
Abstract
The first significant histologic classification of tumors of the central nervous system (CNS) was proposed by Bailey and Cushing in 1926. Thereafter, several major revisions were introduced, and numerous consensus conferences were held. The grading of CNS tumors is an integral part of these revisions and is routinely applied to primary intracranial and spinal tumors. The most widely accepted grading system is the World Health Organization (WHO) classification of tumors, last updated in 2007. Recently the prognostic values of several genetic markers have been validated in several CNS tumors, and the importance of testing them in paraffin-embedded tissue is growing [1]. Rare tumors, lymphomas, paraganglioma, and pituitary adenomas will not be discussed in this concise chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
- Pilocytic Astrocytoma
- Choroid Plexus Carcinoma
- Fibrillary Astrocytoma
- Subependymal Giant Cell Astrocytoma
- Clear Cell Meningioma
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
15.1 Introduction
The first significant histologic classification of tumors of the central nervous system (CNS) was proposed by Bailey and Cushing in 1926. Thereafter, several major revisions were introduced, and numerous consensus conferences were held. The grading of CNS tumors is an integral part of these revisions and is routinely applied to primary intracranial and spinal tumors. The most widely accepted grading system is the World Health Organization (WHO) classification of tumors, last updated in 2007. Recently the prognostic values of several genetic markers have been validated in several CNS tumors, and the importance of testing them in paraffin-embedded tissue is growing [1]. Rare tumors, lymphomas, paraganglioma, and pituitary adenomas will not be discussed in this concise chapter.
15.2 Astrocytoma
Diffusely infiltrating or fibrillary astrocytomas constitute the majority of those encountered in the human central nervous system. The WHO classification determines the grade, on a scale of I–IV, of a diffusely infiltrating astrocytoma according to several microscopic features: cellularity, nuclear atypia, mitotic activity, vascular proliferation, and tumor necrosis. However, grading based on these criteria has been reported to show poor interobserver agreement, especially in stereotactic biopsies [2]. In 1988, Daumas-Duport and colleagues proposed a more reproducible grading system of adult diffusely infiltrating astrocytomas [3], generally known as the St. Anne/Mayo grading system. Assigning equal importance to each of four specific pathologic features – nuclear atypia, mitosis, vascular proliferation, and necrosis – the St. Anne/Mayo grade of a diffusely infiltrating astrocytoma on a scale of 1–4 is determined by the number of feature(s) present plus 1. This system is similar, but not identical to, the WHO classification. For instance, finding one mitotic figure in an otherwise well-differentiated, diffusely infiltrating astrocytoma qualifies it as grade 3 in the St. Anne/Mayo system, but it may still be considered a WHO grade II lesion. On the other hand, a small biopsy showing predominantly necrosis rimmed by a few viable atypical astrocytes without mitosis is diagnosed as glioblastoma (grade IV) by the WHO criteria but remains a St. Anne/Mayo grade 3 lesion. Since both systems are defined by positive features, grading fibrillary astrocytomas based on small biopsies is subject to errors of “undergrading.”
The WHO classification of astrocytic tumors includes the following:
-
Grade I, fibrillary astrocytoma. This tumor is exceedingly rare. It shows a mild increase of cellularity and minimal cytologic atypia without other pathologic features. This grade also includes variants of astrocytoma with specific pathology, radiology, and indolent clinical behavior, i.e., pilocytic astrocytoma and subependymal giant cell astrocytoma.
-
Grade II, well-differentiated or diffuse astrocytoma. This tumor shows a mild to moderate increase of cellularity and nuclear atypia (Fig. 15.1), minimal mitotic activity (usually <2 mitoses per tumor), and no vascular proliferation or tumor necrosis. One variant of WHO grade II diffuse astrocytomas is gemistocytic astrocytoma that has been reported to portend a more aggressive behavior than its lack of mitotic activity suggests. It is composed of neoplastic astrocytes with eccentric nuclei, small nucleoli, and plump, eosinophilic, glassy, and fibrillary cytoplasm with frequent perivascular inflammation and no mitotic figures (Fig. 15.2).
Fig. 15.1 
Astrocytoma. This WHO grade II diffusely infiltrating fibrillary astrocytoma shows mild hypercellularity and mild nuclear atypia
Fig. 15.2 
Gemistocytic astrocytoma. This WHO grade II tumor has moderate atypia with a deceiving lack of detectable mitotic activity (luxol fast blue/HE stain)
-
Grade III, anaplastic or malignant astrocytoma. This tumor has moderate to high cellularity (Fig. 15.3), prominent nuclear atypia, and readily identified mitotic figures. When neither tumor necrosis nor vascular proliferation is recognized, the distinction between grade II and grade III astrocytoma may be subjective. Mitotic count has been the most reliable criterion, with two or more mitoses found per microscopic section in grade III astrocytoma. The use of the MIB-1 labeling index to differentiate more aggressive astrocytomas from less aggressive ones has been suggested [4]; however, result of a recent study [5], significant overlap, and topographic variation of staining limit its practical use, especially in small specimens.
Fig. 15.3 
Anaplastic astrocytoma. Moderate hypercellularity and cytologic atypia are seen in this WHO grade III tumor
-
Grade IV, glioblastoma. This group of tumors includes astrocytomas that not only show nuclear atypia but also exhibit either necrosis (pseudopalisaded or not) (Fig. 15.4) or vascular proliferation (Fig. 15.5) or both. As the old term glioblastoma multiforme implies, the architectural pattern and cytologic details of neoplastic cells are variable. Small cells, clear cells, giant cells with bizarre hyperchromatic nuclei, cells with eosinophilic granular cytoplasm, and spindled cells in fascicles can be seen in various combinations or, less often, in pure forms. Aberrant epithelial (squamous and/or glandular) differentiation is rarely seen in glioblastomas and should not to be mistaken as collision cancer (carcinoma metastatic to glioblastoma), which occurs very rarely. Gliosarcoma is WHO grade IV tumor containing a glioblastoma component and a malignant mesenchymal component derived from vessel walls or associated meninges (Fig. 15.6).
Fig. 15.4 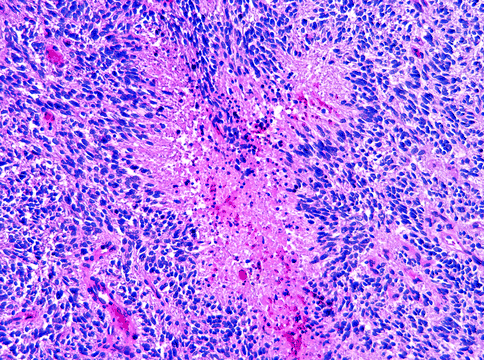
Glioblastoma. This WHO grade IV tumor shows characteristic pseudopalisaded necrosis
Fig. 15.5 
Glioblastoma. Characteristic vascular proliferation is evident in this WHO grade IV tumor
Fig. 15.6 
Gliosarcoma. This WHO grade IV tumor contains GFAP-negative, atypical mesenchymal cells embedded in massive hyalinized stroma






15.2.1 Special Variants of Astrocytoma
In addition to the common variety of infiltrating astrocytomas, several other forms are recognized, including:
-
Pilocytic astrocytoma (PA). The WHO grade I pilocytic astrocytoma is often cystic with an enhanced mural nodule. It is usually well circumscribed but may show microscopic infiltration in the surrounding parenchyma. It often occurs in or near the midline of neuraxis (e.g., cerebellum, hypothalamus, optic nerve) in children and young adults, but cases have been observed in all age groups and at all anatomic locations. The microscopic appearance of PA varies tremendously. Classic features include a biphasic growth pattern (fibrillary compact areas containing piloid cells and fibrillary processes with or without Rosenthal fibers, alternating with loose microcystic areas containing protoplasmic astrocytes in loose mucin [Fig. 15.7]), uniform cells, bland nuclei, eosinophilic granular bodies (Fig. 15.8), bipolar slender fibrillary cytoplasmic processes, and calcospherites. Other characteristics include a mild degree of cellular pleomorphism, nuclear atypia, giant cells with floret-like nuclei, hyalinized blood vessels, perinuclear halos, leptomeningeal infiltration, central infarctive necrosis, vascular proliferation, and palisaded spongioblastoma-like growth pattern. By themselves, necrosis and vascular proliferation are of no prognostic significance. In general, mitotic figures are rare and the MIB-1 labeling index is low (<4 %). Very rare cases of cerebellar PA with malignant features have been reported (WHO grade III) with hypercellularity, high mitotic activity, and pseudopalisaded tumor necrosis [6]. Rarely, a PA may undergo malignant transformation after irradiation, with characteristics of WHO grade III–IV anaplastic astrocytoma or glioblastoma.
Fig. 15.7 
Pilocytic astrocytoma. This WHO grade I tumor shows a fibrillary and loose microcystic biphasic growth pattern
Fig. 15.8 
Pilocytic astrocytoma. Numerous eosinophilic granular bodies can be observed in this WHO grade I tumor
-
Subependymal giant cell astrocytoma (SEGA). This WHO grade I circumscribed ventricular wall tumor is found either incidentally or after presentation with obstructive hydrocephalus. The majority of patients with this tumor have tuberous sclerosis; 6–16 % of patients with this disease develop SEGA. Classic features include a loose or packed collection of large polygonal or elongated cells, with plump or fibrillary eosinophilic cytoplasm in between hyalinized blood vessels (with or without perivascular pseudopalisading) and calcospherites. Some polygonal cells may contain large, round to oval, and ganglionic (neuronal) nuclei with a single, central prominent nucleoli (Fig. 15.9). Mild cellular pleomorphism, binucleation, and occasional mitoses may be seen, but tumor necrosis and vascular proliferation are rare and do not portend malignancy.
Fig. 15.9 
Subependymal giant cell astrocytoma. In this WHO grade I tumor, large neoplastic cells are seen, with round nuclei, central prominent nucleoli, plump eosinophilic cytoplasm, and occasional multinucleation
-
Pleomorphic xanthoastrocytoma (PXA). This WHO grade II tumor usually presents as a circumscribed, often partially cystic, superficial cerebral tumor in children and young adults. Characteristic features include elongated astrocytes, scattered giant cells with large hyperchromatic nuclei, intranuclear pseudoinclusions, vacuolated (lipidized) cytoplasm (Fig. 15.10), scattered eosinophilic granular bodies, perivascular inflammation, and pericellular deposition of reticulin fibers. Most cases are controlled or cured by complete surgical resection. A minority (15–20 %) of PXA manifest tumor necrosis, vascular proliferation, and a higher mitotic activity (>5 mitoses per 10 hpf). These so-called PXA with anaplastic features may have a less favorable prognosis [7]; however, they should not be confused with giant cell glioblastoma.
Fig. 15.10 
Pleomorphic xanthoastrocytoma. Large cells with bizarre hyperchromatic nuclei, intranuclear pseudoinclusions, and vacuolated cytoplasm are hallmarks of this WHO grade II tumor
-
Desmoplastic cerebral astrocytoma of infancy (DCAI) and desmoplastic infantile ganglioglioma (DIGG). These rare WHO grade I massive neoplasms of young children present as large, solid, and cystic cerebral tumors often associated with the dura and leptomeninges. Microscopically, desmoplastic hypocellular zones containing plump fibrillary astrocytes (Fig. 15.11) transform to hypercellular zones of round or spindled cells with atypia and high nucleocytoplasmic ratios (Fig. 15.12). In spite of the readily found mitotic figures and their alarming size, these neoplasms have a good prognosis following resection. The histopathologies of both are similar, except for the presence of small or large cells with neuronal differentiation in DIGG.
Fig. 15.11 
Desmoplastic infantile ganglioglioma. This WHO grade I tumor consists of neoplastic astrocytes with plump fibrillary cytoplasm embedded in sclerotic stroma
Fig. 15.12 
Desmoplastic infantile ganglioglioma. Some areas of this WHO grade I tumor are variably hypercellular and contain small cells with oval or elongated nuclei and scant cytoplasm
-
Rare variants of astrocytoma. This heterogeneous group, not discussed here, includes several entities: protoplasmic astrocytoma, gliomatosis cerebri, chordoid glioma of the third ventricle, granular cell astrocytoma, astroblastoma, pilomyxoid astrocytoma of the suprasellar region in young children and infants, angiocentric glioma, gliofibroma, and sarcoglioma. For grading and characteristic features of these tumors, see specialized neuropathology textbooks and original articles.






15.3 Oligodendroglioma
The WHO classification recognizes two grades of oligodendroglioma: well-differentiated (WHO grade II) and anaplastic (malignant) (WHO grade III). It should be noted that clinical response of many oligodendrogliomas to certain chemotherapeutic agents – such as a PCV regimen (procarbazine, lomustine or CCNU, vincristine) and temozolomide – has been associated with codeletions of chromosomes 1p and 19q found in up to 80 % of grade II and up to two-thirds of grade III oligodendrogliomas.
-
WHO grade II, well-differentiated oligodendroglioma. The classic features of well-differentiated oligodendroglioma include superficial parenchymal (cortical) involvement, infiltrative borders, uniform cells with round nuclei and perinuclear halos (Fig. 15.13), scant cytoplasm without processes, or globular eccentric eosinophilic cytoplasm with fibrillary processes (mini- or microgemistocytes) (Fig. 15.14), a delicate capillary network, associated calcospherites, mucinous microcysts, and occasional hypercellular nodules. Marked nuclear atypia and occasional mitoses may be present. As in infiltrating astrocytomas, mitotic activity has been proposed as a major grading criterion with most grade II lesions showing less than 6 mitoses per 10 hpf [8] and MIB-1 labeling index of <5 %.
Fig. 15.13 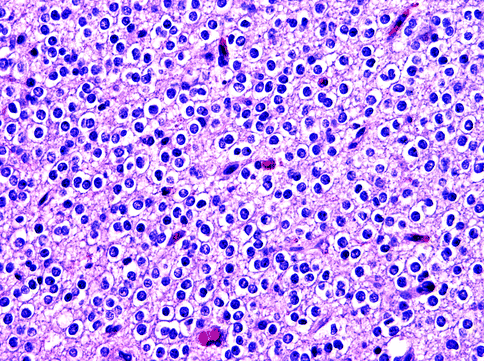
Oligoden-droglioma. This WHO grade II tumor has uniform neoplastic cells with round nuclei and perinuclear halos between delicate capillaries
Fig. 15.14 
Oligoden-droglioma. This WHO grade II tumor contains areas of mini- or microgemistocytes
-
WHO grade III, anaplastic or malignant oligodendroglioma. In addition to the features described above, this tumor shows diffuse hypercellularity, nuclear atypia, cellular pleomorphism, and brisk mitotic activity (Fig. 15.15). Vascular proliferation and tumor necrosis (with or without pseudopalisading) may be observed (Fig. 15.16), but neither feature is a necessary criterion. Some cases may display marked cellular pleomorphism and the formation of giant or elongated cells, making recognition of oligodendrocytic differentiation and distinction from astrocytomas difficult and subjective. Mixed oligoastrocytomas are diagnostic challenges in both classification and grading. A study has concluded that the identification of even a minor (100× microscopic field) component of oligodendroglioma imparts a better prognosis than a pure astrocytoma of the same grade [9]. Since there is no grade IV oligodendroglioma, distinction between anaplastic oligodendroglioma, anaplastic oligoastrocytoma, and glioblastoma can be arbitrary. An alternative is to classify it as WHO grade IV glioblastoma with an oligodendroglioma component, the prognosis of which appears better than that of conventional glioblastoma [10].
Fig. 15.15 
Anaplastic oligodendroglioma. This WHO grade III tumor displays nuclear atypia and a brisk mitotic activity
Fig. 15.16 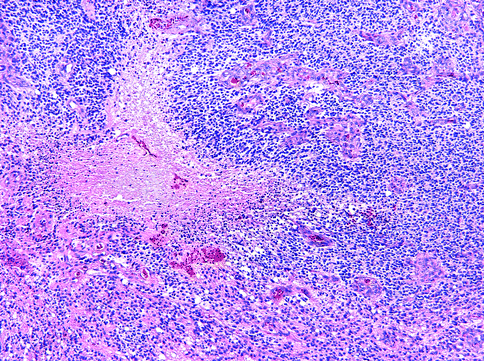
Anaplastic oligodendroglioma. Vascular proliferation and necrosis are sufficient but not necessary features in this WHO grade III tumor




15.4 Ependymoma
Ependymoma arises from ependymal cells lining the cerebrospinal fluid pathways. This tumor occurs in any age group and is found often in the posterior fossa of children and young adults. The WHO classification recognizes several subtypes, graded on a scale from I to III, as follows:
-
WHO grade I subependymoma. This group comprises nodular, well-demarcated, and fibrillary ependymal neoplasms on the walls of ventricles and the central canal (spinal cord). Microscopically, subependymomas are paucicellular and fibrillary, with the formation of microcysts and hyalinized vessels. Neoplastic cells are uniform in size and shape, with small round to oval nuclei, vacuolated or eosinophilic cytoplasm (Fig. 15.17) and tend to be clustered. Scattered cells with large pleomorphic nuclei of a degenerative nature and occasional mitoses may be seen, but necrosis and vascular proliferation is absent. Rare cases of WHO grade II mixed subependymoma and ependymoma containing features of both have been reported.
Fig. 15.17 
Subependymoma. In this WHO grade I tumor, cytologically bland cells and associated microcysts are observed in fibrillary matrix
-
WHO grade I myxopapillary ependymoma. This tumor is almost exclusively found in the conus medullaris/cauda equina region of young adults. Radially arranged, uniform, cuboidal, or elongated glial cells line the surface of papillae with vascular, hyalinized, or myxoid cores (Fig. 15.18). Mucinous vacuoles or microcysts exist between cells. Mitotic activity is low. Local recurrence after resection is uncommon.
Fig. 15.18 
Myxopapillary ependymoma. This WHO grade I tumor contains mucinous pools. The papillary growth pattern is a hallmark of this tumor
-
WHO grade II ependymoma. This often demarcated glial neoplasm shows ependymal differentiation, evidenced by cellular uniformity, varying amounts of fibrillary cytoplasm, and the formation of perivascular pseudorosettes (Fig. 15.19) and true ependymal rosettes (also known as ependymal canals) (Fig. 15.20). The borders are pushing or, less commonly, infiltrative. In general mitotic activity is low. Cystic change, stromal hyalinization, and calcification may be present. Observed multiple patterns of growth and cytology have given rise to four subtypes recognized by WHO: cellular ependymoma, clear cell ependymoma (Fig. 15.21), papillary ependymoma, and tanycytic ependymoma. Features of two or more of these subtypes may be seen focally in a given case, so a subtype is designated only when a pattern predominates (>50 % of the areas examined).
Fig. 15.19 
Ependymoma. Cells forming perivascular pseudorosettes comprise this WHO grade II tumor
Fig. 15.20 
Ependymoma. This area of the WHO grade II tumor contains an ependymal canal
Fig. 15.21 
Clear cell ependymoma. Some cells of this WHO grade II tumor have clear cytoplasm
-
WHO grade III (anaplastic or malignant) ependymoma. In addition to ependymal differentiation, this tumor shows anaplastic features such as hypercellularity, cytologic anaplasia, brisk mitotic activity, pseudopalisaded necrosis, and vascular proliferation (Fig. 15.22). However, necrosis without palisading in a posterior fossa ependymoma is not diagnostic of anaplastic ependymoma. Pseudorosettes are inconspicuous, and true rosettes are hardly seen.
Fig. 15.22 
Anaplastic ependymoma. Necrosis and vascular proliferation are evident in this tumor, as well as brisk mitotic activity and marked cytologic atypia






15.5 Embryonal Tumors, Neuronal Tumors, and Mixed Glioneuronal Tumors
When most constituents in a neuroepithelial neoplasm are poorly differentiated or undifferentiated, it is categorized as a WHO grade IV embryonal tumor or primitive neuroectodermal tumor (PNET). When found in the cerebellum, the pineal gland, and the posterior orbit, PNET is known as medulloblastoma, pineoblastoma, and retinoblastoma, respectively. A neuronal tumor consists of cells with neuronal (ganglionic or neurocytic) differentiation. They include WHO grade I neuronal hamartoma and gangliocytoma, and WHO grade II central (and extraventricular) neurocytoma. Neoplasms containing both mature neuronal and glial components are known as gangliogliomas or mixed glioneuronal tumors (e.g., papillary and rosette-forming variants, not discussed). Less frequently, a neuroepithelial neoplasm consists of a glial component – usually a diffusely infiltrating astrocytoma – and an embryonal component, without the formation of neurons or neurocytes. These are high-grade malignancies of WHO grade III to IV.
15.5.1 Medulloblastoma
These WHO grade IV embryonal neoplasms typically occur in the cerebellum (especially the vermis) of children and young adults. Like other PNET, medulloblastoma tends to spread through cerebrospinal fluid circulation and may metastasize to extraneural sites. Generally regarded as malignant, medulloblastomas show varying degrees of neuronal and glial differentiation in histopathology and immunohistochemistry. Four histologic subtypes are recognized (see below) with apparently different prognosis – patients with desmoplastic medulloblastomas and cerebellar neuroblastomas have a better prognosis than those with classic medulloblastomas, while those with large cell medulloblastomas have a worse prognosis.
-
Classic medulloblastoma. This is the most common form. It consists of diffuse sheets of embryonic cells with round-, oval-, or carrot-shaped hyperchromatic nuclei and scant cytoplasm with possible formation of neuroblastic or Homer-Wright rosettes (Fig. 15.23) and astrocytic differentiation shown by glial fibrillary acidic protein (GFAP) expression. Pale areas of lower cellularity and containing cells with neuronal and astrocytic differentiation may be observed (Fig. 15.24). Neoplastic cells infiltrate the neural parenchyma at interface. Apoptotic bodies and mitotic figures vary in density.
Fig. 15.23 
Medulloblas-toma, classic type. The tumor cells form Homer-Wright (neuroblastic) rosettes
Fig. 15.24 
Medulloblas-toma, classic type. In addition to undifferentiated cells, the tumor displays focal neuronal differentiation
-
Desmoplastic (nodular) medulloblastoma. This tumor tends to be located in the cerebellar hemisphere rather than the vermis. It displays biphasic histology and contains many hypocellular, sometimes confluent nodules between reticulin-rich and hypercellular areas (Fig. 15.25). Cells in nodules are more differentiated (neuronal, neurocytic, and astrocytic), with uniform, round to oval nuclei of varying sizes embedded in a fibrillary, neuropil-like matrix.
Fig. 15.25 
Medulloblas-toma, desmoplastic type. The tumor shows obvious nodularity
-
Medulloblastoma with extensive nodularity and advanced neuronal differentiation (cerebellar neuroblastoma). This rare tumor tends to occur in young children (usually <3 years of age). It displays a strikingly lobular appearance on neuroimaging that corresponds to multiple large nodules on histology. Intranodular neoplastic cells have small, round nuclei and resemble those found in central neurocytomas, accompanied by occasional large neurons.
-
Large cell (anaplastic) medulloblastoma. This tumor accounts for 5–25 % of all medulloblastomas. It consists of cells with large, round, or pleomorphic vesicular nuclei, prominent nucleoli, and abundant cytoplasm (Fig. 15.26). Frequently, there is nuclear molding, cell wrapping, necrosis, and high apoptotic and mitotic activities. Distinction from the highly malignant atypical teratoid/rhabdoid tumor (AT/RT, not discussed) in infants and young children may require the aid of immunohistochemistry.
Fig. 15.26 
Medulloblas-toma, large cell or anaplastic type. The tumor consists of cells with large, round, or pleomorphic vesicular nuclei showing molding or wrapping. The nucleoli are prominent and the cytoplasm more abundant than in classic medulloblastoma




15.5.2 Supratentorial Primitive Neuroepithelial Tumor
This WHO grade IV neoplasm also occurs in children and young adults with a histology (Fig. 15.27) similar to classic medulloblastoma. In general, their cells are poorly differentiated, but some may display divergent differentiation (e.g., neuroblastic, neuronal, astrocytic, ependymal, oligodendrocytic, muscular, and melanocytic). When both neuroblasts and differentiated neurons predominate, the terms cerebral neuroblastoma and ganglioneuroblastoma (Fig. 15.28) may be applied.

Supratentorial PNET. This WHO grade IV tumor shows histology identical to classic medulloblastoma

Ganglioneuro-blastoma, WHO grade IV. The tumor is composed of well-differentiated (right half) and poorly differentiated (left half) areas
15.5.3 Pineal Parenchymal Tumor
The histopathologic hallmark of pineal parenchymal tumors is the pineocytomatous rosettes – small- to medium-size, ill-defined zones of fibrillary processes rimmed by nuclei (Fig. 15.29). Pineocytomatous rosettes are larger and less regular in shape than Homer-Wright rosettes. These tumors include the following:

Pineocytoma. The tumor cells form pineocytomatous rosettes. These structures show vague circular, nuclear arrangements around fibrillary matrix and are larger than Homer-Wright rosettes
-
Pineoblastoma. This WHO grade IV tumor is a poorly differentiated neoplasm in children showing high-grade features expected of PNET.
-
Pineocytoma. This is a slow-growing WHO grade I tumor that affects young adults. It consists of sheets or lobules of small, uniform cells resembling normal pineocytes, arranged between small blood vessels (Fig. 15.30). Rarely, large- or medium-size ganglion cells and mitoses are found.
Fig. 15.30 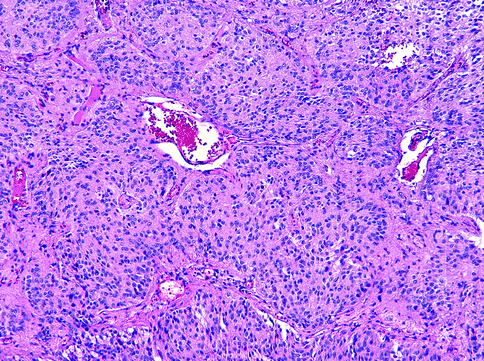
Pineocytoma. The tumor has a lobular structure, and it is difficult at times to distinguish pineocytoma from the normal pineal gland
-
Pineal parenchymal tumor of intermediate differentiation. This WHO grades II–III tumor shows signs of intermediate differentiation and has features that range between those of pineocytomas and pineoblastomas.

15.5.4 Gangliocytoma and Ganglioglioma
Gangliocytoma is a WHO grade I neoplasm of varying cellularity containing numerous differentiated ganglion cells (Fig. 15.31). Typically, this tumor contains hyalinized blood vessels with eosinophilic granular bodies, Rosenthal fibers, calcospherites, and perivascular inflammation. When both dysplastic neurons and neoplastic glia (usually astrocytes) are present, the neoplasm qualifies as a ganglioglioma. The glial component (astrocytic or oligodendrocytic) in a ganglioglioma determines its grade and behavior. It may be well differentiated similar to pilocytic astrocytoma (WHO grade I) (Fig. 15.32), anaplastic (WHO grade III), or, very rarely, indistinguishable from glioblastoma (WHO grade IV).

Gangliocytoma. Large dysplastic ganglion cells admixed with infiltrating lymphocytes and plasma cells are typical of these WHO grade I tumors

Ganglioglioma. This WHO grade I tumor contains dysplastic neurons and atypical astrocytes
15.5.5 Central Neurocytoma
This sharply demarcated WHO grade II neoplasm is typically found in the lateral and third ventricles (near the foramen of Monro) of young adults. It consists of uniform round neoplastic neurocytes with small, round to oval nuclei and pale, granular, eosinophilic, or clear cytoplasm between delicate or hyalinized blood vessels and scattered calcospherites (Fig. 15.33). Isolated or small clusters of larger ganglionic cells and small, ill-defined, and neuropil-like fibrillary zones may be present. Strong synaptophysin and NeuN immunoreactivity is typical and helps its differentiation from oligodendroglioma. There are no histological prognosticators, except that a MIB-1 labeling index of >2–3 % has been associated with a shorter recurrence-free interval. Extraventricular locations and cases with anaplastic features (WHO grade III) have been reported [11].

Central neurocytoma. This WHO grade II neoplasm bears striking resemblance to oligodendroglioma
15.5.6 Dysembryoplastic Neuroepithelial Tumor (DNET)
This WHO grade I glioneuronal neoplasm occurs in children and young adults. It has a supratentorial cortical (especially the temporal lobe) location and is multinodular on neuroimaging and gross examination. Characteristic histology includes prominent nodular growth, a specific glioneuronal element (small oligodendrocyte-like cells decorating delicate columns of axons arranged perpendicular to the cortical surface), small mucinous cysts containing floating neurons, scattered stellate astrocytes with or without brown granular cytoplasmic pigment, and adjacent cortical dysplasia (Fig. 15.34). Rare tumors with identical histology have also been reported in the septum pellucidum and corpus callosum.

Dysembryo-plastic neuroepithelial tumor. This WHO grade I neoplasm contains uniform, bland, and oligodendrocyte-like cells between small mucinous cysts containing floating neurons
15.6 Meningioma
Meningioma is a neoplasm of meningothelial or arachnoid cap cells found in the leptomeninges. It is assigned WHO grades of I to III and is known to display widely variable histopathology. The list of recognized variants of meningiomas continues to change, and at least 13 were described in the 2007 WHO classification. Some variants occur in pure forms (e.g., secretory and clear cell meningiomas), while diagnostic features of others (e.g., chordoid, rhabdoid, and papillary meningiomas) are found only focally. It has been proposed that variant-specific features be observed in >50 % of tumor in order for its designation although this has yet to be accepted universally. WHO grade II atypical meningiomas have a higher risk for local recurrence and malignant transformation after resection/treatment, and WHO grade III malignant (anaplastic) meningiomas have the additional potential of cerebrospinal fluid seeding and distant metastasis. Invasion into the adjacent brain parenchyma may be observed in meningiomas of all grades and is an independent risk factor for local recurrence (hence, the assignment of brain-invasive meningiomas as WHO grade II). Brain invasion is evidenced by an irregular rather than smooth pushing border at the interface (without intervening leptomeninges), with small irregular nests or burrowing tongues of neoplastic meningothelial cells displaced in the parenchyma (Fig. 15.35).

Brain-invasive meningioma. Fingerlike projections of neoplastic meningothelial cells into brain parenchyma without intervening leptomeninges are typical of this WHO grade II tumor
15.6.1 WHO Grade I Benign Meningioma
This group of tumors includes several variants:
-
Meningothelial or syncytial meningioma. Tumors of this group consist of neoplastic meningothelial cells with round, oval, or elongated nuclei, smooth nuclear profiles, dispersed chromatin, indistinct small nucleoli, and occasional intranuclear pseudoinclusions. Various architectural patterns and structures may be formed, most commonly whorls (Fig. 15.36) and syncytia (Fig. 15.37).
Fig. 15.36 
Meningothelial meningioma. Tumor cells form whorls
Fig. 15.37 
Meningothelial meningioma. Streaming neoplastic meningothelial cells display oval nuclei and intranuclear pseudoinclusions in syncytia
-
Fibrous or fibroblastic meningioma. This tumor is composed of slender fibrocyte-like cells between collagen (Fig. 15.38).
Fig. 15.38 
Fibrous meningioma. Elongated fibroblastic cells embedded in collagenous stroma comprise this tumor
-
Psammomatous meningioma. Psammoma bodies are present in most meningiomas, but if they predominate over tumor cells in between, the tumor may be classified as a psammomatous meningioma.
-
Secretory meningioma. This tumor has plump and epithelioid cells in sheets, forming scattered round vacuolar spaces that contain eosinophilic hyaline globules (pseudopsammoma bodies) (Fig. 15.39). Pericytic proliferation around blood vessels may be prominent. Surrounding brain parenchyma is often edematous.
Fig. 15.39 
Secretory meningioma. The tumor contains eosinophilic hyaline globules or “pseudopsammoma” bodies that are PAS positive and CEA immunoreactive
-
Angiomatous meningioma. Numerous, often hyalinized blood vessels between scarce neoplastic cells characterize this tumor (Fig. 15.40). It often coexists with microcystic change and may be associated with surrounding brain edema. It should not be confused with hemangioblastoma or meningeal hemangioma.
Fig. 15.40 
Angiomatous meningioma. The tumor is highly vascular and often contains cells with features of microcystic meningioma
-
Microcystic meningioma. This tumor consists of cells with thin elongated processes between clear or fluid-filled vacuoles and microcysts in a characteristic cobweb pattern (Fig. 15.41). Sometimes cells appear filled with small cytoplasmic vesicles that indent small or large hyperchromatic nuclei. Abundant, often hyalinized, blood vessels are present. It should not be confused with clear cell meningioma.
Fig. 15.41 
Microcystic meningioma. Numerous small vacuoles and larger microcysts between cells typify this tumor – note the characteristic “cobweb” pattern of cytoplasmic processes and peripherally placed nuclei
-
Lymphoplasmacyte-rich meningioma. In this rare variant, the tumor typically contains extensive infiltrates of chronic inflammatory cells.
-
Metaplastic meningioma. This is a rare variant characterized by the formation of bone, cartilage, and/or apparent fat.






15.6.2 WHO Grade II Atypical Meningioma
According the WHO classification, a diagnosis of atypical meningioma should be made if the tumor has an average mitotic rate of ≥4 per 10 hpf or has three or more of the following five features: prominent nucleoli (Fig. 15.42); tumor necrosis; sheet-like, patternless growth; small cells with high nucleocytoplasmic ratios; and hypercellularity (Fig. 15.43). Of these features, only tumor necrosis is objective. Tumor necrosis in meningiomas appears as small or large areas of coagulative necrosis rimmed by a condensed band of cells. It should be distinguished from rare spontaneous central infarction and from infarctive necrosis resulting from presurgical embolization. Occasionally, neoplastic meningothelial cells have enlarged, hyperchromatic nuclei, but these changes are degenerative and are not indicative of true atypia. Two specific variants are designated WHO grade II, due to their atypical clinical behavior:

Atypical meningioma. The WHO grade II tumor contains cells with prominent nucleoli

Atypical meningioma. The tumor appears hypercellular because the small hyperchromatic cells have scant cytoplasm and are compacted into “patternless” sheets
-
Clear cell meningioma. This rare neoplasm is composed almost exclusively of clear cells with glycogen-rich cytoplasm and central small nuclei (Fig. 15.44). Numerous thick collagenous fibers and, sometimes, prominent hyalinization are found in the stroma. Whorls are not apparent, and psammoma bodies are rare. Many are found at the cerebellopontine angle or the cauda equina region in children and young adults. They have a high risk for cerebrospinal fluid seeding and local recurrence after initial treatment.
Fig. 15.44 
Clear cell meningioma. This WHO grade II tumor is composed of bland cells with PAS-positive, diastase-sensitive clear cytoplasm and small, centrally located nuclei between “ropey” collagen
-
Chordoid meningioma. This tumor is rarely found in a pure form. Typically, it is composed of anastomosing cords of epithelioid, sometimes vacuolated, neoplastic cells in a myxoid background (Fig. 15.45).
Fig. 15.45 
Chordoid meningioma. The epithelioid tumor cells form inter-anastomosing cords in a myxoid background


15.6.3 WHO Grade III, Malignant or Anaplastic Meningioma
This group includes meningothelial neoplasms with a very brisk mitotic rate (≥20 per 10 hpf) and other atypical features, or dura-based sarcoma without meningothelial or heterologous differentiation (meningeal sarcomas) (Fig.15.46). The following two histological variants also are known to behave in a malignant fashion:

Meningeal sarcoma. Spindle-shaped tumor cells show marked atypia, a brisk mitotic activity, and no meningothelial differentiation
-
Papillary meningioma. This tumor has a predilection for children and young adults. The hallmark of this neoplasm is the formation of perivascular pseudorosettes in discohesive regions of the neoplasm (Fig. 15.47). Although neoplastic cells display bland cytology (Fig. 15.48), mitoses are readily found.
Fig. 15.47 
Papillary meningioma. The tumor cells line the fibrovascular cores of well-formed papillae
Fig. 15.48 
Papillary meningioma. The papillae are lined by neoplastic cells that still have meningothelial features, such as intranuclear pseudoinclusions (upper left) and whorls (not shown)
-
Rhabdoid meningioma. This variant is rarely found in a pure form, and meningothelial whorls are seen focally. Rhabdoid cells have large vesicular and eccentric nuclei adjacent to globular cytoplasmic bodies composed of intermediate filaments (Fig. 15.49). They are either nested or found in massive sheets.
Fig. 15.49 
Rhabdoid meningioma. The tumor cells have a well-developed cytoplasm that contains globular bodies and eccentric, frequently indented nuclei



15.7 Choroid Plexus Neoplasm
Neoplasms of the choroid plexus epithelium are found in or adjacent to the ventricles, including the cerebellopontine angle. Calcification is common. They present a spectrum spanning from discrete and easily resected benign papillomas to infiltrative high-grade carcinomas with intermediate forms:
-
Choroid plexus papilloma, WHO grade I. This benign neoplasm occurs in patients of all ages. Papillary fronds are lined by a single layer of cuboidal to columnar epithelia over fibrovascular cores. The nuclei of the epithelia are bland, and mitotic activity is low (Fig. 15.50). Focal cytoplasmic clearing may be present. Occasionally, small islands of glial tissue are incorporated in the papillary cores, but brain invasion, cytologic anaplasia, and necrosis are not seen.
Fig. 15.50 
Choroid plexus papilloma. This WHO grade I tumor is composed of cylindrical cells lining papillary structures
-
Atypical choroid plexus papilloma, WHO grade II. This recently described [12] intermediate grade of choroid plexus neoplasms is defined by a mitotic count of ≥2 mitoses per 10 hpf, often accompanied by some of the following atypical features – increased cellularity, nuclear pleomorphism, solid growth, and necrosis (Fig. 15.51).
Fig. 15.51 
Atypical choroid plexus papilloma. This WHO grade II papillary tumor shows hypercellularity and occasional mitotic figures (right lower corner)
-
Choroid plexus carcinoma, WHO grade III. These rare malignancies are found in young children. Histologically, choroid plexus carcinoma consists of papillary structures and solid hypercellular sheets of pleomorphic epithelial cells with readily identified mitoses, associated tumor necrosis, and brain invasion (Fig. 15.52). Poorly differentiated examples may be difficult to distinguish from atypical teratoid/rhabdoid tumor.
Fig. 15.52 
Choroid plexus carcinoma. This WHO grade III tumor invades the brain parenchyma. It has vacuolated cells and contains foci of necrosis



15.8 Hemangioblastoma
This WHO grade I vascular leptomeningeal neoplasm may occur anywhere in the neuraxis but most commonly present as cystic or solid enhancing masses in the posterior fossa. Up to 25 % of the patients with hemangioblastomas have familial (autosomal dominant) von Hippel-Lindau (VHL) disease caused by germ line mutations in the VHL tumor suppressor gene. This tumor consists of abundant capillaries between neoplastic “stromal” cells with round or oval nuclei of varying size, abundant clear or vacuolated, lipidized cytoplasm, and distinctive cellular borders (Fig. 15.53). Occasional stromal cells have giant hyperchromatic nuclei indented by cytoplasmic vacuoles. Mitotic activity is low, and necrosis is rarely seen. Occasionally it may be confused with angiomatous and microcystic meningioma.

Heman-gioblastoma. This tumor is composed of numerous capillaries and sinusoids between vacuolated neoplastic “stromal” cells
15.9 Craniopharyngioma
This WHO grade II tumor typically presents in the form of a circumscribed, often cystic mass in the pituitary fossa and the suprasellar region. It consists of neoplastic, stratified squamous epithelia between supporting stroma that may show old hemorrhage. The epithelia line cystic spaces or form anastomosing sheets with no cytologic features of malignancy. Two morphologic variants have been described – papillary and adamantinomatous. The epithelia in the adamantinomatous variant keratinize and form masses of “wet” keratins that tend to calcify or even ossify (Fig. 15.54). The surrounding gliotic parenchyma often contains prominent Rosenthal fibers.

Craniopharyngioma, adamantinomatous type. Strands and islands of stratified squamous epithelium with a prominent “stellate reticulum” pattern and embedded “wet” keratins that calcify focally
Acknowledgments
The author thanks Dr. Mark A. Edgar at the Emory University for his contribution of Fig. 15.26.
Books and Monographs
Louis DN, Ohgaki H, Weistler OD, Cavenee WK (eds) (2007) WHO classification of tumors of the central nervous system, 4th edn. International Agency for Research on Cancer, Lyon
Love S, Louis DN, Ellison DW (eds) (2008) Greenfield’s neuropathology, 8th edn. Hodder Arnold, London
McLendon RE, Rosenblum MK, Bigner DD (eds) (2006) Russell & Rubinstein’s pathology of tumors of the nervous system, 7th edn. Hodder Arnold, London
Perry A, Brat DJ (eds) (2010) Practical surgical neuropathology: a diagnostic approach. Churchill Livingstone/Elsevier, Philadelphia
Articles
Jansen M, Yip S, Louis DN (2010) Molecular pathology in adult gliomas: diagnostic, prognostic, and predictive markers. Lancet Neurol 9:717–726
Mittler MA, Walters BC, Stopa EG (1996) Observer reliability in histological grading of astrocytoma stereotactic biopsies. J Neurosurg 85:1091–1094
Daumas-Duport C, Scheithauer B, O’Fallon J et al (1988) Grading of astrocytomas. A simple and reproducible method. Cancer 62:2152–2165
McKeever PE, Strawderman MS, Yamini B et al (1998) MIB-1 proliferation index predicts survival among patients with grade II astrocytoma. J Neuropathol Exp Neurol 57:931–936
Lind-Landstrom T, Habberstad AH, Sundstrom S et al (2012) Prognostic values of histologic features in diffuse astrocytomas WHO grade II. Int J Clin Exp Pathol 5:152–158
Rodriguez FJ, Scheithauer BW, Burger PC et al (2010) Anaplasia in pilocytic astrocytoma predicts aggressive behavior. Am J Surg Pathol 34:147–160
Prayson RA, Morris HH 3rd (1998) Anaplastic pleomorphic xanthoastrocytoma. Arch Pathol Lab Med 122:1082–1086
Giannini C, Scheithauer BW, Weaver AL et al (2001) Oligodendrogliomas: reproducibility and prognostic value of histologic diagnosis and grading. J Neuropathol Exp Neurol 60:248–262
Coons SW, Johnson PC, Scheithauer BW et al (1997) Improving diagnostic accuracy and interobserver concordance in the classification and grading of primary gliomas. Cancer 79:1381–1393
Kraus JA, Wenghoefer M, Schmidt MC et al (2000) Long-term survival of glioblastoma multiforme: importance of histopathological reevaluation. J Neurol 247:455–460
Kane AJ, Sughrue ME, Rutkowski MJ et al (2012) Atypia predicting prognosis for intracranial extraventricular neurocytomas. J Neurosurg 116:349–354
Jeibmann A, Hasselblatt M, Gerss J et al (2006) Prognostic implications of atypical histologic features in choroid plexus papilloma. J Neuropathol Exp Neurol 65:1069–1073
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Ma, M.J. (2013). Tumors of the Central Nervous System. In: Damjanov, I., Fan, F. (eds) Cancer Grading Manual. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-34516-6_15
Download citation
DOI: https://doi.org/10.1007/978-3-642-34516-6_15
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-34515-9
Online ISBN: 978-3-642-34516-6
eBook Packages: MedicineMedicine (R0)