langhe SP, Sala FG, Del Moral PM, Fairbanks TJ, Yamada KM, Warburton D, Burns RC, Bellusci S (2005) Dickkopf-1 (DKK1) reveals that fibronectin is a major target of Wnt signaling in branching morphogenesis of the mouse embryonic lung. Dev Biol 277:316–331
Delannet M, Martin F, Bossy B, Cheresh DA, Reichardt LF, Duband JL (1994) Specific roles of the alpha V beta 1, alpha V beta 3 and alpha V beta 5 integrins in avian neural crest cell adhesion and migration on vitronectin. Development 120:2687–2702
Duband JL, Thiery JP (1987) Distribution of laminin and collagens during avian neural crest development. Development 101:461–478
Dutt S, Kleber M, Matasci M, Sommer L, Zimmermann DR (2006) Versican V0 and V1 guide migratory neural crest cells. J Biol Chem 281:12123–12131
Engler AJ, Sen S, Sweeney HL, Discher DE (2006) Matrix elasticity directs stem cell lineage specification. Cell 126:677–689
Erickson CA, Tosney KW, Weston JA (1980) Analysis of migratory behavior of neural crest and fibroblastic cells in embryonic tissues. Dev Biol 77:142–156
Feiner L, Webber AL, Brown CB, Lu MM, Jia L, Feinstein P, Mombaerts P, Epstein JA, Raper JA (2001) Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development 128:3061–3070
Ffrench-Constant C, Hynes RO (1989) Alternative splicing of fibronectin is temporally and spatially regulated in the chicken embryo. Development 106:375–388
Ffrench-Constant C, Van de Water L, Dvorak HF, Hynes RO (1989) Reappearance of an embryonic pattern of fibronectin splicing during wound healing in the adult rat. J Cell Biol 109:903–914
Fontana L, Chen Y, Prijatelj P, Sakai T, Fassler R, Sakai LY, Rifkin DB (2005) Fibronectin is required for integrin alphavbeta6-mediated activation of latent TGF-beta complexes containing LTBP-1. FASEB J 19:1798–1808
Fournier-Thibault C, Blavet C, Jarov A, Bajanca F, Thorsteinsdottir S, Duband JL (2009) Sonic hedgehog regulates integrin activity, cadherin contacts, and cell polarity to orchestrate neural tube morphogenesis. J Neurosci 29:12506–12520
Fuchs S, Herzog D, Sumara G, Buchmann-Moller S, Civenni G, Wu X, Chrostek-Grashoff A, Suter U, Ricci R, Relvas JB et al (2009) Stage-specific control of neural crest stem cell proliferation by the small rho GTPases Cdc42 and Rac1. Cell Stem Cell 4:236–247
Gammill LS, Roffers-Agarwal J (2010) Division of labor during trunk neural crest development. Dev Biol 344:555–565
Gammill LS, Gonzalez C, Gu C, Bronner-Fraser M (2006) Guidance of trunk neural crest migration requires neuropilin 2/semaphorin 3F signaling. Development 133:99–106
Gammill LS, Gonzalez C, Bronner-Fraser M (2007) Neuropilin 2/semaphorin 3F signaling is essential for cranial neural crest migration and trigeminal ganglion condensation. Dev Neurobiol 67:47–56
Giancotti FG, Tarone G (2003) Positional control of cell fate through joint integrin/receptor protein kinase signaling. Annu Rev Cell Dev Biol 19:173–206
Gitler AD, Lu MM, Epstein JA (2004) PlexinD1 and semaphorin signaling are required in endothelial cells for cardiovascular development. Dev Cell 7:107–116
Goddeeris MM, Schwartz R, Klingensmith J, Meyers EN (2007) Independent requirements for Hedgehog signaling by both the anterior heart field and neural crest cells for outflow tract development. Development 134:1593–1604
Graham A, Okabe M, Quinlan R (2005) The role of the endoderm in the development and evolution of the pharyngeal arches. J Anat 207:479–487
Guris DL, Duester G, Papaioannou VE, Imamoto A (2006) Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev Cell 10:81–92
Haack H, Hynes RO (2001) Integrin receptors are required for cell survival and proliferation during development of the peripheral glial lineage. Dev Biol 233:38–55
Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC (2011) Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science 332:361–365
Hari L, Brault V, Kleber M, Lee HY, Ille F, Leimeroth R, Paratore C, Suter U, Kemler R, Sommer L (2002) Lineage-specific requirements of beta-catenin in neural crest development. J Cell Biol 159:867–880
Hari L, Miescher I, Shakhova O, Suter U, Chin L, Taketo M, Richardson WD, Kessaris N, Sommer L (2012) Temporal control of neural crest lineage generation by Wnt/beta-catenin signaling. Development 139:2107–2117
High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA (2008) Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci USA 105:1955–1959
Hirota S, Liu Q, Lee HS, Hossain MG, Lacy-Hulbert A, McCarty JH (2011) The astrocyte-expressed integrin alphavbeta8 governs blood vessel sprouting in the developing retina. Development 138:5157–5166
Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD et al (2011) Noncanonical TGF beta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 332:358–361
Honda H, Oda H, Nakamoto T, Honda Z, Sakai R, Suzuki T, Saito T, Nakamura K, Nakao K, Ishikawa T et al (1998) Cardiovascular anomaly, impaired actin bundling and resistance to Src-induced transformation in mice lacking p130Cas. Nat Genet 19:361–365
Horowitz A, Tkachenko E, Simons M (2002) Fibroblast growth factor-specific modulation of cellular response by syndecan-4. J Cell Biol 157:715–725
Hutson MR, Sackey FN, Lunney K, Kirby ML (2009) Blocking hedgehog signaling after ablation of the dorsal neural tube allows regeneration of the cardiac neural crest and rescue of outflow tract septation. Dev Biol 335:367–373
Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110:673–687
Hynes RO (2009) The extracellular matrix: not just pretty fibrils. Science 326:1216–1219
Hynes RO (2012) Evolution: the evolution of metazoan extracellular matrix. J Cell Biol 196:671–679
Ikeya M, Lee SM, Johnson JE, McMahon AP, Takada S (1997) Wnt signalling required for expansion of neural crest and CNS progenitors. Nature 389:966–970
Jain R, Rentschler S, Epstein JA (2010) Notch and cardiac outflow tract development. Ann N Y Acad Sci 1188:184–190
Jarov A, Williams KP, Ling LE, Koteliansky VE, Duband JL, Fournier-Thibault C (2003) A dual role for Sonic hedgehog in regulating adhesion and differentiation of neuroepithelial cells. Dev Biol 261:520–536
Jones RG, Li X, Gray PD, Kuang J, Clayton F, Samowitz WS, Madison BB, Gumucio DL, Kuwada SK (2006) Conditional deletion of beta1 integrins in the intestinal epithelium causes a loss of Hedgehog expression, intestinal hyperplasia, and early postnatal lethality. J Cell Biol 175:505–514
Kaartinen V, Dudas M, Nagy A, Sridurongrit S, Lu MM, Epstein JA (2004) Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development 131:3481–3490
Kawasaki T, Kitsukawa T, Bekku Y, Matsuda Y, Sanbo M, Yagi T, Fujisawa H (1999) A requirement for neuropilin-1 in embryonic vessel formation. Development 126:4895–4902
Kirby ML, Hutson MR (2010) Factors controlling cardiac neural crest cell migration. Cell Adh Migr 4:609–621
Kodo K, Nishizawa T, Furutani M, Arai S, Yamamura E, Joo K, Takahashi T, Matsuoka R, Yamagishi H (2009) GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc Natl Acad Sci USA 106:13933–13938
Kulesa PM, Gammill LS (2010) Neural crest migration: patterns, phases and signals. Dev Biol 344:566–568
Le Douarin NM, Kalcheim C (1999) The neural crest, vol 36, 2nd edn. Cambridge University Press, Cambridge
Leatherbury L, Gauldin HE, Waldo K, Kirby ML (1990) Microcinephotography of the developing heart in neural crest-ablated chick embryos. Circulation 81:1047–1057
Lepore JJ, Mericko PA, Cheng L, Lu MM, Morrisey EE, Parmacek MS (2006) GATA-6 regulates semaphorin 3C and is required in cardiac neural crest for cardiovascular morphogenesis. J Clin Invest 116:929–939
Li L, Guris DL, Okura M, Imamoto A (2003) Translocation of CrkL to focal adhesions mediates integrin-induced migration downstream of Src family kinases. Mol Cell Biol 23:2883–2892
Liu W, Selever J, Wang D, Lu MF, Moses KA, Schwartz RJ, Martin JF (2004) Bmp4 signaling is required for outflow-tract septation and branchial-arch artery remodeling. Proc Natl Acad Sci USA 101:4489–4494
Liu K, Cheng L, Flesken-Nikitin A, Huang L, Nikitin AY, Pauli BU (2010) Conditional knockout of fibronectin abrogates mouse mammary gland lobuloalveolar differentiation. Dev Biol 346:11–24
Lohela M, Bry M, Tammela T, Alitalo K (2009) VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol 21:154–165
Macatee TL, Hammond BP, Arenkiel BR, Francis L, Frank DU, Moon AM (2003) Ablation of specific expression domains reveals discrete functions of ectoderm- and endoderm-derived FGF8 during cardiovascular and pharyngeal development. Development 130:6361–6374
Martino MM, Hubbell JA (2010) The 12th-14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J 24:4711–4721
Massague J (1998) TGF-beta signal transduction. Annu Rev Biochem 67:753–791
McMahon GA, Dignam JD, Gentry LE (1996) Structural characterization of the latent complex between transforming growth factor beta 1 and beta 1-latency-associated peptide. Biochem J 313(Pt 1):343–351
Mittal A, Pulina M, Hou SY, Astrof S (2010) Fibronectin and integrin alpha 5 play essential roles in the development of the cardiac neural crest. Mech Dev 127:472–484
Miyamoto S, Teramoto H, Gutkind JS, Yamada KM (1996) Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: roles of integrin aggregation and occupancy of receptors. J Cell Biol 135:1633–1642
Moon AM (2006) Mouse models for investigating the developmental basis of human birth defects. Pediatr Res 59:749–755
Moon AM, Guris DL, Seo JH, Li L, Hammond J, Talbot A, Imamoto A (2006) Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev Cell 10:71–80
Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA et al (1999) The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96:319–328
Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC (2003) Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet 33:407–411
Nistala H, Lee-Arteaga S, Smaldone S, Siciliano G, Carta L, Ono RN, Sengle G, Arteaga-Solis E, Levasseur R, Ducy P et al (2010) Fibrillin-1 and −2 differentially modulate endogenous TGF-beta and BMP bioavailability during bone formation. J Cell Biol 190:1107–1121
Park EJ, Ogden LA, Talbot A, Evans S, Cai CL, Black BL, Frank DU, Moon AM (2006) Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development 133:2419–2433
Park EJ, Watanabe Y, Smyth G, Miyagawa-Tomita S, Meyers E, Klingensmith J, Camenisch T, Buckingham M, Moon AM (2008) An FGF autocrine loop initiated in second heart field mesoderm regulates morphogenesis at the arterial pole of the heart. Development 135:3599–3610
Peters JH, Hynes RO (1996) Fibronectin isoform distribution in the mouse. I. The alternatively spliced EIIIB, EIIIA, and V segments show widespread codistribution in the developing mouse embryo. Cell Adhes Commun 4:103–125
Peters JH, Chen GE, Hynes RO (1996) Fibronectin isoform distribution in the mouse. II. Differential distribution of the alternatively spliced EIIIB, EIIIA, and V segments in the adult mouse. Cell Adhes Commun 4:127–148
Pulina MV, Hou SY, Mittal A, Julich D, Whittaker CA, Holley SA, Hynes RO, Astrof S (2011) Essential roles of fibronectin in the development of the left-right embryonic body plan. Dev Biol 354:208–220
Rallis C, Pinchin SM, Ish-Horowicz D (2010) Cell-autonomous integrin control of Wnt and Notch signalling during somitogenesis. Development 137:3591–3601
Ramsdell AF (2005) Left-right asymmetry and congenital cardiac defects: getting to the heart of the matter in vertebrate left-right axis determination. Dev Biol 288:1–20
Rentschler S, Jain R, Epstein JA (2010) Tissue-tissue interactions during morphogenesis of the outflow tract. Pediatr Cardiol 31:408–413
Richarte AM, Mead HB, Tallquist MD (2007) Cooperation between the PDGF receptors in cardiac neural crest cell migration. Dev Biol 306:785–796
Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES et al (2011) Heart disease and stroke statistics–2011 update: a report from the American Heart Association. Circulation 123:e18–e209
Rovasio RA, Delouvee A, Yamada KM, Timpl R, Thiery JP (1983) Neural crest cell migration: requirements for exogenous fibronectin and high cell density. J Cell Biol 96:462–473
Saoncella S, Echtermeyer F, Denhez F, Nowlen JK, Mosher DF, Robinson SD, Hynes RO, Goetinck PF (1999) Syndecan-4 signals cooperatively with integrins in a Rho-dependent manner in the assembly of focal adhesions and actin stress fibers. Proc Natl Acad Sci USA 96:2805–2810
Schwarz Q, Vieira JM, Howard B, Eickholt BJ, Ruhrberg C (2008) Neuropilin 1 and 2 control cranial gangliogenesis and axon guidance through neural crest cells. Development 135:1605–1613
Seo JH, Suenaga A, Hatakeyama M, Taiji M, Imamoto A (2009) Structural and functional basis of a role for CRKL in a fibroblast growth factor 8-induced feed-forward loop. Mol Cell Biol 29:3076–3087
Serini G, Valdembri D, Zanivan S, Morterra G, Burkhardt C, Caccavari F, Zammataro L, Primo L, Tamagnone L, Logan M et al (2003) Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature 424:391–397
Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA (2011) Latent TGF-beta structure and activation. Nature 474:343–349
Sivakumar P, Czirok A, Rongish BJ, Divakara VP, Wang YP, Dallas SL (2006) New insights into extracellular matrix assembly and reorganization from dynamic imaging of extracellular matrix proteins in living osteoblasts. J Cell Sci 119:1350–1360
Stalmans I, Lambrechts D, De Smet F, Jansen S, Wang J, Maity S, Kneer P, von der Ohe M, Swillen A, Maes C et al (2003) VEGF: a modifier of the del22q11 (DiGeorge) syndrome? Nat Med 9:173–182
Stenzel D, Franco CA, Estrach S, Mettouchi A, Sauvaget D, Rosewell I, Schertel A, Armer H, Domogatskaya A, Rodin S et al (2011a) Endothelial basement membrane limits tip cell formation by inducing Dll4/Notch signalling in vivo. EMBO Rep 12:1135–1143
Stenzel D, Lundkvist A, Sauvaget D, Busse M, Graupera M, van der Flier A, Wijelath ES, Murray J, Sobel M, Costell M et al (2011b) Integrin-dependent and -independent functions of astrocytic fibronectin in retinal angiogenesis. Development 138:4451–4463
Stottmann RW, Klingensmith J (2011) Bone morphogenetic protein signaling is required in the dorsal neural folds before neurulation for the induction of spinal neural crest cells and dorsal neurons. Dev Dyn 240:755–765
Stottmann RW, Choi M, Mishina Y, Meyers EN, Klingensmith J (2004) BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development 131:2205–2218
Strachan LR, Condic ML (2004) Cranial neural crest recycle surface integrins in a substratum-dependent manner to promote rapid motility. J Cell Biol 167:545–554
Stuhlmiller TJ, Garcia-Castro MI (2012) FGF/MAPK signaling is required in the gastrula epiblast for avian neural crest induction. Development 139:289–300
Testaz S, Delannet M, Duband J (1999) Adhesion and migration of avian neural crest cells on fibronectin require the cooperating activities of multiple integrins of the (beta)1 and (beta)3 families. J Cell Sci 112(Pt 24):4715–4728
Testaz S, Jarov A, Williams KP, Ling LE, Koteliansky VE, Fournier-Thibault C, Duband JL (2001) Sonic hedgehog restricts adhesion and migration of neural crest cells independently of the Patched- Smoothened-Gli signaling pathway. Proc Natl Acad Sci USA 98:12521–12526
Thomas PS, Kim J, Nunez S, Glogauer M, Kaartinen V (2010) Neural crest cell-specific deletion of Rac1 results in defective cell-matrix interactions and severe craniofacial and cardiovascular malformations. Dev Biol 340:613–625
Todorovic V, Jurukovski V, Chen Y, Fontana L, Dabovic B, Rifkin DB (2005) Latent TGF-beta binding proteins. Int J Biochem Cell Biol 37:38–41
Todorovic V, Frendewey D, Gutstein DE, Chen Y, Freyer L, Finnegan E, Liu F, Murphy A, Valenzuela D, Yancopoulos G et al (2007) Long form of latent TGF-beta binding protein 1 (Ltbp1L) is essential for cardiac outflow tract septation and remodeling. Development 134:3723–3732
Toyoda R, Assimacopoulos S, Wilcoxon J, Taylor A, Feldman P, Suzuki-Hirano A, Shimogori T, Grove EA (2010) FGF8 acts as a classic diffusible morphogen to pattern the neocortex. Development 137:3439–3448
Toyofuku T, Yoshida J, Sugimoto T, Yamamoto M, Makino N, Takamatsu H, Takegahara N, Suto F, Hori M, Fujisawa H et al (2008) Repulsive and attractive semaphorins cooperate to direct the navigation of cardiac neural crest cells. Dev Biol 321:251–262
Trainor PA, Ariza-McNaughton L, Krumlauf R (2002) Role of the isthmus and FGFs in resolving the paradox of neural crest plasticity and prepatterning. Science 295:1288–1291
Turlo KA, Noel OD, Vora R, Larussa M, Fassler R, Hall-Glenn F, Iruela-Arispe ML (2012) An essential requirement for beta1 integrin in the assembly of extracellular matrix proteins within the vascular wall. Dev Biol 365:23–35
Urness LD, Bleyl SB, Wright TJ, Moon AM, Mansour SL (2011) Redundant and dosage sensitive requirements for Fgf3 and Fgf10 in cardiovascular development. Dev Biol 356:383–397
Valdembri D, Caswell PT, Anderson KI, Schwarz JP, Konig I, Astanina E, Caccavari F, Norman JC, Humphries MJ, Bussolino F et al (2009) Neuropilin-1/GIPC1 signaling regulates alpha5beta1 integrin traffic and function in endothelial cells. PLoS Biol 7:e25
Vallejo-Illarramendi A, Zang K, Reichardt LF (2009) Focal adhesion kinase is required for neural crest cell morphogenesis during mouse cardiovascular development. J Clin Invest 119:2218–2230
van den Akker NM, Molin DG, Peters PP, Maas S, Wisse LJ, van Brempt R, van Munsteren CJ, Bartelings MM, Poelmann RE, Carmeliet P et al (2007) Tetralogy of fallot and alterations in vascular endothelial growth factor-A signaling and notch signaling in mouse embryos solely expressing the VEGF120 isoform. Circ Res 100:842–849
Veevers-Lowe J, Ball SG, Shuttleworth A, Kielty CM (2011) Mesenchymal stem cell migration is regulated by fibronectin through alpha5beta1-integrin-mediated activation of PDGFR-beta and potentiation of growth factor signals. J Cell Sci 124:1288–1300
Vincentz JW, McWhirter JR, Murre C, Baldini A, Furuta Y (2005) Fgf15 is required for proper morphogenesis of the mouse cardiac outflow tract. Genesis 41:192–201
Waldo KL, Hutson MR, Stadt HA, Zdanowicz M, Zdanowicz J, Kirby ML (2005) Cardiac neural crest is necessary for normal addition of the myocardium to the arterial pole from the secondary heart field. Dev Biol 281:66–77
Wijelath ES, Murray J, Rahman S, Patel Y, Ishida A, Strand K, Aziz S, Cardona C, Hammond WP, Savidge GF et al (2002) Novel vascular endothelial growth factor binding domains of fibronectin enhance vascular endothelial growth factor biological activity. Circ Res 91:25–31
Wipff PJ, Hinz B (2008) Integrins and the activation of latent transforming growth factor beta1 – an intimate relationship. Eur J Cell Biol 87:601–615
Wurdak H, Ittner LM, Sommer L (2006) DiGeorge syndrome and pharyngeal apparatus development. Bioessays 28:1078–1086
Xu X, Francis R, Wei CJ, Linask KL, Lo CW (2006) Connexin 43-mediated modulation of polarized cell movement and the directional migration of cardiac neural crest cells. Development 133:3629–3639
Zhang J, Lin Y, Zhang Y, Lan Y, Lin C, Moon AM, Schwartz RJ, Martin JF, Wang F (2008) Frs2alpha-deficiency in cardiac progenitors disrupts a subset of FGF signals required for outflow tract morphogenesis. Development 135:3611–3622
Zhang Y, Singh MK, Degenhardt KR, Lu MM, Bennett J, Yoshida Y, Epstein JA (2009) Tie2Cre-mediated inactivation of plexinD1 results in congenital heart, vascular and skeletal defects. Dev Biol 325:82–93
Zou L, Cao S, Kang N, Huebert RC, Shah VH (2012) Fibronectin induces endothelial cell migration through beta1-integrin and Src dependent phosphorylation of fibroblast growth factor receptor-1 at tyrosines 653/654 and 766. J Biol Chem 287(10):7190–7202
Acknowledgments
I would like to thank Glenn Radice, Anne Moon, Jim Weston, and Dongying Chen for their critical comments and helpful suggestions. Studies in my lab are supported by the AHA Scientist Development grant, AHA Innovative Research grant, W.W. Smith Charitable foundation, and NIH NHLBI #5R01HL103920.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Astrof, S. (2013). Interactions Between Neural Crest-Derived Cells and Extracellular Microenvironment During Cardiovascular Development. In: DeSimone, D., Mecham, R. (eds) Extracellular Matrix in Development. Biology of Extracellular Matrix. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-35935-4_5
Download citation
DOI: https://doi.org/10.1007/978-3-642-35935-4_5
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-35934-7
Online ISBN: 978-3-642-35935-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)