
Abstract
With about 25,000 species, fish represent approximately half of all vertebrate species. Yet out of approximately 60 vertebrate genome sequences in the public domain, only six are from fish species, of which two are pufferfish, Tetraodon nigroviridis and Takifugu rubripes. Interest in pufferfish genomes mainly stems from their incredible compactness. Their nuclear DNA content is about eight times smaller than that of human and other mammals, yet their gene content remains similar. This compaction is mainly due to a paucity of repeated sequences such as transposable elements, and a remarkable separation between heterochromatin and euchromatin compartments. Research in pufferfish genomics has led to key findings about human gene position and number. The large evolutionary distance between human and pufferfish enables highly specific genome sequence comparisons to be made, which appear to be mainly restricted to coding sequences. Analysis of the pufferfish genomes has also highlighted the notable stability of their chromosomes with regard to evolutionary rearrangements, which lead to the demonstration that a whole-genome duplication took place in an ancestral fish species about 300 million years ago (Mya). This dramatic event left a striking signature in extant genomes in the form of a clear distribution of duplicated genes in pairs of chromosomes originating from the whole-genome duplication. Experimental biology is not straightforward in pufferfish, especially when compared to other models such as zebrafish or medaka. Yet recent progress in breeding tetraodon in laboratory conditions has now opened new avenues to perform classical genetics and more advanced transgenics experiments with this species.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Bacterial Artificial Chromosome
- Bacterial Artificial Chromosome Clone
- Phylogenetic Footprinting
- Fish Genome
- Aquarium Trade
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
5.1 Introduction
With the completion of the genomes of six ray-finned fish the stage has been set for a renaissance in the comparative genomics of a group that encompasses half of all vertebrate species. Two smooth pufferfish (the green spotted pufferfish Tetraodon nigroviridis and the Japanese pufferfish Takifugu rubripes) are key players in the cast of newly sequenced fish genomes. Although the spotlight is currently on only a handful of completed genomes from this group the power of comparative approaches means that these sequenced genomes will play an ever greater role in shedding light on the biology of the multiple other fish species that remain in the murky unsequenced depths.
The prominence of pufferfish in the public imagination is a consequence of their use in the popular practice of aquarium keeping (in the case of T. nigroviridis) or in gastronomy (in the case of T. rubripes). Despite this familiarity, important aspects of pufferfish biology remain unknown, including those relating to their physiology, reproduction, and lifestyle.
The genomics revolution has redirected interest in both T. rubripes and T. nigroviridis to their potential as model organisms for genome sequencing (Brenner et al. 1993; Crnogorac-Jurcevic et al. 1997). This interest was provoked by a study predating the genomic era that determined the haploid nuclear DNA content of about 300 fish species and established the compact nature of the pufferfish genome (Hinegardner 1968). Hinegardner’s work showed that the haploid genome of fish belonging to the order Tetraodontiformes (including both T. rubripes and T. nigroviridis) is less than 500 Mb in size. This finding proved key to propelling both pufferfish into the genomics arena.
5.2 Pufferfish Divergence Time, Taxonomy, and Ecology
T. rubripes and T. nigroviridis belong to the superfamily Tetraodontidea (the “puffers”) and at a higher systematic level to the order Tetraodontiformes, which also includes the spiny pufferfish (Diodontidae), sunfishes (Molidae), boxfishes (Ostraciidae), and triggerfishes (Balistidae). The Order Tetraodontiformes includes approximately 350 species exhibiting extensive diversity in morphology and way of life (Tyler and Holcroft 2007). The divergence of fugu and tetraodon represents the divergence of the crown group of the Tetraodontidae. However, estimates of the time of this divergence based on molecular and paleontological data have shown discrepancies owing to the incompleteness of the fossil record.
The first estimate for this date came from an analysis of 376 bp of CytB sequence from T. rubripes and T. nigroviridis, which suggested that these lineages diverged as recently as 18–30 million years ago (Mya) (Crnogorac-Jurcevic et al. 1997). The completion of the mitochondrial genome of T. nigroviridis provided more extensive sequence data and was the basis for a new divergence date estimate of 85 Mya using an approach that did not assume the operation of a molecular clock (Yamanoue et al. 2006). Finally, a comprehensive synthesis of molecular and paleontological evidence from several animal model organisms has constrained the fugu–tetraodon divergence date to between 32.3 and 56.0 million years (Myr) (Benton and Donoghue 2007).
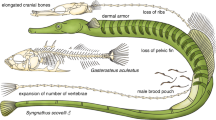
The evolutionary diversification of the Tetraodontidae is largely characterized by the reduction, simplification, or loss of morphological structures. In pufferfish, this is manifested in the absence of ribs and pelvic fins from the skeleton and the fusion of bones in the cranium and jaw. The fact that this tendency mirrors the trend towards pufferfish genome reduction has prompted speculation that the simplified pufferfish body plan has its origin in the recent reduction of genetic complexity in this lineage. This has been investigated in the context of Hox cluster evolution (see Sect. 5.14) (Amores et al. 2004).
T. rubripes (commonly known as the tiger pufferfish or “torafugu”) is a marine fish native to the Sea of Japan, East China Sea, and Yellow Sea where it reaches lengths of up to 70 cm and may attain a weight of several kilograms. Fugu was originally proposed as a model organism for genomics because of its compact genome. However, its potential as a laboratory model is limited by its relatively large size and the fact that it contains high doses of tetrodotoxin, a highly potent neurotoxin. These considerations impose practical limitations on fugu’s experimental use since large tanks of seawater are necessary to maintain fish and strict regulations are imposed on the importation of live specimens or frozen samples into non-Asian countries.
The green spotted puffer T. nigroviridis is a small fish (attaining lengths of up to 17 cm) native to the rivers and streams of South-East Asia. Although commonly referred to as the freshwater pufferfish, tetraodon’s ecological range extends not only to estuaries and mangrove swamps but also occasionally to the sea. Tetraodon’s status as a genomic model organism derives from its small size, ease of maintenance in a freshwater aquarium and its broad availability (Crnogorac-Jurcevic et al. 1997).
In the aquarium trade in particular, T. nigroviridis has frequently been confused with the closely related species Tetraodon fluviatilis, largely as a consequence of their morphological similarities (Eschmeyer 1998; Ebert 2001). Nineteenth-century taxonomists reported the identification of both pufferfish at approximately the same time yet T. fluviatilis possesses three distinctive large patches on its back in addition to numerous smaller spots, while T. nigroviridis displays only homogeneous small black spots. The confusion has now receded both in the aquaria trade and in the scientific literature, thanks in part to the work of Deckers (Dekkers 1975). To clarify things further, molecular markers have been identified that allow these species to be differentiated (http://www.genoscope.cns.fr).
Finally, using the tetraodon sequence to estimate levels of polymorphism in this species allows an estimate of the short-term effective population size (N e) of this species. This quantity is of importance not only in the field of evolutionary genetics but can also provide ecological insights by allowing an estimate of N (the total number of breeding adults or census size) by virtue of the fact that, in vertebrates, N is on average tenfold greater than N e (Frankham 1995).
Levels of tetraodon polymorphism (excluding indels) are estimated to range from 0.1 to 0.4%. Using this fact and the upper and lower bounds of the estimated date of fugu–tetraodon divergence, N e can be placed in the range ~90,000–520,000 for tetraodon. Therefore, assuming an effective population size of the order of 105, a reasonable estimate of the census population size for tetraodon is ~106.
5.3 Tetraodon Laboratory Maintenance
Of the two pufferfish species covered in this chapter, fugu is by far the more difficult to maintain in a controlled laboratory environment for the reasons outlined in the previous section, and thus will not be described here. Tetraodon, on the other hand, is a popular ornamental fish for home-based aquariums and can easily be purchased or ordered from the local aquaria specialist. As alluded to above, until recently, T. nigroviridis (“green spotted pufferfish”) was often mistakenly sold under the name T. fluviatilis in the aquaria trade. The latter is a different species and is in fact rarely found in aquarium shops. The following reference contains abundant descriptions and pictures of the different known species of fresh and brackish water pufferfish (Ebert 2001).
Maintaining live T. nigroviridis in the laboratory for short periods of time (up to 2 weeks) is sometimes required before tissue preparation, for example for RNA or DNA extraction or tissue preparation. Basic guidelines are provided below. For longer periods of laboratory maintenance, for example if breeding experiments are required, a more elaborate set up may be needed, described in details in Watson et al. 2009 (see Sect. 5.3.2).
Most individuals that are purchased through the ornamental fish market are likely to be juveniles, i.e., 2–5 cm in length. When choosing tetraodons, ensure that the animals are healthy: the skin under their stomach and along their sides must be white without any dark patches, they must swim actively with no signs of rigidity in their tails. It is frequent that fins are temporarily damaged through nipping from other fish, especially during the stress of transportation, and this should not be a problem as fins will regrow. As juveniles, they are likely to be sold in 26–28°C freshwater and can be kept in these conditions for a few days in the laboratory. Tetraodons are sturdy animals and do not require specific precautions. If they are sold in brackish water, they can nevertheless return to fresh water in the lab after a short acclimatization period of a few minutes.
5.3.1 Keeping Tetraodons in the Lab for up to 2 Weeks
Equipment (for 1 to approximately 10 small animals <5 cm):
-
A 40–60 L aquarium tank
-
A small electric heater
-
A small water pump
No water filters, lights, sand, salt, chemicals, etc., are necessary if the fish are healthy and only need to be maintained in the lab for a few days.
-
1.
A few days in advance of purchasing the fish, fill the tank with tap water. Heat the water to 26–28°C using the heater and put the water pump in place.
-
2.
The fish are generally sold in plastic bags. Float a bag on the surface of the aquarium water, slice it open in one or two places and wait for the fish to exit the bag. If the bag contains brackish water, first make smaller openings in the bag to wait for the salt concentration to equilibrate gradually before releasing the fish in the aquarium (a few minutes).
-
3.
After releasing the fish in the water, they need to be fed about once every 2–3 days with frozen mussel flesh or frozen blood worms (purchased in the aquarium shop). Tetraodons are carnivores and only eat fresh or frozen foods. They will refuse dry pellets or flakes.
If the fish need to remain in the aquarium for a longer period of time (several weeks/months/years), a slightly more elaborate system is required. Briefly, between 10 and 15 g of sea salt per liter of tap water needs to be added (mixed in the water before adding to the aquarium), a filtering system is required for the water pump, and regular water changes (e.g., 20% every 2 weeks) must be performed. Tetraodon’s teeth grow continuously during its lifetime and are filed, in their natural environment, through the regular need to break open and grind small crabs, snails, and shellfish. In the laboratory food needs to be adapted to this requirement, otherwise the teeth will grow to an extent that will prevent the fish feeding properly. In our laboratory, we buy live mussels and other small shellfish, and store them frozen. When needed, we defreeze the required amount, and preopen each shell before placing them in the aquarium.
5.3.2 Tetraodon Reproduction and Breeding
Many aquarium hobbyists have reportedly tried in the last decades to breed tetraodon, as well as several laboratories engaged in genome research with the tetraodon genome, including ourselves while at Genoscope, the French national sequencing center. All along however, hopes to reach this objective rested with the aquaria trade, which increasingly requires first that the trade does not deplete natural habitats and second, some guarantees that the animals are free of contamination, in good health and already acclimatized to captivity. To this end, efforts are being made to breed ornamental fish closer to their retail network rather than relying on the importation of wild fish from Singapore as is the case for tetraodon. In the case of tetraodon, success finally struck in Florida, where a group working in collaboration with the local aquaria trade developed a protocol to breed tetraodon (Watson et al. 2009). Fecundation takes place externally and several thousand embryos successfully reach adulthood per female. This is no small feat given that wild tetraodons are euryhaline and live in the mangroves of Indonesia, Malaysia, or Singapore. The wide range of salinity and habitats that they can tolerate made it difficult to predict the optimal condition required to breed tetraodon in captivity. No sexual dimorphism related to anatomy, weight, or color is apparent.
5.4 Sequencing and Assembly
The major difference between the draft assemblies of the two sequenced pufferfish is that assembly of the fugu genome was carried out exclusively by a whole-genome shotgun strategy without resort to physical mapping whereas a key feature of the tetraodon draft sequence is the physical anchoring to chromosomes of roughly two-thirds of its assembly. This has been of central importance to long-range studies of pufferfish genome structure and particularly in deciphering evidence relevant to the question of whether the ancestral teleost underwent whole-genome duplication (WGD).
5.4.1 Fugu Sequencing and Assembly
In November 2000, the International Fugu Genome Consortium of four partners (the Institute of Molecular and Cell Biology in Singapore, the Joint Genome Institute, the Human Genome Mapping Project, and the Institute for Systems Biology) was founded and charged with the generation and subsequent annotation of a draft sequence of the fugu genome. A whole-genome shotgun strategy was employed and in 2002 the analysis of version 2 of its assembly was published. Thus, fugu became the second vertebrate genome to be sequenced at draft level following on from the publication of the human draft sequence in 2001 (Aparicio et al. 2002). Notably, fugu was sequenced at the cost of only $12 million while the cost of the human genome project exceeded this by more than an order of magnitude thus vindicating the premise of the fugu genome project both with regard to the choice of organism and sequencing strategy.
The genomic DNA from the testis of a single fugu individual was used to construct shotgun libraries. Although the use of a single fish reduces the impact of polymorphism on the assembly process levels of polymorphism were nevertheless found to be roughly fourfold higher than that of human (0.4% of nucleotide sites were found to be polymorphic in the JGI assembly) (Aparicio et al. 2002).
Sequence reads were assembled into scaffolds using the JAZZ software. The initial assembly (version 2) consisted of 12,400 scaffolds covering 320 Mb of the genome and attained 5.6-fold redundant genome sequence coverage. Extrapolating from this led to an estimated total genome size of 365 Mb.
The current fugu assembly (version 4, released in October 2004) has progressed to ~8.7-fold coverage of the genome with a reduced count of roughly 7,200 scaffolds. This version produced an increase in the estimate of the genome size to 400 Mb of which 393 Mb are represented in the assembly.
5.4.2 Tetraodon Sequencing and Assembly
The tetraodon genome project was initiated by Genoscope and sequencing was undertaken as part of a collaboration with the Whitehead Center for Genome Research (now the Broad Institute). A whole-genome shotgun (WGS) approach was applied to shotgun libraries prepared from the genomic DNA of two individuals (Jaillon et al. 2004). Notably, polymorphism levels were unexpectedly high (1–2% including indels) and this proved to be a complication in the assembly process. Random paired-end sequences of plasmid and bacterial artificial chromosome (BAC) clone libraries yielded 8.3-fold redundant coverage of the genome. Inputting this to the Arachne assembly program generated about 50,000 contigs covering 312 Mb (92% of the genome). These were further connected by Arachne to form ~25,000 supercontigs covering 342 Mb including gaps. Tetraodon supercontigs were then linked into ultracontigs using physical mapping data from BAC clone fluorescence in situ hybridization (FISH) experiments, BAC clone fingerprinting and library hybridization, and from alignments to the fugu assembly. The tetraodon assembly exhibits extensive continuity at the level of N50 ultracontig size: half of the assembly is contained in ultracontigs of 7.62 Mb or greater. By contrast, half of the fugu assembly (version 4) is contained in scaffolds of 858 kilobases (kb) or greater. Thus, the continuity of the tetraodon assembly exceeds that of fugu almost ninefold. The largest ultracontigs were anchored to tetraodon chromosomes by FISH of BAC probes on metaphase chromosomes. This allowed the creation of a total of 128 ultracontigs representing ~80% of the genome and furthermore allowed the assignment of the 39 largest ultracontigs to tetraodon chromosomes, thus anchoring 64% of the assembly to chromosomes. Crucially, this has provided the first overview of a fish genome on a chromosomal scale and this, in turn, has permitted long-range studies of genome structure, particularly with respect to the postulated fish-specific whole-genome duplication.
5.5 Pufferfish Karyotype
Two studies have independently established the karyotype of tetraodon as consisting of 21 biarmed chromosomes of which roughly half are metacentric or submetacentric and half subtelocentric (Grutzner et al. 1999; Fischer et al. 2000). In contrast, fugu possesses 22 pairs of chromosomes (Miyaki et al. 1995). The availability of pufferfish genetic maps and whole-genome sequence now permits a more comprehensive enumeration of the rearrangements that took place between the two genomes since their divergence (see Sect. 5.8).
5.6 Features of the Pufferfish Proteome
With the completion of the draft sequence of the fugu genome and the availability of the human draft sequence came the first opportunity to compare the protein repertoire of two vertebrate species and to investigate proteome evolution across the full breadth of vertebrate evolution.
In this comparison, sequence similarity was used successfully to detect fugu homologs for 75% of human proteins (Aparicio et al. 2002). However, one quarter of human proteins remained without a detectable fugu homolog. These putatively human-specific proteins were predominantly associated with immune and hematopoietic functions with immune cytokines representing the most notable apparent absence from the fugu proteome. This led to the hypothesis that fish may lack these genes and that this family (that includes hormones and interleukins) may have evolved de novo in the tetrapod lineage.
In the light of this result, much effort was focused on detecting these genes, in particular, type I helical cytokines and their receptors in the second sequenced pufferfish, tetraodon (Jaillon et al. 2004). This analysis revealed 30 tetraodon genes in this category, including representatives of all known mammalian families. The apparent absence of fugu homologs of these genes may be due to the fast evolution and thus low sequence conservation of immunity genes, combined with the conservative similarity thresholds used in the analysis.
Few major points of functional difference between the human and tetraodon proteomes were revealed at the relatively coarse level of Gene Ontology term representations (Jaillon et al. 2004). However, protein domain enumeration using the InterPro classification revealed that domains concerned with sodium transport are more abundant in fish in an apparent reflection of their salt-water habitat. Strikingly, KRAB box transcriptional repressors involved in chromatin-mediated gene regulation were found to be completely absent from the fish proteome although the mammalian proteome has hundreds of representatives. Conversely, the diversity of collagen molecules is much greater in fish than in mammals. Similarly, purine nucleosidases are more abundant in fish and this is exemplified by an allantoin pathway for purine degradation found in tetraodon but absent from human.
In one sense the absence of clear functional differences between human and fish from comparison of their complete gene catalogs is surprising. However, it should be noted that this analysis is dependent on the completeness of gene annotation in human and tetraodon. Notably, the process of gene annotation in tetraodon was based on either homology to human proteins or tetraodon cDNA evidence. Therefore, among fish-specific genes those expressed at low levels are likely to be poorly represented among annotated tetraodon genes and this category of genes may be a significant source of fish-specific functions. A new annotation (v8.2) from Genoscope is now available at the Ensembl database and incorporates a much wider range of resources including more than two million mRNAs and EST sequence from several fish species.
5.7 Evidence for a Whole-Genome Duplication in Fish
Before the entry of the field of fish biology into the genomic era evidence for a putative fish-specific whole-genome duplication was limited to the apparent excess of duplicated fish genes for several different gene families (Wittbrodt et al. 1998; Meyer and Malaga-Trillo 1999). The most compelling of these anecdotal lines of evidence came from the number and distribution of Hox clusters in fish (Amores et al. 1998). The discovery in zebrafish of seven Hox clusters suggests that fish have sustained an increase in Hox cluster number compared to the archetypal mammalian repertoire of four Hox clusters. A particular “smoking gun” implicating whole-genome duplication came from the spatial distribution of these clusters (each of which maps to a different zebrafish chromosome). This arrangement of Hox clusters is consistent with WGD in the fish lineage and subsequent loss of a single Hox cluster in zebrafish.
However, more convincing support for the whole-genome duplication hypothesis had to await the completion of the genomes of both pufferfish species. Whole-genome analyses using both tree-based and map-based approaches galvanized support for a fish-specific whole-genome duplication (Christoffels et al. 2004; Jaillon et al. 2004; Vandepoele et al. 2004). The most compelling case for fish-specific duplication came from an analysis of the tetraodon genome that was made possible by the availability of long-range contiguous sequence in the genome assembly (Jaillon et al. 2004). This allowed the detection in tetraodon of two genomic properties that are tell-tale signatures of whole-genome duplication. These signatures persist despite the frequent loss of gene duplicates; a process that returns the majority of duplicated genes to single copy.
The first signature is provided by the direct pairing of paralogous sister regions within tetraodon chromosomes using the minority of genes that are retained in duplicate. A second and more powerful signature comes from considering the human orthologs of the majority of genes that have reverted to single copy in tetraodon. The human genome is itself paleopolyploid as a consequence of the postulated two rounds of genome duplication in early vertebrate evolution (Dehal and Boore 2005). However, since no additional round of whole-genome duplication has occurred in tetrapods since the fish/tetrapod split, mammals are suitable outgroups for investigating the fish-specific genome duplication. This comparison revealed a striking pattern in which pairs of paralogous chromosomes in tetraodon can be indirectly paired by projecting them onto a single orthologous human chromosome. Since each human chromosomal region exhibits conserved synteny with two sister regions in fish this 1 to 2 mapping is referred to as a “double conserved synteny” (DCS) map. The power of this indirect approach lies in its ability to unveil paralogous relationships between sister chromosomes in tetraodon whose pairwise resemblance has been eroded with the passage of time as a consequence of the progressive loss of gene duplicates.
Using this approach, a substantial portion of the tetraodon genome assembly could be incorporated into the DCS map. This can be appreciated from the fact that approximately 40% of tetraodon genes with a detectable mammalian ortholog could be mapped to DCS blocks.
5.8 Chromosome Evolution: Insights from the Resurrected Ancestral Teleost
The completion of the tetraodon genome permitted an ancestral genome reconstruction that for the first time shed light on the karyotype of the ancestral teleost (Jaillon et al. 2004). The availability of a double conserved synteny map consisting of paired sister chromosomes or chromosome segments was again instrumental in this analysis. It soon became apparent that the DCS blocks determined in the analysis of whole-genome duplication showed a distribution pattern consistent with the existence of 12 ancestral chromosomes. The modal value of the haploid chromosomal complement in teleosts is 24 in further support of the conclusion that the ancestral teleost possessed 12 chromosomes prior to the whole-genome duplication in this lineage. Moreover, it seems reasonable to assume that the ancestral teleost karyotype is a close approximation of that of the ancestral bony vertebrate (Osteichthyes) since these are separated by a short evolutionary period of perhaps 50 Myr.
Analysis of interspecific synteny conservation (between human and both the inferred ancestral teleost genome and the modern tetraodon genome) and of intraspecific synteny conservation (between the duplicated regions of the tetraodon genome) combined to shed light on the nature of chromosomal rearrangement in teleosts (Jaillon et al. 2004).
The interspecific comparison demonstrated that tetraodon and human show considerable conservation of synteny, implying that chromosomal integrity has been disrupted relatively rarely by interchromosomal rearrangements (Jaillon et al. 2004). Additionally, since fully 8 out of 12 DCS blocks consist of only two current tetraodon chromosomes interleaved on a single ancestral chromosome this indicates that the period since the whole-genome duplication has also been characterized by a low rate of interchromosomal exchange. Moreover, it appears that as few as ten large interchromosomal rearrangements separate the ancestral teleost genome from the present-day tetraodon genome. Strikingly, 11 tetraodon chromosomes have not been rearranged in this way.
Furthermore, the intraspecific comparison between duplicate regions of the tetraodon genome revealed extensive scrambling of gene order among paralogous genes (Jaillon et al. 2004). Therefore, although interchromosomal rearrangement has been relatively rare in teleosts, the chromosomal landscape has been extensively reshaped by intrachromosomal inversions.
More recently, the construction of a fugu genetic map and its comparison with the tetraodon genome assembly has confirmed these conclusions and has revealed the nature of more recent rearrangements since these species diverged (Kai et al. 2005, 2011). First, the extent of conserved synteny between fugu and zebrafish was found to be similar to that of human and mouse despite the fact that the fish lineages diverged earlier than the mammalian split. This supports the idea that interchromosomal exchange has been less frequent in teleost fish than in the mammalian lineage. Second, the conservation of synteny between fugu and tetraodon was quantified by constructing an Oxford grid associating fugu linkage groups with tetraodon chromosomes (Kai et al. 2005). This allowed the assignment of individual fugu scaffolds to orthologous segments of the tetraodon genome. Out of a total of 152 such segments distributed among 22 fugu linkage groups, only six showed evidence of relocation. This result testifies to the rarity of interchromosomal exchange since the divergence of fugu and tetraodon.
Comparison of the fugu genetic map with the physical assembly of the tetraodon genome has been informative in describing the small number of interchromosomal exchanges that have occurred since these species diverged. These include at least two interchromosomal rearrangements each of which may correspond to either a chromosome fission in fugu or a chromosome fusion in tetraodon. These rearrangements can be further clarified with reference to the reconstructed ancestral teleost genome. Combining these analyses points to two chromosomal fusions since the fugu–tetraodon split as underlying the creation of tetraodon chromosomes one and three.
A particularly noteworthy case of interchromosomal rearrangement has been highlighted by the availability of a chromosomally anchored genome assembly for tetraodon and of a genetic map for fugu. Moreover, the reconstructed ancestral teleost genome has served to shed further light on the nature of this rearrangement. It is apparent from comparison of these contemporary and ancestral maps that both pufferfish species deviate from the canonical vertebrate Hox cluster arrangement whereby each cluster is typically located on a different chromosome. The chromosomally anchored portion of the tetraodon genome assembly reveals that the HoxBb and HoxDa clusters both map to chromosome 2. Similarly, the fugu genetic map reveals that these clusters are also linked in fugu, both residing on Linkage Group 1 (Kai et al. 2005) implying that this rearrangement occurred prior to fugu–tetraodon divergence. The fact that this was a chromosomal fusion was revealed by the reconstruction of the ancestral teleost genome. Moreover, the observation that the HoxBb and HoxDa clusters map to different linkage groups in both medaka and zebrafish means that this chromosomal fusion must have occurred in an ancestor of modern pufferfish following the divergence of the zebrafish and medaka lineages (Kai et al. 2005). Medaka genomics and functional genomics are covered in more depth in Chap. 6.
In summary, these analyses have pointed to a surprising degree of resistance on the part of the pufferfish genome to the disruptive impact of interchromosomal rearrangements, particularly when compared to mammalian genomes. One possibility is that translocations have been more frequent in mammals as a result of the explosion in the number of transposable elements in this lineage following the divergence from teleosts (Jaillon et al. 2004). An alternative explanation is that the infrequency of such rearrangements in pufferfish is due to the fact that, in this gene-dense genome, rearrangement breakpoints are more likely to have highly deleterious gene-disrupting consequences.
However, neither repeat content nor gene density is likely to provide complete explanations of variation in the rearrangement rate of fish genomes. This is suggested by the observation that, in the past 300 Myr, the medaka genome has remained untouched by major interchromosomal rearrangements. This extraordinary genome stability stands in contrast to two features of the medaka genome. Compared to pufferfish, the medaka genome is more repeat-rich (17% of its genome is repetitive) and less gene-dense (it has approximately the same number of genes as pufferfish in a genome roughly twice as large) (Kasahara et al. 2007). This raises the possibility that other potential determinants of fish genome rearrangement rate remain to be uncovered.
5.9 Pufferfish Aids Functional Annotation of the Human Genome
Comparative genomics has the power to filter the abundant chaff of nonfunctional sequence in the vertebrate genome from the valuable wheat of protein-coding exons and regulatory elements. This filtering process, known as “phylogenetic footprinting” (Tagle et al. 1988), can be carried out by interspecies comparisons at a range of evolutionary distances that represent differing grades of stringency.
Crucially, any choice of compared species represents a trade-off between the two competing requirements of specificity and sensitivity. The need for specificity to provide optimal discrimination between functional and nonfunctional regions mitigates in favor of distant sequence comparisons (Boffelli et al. 2004). In this regard, comparisons between mammalian and teleost genomes are appealing since they represent a broad phylogenetic span that allows a high degree of specificity in detection of functional regions. At this scale sequence conservation persists despite 900 million years of divergent evolution since the tetrapod and fish lineages split ~450 Mya. Therefore, it seems reasonable to assume that this ancient conservation is a product of selective constraint’s opposition to the eroding effects of mutations that have caused neutrally evolving regions to diverge beyond recognition. However, with such distant comparisons the risk of missing functional regions that have emerged as lineage-specific innovations is greatly increased (Cooper and Brown 2008).
Of the sequenced teleosts, the pufferfish genome possesses a further advantage in phylogenetic footprinting studies with human. Apart from providing the discriminatory power of the long phylogenetic distance common to all teleost-mammal comparisons, the compact nature of the pufferfish genome means that it is significantly depleted of nonfunctional sequences. This reduces the probability that a candidate functional region generated by phylogenetic footprinting will prove to be a false positive in subsequent in vitro assays.
In the few years since their completion both pufferfish genomes have become valuable tools in pinpointing functional regions of the human genome by phylogenetic footprinting. In this regard, the availability of the tetraodon genome has contributed to improving the annotation of many human gene models through the application of the exon-finding tool “Exofish” (for Exon Finding by Sequence Homology) (Fischer et al. 2000). Conversely, the fugu genome has proven to be a popular choice in studies using noncoding sequence conservation between mammals and fish to pinpoint regulatory sequences (Woolfe et al. 2005).
5.9.1 Exofish: A Reappraisal of Human Gene Count
One of the primary motivations for sequencing the tetraodon genome was to facilitate the identification of genes in the human genome. To this end, the Exofish tool was developed to aid human gene annotation. Exofish makes use of the BLAST program and recovers exons among evolutionarily conserved regions (“ecores”) in human–pufferfish comparisons. At this phylogenetic distance most nonfunctional sequence, including most intronic and intergenic sequence, shows no detectable similarity whereas most exonic sequence is expected to be conserved by selective constraint.
In what has been described as the first large-scale comparison of vertebrate genomes (Heilig et al. 2003), Exofish was used to estimate the number of human protein-coding genes through a comparison of the tetraodon and human genomes (Fischer et al. 2000). The rationale for this choice of species rests on the fact that human and fish share most core vertebrate traits and functions and possess most of the same developmental pathways, organs, and physiology. It is therefore likely that genes shared between these species constitute the core vertebrate gene repertoire. Although gene duplication and de novo gene creation are likely to have expanded this gene set since the tetrapod and fish lineages diverged only the latter case (expected to be negligible in number) is beyond the scope of homology-based gene finding.
The first step in the Exofish study used a small benchmark set of known human genes and their fugu orthologs to establish BLAST search parameters that maximize the sensitivity and specificity of exon coverage among the retrieved ecores. When tested on a larger set of human genes represented by 4,888 complete cDNAs these parameters were found to detect 70% of genes each of which was represented by an average of 3.2 ecores corresponding to exons. Following this benchmarking, Exofish was run on the available partial sequence for both genomes (corresponding at that time to 33% of the tetraodon genome and 42% of the human genome) and the observed number of ecores was subsequently extrapolated to the entire genome. This produced a human gene number estimate of between 28,000 and 34,000 in human. In doing so, this study contributed to an overturning of the conventional wisdom that the human genome contained between 50,000 and 90,000 genes.
At the time, this result was surprising in that it brought the estimated number of human genes close to that of the fly and nematode whose genomes had also been sequenced. This result provoked a reappraisal of the assumption that human complexity could be explained as a simple function of an increase in gene number. Notably, the conclusion was supported by a similarly low estimate from a simultaneously published study using a different approach (Ewing and Green 2000). Both of these results contrasted with a third contemporaneous study that continued to support a higher estimate of 120,000 human genes (Liang et al. 2000). However, it is notable that a subsequent correction reduced this estimate by one half.
The fundamental conclusions of the initial Exofish result were shown to be robust with the completion of the tetraodon genome in 2004. Using the 99% complete human genome and a tetraodon assembly that covered more than 90% of the euchromatic fraction of the genome the number of human genes was reevaluated with Exofish. Crucially, the availability of manual gene annotation for five “finished” human chromosomes enabled the updating of the expected number of ecores per gene. First the number of ecores for the entire human genome was obtained and then corrected based on the known rate of occurrence of ecores in pseudogenes. This analysis placed the human gene count in the range between 22,500 and 29,000. Furthermore, Exofish permitted an estimate of the count of human pseudogenes in the range between 13,000 and 19,000.
These Exofish-based estimates have been corroborated by the more recent emergence of a consensus of 20,000–25,000 for the human protein-coding gene count since the finishing of the human genome sequence in 2004 (Churchill et al. 2004).
5.9.2 Expanding the Human Gene Catalog
The ecores detected by Exofish have been an important aid in the mammoth task of annotating the full complement of human protein-coding genes. At the time of the completion of the tetraodon genome approximately 14,500 ecores conserved between pufferfish and human were found to lie outside of human gene annotations. These were then processed to generate 904 novel human gene predictions (Jaillon et al. 2004). These genes are likely to have remained undetected because of their relatively small size. It was discovered that 60% of these gene predictions showed evidence of expression thus providing strong endorsement for the comparative approach to the detection of novel human genes using pufferfish. In addition, the Exofish approach using tetraodon sequence was applied in the manual curation of 11 human chromosomes (Dunham et al. 1999, 2004; Hattori et al. 2000; Deloukas et al. 2001, 2004; Heilig et al. 2003; Mungall et al. 2003; Humphray et al. 2004; Argmann et al. 2005; Gregory et al. 2006; Smyth et al. 2006).
5.9.3 Phylogenetic Footprinting of the Human Genome: Using Pufferfish Minimizes False Positives
Recently, the pufferfish genome has been successfully used in whole-genome phylogenetic footprinting studies directed at the discovery of regulatory elements. Even prior to the completion of vertebrate genome sequencing, single gene analyses provided a “proof of principle” supporting this approach. An early illustration of its potential came from studies that successfully identified regulatory elements for the Hox genes (Aparicio et al. 1995; Popperl et al. 1995). For example, a noncoding region that is conserved in human was discovered in sequence flanking the fugu Hox4b gene and was shown to exhibit enhancer activity in transgenic mice (Aparicio et al. 1995).
These results spurred on genome-wide studies of potential regulatory regions that compare the human and pufferfish genomes in order to compile the complete repertoire of their conserved noncoding elements (CNEs). Among these efforts, one pioneering study detected 1,400 CNEs in a whole-genome comparison of fugu and human (Woolfe et al. 2005). This CNE set was clustered by physical proximity on the chromosome with each cluster of CNEs likely to represent a modular array of regulatory sequences. Next, the closest human gene was designated as the likely regulatory target of each CNE cluster. Using this approach, more than 90% of CNE clusters were shown to lie within 500 kb of a human gene functioning in either transcriptional regulation or development (so-called trans-dev genes). Strikingly, fully 23 out of a sample of 25 CNEs associated with four different developmental gene clusters were found to promote GFP reporter expression in a tissue-specific manner in zebrafish embryos.
Having established the regulatory activity of these fugu–human CNEs the next question addressed whether the in situ expression pattern directed by these elements corresponds to that of their presumed target genes. The CNEs associated with PAX6 and SOX21 were found to direct GFP expression patterns that were in good agreement with the endogenous expression of the “associated” gene, thus supporting the assumption that these are the true “target” genes of the tested CNE. However, some CNEs in the vicinity of HLXB9 and SHH drove reporter gene expression in tissues that were not typical of the associated gene’s expression domain. One possible explanation for this discrepancy is that the assumption that the nearest gene to a given regulatory element must necessarily be its target is not always correct. In fact it has been demonstrated that tissue-specific enhancers can regulate their target genes over long genomic intervals and that these intervals frequently encompass many so-called bystander genes that typically are of unrelated function and are broadly expressed (Goode et al. 2005; Kikuta et al. 2007).
More recently, a larger scale study investigated the function of a sample of over 3,100 conserved noncoding elements detected in a comparison of the fugu and human genomes (Pennacchio et al. 2006). The human sequence of 137 of these CNEs was tested for enhancer activity at embryonic day 11.5 in a transgenic mouse assay. Of these, 57 elements (41%) were shown to reproducibly function as transcriptional enhancers in a tissue-specific manner. Interestingly, a large fraction of fugu–human CNEs were found to also be ultraconserved among human, mouse, and rat and of these 61% functioned as enhancers. Moreover, a majority of these enhancers activated expression in the developing nervous system, in agreement with previous results that have suggested human–fish CNEs are enriched for these functions (Woolfe et al. 2005).
This study also sought to test whether the detected CNEs recapitulate the endogenous expression of their presumed target but in this case did so by considering a single putative target gene. Of 23 CNEs in a gene desert flanking the SALL1 gene, seven were shown to recapitulate its expression domain. This observation echoes that of Woolfe et al. and raises the possibility that many of these elements regulate the expression of more distant genes or that the specificity of some enhancers is contingent on their simultaneous presence in the regulatory module, a situation that is not replicated in such assays.
Finally, the fugu genome has aided in the detection of putative regulatory regions within the human Hox clusters in a study that has pointed to the possible untapped potential of pufferfish in functional studies (Gregory et al. 2006). Numerous human–fugu CNEs extend throughout the intergenic regions between Hox genes and into flanking sequence. Notably, the arrangement of regulatory elements in these loci could impose a significant degree of selective constraint that preserves gene neighborhoods from disruption by rearrangements (Goode et al. 2005; Gregory et al. 2006).
5.10 Streamlining the Genome: Depletion of Transposable Elements and Pseudogenes
The key property underlying the streamlining of pufferfish genomes is the striking depletion of repetitive DNA associated with transposable elements. Less than 10% of the genome assemblies of tetraodon and fugu consist of repetitive DNA in marked contrast to the preponderance of these sequences in mammals where they make up nearly half of the genome (Consortium 2001). In fact it was this property that lead to the original proposal to sequence the pufferfish genome as a low-cost route to capturing the full catalog of vertebrate genes (Brenner et al. 1993).
Notably, the scarcity of transposable elements in pufferfish is not a consequence of their quiescence. These elements show evidence of recent activity (Bouneau et al. 2003) and represent a wide diversity of families each with relatively few representatives (Aparicio et al. 2002; Jaillon et al. 2004). An illustration of this comes from contrasting the degree of retrotransposon diversity in mammalian and fish genomes. Although the fugu and tetraodon genomes contain 23 and 16 retrotransposon clades, respectively, only six clades are found in mammalian genomes (Bouneau et al. 2003). Therefore, the genomes of mammals and fish display two starkly contrasting properties with respect to transposable elements. First, in the case of pufferfish, the increased diversity of retrotransposons is paradoxically achieved with copy numbers that are an order of magnitude lower than those of mammals. Conversely, in mammals, the great abundance of transposable elements (e.g., constituting 45% of the human genome) is attained through the massive amplification of only a handful of families.
A further striking feature of pufferfish TEs is their highly nonrandom spatial distribution in the genome. TEs are depleted from the euchromatin of tetraodon and are enriched in the heterochromatic short arms of 10 subtelocentric chromosomes, a pattern that is replicated by pufferfish pseudogenes (see below) (Dasilva et al. 2002). Since only the euchromatic portion of the pufferfish genome has been sequenced it remains possible that the global TE content of the pufferfish genome has been slightly underestimated.
The distinctive spatial arrangement of TEs in the tetraodon genome relative to that of the mammalian genome extends beyond their heterochromatic compartmentalization. A further intriguing difference relates to the distribution of the minority of TEs that are located in the euchromatin. Rather than exhibiting the enrichment of LINEs and LTRs (long terminal repeats) in AT-rich regions and SINEs in GC-rich regions typical of mammalian genomes (Consortium 2001; Gregory et al. 2002), the analysis of the tetraodon genome showed that its TEs show exactly the opposite bias. The reasons for these differences between pufferfish and mammals are unknown, but the preferred integration site for LINEs and the nucleotide composition of the individual elements may play a role in shaping their ultimate distribution in the genome.
At a shorter evolutionary timescale, interesting differences also become apparent when comparing the TE profiles of both sequenced pufferfish genomes. There are several TE families that are present in only one of the two pufferfish species indicating that recent lineage-specific TE activity has continued to shape these genomes since their divergence (Bouneau et al. 2003).
An alternative perspective on the evolution of nonfunctional DNA in the pufferfish genome is provided by pseudogenes which, like TEs, are depleted relative to their abundance in vertebrate genomes. Mammalian genomes could potentially contain as many pseudogenes as functional genes (Torrents et al. 2003) and the appearance of processed pseudogenes coincides with bursts of TE activity (Bentley et al. 2008). Pseudogenes are either created by the genomic integration of reverse-transcribed mRNA (in which case they are termed “processed” pseudogenes) or by duplication of genomic DNA (“unprocessed” pseudogenes). The rarity of pufferfish pseudogenes, at least in euchromatin (see below), is likely to have two causes. First, the paucity of processed pseudogenes may be due to the relative quiescence of LINE activity in pufferfish since LINE-encoded enzymes are required to catalyze retrotranscription. Second, both processed and unprocessed pseudogenes might be depleted by a high neutral rate of deletion in this lineage.
The conventional wisdom that pufferfish genomes are almost devoid of pseudogenes has been reassessed following the discovery of two pseudogene families that are restricted to the heterochromatic fraction of the genome (Dasilva et al. 2002). The first of these is an unprocessed pseudogene with a chimeric structure composed of parts of two active tetraodon genes (the homologs of human EZH2 and TRAPα). Both parental genes exist in the tetraodon genome and it appears that these gave rise to the Trapeze pseudogene following their duplication and fusion. The resultant pseudogene consists of eight exons from EZH2 at its 5′ end, three exons from TRAPα at its 3′ end and an intervening fused exon derived from both genes. This chimeric pseudogene has been amplified to an estimated 50 copies in the genome and appears to colocalize with TEs in the short heterochromatic arms of subtelocentric chromosomes.
The second pseudogene found at high abundance in tetraodon (named “iSET”) is an intronless sequence homologous to the last 4 exons of tetraodon EZH2. Although this points to the fact that iSET is a processed pseudogene originally created by retrotransposition, its subsequent amplification appears to have been achieved by segmental DNA duplication. This sequence has attained a copy number of approximately 240 in the genome and interestingly also has a heterochromatic location in the vicinity of Trapeze and of TEs. It is intriguing that two distinct pseudogenes, derived from the same source gene (EZH2) but generated by different mechanisms, are found as near neighbors in the same genomic compartment.
A clue to explaining the compaction of the pufferfish genome has come from a study of the evolution of genome size in smooth tetraodontids since their divergence from the spiny diodontid pufferfish (Neafsey and Palumbi 2003). In the 50–70 Mya since these lineages diverged, the pufferfish genome has undergone a significant size change: the smooth tetraodontid puffers have a haploid genome size of ~400 Mb, which is half that of their spiny diodontid cousins (haploid size ~800 Mb). Moreover, because the sunfish, Mola mola (an outgroup to these groups) possesses a genome similar in size to that of the spiny pufferfish this implies that the genome size difference between contemporary pufferfish is due to a process of genome compaction in the smooth pufferfish lineage. By examining the neutral accumulation of insertions and deletions (collectively termed “indels”) in defunct non-LTR retrotransposons, it was confirmed that both pufferfish lineages are subject to a mutational process biased towards DNA loss (Neafsey and Palumbi 2003). This is as a consequence of deletions that are larger and more frequent than insertions. This is consistent with the results of DaSilva et al. Both studies show that, in pufferfish, a greater deletion bias prevails than that seen in mammals: deletions in pufferfish averaging 7 bp (Dasilva et al. 2002) to 19 bp (Neafsey and Palumbi 2003) are larger than deletions in mammals (3.2 bp on average) (Graur et al. 1989).
Although such differences in small-scale deletion bias appear to account for the eightfold compaction of the pufferfish genome relative to the human genome this factor does not account for the twofold size difference between the genomes of smooth and spiny pufferfish. The similar indel profiles for smooth and spiny pufferfish at the observed length scale imply that the difference in their genome sizes is due to a difference in indel profiles at a larger scale. Therefore, smooth pufferfish should exhibit either a higher rate of large deletions or a lower rate of small insertions compared to spiny pufferfish. Given that large deletions are expected to be more deleterious than large insertions (particularly in a genome of high gene density) Neafsey et al. speculate that a reduced rate of large insertions in smooth pufferfish is the more plausible explanation.
Overall, the profiles of TEs and pseudogenes in the pufferfish genome are reminiscent of those in other compact metazoan genomes (e.g., Drosophila and Arabidopsis). In the Drosophila genome, for example, TEs exhibit low overall abundance and a high diversity of families, each of which has few representatives (Bartolome et al. 2002). This is coupled with a bias in the spatial distribution of Drosophila TE families towards heterochromatic regions. On the face of it, it would appear that similar mutational or selective forces have shaped the transposon populations of these two genomes. However, such a TE profile can have either a mutational or a selective explanation. For example, the enrichment of TEs in heterochromatin may be the result of a mutational bias owing to preferential insertion of TEs into regions that are already TE rich. Conversely, purifying selection may oppose the deleterious effects of TE insertion events that disrupt genes or that promote ectopic recombination. Using population genetic data to disentangle these two alternatives has shown that TE insertion in tetraodon accords with neutral expectations in contrast to Drosophila where TE activity bears the hallmarks of purifying selection (Neafsey and Palumbi 2003). These contrasting results bear testament to the fact that a single genomic property (compaction) can be a consequence of two very different potential causes (mutational or selective).
There is evidence that the genome-wide tendency towards compaction in pufferfish is likely to have had an impact on the degree of retention of duplicate genes. Analysis of the fugu genome has revealed that although the ancestral teleost genome was expanded by an ancient whole-genome duplication, the modern pufferfish genome contains fewer young gene duplicates than expected (Vandepoele et al. 2004). This may be a consequence of either a recent reduction in the rate of gene duplication or an increased rate of duplicate gene loss in the early neutral phase of duplicate gene evolution. Although these two factors are not mutually exclusive it is notable that the latter possibility is consistent with the high rate of loss of neutrally evolving nonfunctional DNA described earlier.
Finally, it is interesting to note that although there is a universal tendency towards streamlining in the pufferfish genome some genes manage to buck this trend (Aparicio et al. 2002). Analysis of the fugu genome revealed the existence of “giant” genes whose loci span a larger genomic interval in fugu than in human due to the larger size of their introns. In fact, a total of 571 fugu gene loci were found to be at least 30% larger than their human orthologs. Most strikingly, the giant musashi gene spans ~176 kb in fugu but <50 kb in human.
5.11 What Explains the Wealth of Fish Species?
It has been postulated that whole-genome duplication may play a role in fuelling the process of speciation through at least two theoretical mechanisms which could underlie this association. Firstly, the genetic raw material provided by WGD could fuel adaptive radiation into novel nonoverlapping ecological niches among post-WGD descendent lineages (Ohno 1970; Meyer and Malaga-Trillo 1999; Otto and Whitton 2000). Alternatively, an increased rate of speciation can proceed neutrally as a consequence of duplicate gene loss. If there is reciprocal loss of duplicate genes among post-duplication lineages this can lead to reproductive isolation in accordance with a Bateson–Dobzhansky–Mueller speciation model (Werth and Windham 1991; Lynch and Conery 2000; Deloukas et al. 2001). This phenomenon was first demonstrated in the context of the well-known whole-genome duplication in yeast by genome comparisons between multiple pre- and post-duplication yeast species (Scannell et al. 2006). This study relied on the confident distinction between orthology and paralogy among one-to-one interspecies homologs in post-duplication yeast species. The analysis was based on the recognition that, not all one-to-one interspecies homologs in post-duplication yeast species are orthologs (genes whose lineages diverged as a result of speciation) but that, as a consequence of reciprocal gene loss, some are paralogs (genes whose lineages diverged following gene duplication). In the case of yeast, the strong conservation of gene order among species meant that the distinction between homology types was aided by synteny-based inferences of orthology. In contrast, the high rate of chromosomal inversions in fish species means that local gene order is disrupted, thus complicating the use of conserved synteny in discerning paralogs from orthologs. This complication has been overcome in an elegant study that succeeded in detecting reciprocal gene loss in post-WGD fish species and has shown that these occur at a rate comparable to that observed in yeast (Semon and Wolfe 2007).
Although the fish-specific whole-genome duplication provides a seductive explanation for the incredible species richness of the teleost fish, establishing a causal connection is not straightforward. As a minimum this assertion requires the demonstration of a close temporal correlation between the WGD and the teleost radiation although such a correlation is not sufficient to infer causality. Currently, circumstantial evidence provides only ambiguous support for an association between WGD and speciation. The example of rich species diversity in tetraploid lineages such as Salmonids is negated by the counterexample of polyploid amphibians and reptiles that do not show such diversity (Venkatesh 2003). Similarly, the case for duplication-driven speciation is not clearly supported by the Cambrian explosion which appears to have occurred a period of genomic quiescence in the interval between the first two rounds of WGD (Miyata and Suga 2001).
Notably, it has been suggested that the apparent correlation between the fish-specific WGD and increased species richness is an artifact of incomplete taxon sampling that disappears when extinct lineages are considered (Donoghue and Purnell 2005). Furthermore, it has been noted that one group that accounts for much of teleost diversity, the acanthopterygians, underwent a species radiation as recently as 55 Mya without experiencing an additional WGD (Mulley and Holland 2004). Finally, a further study has also suggested that, rather than coinciding with the WGD, the teleost radiation significantly postdates it (Hurley et al. 2007).
If the WGD itself is not responsible for the extraordinary diversity of fish species this raises the question of what other aspect of fish genome evolution could be responsible. The high rate of intrachromosomal rearrangement described in fish may provide an explanation in the context of the chromosomal speciation hypothesis (White 1978). In this context, the fixation of different chromosome rearrangements in distinct populations is postulated as a lineage-splitting force. The suppression of recombination in rearranged regions may construct a genetic barrier by reducing gene flow between the incipient species. However, multiple studies of diverse eukaryote species have produced comparatively little evidence in support of the chromosomal speciation hypothesis (Coghlan et al. 2005). Here again even the demonstration of a strong temporal association between increased rates of rearrangement and speciation would not constitute proof of a causal relationship.
5.12 Sex Determination in Pufferfish
Teleost fish display a kaleidoscopic array of modes of sex determination. Genetic sex determination mechanisms encompass male heterogamety, female heterogamety, autosomal and polygenic mechanisms. Furthermore, sex determination in fish can also have environmental influences including temperature and pH of water and fish density (Volff 2005).
This diversity of sex-determination mechanisms implies that there is no master sex determining gene operating universally in teleosts equivalently to the male-determining gene Sry in mammals and other vertebrates. The identification of DMY in medaka constitutes the first discovery of a master sex determining gene in fish (Nanda et al. 2002). This gene, located on the Y chromosome, arose by duplication of the autosomal gene dmrt1. The high level of sequence similarity between these duplicates suggests that this is a recent event. A search in both sequenced pufferfish genomes established that DMY is absent from these genomes. Moreover, phylogenetic analysis has confirmed that this duplication took place specifically in the medaka lineage after divergence from the other percomorphs (Lutfalla et al. 2003). A second analysis established that DMY is absent even from medaka’s closest relatives (Kondo et al. 2003). More recently, linkage mapping in stickleback has localized its sex-determining locus to a nascent Y-chromosome (Peichel et al. 2004).
The plasticity of sex determination in fish is further emphasized by the discovery of a sex-determination locus in fugu that has evolved independently of those in medaka and stickleback (Kikuchi et al. 2007). This study exploited the strong conservation of synteny in pufferfish in an approach integrating the genetic map of T. rubripes and the physical map of tetraodon. Notably, the orthologs of genes flanking the sex-determining locus of fugu were not found to map to the medaka and stickleback sex chromosomes. This is evidence for the independent and relatively recent emergence of sex determination in the pufferfish lineage. In summary, it appears that the sex-determination pathway in fish is highly plastic and evolves very rapidly. Moreover, the diversity of sex-determination mechanisms may have its origin in this plasticity.
5.13 Pufferfish as a Tool for Comparative Biology
In the recent past, the completed pufferfish genomes have begun to serve as informative points of reference for vertebrate biology in general and for fish biology, in particular. In the latter context, it is noteworthy that many fish species of evolutionary, environmental, or commercial importance remain poorly served by genomics projects. Therefore, genomic studies of such species are dependent on the ability to transfer both positional and functional genomic information from sequenced reference genomes (e.g., pufferfish) by means of comparative genomics. For example, sea bream, sea bass, rainbow trout and salmon are among the fish species of importance in aquaculture that now lie within close evolutionary range of sequenced pufferfish. This is also the case for groups of evolutionary interest that are currently unserved by genomics projects such as guppies, cichlids, and Xiphophorus.
The transfer of positional information from model organisms, whose genomes are sequenced to other, as yet poorly characterized, genomes is termed comparative mapping. If significant synteny conservation exists between the studied species this allows the confident assertion that a pair of linked genes (or markers) of interest in the reference organism is also linked in the nonmodel organism.
Conversely, the transfer of functional genomic information is usually carried out between pairs of finished genomes and exploits high-quality gene annotation in one genome to describe gene function in the second. This strategy relies on the confident assertion that the genes considered are orthologs. Despite the power of this approach, a caveat should be raised relating to occasional departures from the dogma that orthologous genes share the same functions. Therefore, the demonstration of orthology is not always sufficient for the projection of functional information between orthologs. Among such departures are cases of partitioning of functions between duplicate genes that proceed differentially between lineages, a phenomenon that is particularly relevant in the case of paleopolyploid genomes (Cresko et al. 2003).
5.13.1 Pufferfish as a Reference for Comparative Mapping
It was initially hoped that the transfer of genomic information from pufferfish could be extended as far as the human genome and that conserved synteny would expedite the process of physical mapping of human genes, then a priority in the Human Genome Project (Brenner et al. 1993). However, prior to the completion of the fugu genome studies of individual loci (e.g., the Surfeit locus) showed that there is limited long range conservation of gene order between fugu and human (Gilley et al. 1997). Despite the demonstration that genes belonging to the mammalian Surfeit locus are rearranged in fish, this study highlighted the annotation potential of pufferfish since the exon–intron structures of the constituent genes are conserved between fish and mammals. The lack of conserved gene order can be understood in the context of two distinct aspects of fish genome evolution. First, gene order is disrupted by a high rate of intrachromosomal rearrangements (i.e., inversions) in fish chromosomes (Jaillon et al. 2004).
Second, despite the scarcity of interchromosomal rearrangements across this timescale, microsynteny is disrupted by patterns of duplicate gene loss subsequent to the fish-specific whole-genome duplication. This is highlighted in the case of the Parahox genes which, although clustered in most vertebrates, have redistributed between paralogous sister regions in fish following the whole-genome duplication (Mulley et al. 2006).
Despite this, comparative mapping remains a promising tool for fish-specific studies since positional information can be transferred from model species (e.g., pufferfish) to other (nonmodel) fish species. A demonstration of this potential came from an effort to create a genetic map for the bullhead, Cottus gobio (Stemshorn et al. 2005). A panel of 171 microsatellite markers was used to construct a genetic map of Cottus consisting of 20 linkage groups. The flanking sequences of these microsatellites were used as queries in sequence similarity searches of the tetraodon genome. In this manner, 77 of these marker flanks detected a tetraodon homolog. Approximately two-thirds of the assembled tetraodon genome is assigned to physical chromosomal locations thus enabling an assessment of conservation of synteny between these species. Most markers from a single linkage group in Cottus were found to possess homologs mapping to a single tetraodon chromosome. This strong conservation of synteny coupled with the long-range contiguity of the tetraodon assembly means that the tetraodon genome will serve as an important genomic reference for species for which no genome sequencing projects are currently underway. Furthermore, it is worth noting that the comparative mapping process between Cottus and tetraodon exploits not only conserved synteny but also the significant noncoding sequence conservation between these species since microsatellites are predominantly restricted to noncoding regions. On the other hand, the reduction in conservation of gene order owing to intrachromosomal rearrangement may limit the cross-species transfer of positional information between these species to some extent.
A second example illustrating the utility of the tetraodon genome in comparative mapping comes from a study integrating a total of 74 microsatellite markers and 428 ESTs on a preexisting radiation hybrid (RH) map of gilthead sea bream (Sparus aurata), a species of both commercial and evolutionary interest (Sarropoulou et al. 2007). Extensive conserved synteny was clearly demonstrable between the species in this study and this provided a shortcut for the mapping of ESTs to the sea-bream RH map by first mapping them in silico to their homolog in the tetraodon genome.
A further potential benefit of the comparative mapping approach is in facilitating the transfer of positional information following quantitative trait loci (QTL) mapping to pinpoint traits of interest in the nonmodel organism. Specifically, markers linked to a QTL can be projected through homology onto the tetraodon genome thus “zooming in” on candidate genes annotated in pufferfish. This strategy promises to open up lines of inquiry into various traits of interest including disease resistance, sex determination and growth in commercially important but, as yet, unsequenced fish.
5.13.2 Pufferfish as a Model for Functional Annotation
In addition to facilitating positional cloning by comparative mapping, completed fish genomes can also serve as an important tool for the homology-based transfer of functional annotation between homologous genes in fish and tetrapods or between homologs in different fish species. In this context it is important to consider the impact of frequent gene duplication and subfunctionalization in fish genomes. In this respect it is noteworthy that gene duplicates are a prevalent feature of fish genomes primarily as a consequence of the whole-genome duplication early in teleost evolution. Moreover, many of these duplicates are likely to have undergone subfunctionalization, a mechanism postulated to be a major determinant of gene duplicate preservation (Force et al. 1999).
The functional characterization of a multifunctional human gene can benefit from the fact that a single-copy human gene may possess two fish coorthologs that have undergone subfunctionalization (Amores et al. 2004). In mammalian models, the characterization of such genes is impeded by the confounding effects of pleiotropy. However, by exploiting the reduction in pleiotropy associated with the frequent duplication and subfunctionalization of genes in fish, individual subfunctions can be disentangled through the investigation of each duplicate in turn. This can be achieved either through mutagenesis of individual fish duplicates in a coorthologous pair or through the detection of conserved noncoding regions that may represent regulatory regions associated with each subfunction. Although the former strategy is currently only feasible in Zebrafish the latter approach is feasible using any sequenced fish genome (including tetraodon and fugu).
5.14 Early Pitfalls of Initial Pufferfish Genome Draft Assemblies
After the completion of the human genome, fugu was the second vertebrate to have its genome sequenced. The WGS strategy used represented a considerable technical advance and yielded a preliminary version of the pufferfish gene catalogue. Nevertheless, on subsequent completion of the genome of its close relative, tetraodon, differences between these two species of fish emerged that appeared particularly anomalous. First, the G + C rich component present in the tetraodon and mammalian genomes was not represented in the fugu assembly. One possible explanation is that underrepresentation of the GC-rich fraction of the fugu genome at some stage of the cloning, sequencing or assembly process (Jaillon et al. 2004). Second, the absence of type I cytokines and their receptors from the fugu genome was surprising given their ubiquity among other vertebrate species but this subsequently proved to be due to difficulties in annotating these genes in fugu.
Two anecdotal examples illustrate how biologically important but erroneous conclusions based on the initial assemblies of the fugu and tetraodon genomes were ultimately corrected with improvements in the accuracy of both these assemblies. This is most apparent in the case of the enumeration of pufferfish Hox clusters. Prior to the availability of whole pufferfish genomes an apparent dearth of Hox clusters in pufferfish was proposed as a causal explanation for the simplified morphology of these fish (Aparicio et al. 1997; Holland 1997; Meyer and Malaga-Trillo 1999; Snell et al. 1999; Aparicio 2000; Naruse et al. 2000). The completion of these genomes provided the opportunity to allow a definitive census of pufferfish Hox clusters. This analysis proposed that pufferfish is actually not depleted of Hox clusters (thus refuting a causal connection between Hox cluster count and morphological simplification) and further suggested that fugu possesses a third copy of the HoxA cluster, provisionally named HoxAc (Amores et al. 2004). However, it emerged subsequently that this cluster represented a BAC sequence from Tilapia that was present in the second Fugu assembly but removed by the time of the third assembly (Gregory et al. 2006).
The second example relates to “numts,” sequences of mitochondrial origin found integrated in the nuclear genomes of many eukaryotes but previously thought to be absent from teleost genomes. A reappraisal of the surprising discovery of several recent numts in initial releases of both pufferfish genomes provides a further illustration of improvements in these genome assemblies (Antunes and Ramos 2005). This anomaly was clarified with the release of version 4 of the fugu assembly that showed improved accuracy due to increased sequence coverage. A search of this assembly revealed that the originally reported numts all mapped to a single scaffold corresponding to the fugu mitochondrial sequence. Thus, the apparent existence of recent numts in the fugu genome is an artifact of the misassembly of mitochondrial sequences with nuclear sequences in the previous assembly (Lee et al. 2006). However, in the case of tetraodon, Venkatesh et al. were not able to exclude the possibility that recent numts exist in the nuclear genome because the tetraodon mitochondrial genome had not been completed at the time of their study (Venkatesh 2003). Nevertheless, the possibility that recent tetraodon numts are also assembly artifacts is consistent with the fact that none of the scaffolds bearing these sequences were contained in chromosomally mapped ultracontigs.
In tetraodon, two potentially important artifacts in the published assembly and annotation should be mentioned. The first is the suspicious dearth of introns that are a multiple of three in length and that lack an in-frame stop codon. This was subsequently found to be caused by the general tendency of the Gaze annotation program to incorporate such introns, especially short ones, in the coding sequence of predicted genes. This has now been addressed in the latest annotation release (v8.2). The second artifact comes from an ultracontig misassembly on chromosome 1, which incorporates a large region from chromosome 6. This has also been corrected in the most recent assembly release (version 8) that integrates a new 6× fosmid clone coverage.
5.15 Conclusions and Perspectives
The contributions of pufferfish to biological research have so far been primarily genomics as opposed to experimental. The two key factors that explain the important contributions of these genomes to our understanding of vertebrate genome evolution and biology are their compactness, leading to early access to their sequence, and their large evolutionary distance to mammals, a property that intensifies the contrast when identifying functional sequences in genome alignments. It is therefore important to monitor improvements in the quality and volume of pufferfish genomic resources. As noted here, the tetraodon assembly is the result of a two-staged procedure where sequences from one fish specimen were assembled first, and sequences from a second specimen were layered on top (Jaillon et al. 2004). The full potential of the 8× tetraodon shotgun dataset is therefore not realized, as this would most likely require that both datasets be assembled simultaneously. Progress in dealing with highly polymorphic shotgun sequence data, especially by the developers of the ARACHNE software (Jaffe et al. 2003), lead us to believe that a new, hopefully more contiguous, tetraodon assembly is not far from completion.
However, to assert that neither pufferfish species will make a contribution to experimental biology may give hostage to fortune, particularly with the recent possibility to breed tetraodon at will in a controlled laboratory environment (Watson et al. 2009; see Sect. 5.4). This imemdiately opens several new perspectives: regular access to embryonnic tissues, the possibility to study tetraodon development, and potentially to perform transgenic experiments. Here also, the latter technology may greatly benefit from compact genomic DNA. A concrete illustration of this comes from considering the use of pufferfish in detecting 59 putative regulatory elements in a region of 741 kb in the human HoxD cluster (Gregory et al. 2006). Some of these are located in the global control region (GCR) and may be involved in the coordinated control of this locus. A genomic clone suitable for a comprehensive functional assay of this control region and that included all 59 of these putative regulatory elements would be difficult to obtain from a mammalian source. However, in fugu the fact that these putative regulatory elements are compacted into a stretch of 74 kb means that a fugu BAC encompassing this region could be used in transgenic experiments in zebrafish and mammals (Gregory et al. 2006). Therefore, the fact that fugu has distilled the regulatory information controlling this locus into a region one tenth the size of that in human highlights an, as yet, relatively unexploited role for pufferfish in functional studies. More generally, since 75% of intergenic regions are less than 6 kb in length in tetraodon, it becomes possible to clone an entire intergenic spacer in a reporter construct thus enabling the simultaneous assaying of entire cis-regulatory modules in pufferfish.
A more distant prospect is the potential offered by tetraodon or fugu in the field of synthetic biology. Such potential would almost certainly belong to the realm of speculation were it not for recent precipitous advances in genome construction technology that, within a few years, has already progressed from the synthesis of viral to bacterial genomes. This forward step was made possible by the choice of the tractable minimal bacterial genome of Mycoplasma genitalium (Gibson et al. 2008). If progress in this technology matches that seen in the field of genome sequencing, which advanced from the first bacterial genome sequence in 1995 (Fleischmann et al. 1995) to the landmark sequencing of the human genome in 2001 (Consortium 2001), the leap to synthesizing vertebrate genomes should occur in the near future. When this leap is eventually made, compact pufferfish genomes could provide the crucial stepping stone.
References
Amores A, Force A, Yan YL, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang YL, Westerfield M, Ekker M, Postlethwait JH (1998) Zebrafish Hox clusters and vertebrate genome evolution. Science 282:1711
Amores A, Suzuki T, Yan YL, Pomeroy J, Singer A, Amemiya C, Postlethwait JH (2004) Developmental roles of pufferfish Hox clusters and genome evolution in ray-fin fish. Genome Res 14:1
Antunes A, Ramos MJ (2005) Discovery of a large number of previously unrecognized mitochondrial pseudogenes in fish genomes. Genomics 86:708
Aparicio S (2000) Vertebrate evolution: recent perspectives from fish. Trends Genet 16:54
Aparicio S, Morrison A, Gould A, Gilthorpe J, Chaudhuri C, Rigby P, Krumlauf R, Brenner S (1995) Detecting conserved regulatory elements with the model genome of the Japanese puffer fish, Fugu rubripes. Proc Natl Acad Sci USA 92:1684
Aparicio S, Hawker K, Cottage A, Mikawa Y, Zuo L, Venkatesh B, Chen E, Krumlauf R, Brenner S (1997) Organization of the Fugu rubripes Hox clusters: evidence for continuing evolution of vertebrate Hox complexes. Nat Genet 16:79
Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit AF, Sollewijn Gelpke MD, Roach J, Oh T, Ho IY, Wong M, Detter C, Verhoef F, Predki P, Tay A, Lucas S, Richardson P, Smith SF, Clark MS, Edwards YJ, Doggett N, Zharkikh A, Tavtigian SV, Pruss D, Barnstead M, Evans C, Baden H, Powell J, Glusman G, Rowen L, Hood L, Tan YH, Elgar G, Hawkins T, Venkatesh B, Rokhsar D, Brenner S (2002) Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 25:25
Argmann CA, Chambon P, Auwerx J (2005) Mouse phenogenomics: the fast track to “systems metabolism”. Cell Metab 2:349
Bartolome C, Maside X, Charlesworth B (2002) On the abundance and distribution of transposable elements in the genome of Drosophila melanogaster. Mol Biol Evol 19:926
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu X, Zhang L, Alam MD, Anastasi C, Aniebo IC, Bailey DM, Bancarz IR, Banerjee S, Barbour SG, Baybayan PA, Benoit VA, Benson KF, Bevis C, Black PJ, Boodhun A, Brennan JS, Bridgham JA, Brown RC, Brown AA, Buermann DH, Bundu AA, Burrows JC, Carter NP, Castillo N, Chiara ECM, Chang S, Neil Cooley R, Crake NR, Dada OO, Diakoumakos KD, Dominguez-Fernandez B, Earnshaw DJ, Egbujor UC, Elmore DW, Etchin SS, Ewan MR, Fedurco M, Fraser LJ, Fuentes Fajardo KV, Scott Furey W, George D, Gietzen KJ, Goddard CP, Golda GS, Granieri PA, Green DE, Gustafson DL, Hansen NF, Harnish K, Haudenschild CD, Heyer NI, Hims MM, Ho JT, Horgan AM, Hoschler K, Hurwitz S, Ivanov DV, Johnson MQ, James T, Huw Jones TA, Kang GD, Kerelska TH, Kersey AD, Khrebtukova I, Kindwall AP, Kingsbury Z, Kokko-Gonzales PI, Kumar A, Laurent MA, Lawley CT, Lee SE, Lee X, Liao AK, Loch JA, Lok M, Luo S, Mammen RM, Martin JW, McCauley PG, McNitt P, Mehta P, Moon KW, Mullens JW, Newington T, Ning Z, Ling Ng B, Novo SM, O’Neill MJ, Osborne MA, Osnowski A, Ostadan O, Paraschos LL, Pickering L, Pike AC, Chris Pinkard D, Pliskin DP, Podhasky J, Quijano VJ, Raczy C, Rae VH, Rawlings SR, Chiva Rodriguez A, Roe PM, Rogers J, Rogert Bacigalupo MC, Romanov N, Romieu A, Roth RK, Rourke NJ, Ruediger ST, Rusman E, Sanches-Kuiper RM, Schenker MR, Seoane JM, Shaw RJ, Shiver MK, Short SW, Sizto NL, Sluis JP, Smith MA, Ernest Sohna Sohna J, Spence EJ, Stevens K, Sutton N, Szajkowski L, Tregidgo CL, Turcatti G, Vandevondele S, Verhovsky Y, Virk SM, Wakelin S, Walcott GC, Wang J, Worsley GJ, Yan J, Yau L, Zuerlein M, Mullikin JC, Hurles ME, McCooke NJ, West JS, Oaks FL, Lundberg PL, Klenerman D, Durbin R, Smith AJ (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456:53
Benton MJ, Donoghue PC (2007) Paleontological evidence to date the tree of life. Mol Biol Evol 24:26
Boffelli D, Nobrega MA, Rubin EM (2004) Comparative genomics at the vertebrate extremes. Nat Rev Genet 5:456
Bouneau L, Fischer C, Ozouf-Costaz C, Froschauer A, Jaillon O, Coutanceau JP, Korting C, Weissenbach J, Bernot A, Volff JN (2003) An active non-LTR retrotransposon with tandem structure in the compact genome of the pufferfish Tetraodon nigroviridis. Genome Res 13:1686
Brenner S, Elgar G, Sandford R, Macrae A, Venkatesh B, Aparicio S (1993) Characterization of the pufferfish (Fugu) genome as a compact model vertebrate genome. Nature 366:265
Christoffels A, Koh EG, Chia JM, Brenner S, Aparicio S, Venkatesh B (2004) Fugu genome analysis provides evidence for a whole-genome duplication early during the evolution of ray-finned fishes. Mol Biol Evol 21:1146
Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W, Bleich A, Bogue M, Broman KW, Buck KJ, Buckler E, Burmeister M, Chesler EJ, Cheverud JM, Clapcote S, Cook MN, Cox RD, Crabbe JC, Crusio WE, Darvasi A, Deschepper CF, Doerge RW, Farber CR, Forejt J, Gaile D, Garlow SJ, Geiger H, Gershenfeld H, Gordon T, Gu J, Gu W, de Haan G, Hayes NL, Heller C, Himmelbauer H, Hitzemann R, Hunter K, Hsu HC, Iraqi FA, Ivandic B, Jacob HJ, Jansen RC, Jepsen KJ, Johnson DK, Johnson TE, Kempermann G, Kendziorski C, Kotb M, Kooy RF, Llamas B, Lammert F, Lassalle JM, Lowenstein PR, Lu L, Lusis A, Manly KF, Marcucio R, Matthews D, Medrano JF, Miller DR, Mittleman G, Mock BA, Mogil JS, Montagutelli X, Morahan G, Morris DG, Mott R, Nadeau JH, Nagase H, Nowakowski RS, O’Hara BF, Osadchuk AV, Page GP, Paigen B, Paigen K, Palmer AA, Pan HJ, Peltonen-Palotie L, Peirce J, Pomp D, Pravenec M, Prows DR, Qi Z, Reeves RH, Roder J, Rosen GD, Schadt EE, Schalkwyk LC, Seltzer Z, Shimomura K, Shou S, Sillanpaa MJ, Siracusa LD, Snoeck HW, Spearow JL, Svenson K, Tarantino LM, Threadgill D, Toth LA, Valdar W, de Villena FP, Warden C, Whatley S, Williams RW, Wiltshire T, Yi N, Zhang D, Zhang M, Zou F (2004) The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet 36:1133
Coghlan A, Eichler EE, Oliver SG, Paterson AH, Stein L (2005) Chromosome evolution in eukaryotes: a multi-kingdom perspective. Trends Genet 21:673
Consortium IHGS (2001) Initial sequencing and analysis of the human genome. Nature 409:860
Cooper GM, Brown CD (2008) Qualifying the relationship between sequence conservation and molecular function. Genome Res 18:201
Cresko WA, Yan YL, Baltrus DA, Amores A, Singer A, Rodriguez-Mari A, Postlethwait JH (2003) Genome duplication, subfunction partitioning, and lineage divergence: Sox9 in stickleback and zebrafish. Dev Dyn 228:480
Crnogorac-Jurcevic T, Brown JR, Lehrach H, Schalkwyk LC (1997) Tetraodon fluviatilis, a new puffer fish model for genome studies. Genomics 41:177
Dasilva C, Hadji H, Ozouf-Costaz C, Nicaud S, Jaillon O, Weissenbach J, Roest Crollius H (2002) Remarkable compartmentalization of transposable elements and pseudogenes in the heterochromatin of the Tetraodon nigroviridis genome. Proc Natl Acad Sci USA 99:13636
Dehal P, Boore JL (2005) Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol 3:e314
Dekkers WJ (1975) Review of the asiatic freshwater puffers of the genus Tetraodon Linnaeus, 1758 (pisces, tetraodontiformes, tetraodontidae). Bijdragen tot de Dierkunde 45:88
Deloukas P, Matthews LH, Ashurst J, Burton J, Gilbert JG, Jones M, Stavrides G, Almeida JP, Babbage AK, Bagguley CL, Bailey J, Barlow KF, Bates KN, Beard LM, Beare DM, Beasley OP, Bird CP, Blakey SE, Bridgeman AM, Brown AJ, Buck D, Burrill W, Butler AP, Carder C, Carter NP, Chapman JC, Clamp M, Clark G, Clark LN, Clark SY, Clee CM, Clegg S, Cobley VE, Collier RE, Connor R, Corby NR, Coulson A, Coville GJ, Deadman R, Dhami P, Dunn M, Ellington AG, Frankland JA, Fraser A, French L, Garner P, Grafham DV, Griffiths C, Griffiths MN, Gwilliam R, Hall RE, Hammond S, Harley JL, Heath PD, Ho S, Holden JL, Howden PJ, Huckle E, Hunt AR, Hunt SE, Jekosch K, Johnson CM, Johnson D, Kay MP, Kimberley AM, King A, Knights A, Laird GK, Lawlor S, Lehvaslaiho MH, Leversha M, Lloyd C, Lloyd DM, Lovell JD, Marsh VL, Martin SL, McConnachie LJ, McLay K, McMurray AA, Milne S, Mistry D, Moore MJ, Mullikin JC, Nickerson T, Oliver K, Parker A, Patel R, Pearce TA, Peck AI, Phillimore BJ, Prathalingam SR, Plumb RW, Ramsay H, Rice CM, Ross MT, Scott CE, Sehra HK, Shownkeen R, Sims S, Skuce CD, Smith ML, Soderlund C, Steward CA, Sulston JE, Swann M, Sycamore N, Taylor R, Tee L, Thomas DW, Thorpe A, Tracey A, Tromans AC, Vaudin M, Wall M, Wallis JM, Whitehead SL, Whittaker P, Willey DL, Williams L, Williams SA, Wilming L, Wray PW, Hubbard T, Durbin RM, Bentley DR, Beck S, Rogers J (2001) The DNA sequence and comparative analysis of human chromosome 20. Nature 414:865
Deloukas P, Earthrowl ME, Grafham DV, Rubenfield M, French L, Steward CA, Sims SK, Jones MC, Searle S, Scott C, Howe K, Hunt SE, Andrews TD, Gilbert JG, Swarbreck D, Ashurst JL, Taylor A, Battles J, Bird CP, Ainscough R, Almeida JP, Ashwell RI, Ambrose KD, Babbage AK, Bagguley CL, Bailey J, Banerjee R, Bates K, Beasley H, Bray-Allen S, Brown AJ, Brown JY, Burford DC, Burrill W, Burton J, Cahill P, Camire D, Carter NP, Chapman JC, Clark SY, Clarke G, Clee CM, Clegg S, Corby N, Coulson A, Dhami P, Dutta I, Dunn M, Faulkner L, Frankish A, Frankland JA, Garner P, Garnett J, Gribble S, Griffiths C, Grocock R, Gustafson E, Hammond S, Harley JL, Hart E, Heath PD, Ho TP, Hopkins B, Horne J, Howden PJ, Huckle E, Hynds C, Johnson C, Johnson D, Kana A, Kay M, Kimberley AM, Kershaw JK, Kokkinaki M, Laird GK, Lawlor S, Lee HM, Leongamornlert DA, Laird G, Lloyd C, Lloyd DM, Loveland J, Lovell J, McLaren S, McLay KE, McMurray A, Mashreghi-Mohammadi M, Matthews L, Milne S, Nickerson T, Nguyen M, Overton-Larty E, Palmer SA, Pearce AV, Peck AI, Pelan S, Phillimore B, Porter K, Rice CM, Rogosin A, Ross MT, Sarafidou T, Sehra HK, Shownkeen R, Skuce CD, Smith M, Standring L, Sycamore N, Tester J, Thorpe A, Torcasso W, Tracey A, Tromans A, Tsolas J, Wall M, Walsh J, Wang H, Weinstock K, West AP, Willey DL, Whitehead SL, Wilming L, Wray PW, Young L, Chen Y, Lovering RC, Moschonas NK, Siebert R, Fechtel K, Bentley D, Durbin R, Hubbard T, Doucette-Stamm L, Beck S, Smith DR, Rogers J (2004) The DNA sequence and comparative analysis of human chromosome 10. Nature 429:375
Donoghue PC, Purnell MA (2005) Genome duplication, extinction and vertebrate evolution. Trends Ecol Evol 20:312
Dunham I, Dewar K, Kim U-J, Ross M (1999) Bacterial cloning systems. In: Birren B, Green ED, Klapholz S, Myers RM, Riethman H, Roskams J (eds) Genome analysis: a laboratory manual series, vol 3, Cloning systems. Cold Spring Harbor, New York, pp 1–86
Dunham A, Matthews LH, Burton J, Ashurst JL, Howe KL, Ashcroft KJ, Beare DM, Burford DC, Hunt SE, Griffiths-Jones S, Jones MC, Keenan SJ, Oliver K, Scott CE, Ainscough R, Almeida JP, Ambrose KD, Andrews DT, Ashwell RI, Babbage AK, Bagguley CL, Bailey J, Bannerjee R, Barlow KF, Bates K, Beasley H, Bird CP, Bray-Allen S, Brown AJ, Brown JY, Burrill W, Carder C, Carter NP, Chapman JC, Clamp ME, Clark SY, Clarke G, Clee CM, Clegg SC, Cobley V, Collins JE, Corby N, Coville GJ, Deloukas P, Dhami P, Dunham I, Dunn M, Earthrowl ME, Ellington AG, Faulkner L, Frankish AG, Frankland J, French L, Garner P, Garnett J, Gilbert JG, Gilson CJ, Ghori J, Grafham DV, Gribble SM, Griffiths C, Hall RE, Hammond S, Harley JL, Hart EA, Heath PD, Howden PJ, Huckle EJ, Hunt PJ, Hunt AR, Johnson C, Johnson D, Kay M, Kimberley AM, King A, Laird GK, Langford CJ, Lawlor S, Leongamornlert DA, Lloyd DM, Lloyd C, Loveland JE, Lovell J, Martin S, Mashreghi-Mohammadi M, McLaren SJ, McMurray A, Milne S, Moore MJ, Nickerson T, Palmer SA, Pearce AV, Peck AI, Pelan S, Phillimore B, Porter KM, Rice CM, Searle S, Sehra HK, Shownkeen R, Skuce CD, Smith M, Steward CA, Sycamore N, Tester J, Thomas DW, Tracey A, Tromans A, Tubby B, Wall M, Wallis JM, West AP, Whitehead SL, Willey DL, Wilming L, Wray PW, Wright MW, Young L, Coulson A, Durbin R, Hubbard T, Sulston JE, Beck S, Bentley DR, Rogers J, Ross MT (2004) The DNA sequence and analysis of human chromosome 13. Nature 428:522
Ebert E (2001) The puffers of fresh and brackish waters. Aqualog animalbook GmbH, Rodgau
Eschmeyer WN (1998) Catalog of fishes. California Academy of Sciences, San Francisco, CA
Ewing B, Green P (2000) Analysis of expressed sequence tags indicates 35,000 human genes. Nat Genet 25:232
Fischer C, Ozouf-Costaz C, Roest Crollius H, Dasilva C, Jaillon O, Bouneau L, Bonillo C, Weissenbach J, Bernot A (2000) Karyotype and chromosomal localization of characteristic tandem repeats in the pufferfish Tetraodon nigroviridis. Cytogenet Cell Genet 88:50
Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM et al (1995) Whole-genome random sequencing and assembly of Haemophilus influenzae. Science 269:496
Force A, Lynch M, Pickett FB, Amores A, Yan Y, Postlethwait J (1999) Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151:1531
Frankham R (1995) Conservation genetics. Annu Rev Genet 29:305
Gibson DG, Benders GA, Andrews-Pfannkoch C, Denisova EA, Baden-Tillson H, Zaveri J, Stockwell TB, Brownley A, Thomas DW, Algire MA, Merryman C, Young L, Noskov VN, Glass JI, Venter JC, Hutchison CA 3rd, Smith HO (2008) Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319:1215
Gilley J, Armes N, Fried M (1997) Fugu genome is not a good mammalian model. Nature 385:305
Goode DK, Snell P, Smith SF, Cooke JE, Elgar G (2005) Highly conserved regulatory elements around the SHH gene may contribute to the maintenance of conserved synteny across human chromosome 7q36.3. Genomics 86:172
Graur D, Shuali Y, Li WH (1989) Deletions in processed pseudogenes accumulate faster in rodents than in humans. J Mol Evol 28:279
Gregory SG, Sekhon M, Schein J, Zhao S, Osoegawa K, Scott CE, Evans RS, Burridge PW, Cox TV, Fox CA, Hutton RD, Mullenger IR, Phillips KJ, Smith J, Stalker J, Threadgold GJ, Birney E, Wylie K, Chinwalla A, Wallis J, Hillier L, Carter J, Gaige T, Jaeger S, Kremitzki C, Layman D, Maas J, McGrane R, Mead K, Walker R, Jones S, Smith M, Asano J, Bosdet I, Chan S, Chittaranjan S, Chiu R, Fjell C, Fuhrmann D, Girn N, Gray C, Guin R, Hsiao L, Krzywinski M, Kutsche R, Lee SS, Mathewson C, McLeavy C, Messervier S, Ness S, Pandoh P, Prabhu AL, Saeedi P, Smailus D, Spence L, Stott J, Taylor S, Terpstra W, Tsai M, Vardy J, Wye N, Yang G, Shatsman S, Ayodeji B, Geer K, Tsegaye G, Shvartsbeyn A, Gebregeorgis E, Krol M, Russell D, Overton L, Malek JA, Holmes M, Heaney M, Shetty J, Feldblyum T, Nierman WC, Catanese JJ, Hubbard T, Waterston RH, Rogers J, de Jong PJ, Fraser CM, Marra M, McPherson JD, Bentley DR (2002) A physical map of the mouse genome. Nature 418:743
Gregory SG, Barlow KF, McLay KE, Kaul R, Swarbreck D, Dunham A, Scott CE, Howe KL, Woodfine K, Spencer CC, Jones MC, Gillson C, Searle S, Zhou Y, Kokocinski F, McDonald L, Evans R, Phillips K, Atkinson A, Cooper R, Jones C, Hall RE, Andrews TD, Lloyd C, Ainscough R, Almeida JP, Ambrose KD, Anderson F, Andrew RW, Ashwell RI, Aubin K, Babbage AK, Bagguley CL, Bailey J, Beasley H, Bethel G, Bird CP, Bray-Allen S, Brown JY, Brown AJ, Buckley D, Burton J, Bye J, Carder C, Chapman JC, Clark SY, Clarke G, Clee C, Cobley V, Collier RE, Corby N, Coville GJ, Davies J, Deadman R, Dunn M, Earthrowl M, Ellington AG, Errington H, Frankish A, Frankland J, French L, Garner P, Garnett J, Gay L, Ghori MR, Gibson R, Gilby LM, Gillett W, Glithero RJ, Grafham DV, Griffiths C, Griffiths-Jones S, Grocock R, Hammond S, Harrison ES, Hart E, Haugen E, Heath PD, Holmes S, Holt K, Howden PJ, Hunt AR, Hunt SE, Hunter G, Isherwood J, James R, Johnson C, Johnson D, Joy A, Kay M, Kershaw JK, Kibukawa M, Kimberley AM, King A, Knights AJ, Lad H, Laird G, Lawlor S, Leongamornlert DA, Lloyd DM, Loveland J, Lovell J, Lush MJ, Lyne R, Martin S, Mashreghi-Mohammadi M, Matthews L, Matthews NS, McLaren S, Milne S, Mistry S, Moore MJ, Nickerson T, O’Dell CN, Oliver K, Palmeiri A, Palmer SA, Parker A, Patel D, Pearce AV, Peck AI, Pelan S, Phelps K, Phillimore BJ, Plumb R, Rajan J, Raymond C, Rouse G, Saenphimmachak C, Sehra HK, Sheridan E, Shownkeen R, Sims S, Skuce CD, Smith M, Steward C, Subramanian S, Sycamore N, Tracey A, Tromans A, Van Helmond Z, Wall M, Wallis JM, White S, Whitehead SL, Wilkinson JE, Willey DL, Williams H, Wilming L, Wray PW, Wu Z, Coulson A, Vaudin M, Sulston JE, Durbin R, Hubbard T, Wooster R, Dunham I, Carter NP, McVean G, Ross MT, Harrow J, Olson MV, Beck S, Rogers J, Bentley DR, Banerjee R, Bryant SP, Burford DC, Burrill WD, Clegg SM, Dhami P, Dovey O, Faulkner LM, Gribble SM, Langford CF, Pandian RD, Porter KM, Prigmore E (2006) The DNA sequence and biological annotation of human chromosome 1. Nature 441:315
Grutzner F, Lutjens G, Rovira C, Barnes DW, Ropers HH, Haaf T (1999) Classical and molecular cytogenetics of the pufferfish Tetraodon nigroviridis. Chrom Res 7:655
Hattori M, Fujiyama A, Taylor TD, Watanabe H, Yada T, Park HS, Toyoda A, Ishii K, Totoki Y, Choi DK, Groner Y, Soeda E, Ohki M, Takagi T, Sakaki Y, Taudien S, Blechschmidt K, Polley A, Menzel U, Delabar J, Kumpf K, Lehmann R, Patterson D, Reichwald K, Rump A, Schillhabel M, Schudy A, Zimmermann W, Rosenthal A, Kudoh J, Schibuya K, Kawasaki K, Asakawa S, Shintani A, Sasaki T, Nagamine K, Mitsuyama S, Antonarakis SE, Minoshima S, Shimizu N, Nordsiek G, Hornischer K, Brant P, Scharfe M, Schon O, Desario A, Reichelt J, Kauer G, Blocker H, Ramser J, Beck A, Klages S, Hennig S, Riesselmann L, Dagand E, Haaf T, Wehrmeyer S, Borzym K, Gardiner K, Nizetic D, Francis F, Lehrach H, Reinhardt R, Yaspo ML (2000) The DNA sequence of human chromosome 21. Nature 405:311
Heilig R, Eckenberg R, Petit JL, Fonknechten N, Da Silva C, Cattolico L, Levy M, Barbe V, de Berardinis V, Ureta-Vidal A, Pelletier E, Vico V, Anthouard V, Rowen L, Madan A, Qin S, Sun H, Du H, Pepin K, Artiguenave F, Robert C, Cruaud C, Bruls T, Jaillon O, Friedlander L, Samson G, Brottier P, Cure S, Segurens B, Aniere F, Samain S, Crespeau H, Abbasi N, Aiach N, Boscus D, Dickhoff R, Dors M, Dubois I, Friedman C, Gouyvenoux M, James R, Mairey-Estrada B, Mangenot S, Martins N, Menard M, Oztas S, Ratcliffe A, Shaffer T, Trask B, Vacherie B, Bellemere C, Belser C, Besnard-Gonnet M, Bartol-Mavel D, Boutard M, Briez-Silla S, Combette S, Dufosse-Laurent V, Ferron C, Lechaplais C, Louesse C, Muselet D, Magdelenat G, Pateau E, Petit E, Sirvain-Trukniewicz P, Trybou A, Vega-Czarny N, Bataille E, Bluet E, Bordelais I, Dubois M, Dumont C, Guerin T, Haffray S, Hammadi R, Muanga J, Pellouin V, Robert D, Wunderle E, Gauguet G, Roy A, Sainte-Marthe L, Verdier J, Verdier-Discala C, Hillier L, Fulton L, McPherson J, Matsuda F, Wilson R, Scarpelli C, Gyapay G, Wincker P, Saurin W, Quetier F, Waterston R, Hood L, Weissenbach J (2003) The DNA sequence and analysis of human chromosome 14. Nature 421:601
Hinegardner R (1968) Evolution of cellular DNA content in teleost fishes. Am Nat 102:517
Holland PW (1997) Vertebrate evolution: something fishy about Hox genes. Curr Biol 7:R570
Humphray SJ, Oliver K, Hunt AR, Plumb RW, Loveland JE, Howe KL, Andrews TD, Searle S, Hunt SE, Scott CE, Jones MC, Ainscough R, Almeida JP, Ambrose KD, Ashwell RI, Babbage AK, Babbage S, Bagguley CL, Bailey J, Banerjee R, Barker DJ, Barlow KF, Bates K, Beasley H, Beasley O, Bird CP, Bray-Allen S, Brown AJ, Brown JY, Burford D, Burrill W, Burton J, Carder C, Carter NP, Chapman JC, Chen Y, Clarke G, Clark SY, Clee CM, Clegg S, Collier RE, Corby N, Crosier M, Cummings AT, Davies J, Dhami P, Dunn M, Dutta I, Dyer LW, Earthrowl ME, Faulkner L, Fleming CJ, Frankish A, Frankland JA, French L, Fricker DG, Garner P, Garnett J, Ghori J, Gilbert JG, Glison C, Grafham DV, Gribble S, Griffiths C, Griffiths-Jones S, Grocock R, Guy J, Hall RE, Hammond S, Harley JL, Harrison ES, Hart EA, Heath PD, Henderson CD, Hopkins BL, Howard PJ, Howden PJ, Huckle E, Johnson C, Johnson D, Joy AA, Kay M, Keenan S, Kershaw JK, Kimberley AM, King A, Knights A, Laird GK, Langford C, Lawlor S, Leongamornlert DA, Leversha M, Lloyd C, Lloyd DM, Lovell J, Martin S, Mashreghi-Mohammadi M, Matthews L, McLaren S, McLay KE, McMurray A, Milne S, Nickerson T, Nisbett J, Nordsiek G, Pearce AV, Peck AI, Porter KM, Pandian R, Pelan S, Phillimore B, Povey S, Ramsey Y, Rand V, Scharfe M, Sehra HK, Shownkeen R, Sims SK, Skuce CD, Smith M, Steward CA, Swarbreck D, Sycamore N, Tester J, Thorpe A, Tracey A, Tromans A, Thomas DW, Wall M, Wallis JM, West AP, Whitehead SL, Willey DL, Williams SA, Wilming L, Wray PW, Young L, Ashurst JL, Coulson A, Blocker H, Durbin R, Sulston JE, Hubbard T, Jackson MJ, Bentley DR, Beck S, Rogers J, Dunham I (2004) DNA sequence and analysis of human chromosome 9. Nature 429:369
Hurley IA, Mueller RL, Dunn KA, Schmidt EJ, Friedman M, Ho RK, Prince VE, Yang Z, Thomas MG, Coates MI (2007) A new time-scale for ray-finned fish evolution. Proc Biol Sci 274:489
Jaffe DB, Butler J, Gnerre S, Mauceli E, Lindblad-Toh K, Mesirov JP, Zody MC, Lander E (2003) Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res 13:91
Jaillon O, Aury JM, Brunet F, Petit JL, Stange-Thomann N, Mauceli E, Bouneau L, Fischer C, Ozouf-Costaz C, Bernot A, Nicaud S, Jaffe D, Fisher S, Lutfalla G, Dossat C, Segurens B, Dasilva C, Salanoubat M, Levy M, Boudet N, Castellano S, Anthouard V, Jubin C, Castelli V, Katinka M, Vacherie B, Biemont C, Skalli Z, Cattolico L, Poulain J, De Berardinis V, Cruaud C, Duprat S, Brottier P, Coutanceau JP, Gouzy J, Parra G, Lardier G, Chapple C, McKernan KJ, McEwan P, Bosak S, Kellis M, Volff JN, Guigo R, Zody MC, Mesirov J, Lindblad-Toh K, Birren B, Nusbaum C, Kahn D, Robinson-Rechavi M, Laudet V, Schachter V, Quetier F, Saurin W, Scarpelli C, Wincker P, Lander ES, Weissenbach J, Roest Crollius H (2004) Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431:946
Kai W, Kikuchi K, Fujita M, Suetake H, Fujiwara A, Yoshiura Y, Ototake M, Venkatesh B, Miyaki K, Suzuki Y (2005) A genetic linkage map for the tiger pufferfish, Takifugu rubripes. Genetics 171:227
Kai W, Kikuchi K, Tohari S, Chew AK, Tay A, Fujiwara A, Hosoya S, Suetake H, Naruse K, Brenner S, Suzuki Y, Venkatesh B (2011) Integration of the genetic map and genome assembly of fugu facilitates insights into distinct features of genome evolution in teleosts and mammals. Genome Biol Evol 3:424
Kasahara M, Naruse K, Sasaki S, Nakatani Y, Qu W, Ahsan B, Yamada T, Nagayasu Y, Doi K, Kasai Y, Jindo T, Kobayashi D, Shimada A, Toyoda A, Kuroki Y, Fujiyama A, Sasaki T, Shimizu A, Asakawa S, Shimizu N, Hashimoto S, Yang J, Lee Y, Matsushima K, Sugano S, Sakaizumi M, Narita T, Ohishi K, Haga S, Ohta F, Nomoto H, Nogata K, Morishita T, Endo T, Shin IT, Takeda H, Morishita S, Kohara Y (2007) The medaka draft genome and insights into vertebrate genome evolution. Nature 447:714
Kikuchi K, Kai W, Hosokawa A, Mizuno N, Suetake H, Asahina K, Suzuki Y (2007) The sex-determining locus in the tiger pufferfish, Takifugu rubripes. Takifugu rubripes. Genetics 175:2039
Kikuta H, Laplante M, Navratilova P, Komisarczuk AZ, Engstrom PG, Fredman D, Akalin A, Caccamo M, Sealy I, Howe K, Ghislain J, Pezeron G, Mourrain P, Ellingsen S, Oates AC, Thisse C, Thisse B, Foucher I, Adolf B, Geling A, Lenhard B, Becker TS (2007) Genomic regulatory blocks encompass multiple neighboring genes and maintain conserved synteny in vertebrates. Genome Res 17:545
Kondo M, Nanda I, Hornung U, Asakawa S, Shimizu N, Mitani H, Schmid M, Shima A, Schartl M (2003) Absence of the candidate male sex-determining gene dmrt1b(Y) of medaka from other fish species. Curr Biol 13:416
Lee AP, Koh EG, Tay A, Brenner S, Venkatesh B (2006) Highly conserved syntenic blocks at the vertebrate Hox loci and conserved regulatory elements within and outside Hox gene clusters. Proc Natl Acad Sci USA 103:6994
Liang F, Holt I, Pertea G, Karamycheva S, Salzberg SL, Quackenbush J (2000) Gene Index analysis of the human genome estimates approximately 120,000 genes. Nat Genet 25:239
Lutfalla G, Roest Crollius H, Brunet F, Laudet V, Robinson-Rechavi M (2003) Inventing a sex-specific gene: a conserved role of DMRT1 in teleost fishes plus a recent duplication in the medaka Oryzias latipes resulted in DMY. J Mol Evol 57:S148
Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290:1151
Meyer A, Malaga-Trillo E (1999) Vertebrate genomics: more fishy tales about Hox genes. Curr Biol 9:R210
Miyaki K, Tabeta O, Kayano H (1995) Karyotypes in six species of pufferfishes Takifugu (Tetraodondontidae, Tetraodontiformes). Fish Sci 61:594
Miyata T, Suga H (2001) Divergence pattern of animal gene families and relationship with the Cambrian explosion. Bioessays 23:1018
Mulley J, Holland P (2004) Comparative genomics: small genome, big insights. Nature 431:916
Mulley JF, Chiu CH, Holland PW (2006) Breakup of a homeobox cluster after genome duplication in teleosts. Proc Natl Acad Sci USA 103:10369
Mungall AJ, Palmer SA, Sims SK, Edwards CA, Ashurst JL, Wilming L, Jones MC, Horton R, Hunt SE, Scott CE, Gilbert JG, Clamp ME, Bethel G, Milne S, Ainscough R, Almeida JP, Ambrose KD, Andrews TD, Ashwell RI, Babbage AK, Bagguley CL, Bailey J, Banerjee R, Barker DJ, Barlow KF, Bates K, Beare DM, Beasley H, Beasley O, Bird CP, Blakey S, Bray-Allen S, Brook J, Brown AJ, Brown JY, Burford DC, Burrill W, Burton J, Carder C, Carter NP, Chapman JC, Clark SY, Clark G, Clee CM, Clegg S, Cobley V, Collier RE, Collins JE, Colman LK, Corby NR, Coville GJ, Culley KM, Dhami P, Davies J, Dunn M, Earthrowl ME, Ellington AE, Evans KA, Faulkner L, Francis MD, Frankish A, Frankland J, French L, Garner P, Garnett J, Ghori MJ, Gilby LM, Gillson CJ, Glithero RJ, Grafham DV, Grant M, Gribble S, Griffiths C, Griffiths M, Hall R, Halls KS, Hammond S, Harley JL, Hart EA, Heath PD, Heathcott R, Holmes SJ, Howden PJ, Howe KL, Howell GR, Huckle E, Humphray SJ, Humphries MD, Hunt AR, Johnson CM, Joy AA, Kay M, Keenan SJ, Kimberley AM, King A, Laird GK, Langford C, Lawlor S, Leongamornlert DA, Leversha M, Lloyd CR, Lloyd DM, Loveland JE, Lovell J, Martin S, Mashreghi-Mohammadi M, Maslen GL, Matthews L, McCann OT, McLaren SJ, McLay K, McMurray A, Moore MJ, Mullikin JC, Niblett D, Nickerson T, Novik KL, Oliver K, Overton-Larty EK, Parker A, Patel R, Pearce AV, Peck AI, Phillimore B, Phillips S, Plumb RW, Porter KM, Ramsey Y, Ranby SA, Rice CM, Ross MT, Searle SM, Sehra HK, Sheridan E, Skuce CD, Smith S, Smith M, Spraggon L, Squares SL, Steward CA, Sycamore N, Tamlyn-Hall G, Tester J, Theaker AJ, Thomas DW, Thorpe A, Tracey A, Tromans A, Tubby B, Wall M, Wallis JM, West AP, White SS, Whitehead SL, Whittaker H, Wild A, Willey DJ, Wilmer TE, Wood JM, Wray PW, Wyatt JC, Young L, Younger RM, Bentley DR, Coulson A, Durbin R, Hubbard T, Sulston JE, Dunham I, Rogers J, Beck S (2003) The DNA sequence and analysis of human chromosome 6. Nature 425:805
Nanda I, Kondo M, Hornung U, Asakawa S, Winkler C, Shimizu A, Shan Z, Haaf T, Shimizu N, Shima A, Schmid M, Schartl M (2002) A duplicated copy of DMRT1 in the sex-determining region of the Y chromosome of the medaka, Oryzias latipes. Proc Natl Acad Sci USA 99:11778
Naruse K, Fukamachi S, Mitani H, Kondo M, Matsuoka T, Kondo S, Hanamura N, Morita Y, Hasegawa K, Nishigaki R, Shimada A, Wada H, Kusakabe T, Suzuki N, Kinoshita M, Kanamori A, Terado T, Kimura H, Nonaka M, Shima A (2000) A detailed linkage map of medaka, Oryzias latipes: comparative genomics and genome evolution. Genetics 154:1773
Neafsey DE, Palumbi SR (2003) Genome size evolution in pufferfish: a comparative analysis of diodontid and tetraodontid pufferfish genomes. Genome Res 13:821
Ohno S (1970) Evolution by gene duplication. Allen and Unwin, London
Otto SP, Whitton J (2000) Polyploid incidence and evolution. Annu Rev Genet 34:401
Peichel CL, Ross JA, Matson CK, Dickson M, Grimwood J, Schmutz J, Myers RM, Mori S, Schluter D, Kingsley DM (2004) The master sex-determination locus in threespine sticklebacks is on a nascent Y chromosome. Curr Biol 14:1416
Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, Plajzer-Frick I, Akiyama J, De Val S, Afzal V, Black BL, Couronne O, Eisen MB, Visel A, Rubin EM (2006) In vivo enhancer analysis of human conserved non-coding sequences. Nature 444:499
Popperl H, Bienz M, Studer M, Chan SK, Aparicio S, Brenner S, Mann RS, Krumlauf R (1995) Segmental expression of Hoxb-1 is controlled by a highly conserved autoregulatory loop dependent upon exd/pbx. Cell 81:1031
Sarropoulou E, Franch R, Louro B, Power DM, Bargelloni L, Magoulas A, Senger F, Tsalavouta M, Patarnello T, Galibert F, Kotoulas G, Geisler R (2007) A gene-based radiation hybrid map of the gilthead sea bream Sparus aurata refines and exploits conserved synteny with Tetraodon nigroviridis. BMC Genomics 8:44
Scannell DR, Byrne KP, Gordon JL, Wong S, Wolfe KH (2006) Multiple rounds of speciation associated with reciprocal gene loss in polyploid yeasts. Nature 440:341
Semon M, Wolfe KH (2007) Reciprocal gene loss between Tetraodon and zebrafish after whole genome duplication in their ancestor. Trends Genet 23:108
Smyth IM, Wilming L, Lee AW, Taylor MS, Gautier P, Barlow K, Wallis J, Martin S, Glithero R, Phillimore B, Pelan S, Andrew R, Holt K, Taylor R, McLaren S, Burton J, Bailey J, Sims S, Squares J, Plumb B, Joy A, Gibson R, Gilbert J, Hart E, Laird G, Loveland J, Mudge J, Steward C, Swarbreck D, Harrow J, North P, Leaves N, Greystrong J, Coppola M, Manjunath S, Campbell M, Smith M, Strachan G, Tofts C, Boal E, Cobley V, Hunter G, Kimberley C, Thomas D, Cave-Berry L, Weston P, Botcherby MR, White S, Edgar R, Cross SH, Irvani M, Hummerich H, Simpson EH, Johnson D, Hunsicker PR, Little PF, Hubbard T, Campbell RD, Rogers J, Jackson IJ (2006) Genomic anatomy of the Tyrp1 (brown) deletion complex. Proc Natl Acad Sci USA 103:3704
Snell EA, Scemama JL, Stellwag EJ (1999) Genomic organization of the Hoxa4-Hoxa10 region from Morone saxatilis: implications for Hox gene evolution among vertebrates. J Exp Zool 285:41
Stemshorn KC, Nolte AW, Tautz D (2005) A genetic map of Cottus gobio (Pisces, Teleostei) based on microsatellites can be linked to the physical map of Tetraodon nigroviridis. J Evol Biol 18:1619
Tagle DA, Koop BF, Goodman M, Slightom JL, Hess DL, Jones RT (1988) Embryonic epsilon and gamma globin genes of a prosimian primate (Galago crassicaudatus). Nucleotide and amino acid sequences, developmental regulation and phylogenetic footprints. J Mol Biol 203:439
Torrents D, Suyama M, Zdobnov E, Bork P (2003) A genome-wide survey of human pseudogenes. Genome Res 13:2559
Tyler JC, Holcroft N (2007) Tetraodontiformes. triggerfishes, boxfishes, puffers (fugu), molas and allies. http://tolweb.org/Tetraodontiformes/52153
Vandepoele K, De Vos W, Taylor JS, Meyer A, Van de Peer Y (2004) Major events in the genome evolution of vertebrates: paranome age and size differ considerably between ray-finned fishes and land vertebrates. Proc Natl Acad Sci USA 101:1638
Venkatesh B (2003) Evolution and diversity of fish genomes. Curr Opin Genet Dev 13:588
Volff JN (2005) Genome evolution and biodiversity in teleost fish. Heredity 94:280
Watson CA, Hill JE, Graves JS, Wood AL, Kilgore KH (2009) Use of a novel induced spawning technique for the first reported captive spawning of Tetraodon nigroviridis. Mar Genomics 2:143–146
Werth CR, Windham MD (1991) A model for divergent, allopatric speciation of polyploid pteridophytes resulting from silencing of duplicate-gene expression. Am Nat 137:515
White MJD (1978) Modes of speciation. W.H. Freeman, San Francisco
Wittbrodt J, Meyer A, Schartl M (1998) More genes in fish? Bioessays 20:511
Woolfe A, Goodson M, Goode DK, Snell P, McEwen GK, Vavouri T, Smith SF, North P, Callaway H, Kelly K, Walter K, Abnizova I, Gilks W, Edwards YJ, Cooke JE, Elgar G (2005) Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol 3:e7
Yamanoue Y, Miya M, Inoue JG, Matsuura K, Nishida M (2006) The mitochondrial genome of spotted green pufferfish Tetraodon nigroviridis (Teleostei: Tetraodontiformes) and divergence time estimation among model organisms in fishes. Genes Genet Syst 81:29
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Cusack, B., Crollius, H.R. (2012). Punching Above Their Weight: The Compact Genomes of Pufferfishes. In: Denny, P., Kole, C. (eds) Genome Mapping and Genomics in Laboratory Animals. Genome Mapping and Genomics in Animals, vol 4. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-31316-5_5
Download citation
DOI: https://doi.org/10.1007/978-3-642-31316-5_5
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-31315-8
Online ISBN: 978-3-642-31316-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)