
Abstract
Bioinformatics typically treats genomes as linear DNA sequences, with features annotated upon them. In the nucleus, genomes are arranged in space to form three-dimensional structures at several levels. The three-dimensional organization of a genome contributes to its activity, affecting the accessibility and regulation of its genes, reflecting the particular cell type and the epigenetic state of the cell.
The majority of the cell cycle occurs during interphase. During metaphase and meiosis, chromosomes are highly condensed. By contrast, interphase chromosomes are difficult to visualize by direct microscopy. Several attempts have been made to understand the nature of metaphase chromosomes and genome structures. Approaches to indirectly derive the spatial proximity of portions of a genome have been devised and applied (Fig. 8.1, Table 8.1).

This chapter reviews these approaches briefly and examines early methods used to investigate the structure of a genome from these data. This research involves taking experimental data, processing them with variants of existing bioinformatic DNA sequence analyses, then analyzing the proximity data derived using biophysical approaches. This chapter emphasizes the background to the biological science and the latter, biophysics-oriented, analyses. The processing of the genomic data is outlined only briefly, as these approaches draw on established bioinformatic methods covered elsewhere in this Handbook. The main focus is on the methods used to derive three-dimen sional (3-D) structural information from the interaction data.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Interaction Data
- Interaction Frequency
- Monte Carlo Step
- Inverse Polymerase Chain Reaction
- Chromosome Conformation Capture
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
The Structure of Chromosomes, Genomes, and Nuclei
For readers new to the biology of the cell nucleus, this section gives a very brief overview of current knowledge of genome structure. This description best relates to animal genomes; each of the major classes of life have some differences that are not examined here. Those working on computational methods in this area are strongly encouraged to develop a full and detailed understanding of the structure of DNA within the eukaryotic nucleus. This is a substantial topic with several textbooks devoted to it.
The description below roughly works in order from the smallest structural elements to the largest, as outlined in Fig. 8.1, aiming to note key terms and structural organizations that can serve as starter keywords for researchers venturing into this arena to explore further.
DNA bases can be decorated with a range of covalently bonded additions. A considerable number of these extra or alternative DNA bases are known, with their distribution differing in different classes of life. While many covalent modifications of DNA bases have been identified, most are rare. The best known base modification, and the dominant modification found in eukaryotes, is methylation of cytosine (5-hydroxymethylcytosine), typically in CpG steps.
These can denote epigenetic states. DNA methylation (or other modifications) can impede the binding of DNA-binding proteins, or serve as recruiting points for protein–DNA complexes and through this define the state of the gene, accessible to be used or not. (Note how this differs from the classical control of the rate of transcription of a gene.)
Little of the DNA in a cell is naked. The most consistently exposed DNA in a genome is near the start of a gene and the immediate promoter; the majority of the remainder of the DNA in the nucleus is bound into nucleosome–DNA complexes. Coding regions (both introns and exons) have nucleosomes that are displaced and reassembled as the DNA passes the transcriptional machinery.
Nucleosomes are made from an octamer of histone proteins (composed of two of each of the H2A, H2B, H3, and H4 proteins) around which approximately 147 base pairs of DNA is wrapped, taking approximately 1 and 7/8ths turns around the histone octamer. A so-called linker, typically 80 base pairs and usually bound by histone H1 or its isoforms, spans each bound octamer. The typical linker length varies between different species.
Histones have a compact core with extended, disordered N- and C-terminal tails that protrude from the histone octamer core of nucleosomes. These exposed tails are covalently modified in what has been termed the histone code.
Different histone modifications have been associated with different transcriptional states, in particular if chromatin containing the gene is open or closed. In active genes, the chromatin is open (euchromatin) with the nucleosome-bound DNA forming a relatively extended structure. In closed chromatin (heterochromatin), typically located near the periphery of the nucleus, nucleosomes are packed against one another to condense unused portions of the genome. A number of models of higher-order structures – so-called nucleosomal array models – have been proposed. The detailed models proposed for these higher-order structures are somewhat controversial, as is the extent to which they are well defined in vivo. These arrays of nucleosomes are considered to form the extended fiber, 30 nm fiber, and higher-order packing of DNA, such as that seen in densely packed chromatin.
In interphase, the active or growth phase of the cell cycle, the packing of chromatin varies along the length of the chromosomes, reflecting the accessibility of each portion of the chromosome to gene expression.
The genome is anchored to a number of substrates in the periphery of the nucleus (e.g., DNA–lamina interactions), the nuclear matrix, and complexes associated with the genome. Insulator or boundary elements (two separable but related activities) are among the attachment points.
Loops within chromosomes form, determined by DNA methylation, histone modification and the binding proteins that organize the genome into higher-order structural units. These loops affect how regulatory factors act on the genes, for example, depending on if the regulatory element is in the same DNA loop as the gene it might regulate, or not.
Related to this is the well-established observation that many regulatory sites are tens, or even hundreds, of thousands of bases away from the genes they regulate. While they might be distant in linear sequence, they may be in close proximity in space. Thus, we can think of linear (along the genome) and spatial (through space) distances with respect to the genome. The methods described in this chapter identify pairs (or more) of loci that have large linear but short spatial distances.
At a higher level, chromosome territories define large portions of a chromosome that tend to occupy one region of the volume of the nucleus. (That is, regions of chromosomes tend to occupy their own space in the nucleus rather than mixing or tangling with other chromosomes or other parts of the same chromosome.)
In addition to these layers, or hierarchy, of structure are specialized regions of the genome, such as telomeric regions, ribosomal DNA (e.g., nucleolus), and a number of nuclear bodies. Each of these regions have specialized genome structures.
These observations have been informed, in part, by the techniques reviewed in this chapter. A key question is how the three-dimensional organization of the genome relates to control of gene expression. Related questions are how genome structures define what genes are made accessible to be used in a particular cell type and how or if alterations in these structures impact on disease.
Careful interpretation of the interaction data is important. One point to remember as you read about these methods is that the structure of a genome is dynamic, changing over time, varying with environmental conditions and cell type. Another is that, while the regions of chromatin between complexes bound to a matrix or lamina or other substrate may have structural elements and structural properties and be (relatively) well defined, the loop regions may not have any particularly well-defined structure.
The remainder of this chapter proceeds by outlining the main experimental techniques used, briefly examining the data analysis requirements of these methods (Table 8.1), then exploring the methods used to derive spatial information from the proximity data. It closes by briefly mentioning complementary methods and thoughts for the future.
An important complementary field to this endeavor is (theoretical) polymer modeling, which aims to build models of chromatin structure from an understanding of polymer physics. While this strongly impacts on what is presented here, this is not covered. Readers approaching this field would benefit from gaining an understanding of the polymer science and biophysical aspects, as this field is a translational field, moving from linear sequence-based genomics to a three-dimensional, biophysical genome (or four-dimensional if time is considered).
Testing for Interacting Chromatin Using Chromosome Conformation Capture
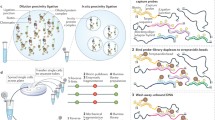
When applied to a modest number of loci, chromosome conformation capture (3C or CCC) can identify interacting loci. Initially developed in 2002 [8.2], a range of variants have since been developed aiming to allow high-throughput processing (e.g., 5C [8.3] or Hi-C [8.4]) or relating chromatin-associated proteins to chromosome conformation (e.g., 4C [8.5] or ChIP-PET (chromatin immunoprecipitation using paired-end tags)) (Fig. 8.2). Some sources point to the work of Cullen et al. [8.6] as an early precursor of these methods, with its proximity ligation concept.

A schematic outline of the conceptual differences in the different chromosome conformation capture (CCC) methods published at the time of writing. (Further differences occur throughout each method.) All CCC methods rely on cross-linking spatially adjacent chromatin, breaking the linearly adjacent DNA (either through sonication or use of restriction enzymes), ligating the ends formed, then sequencing across these ends or screening against gene arrays if desired and appropriate. ChIP-PET uses sonication to fragment the cross-linked DNA, then immunoprecipitation to extract the cross-linked material, before ligating the ends, isolating and purifying the DNA from chromatin, then sequencing it. The immunoprecipitation selects from the sample particular proteins of interest, thus the method is able to investigate the interactions formed by particular proteins. Hi-C uses restriction enzymes (HindIII) to cleave the cross-linked DNA. The sticky ends are filled in with biotinylated bases, then ligated. After fragmenting the DNA using sonication, the ligated ends are isolated using using streptavidin-coated beads and the DNA is sequenced. A particular value of this approach is that it scales well. The 3C, 4C, and 5C methods mainly differ in setting up different later stages, enabling high-throughput analysis. Note in particular that, for 5C, the DNA sequenced is ligation-mediated annealed (LMA) copy. As 5C uses standardized primers, this scales better than 3C or 4C. (These standardized primer sites are depicted in the figure as tails on the LMA copy)
This section outlines the conceptual basis of these methods. Experimental details are left for the interested reader to pursue via the references supplied.
These methods do not determine a structure, as such, but identify interactions between different portions of the genome through DNA-bound proteins. With these in hand, one can infer what modeled structures might be consistent with the interactions observed.
A common feature of these techniques of note for a computational biologist or bioinformatician is that, the final 3-D analysis aside, the data are familiar DNA sequencing products. This means that much of the initial data processing has similarities to other (high-throughput) sequence analysis projects.
For all of the methods described, the first step is to cross-link the DNA-bound proteins. Usually formaldehyde-based cross-linking is used. This yields a portion of DNA, its bound proteins from one portion of the genome cross-linked to a portion of DNA, and its bound proteins from another portion of the genome. The different portions may lie on the same chromosome or different chromosomes. The cross-linked complex may capture more than two protein–DNA complexes.
Next, excess DNA from around the complexed protein–DNA structure is removed. For the 3C method, restriction endonucleases (REs) are used. REs of different cutting frequency can be chosen. REs with smaller DNA recognition sites will typically cut the genome into more pieces of smaller size. Alternatively, sonication can be used, as in the ChIP-PET and Hi-C protocols. (Sonication is ultrasound vibration of the mixture aimed at physically fracturing the DNA.)
The free ends of the DNA strands from the complexes are then ligated. Conditions used are chosen to favor ligation within each protein–DNA complex, rather than joining different protein–DNA complexes together (e.g., by using highly dilute solutions of the ligation agent). This favoring of ligating complexes held together by protein–DNA interactions, over those that are not, is key to the approach.
The cross-links are reversed (broken), protein removed, DNA extracted and purified, and the DNA ligation products quantified (e.g., by polymerase chain reaction (PCR)) to measure the frequency at which the different interactions occur.
Which sequencing and quantitation methods can be applied depends on how the DNA is cut and ligated. A number of different approaches have been developed with a view to automating the sequencing of the joins (if possible) to allow the material to be sequenced en masse or profiled against a gene array. The cutting method also affects the number of complexes found and the extent to which complexes of weaker interactions are retained in the sample, as discussed below.
Adding chromatin immunoprecipitation (ChIP) biases the sample to contain a particular protein by using an antibody for the particular protein to select complexes with the protein.
Figure 8.1 shows the key steps discussed above, using as an illustration a complex of only a pair of DNA fragments. In practice, complexes can be of several protein–DNA complexes cross-linked, with more complex DNA products to be considered. To the left are the key steps; arrayed along the bottom are the different methods.
Estimates of the typical distance between loci is derived from the argument that the frequency of ligation is approximately inversely proportional to the typical distance of separation of the ligated elements.
The different techniques have different merits.
Because 3C DNA sequences from specific cut ends, for any one sequencing reaction it can only investigate one particular ligation. 3C is also limited to intramolecular interactions, i.e., within one chromosome, whereas the variants can also examine interactions between chromosomes (needed for whole-genome studies).
4C ensures the ligated ends form circular products. Any one end is circularized with all the other ends in the complex, resulting in a mixture of circular products. As a result, one sequencing reaction can investigate all the ligation products of a given end.
5C, with its carbon-copy step that adds universal sequencing primers, can capture all combinations of the ligated ends into the sequencing mixture, with the result that all (many) pairs of ligated ends can be sequenced from one sequencing mixture.
In addition, to add selection for a particular protein in the complexes examined, the ChIP-based variants can, in principle, be more sensitive, too.
Below, each of the methods are individually examined.
Chromosome Conformation Capture for Case Studies: 3C
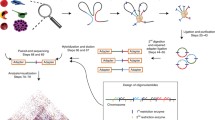
Conceptually, the idea behind 3C is straightforward. Chromosomal DNA has many proteins associated with it, in particular nucleosomes. Formaldehyde (methanal, CH2O) is a small, reactive and diffusible chemical that can cross-link nearby protein (and DNA) amino groups through formation of a –CH2– linkage. Once cross-linked, the DNA in the protein–DNA complexes is cleaved using restriction enzymes with known specificity. The cleaved ends of the DNA in the protein–DNA complexes are then ligated (joined) under dilute conditions that favor ligation of DNA ends from the same protein–DNA complex. The ligated DNA is isolated, amplified (e.g., via PCR), and sequenced. The sequences obtained are mapped onto a reference genome sequence to identify the two portions of the genome that the formaldehyde-based reaction cross-linked. The frequency with which a particular pair of sequences is found to be cross-linked is considered to reflect their proximity in space in the nucleus. (In the opinion of the author, this founding assumption would benefit from closer examination.) From this, spatial models of (a region of) the genome are constructed that satisfy the proximity measures derived (Fig. 8.3).

Representative examples of spatial models derived from CCC studies are shown. (a) Three-dimensional (3-D) model of the yeast genome (after Fig. 5a [8.7]). (b) 3-D model of the human α-globin locus (after Fig. 2b [8.8]). (c) Structure around the TMEM8 gene of chicken erythroid cells under two different salt conditions (after Fig. 5 [8.9]). (d) Schematic of the structures of the mouse Hox gene clusters (after Fig. 3 [8.10]). (e) 3-D model of human chromosome 14 (after Fig. 10 [8.11])
A key limitation of the original 3C protocol is that each annealed sequence pair must be individually amplified, through PCR, using specific primer pairs from the target sequence and potential interacting sequences. (Because this targets specific sequences, the method is applied to previously known or suspected sites, rather than as a screen to locate interacting sites.) Furthermore, each amplification needs to be individually controlled. As a consequence this approach is not practicable for studies involving a large number of sites. Typically it is used for regions up to a several hundred kilobases in size to explore the finer structure of particular model genes or genetic loci.
Another issue is that the high level of local noise means that results are of low resolution. The number of nonspecific interactions is related to the inverse square of the linear distance separating two loci. As a consequence, putative interactions between sites of less than some distance (e.g., 100 kb) are typically discounted.
The 3C approach has been applied to a number of individual genes, revealing the looping structure of the gene under different methylation states or cell types.
Finding All Interactions with a Single Locus: 4C
Four laboratories independently developed variants of what are now grouped under the label 4C, circular chromosome conformation capture (circular 3C: Zhao et al. [8.5]; 3C-on-chip: Simonis et al. [8.12]; open-ended 3C: Wurtele and Chartrand [8.13]; olfactory receptor 3C: Lomvardas et al. [8.14]).
This approach determines the interactions formed by a single site or locus. The 3C products are circularized (e.g., using a frequent-cutting restriction enzyme followed by ligation), selectively amplified using inverse PCR, then passed on to microarray analysis or (deep) sequencing. Inverse PCR uses only a single primer, rather than two, allowing amplification then sequencing of all of the sequences present on the other side of the ligation from a known sequence (unique to the chosen site being studied).
Towards High-Throughput Chromosome Conformation Capture: 5C
A high-throughput variant of 3C, this method enables larger-scale screening of many potential interactions in parallel (see [8.15] for an introduction). The key innovation is that, once the 3C ligation step is complete, probes with standardized PCR primers (T7 and its complement T3) are hybridized to the ligated region. This enables quantitative capture of all the individual ligations within DNA fragments with standardized primers at either end.
This process, known as ligation-mediated amplification (LMA), creates a copy of the original 3C library that is based on standard primers, allowing many genome interactions to be surveyed at once in parallel. Because the copied library is amplified, the products can be examined using (so-called) deep sequencing or microarray analysis. Furthermore, all pairs of ligation products can be examined to generate a matrix of all interacting loci and their frequencies without many rounds of analysis.
Deep sequencing may be preferred over gene arrays, as it is more capable of handling very large datasets. (The complexity of the datasets increases exponentially with the number of primers.)
The automation and large scale of this approach make it useful to look at much larger portions of chromosomes (e.g., Mbps in length) or explore 3-D structure or complex interactions (e.g., promoter–enhancer interactions [8.16]).
Adding Paired-End Tag Sequencing: 6C
6C and some other variants, e.g., ChIP-loop (also called ChIP-3C) and ChIA-PET (chromatin interaction analysis using paired-end tag sequencing, e4C), combine 3C with chromatin immunoprecipitation (ChIP) through first enriching the chromatin for the protein of interest before ligation. ChIP is a popular way of identifying the location of protein–DNA complexes, genome-wide, annotating the linear sequence of the genome with the positions of the protein–DNA interactions identified. Through applying chromosome conformation capture to ChIP-seq libraries, the long-range interactions with which the protein–DNA complexes are associated may be investigated.
Fullwood and Ruan [8.17] provide good coverage of the issues associated with these approaches. In their discussion, they recommend sonication of the cross-linked product, rather than use of restriction enzymes (REs), arguing that 3C and 4C methods are noisy (capturing many nonspecific chromatin interactions) and that sonication shakes apart weak or nonspecific interactions, reducing the noise in the data. (It would be interesting to see if computational methods that combine both levels of interactions can be put to good effect.) Fraser et al. argue that addition of ChIP reduces the noise further and makes the results specific for the protein of interest.
High-Throughput Chromosome Conformation Capture: Hi-C
Used to examine whole genome structure, this method labels the ligated regions of 3C constructs with biotin before shearing the DNA, then capturing the biotin-labeled fragments using streptavidin beads. The captured DNA is then sequenced. The biotin is incorporated by choosing restriction enzymes that leave a 5′ overhang that is filled in using nucleotides that include a biotinylated nucleotide. The blunt-ended fragments are ligated.
This approach collects only the ligated products (i.e., those labeled with biotin) for sequencing. Furthermore, shearing the DNA does not restrict the interactions to DNA regions containing the sites of the restriction enzymes used in other variants.
The particular value of this method is that it can be applied to large datasets, i.e., whole genomes.
Processing Chromosome Conformation Capture Sequence Data
As the focus of this chapter is the methods to construct models of three-dimensional structures of genomes, this section is limited to lightly covering conceptual issues of the main steps typically involved in a CCC-based study, raising a few of the key issues and pointing to some of the software developed to address them. Excellent coverage of the public tools for working with 3C data can be found in Fraser et al. [8.15] (Table 8.2).
It is useful to first appreciate the limitations of the experimental elements that impact on the computational work. As one example, Fullwood and Ruan [8.17] point out that the use of restriction enzymes (versus sonication) is not truly genome-wide, as it is biased to where the RE sites are in the genome. Also, there is the issue of eliminating oversampling of the same sequence by removal of nonunique sequences, as the methods in use rely on multiple overlapping unique signals to correct oversampling. (Likewise for clustering methods.)
In the case of RE-based digestion of the isolated cross-linked DNA, a first step is to choose which REs to use. RE digest patterns are readily generated using a wide variety of software. The choice of REs balances the frequency of cutting, fragment size, evenness of cutting across the studied (portion of the) genome, where they cut (especially with respect to low-complexity regions), etc. Advice (e.g., Fraser et al.) suggests that is that it is useful at this point to create files of all annotation data, e.g., locations of genes and other features, for reference through the project.
Next is primer design. In the case of the 5C method, with its carbon-copy step, the 5C forward and reverse primers need to be designed. The Dostie lab offer software via a WWW interface for this purpose (Table 8.2). One typical concern is avoiding primers that are homologous to low-complexity regions within the genome (region) under investigation. Another is to determine the uniqueness of the primers (e.g., via BLAST searches or a hash lookup). For other CCC methods, except those using sonication to break the DNA, similar needs for primer design have to be addressed.
If a gene array is to be used to (semi)quantitate the frequency of the interaction events, this will need to be designed. One approach, used in the Dostie labʼs 5CArrayBuilder program, is to determine a series of probes of increasing length, where the shorter length is used to assess the background signal when calculating interaction frequencies.
Once the raw sequencing or array data are available, interaction frequencies for each set of interacting loci are derived. This considers background noise and controls; For example, using different length probes for each loci, all probe interaction frequencies that are too close to the background probe interaction frequencies can be discarded and the useful values averaged. Suitable quality control steps might (and should) be undertaken, e.g., serial dilution and PCR amplification of a sample of the probes.
As one can see, with the exception of the determination of the interaction frequencies, these steps are variants on existing molecular-genetics problems and largely do not introduce any new concepts. Further details on these types of tasks can be found elsewhere in this Handbook.
During the course of writing this chapter, Yaffe and Tanay presented an examination of factors biasing the initial data in Hi-C experiments, offering an (in their words) integrated probabilistic model for analyzing this data [8.24]. They draw attention in particular to ligation products that are likely to have arisen from nonspecific cleavage sites rather than restriction fragment ends, the length of restriction fragments (with respect to ligation efficiency), the nucleotide composition of the genome (chromosome) being investigated, and issues with uniquely mapping the interactions back onto the reference genome (chromosome) sequence.
For 5C data, the My5C server [8.16] offers primer design facilities, in several different ways. (See Table 8.2 for related software and access to them.) Given the large genomic regions covered, primer design needs to be automated.
My5C also provides online generation of heatmaps of interaction data, which can be manually examined for particular interactions. Comparisons of two heatmaps is provided, using difference, ratio, or log ratio comparisons. Smoothing functions can be applied to examine larger interaction patterns, e.g., averaging over a sliding window. Similarly, sliding window plots of the data can be examined.
My5C interacts with the UCSC genome browser, in the sense that data can be moved between the two, with genome annotation data placed on the My5C data and custom tracks prepared to be presented in the UCSC browser alongside genome data.
Although My5C offers to generate pairwise interaction data that Cytoscape (used to visualize complex networks) can present, this treats these data as static, whereas in practice they are averages over different cells and potentially different chromatin conformations.
Calculating Genomic Loci Proximity or Interaction Data
General Observations
The previous section dealt with processing steps that have much in common with other high-throughout genomic studies, with familiar issues of primer design, PCR amplification, sequencing error, etc.
These data yield collections of loci which have been identified as (putative) sites of interactions. It is assumed that the frequency with which a particular loci pair is observed reflects the relative proximity or opportunity to interact of that loci pair compared with other loci pairs in the dataset. (Note that proximity and opportunity to interact are not synonyms but alternative ways of viewing interaction frequencies. Consider that two loci can be spatially close, but not interact because they are constrained; by contrast, two loci that are typically well separated spatially might occasionally interact through large-scale movements.)
As the data are from a population of cells, and accepting that chromosomes are (perhaps highly) flexible structures, one may not be able to, or perhaps cannot, derive a meaningful single structure from these interaction data.
Related to this is that the methods deriving structural models from interaction data are probabilistic and yield an ensemble of possible models, rather than a single model. This is familiar ground to those working in molecular modeling and other probabilistic areas (e.g., phylogenetics), but may be new to those working on DNA sequence data. In some senses, the resulting models are perhaps best considered to represent structural properties, rather than structures per se.
Interactions with very low interaction frequencies have very few constraints in modeling. These points should not be overinterpreted in the final models. It may be possible to infer that they are likely to be distant from the other points, provided false-negative rates are low.
Models derived may reflect cell growth conditions (or cell type). In additional to computational and experimental protocol repeatability, there is biological repeatability to consider (Fraser et al. report that they are investigating this issue [8.15]). This requires that all environmental factors, reagents, and cell conditions be standardized (e.g., the point on the growth curve, synchronizing cell growth, etc.).
My5C, 5C3D, and Microcosm (Dostie et al.)
Dostie and colleagues have developed software to identify chromatin conformation signatures in 5C data [8.1], which they have applied to their work on the HoxA gene cluster. Their overall approach is to generate a series of randomized starting conformations, which they minimize against the root-mean-square (RMS) deviation of their interloci data, being the inverse of the interaction frequencies. The computational tools are presented online on the My5C website (Table 8.2).
Fraser et al. [8.15] describe the Dostie groupʼs program 5C3D, which performs a gradient descent minimization approach. Euclidian distances are set to be the inverse of the interaction frequencies. Starting models are a chain set on N points randomly distributed within a cube. Gradient descent is applied in a conventional way to minimize the overall difference in the distance matrix and the Euclidian distances until convergence is achieved.
While this approach is perhaps applicable to smaller models with few interaction points, and will be very fast to execute, it is likely to be too naïve to tackle larger-scale models or genome-wide modeling. In particular, gradient descent used alone is well known to become trapped away from the global minima if local minima are present. Having said this, this general approach may be of use to range-find complex models using a reduced set of interactions as the method will be fast.
Their approach concludes with a separate inference of the best fitting model(s) using their Microcosm program. (Their description is not especially clear, but this appears to be a best-fit procedure by inspecting if spheres of appropriate size placed around each interaction locus capture most of the data points of collections of data selected at random from the original interaction frequency data, under the distributions associated with the interaction frequency of each locus. The size of the spheres chosen appears to be arbitrary in that is it set by the user by manual experiment.)
The local density of interactions is represented and plotted. This approach can also be used to present a comparison of two models as a graph with assessment by deriving p-values for the differences (being the probability of incorrectly predicting a difference, assuming normally distributed differences).
Modeling the Yeast Genome
A model of the yeast genome [8.7] is based on a simple polymer model where each 130 bp of DNA is set to occupy 1 nm with chromosomes treated as a string with 10 kb of these 1 nm units assigned to each RE fragment from the experiment. The model was then constructed by minimizing these 10 kb fragments against the distances derived from the experiments.
This mixes something that might otherwise be close to a pure experimental approach with one that draws on past work on chromatin structure models and polymer theory. Given the noisiness of the data over shorter linear distances, it seems worth exploring incorporation of some model for structure over the shorter distances; further studies on appropriate models for the shorter distances will be useful.
Using the Integrative Modeling Platform
Baù and Marti-Renom [8.25] suggest adopting a modeling platform (IMP) intended for protein assemblies (Table 8.2), illustrating their example by examining the 3-D structure of the α-globin locus, which corresponds to the Enm008 ENCODE region.
Before describing this work, it should be noted that the concepts used are based on those used for molecular simulations, e.g., of proteins. These will be new and different concept for those whose work resolves solely around DNA sequences.
The IMP software allows users to develop a representation of spatial data and a scoring scheme, and then generates models fitting the criteria of the model, as well as providing some analysis facilities.
Initial data are normalized to generate an interaction matrix. Nominally each RE site is where, or close to where, the interactions occur. Baù and Marti-Renom [8.25] represented the α-globin locus as a polymer of 70 particles, one for each HindIII RE fragment, using a sphere with an exclusion volume proportional to the size of the particle. This was modeled in part on a canonical 30 nm fiber, with length to base-pair ratio of 0.01 nm/base. (There is some controversy over the extent to which 30 nm fiber is present in vivo.)
This step is a common theme in these modeling efforts: how to simplify the initial model (i.e., every base pair) in a way that reduces the complexity of the modeling without incorrectly representing the data so as to render the model void. Compared with molecular modeling, one may think of these fragments as being the residue, or repeated unit, used for the modeling.
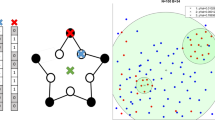
Calculating the energy (cost, favorability) of a model draws on polymer simulation norms, with neighbor and nonneighbor interaction properties and restraints being defined. (One can consider these restraints to be springs attached to a point, restraining the residue if it is pulled away from the point.)
Neighboring (adjacent) residues were constrained to have an approximate distance between them, to lie within what was considered a reasonable range of the distribution of interaction distances derived from the interaction frequencies (i.e., upper- and lower-bound harmonic restraints were applied to the distances of neighboring residues based on the interaction data).
Neighbors with no interaction data were set to remain bound to their neighboring residue through an upper-bound harmonic constraint.
Nonneighboring residues were defined to be at least a minimum distance apart, using a lower-bound harmonic (essentially defining a sphere of exclusion that defines the volume occupied by each residue).
Modeling seeks to minimize the sum of the violations of the individual restraints using simulated annealing, with a subset of positions in each Monte Carlo step moved in probabilistic proportion to the objective function score of the model before and after the move, given the temperature of the system. (Higher temperatures allow greater dynamic movement.) Optimization used 500 Monte Carlo steps with 5 steps of local optimization (minimization) from 50000 random starting models, yielding 50000 potential models of the α-globin locus.
While more detailed, and perhaps involving more labor, this approach builds on many years of developments in molecular modeling (but see the discussion in the following section for possible caveats). Discussion of the analysis and interpretation of the ensembles generated can be found in the next section.
Interpreting the Data
An issue common to these methods is that they identify interactions observed in a (large) population of cells. It is an average picture that is obtained, not data specific to any one cell or one cell type within that population.
By contrast, microscopy studies including methods such as FISH (fluorescence in situ hybridization) that use single cells, may only represent a subset of the interactions of that one cell through time but are able to provide absolute measures of distances – as opposed to inferred probabilities of proximity from population studies – and some idea of the true frequency with which (subsets of) the interactions occur. Subsets are important in that some subgroups of interactions may work as a unit in the presence or absence of specific transcription factors.
Another way of looking at this is to compare with what is used for protein nuclear magnetic resonance (NMR) data, which similarly uses distance restraints to derive structural models. A key difference is that secondary structural elements in proteins are relatively stable. While the detailed interactions with a particular side-chain might differ in different individual proteins in the sample, the overall relationship of the secondary structural elements, or fold, of the protein will be relatively constant (breathing motions excepted). Furthermore, these stable structures show many interactions that in effect triangulate the positions of the structural features, leading to relatively robust structural models.
By contrast, the chromatin structure equivalent of protein secondary structural elements – the different nucleosome array conformations – may be too flexible to sustain stable structures in the way that protein secondary structures do. If so, there may be no real way to compute an overall structure for the genome, as it would be a constantly moving target within any one cell, and/or varying from cell to cell. However, one could still examine properties of the structure.
Likewise, you might identify the tethers, or anchoring points, of loop structures that are defined by well-bound protein complexes and hence stable as a complex forming the base of a loop, even if the body of the loop itself does not form a stable structure (and hence interactions that the portions of the loop make with the rest of the genome vary constantly over time).
Thus, although it is tempting to compare this problem with protein modeling (e.g., using NMR restraints), as, for example, Marti-Renom and Mirny [8.26] do, caution is needed not to take such comparison too far.
As these methods generate ensembles of models, the collection of models generated need to be examined as a dataset itself. A simple strategy, such as that taken in Baù et al. [8.8], is to take the better models generated (the better 10000 of the 50000 in total in their case) and apply cluster analysis to identify common themes in the models generated. In these authorsʼ case, a Markov cluster algorithm was used based on rigid-body superimposition, but other approaches might be adopted.
Their 10000 models yielded 393 clusters, the largest two clusters having 483 and 314 models, respectively, revealing a dumbbell-shaped structure with two relatively globular domains spanned by an extended domain (Fig. 8.3b).
This author has some concern over the very large number of clusters reported. This would suggest either that the cluster criteria are too fine, splitting models into clusters that might otherwise resemble a larger group, or that the modeling process was unable to yield consistent models. One possible avenue that future groups might explore is to use structure comparisons that allow for movement, i.e., non-rigid-body comparison. Another approach would be to identify structural motifs or domains that can be found within the simulation models. (For example, without examining Baù and Marti-Renomʼs data first-hand, it is difficult to know if the actual results are that there are two relatively globular domains spanned by an extended segment, but that the conformation within each globular domain taken over all the models is not well defined. Using a mixture of examining domains and non-rigid-body comparisons might resolve this.)
Considerable effort has been expended on methods such as this for protein structure comparison over many years; it would be worth those working in the genome structure field examining the possible ways of comparing the ensembles of structures more closely.
Different subclasses of solutions are not in themselves errors or the result of a lack of constraints – they may indicate different structure in different transcription states, or different subtypes of cells in the population studied.
These concerns indicate a need to examine carefully if the data are adequately explored in the simulations (e.g., the degree to which the constraints have been tested). Independent verification by experimental data may assist, e.g., FISH experiments.
Sanyal et al. [8.27] point out that current methods, despite being able to collect data over whole genomes, are perhaps limited to domains of several Mb, as the larger structures are expected to be dynamic in nature and vary between cells. Related to this concern is that, to model whole genomes computationally, very large numbers of long-range interactions would be required to yield stable (meaningful) structural models.
Future Prospects
While an exciting area, there is clearly a lot to do.
Annotation of the interaction data and three-dimensional aspects of genome structure will be required. Reed et al. [8.28] briefly touch on the then upcoming need for this. This may require tools beyond the current approach of annotating genomes using linear tracks.
Visualization tools are needed to present the interactions and the resulting three-dimensional models. Heat maps and circular plots (Table 8.2) offer static presentations of interaction data. While giving overviews of the datasets, these static plots have limited capacity to represent the scale and complexity of the data, which will likely involve careful examination of patterns and subgroups within the datasets. Thus, we can expect further development of tools to aid examination of these datasets.
An initial tool to visualize genome models in three dimensions is Genome3D [8.18]; we can expect further development in this direction.
The data analysis itself will further evolve.
With larger datasets, higher resolution might be possible provided that background noise does not prove problematic. Optimistically, it may be possible to detect if 30 nm fiber-like structures are present in vivo, although if there are too many short-range random collisions, this nonspecific background noise may make detection of smaller-scale regularities such as a 30 nm structure difficult or impossible [8.29]. Developing methods that cope with noisy data might be one useful avenue for exploration. Related to this may be further examination of what controls might be applied to these data.
Another avenue might be approaches to compare different datasets, say from different cell types or stages of the cell cycle.
Better support for, and understanding of, the main underlying premise – that the frequency of interactions between two loci is inversely proportional to their proximity in space – would be useful. As far as the author is aware, at the time of writing, few imaging studies are available to offer support for this premise (e.g., Lieberman-Aiden et al. [8.4] and Miele et al. [8.30]).
The lack of (high-resolution) true-positive truth sets against which these computational methods might be compared is a concern. While one can test for self-consistency, there needs to be some way to assess the accuracy of the models generated and what can be inferred from the interaction data.
To conclude, an important overriding conclusion is that biophysics matters.
While you can argue about the specifics [8.31], it should be clear to anyone perusing this field that this work involves a shift to a physical genome: physical both in the sense of dealing with a physical substrate (rather than abstract information) and in the sense of physics.
Abbreviations
- 3-D:
-
three-dimensional
- 3C:
-
chromosome conformation capture
- BLAST:
-
basic local alignment search tool
- CCC:
-
chromosome conformation capture
- ChIA-PET:
-
chromatin interaction analysis using paired-end tag sequencing
- ChIP-3C:
-
ChIP-loop
- ChIP:
-
chromatin immunoprecipitation
- DNA:
-
deoxyribonucleic acid
- FISH:
-
fluorescence in situ hybridization
- LMA:
-
ligation-mediated amplification
- LMA:
-
ligation-mediated annealed
- NMR:
-
nuclear magnetic resonance
- PCR:
-
polymerase chain reaction
- PET:
-
positron emission tomography
- RE:
-
restriction endonuclease
- RE:
-
restriction enzyme
- RMS:
-
root-mean-square
- UCSC:
-
University of California Santa Cruz
- log:
-
logistic regression
References
J. Fraser, M. Rousseau, S. Shenker, M.A. Ferraiuolo, Y. Hayashizaki, M. Blanchette, J. Dostie: Chromatin conformation signatures of cellular differentiation, Genome Biol. 10, R37 (2009)
J. Dekker, K. Rippe, M. Dekker, N. Kleckner: Capturing chromosome conformation, Science 295, 1306–1311 (2002)
J. Dostie, T.A. Richmond, R.A. Arnaout, R.R. Selzer, W.L. Lee, T.A. Honan, E.D. Rubio, A. Krumm, J. Lamb, C. Nusbaum, R.D. Green, J. Dekker: Chromosome conformation capture carbon copy (5C): A massively parallel solution for mapping interactions between genomic elements, Genome Res. 16, 1299–1309 (2006)
E. Lieberman-Aiden, N.L. van Berkum, L. Williams, M. Imakaev, T. Ragoczy, A. Telling, I. Amit, B.R. Lajoie, P.J. Sabo, M.O. Dorschner, R. Sandstrom, B. Bernstein, M.A. Bender, M. Groudine, A. Gnirke, J. Stamatoyannopoulos, L.A. Mirny, E.S. Lander, J. Dekker: Comprehensive mapping of long-range interactions reveals folding principles of the human genome, Science 326, 289–293 (2009)
Z. Zhao, G. Tavoosidana, M. Sjölinder, A. Göndör, P. Mariano, S. Wang, C. Kanduri, M. Lezcano, K.S. Sandhu, U. Singh, V. Pant, V. Tiwari, S. Kurukuti, R. Ohlsson: Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions, Nat. Genet. 38, 1341–1347 (2006)
K.E. Cullen, M.P. Kladde, M.A. Seyfred: Interaction between transcription regulatory regions of prolactin chromatin, Science 261, 203–206 (1993)
Z. Duan, M. Andronescu, K. Schutz, S. McIlwain, Y. Kim, C. Lee, J. Shendure, S. Fields, C.A. Blau, W.S. Noble: A three-dimensional model of the yeast genome, Nature 465, 363–367 (2010)
D. Baù, A. Sanyal, B.R. Lajoie, E. Capriotti, M. Byron, J.B. Lawrence, J. Dekker, M.A. Marti-Renom: The three-dimensional folding of the α-globin gene domain reveals formation of chromatin globules, Nat. Struct. Mol. Biol. 18, 107–114 (2011)
A.A. Gavrilov, I.S. Zukher, E.S. Philonenko, S.V. Razin, O.V. Iarovaia: Mapping of the nuclear matrix-bound chromatin hubs by a new M3C experimental procedure, Nucleic Acids Res. 38, 8051–8060 (2010)
D. Noordermeer, M. Leleu, E. Splinter, J. Rougemont, W. De Laat, D. Duboule: The dynamic architecture of hox gene clusters, Science 334, 222–225 (2011)
M. Rousseau, J. Fraser, M.A. Ferraiuolo, J. Dostie, M. Blanchette: Three-dimensional modeling of chromatin structure from interaction frequency data using Markov chain Monte Carlo sampling, BMC Bioinformatics 12, 414 (2011)
M. Simonis, P. Klous, E. Splinter, Y. Moshkin, R. Willemsen, E. de Wit, B. van Steensel, W. de Laat: Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C), Nat. Genet. 38, 1348–1354 (2006)
H. Wurtele, P. Chartrand: Genome-wide scanning of HoxB1-associated loci in mouse ES cells using and open-ended chromosome conformation capture methodology, Chromosome Res. 14, 477–495 (2006)
S. Lomvardas, G. Barnea, D.J. Pisapia, M. Mendelsohn, J. Kirkland, R. Axel: Interchromosomal interactions and olfactory receptor choice, Cell 126, 248–250 (2006)
J. Fraser, M. Rousseau, M. Blanchette, J. Dostie: Computing Chromosome Conformation. In: Computational Biology of Transcription Factor Binding, Methods in Molecular Biology, Vol. 674, ed. by I. Ladunga (Humana, Totowa 2010) pp. 251–268
B.R. Lajoie, L.N. van Berkum, A. Sanyal, J. Dekker: My5C: Web tools for chromosome conformation capture studies, Nat. Methods 6, 690–691 (2009)
M.J. Fullwood, Y.J. Ruan: ChIP-Based methods for the identification of long-range chromatin interactions, Cell. Biochem. 107, 30–39 (2009)
T.M. Asbury, M. Mitman, J. Tang, W.J. Zheng: A viewer-model framework for integrating and visualizing multi-scale epigenomic information within a three-dimensional genome, BMC Bioinformatics 11, 444 (2010)
D. Russel, K. Lasker, B. Webb, J. Velázquez-Muriel, E. Tjioe, D. Schneidman-Duhovny, B. Peterson, A. Sali: Putting the pieces together: Integrative modeling platform software for structure determination of macromolecular assemblies, PLoS Biology 10(1), e1001244 (2012)
N. Darzentas: Circoletto: Visualizing sequence similarity with Circos, Bioinformatics 26, 2620–2621 (2010)
M. Krzywinski, J. Schein, I. Birol, J. Connors, R. Gascoyne, D. Horsman, S.J. Jones, M.A. Marra: Circos: An information aesthetic for comparative genomics, Genome Res. 19, 1639–1645 (2009)
T.M. OʼBrien, A.M. Ritz, B.J. Raphael, D.H. Laidlaw: Gremlin: An interactive visualization model for analyzing genomic rearrangements, IEEE Trans. Vis. Comput. Graph. 16, 918–926 (2010)
S. Fröhler, C. Dieterich: 3PD: Rapid design of optimal primers for chromosome conformation capture assays, BMC Bioinformatics 10, 635 (2009)
E. Yaffe, A. Tanay: Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture, Nat. Genet. 43, 1059–1063 (2011)
D. Baù, M.A. Marti-Renom: Structural determination of genomic domains by satisfaction of spatial restraints, Chromosome Res. 19, 25–35 (2011)
M.A. Marti-Renom, L.A. Mirny: Bridging the resolution gap in structural modeling of 3D genome organization, PLoS Comput. Biol. 7, 2125 (2011)
A. Sanyal, D. Baù, M.A. Martí-Renom, J. Dekker: Chromatin globules: A common motif of higher order chromosome structure?, Curr. Opin. Cell Biol. 23, 325–331 (2011)
J.L. Reed, I. Famili, I. Thiele, B.O. Palsson: Towards multidimensional genome annotation, Nat. Rev. Genet. 7, 130–141 (2006)
P.J. Shaw: Mapping chromatin conformation, F1000, Biology Rep. 2, 18 (2010)
A. Miele, J. Dekker: Mapping cis- and trans-chromatin interaction networks using chromosome conformation capture (3C). In: The Nucleus. Volume 2: Chromatin, Transcription, Envelope, 105 Proteins, Dynamics, and Imaging, Methods in Molecular Biology, Vol. 468, ed. by R. Hancock (Humana, Totowa 2008)
J. Langowski: Chromosome conformation by crosslinking: Polymer physics matters, Nucleus 1, 37–39 (2010)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag
About this chapter
Cite this chapter
Jacobs, G.H. (2014). Exploring the Interactions and Structural Organization of Genomes. In: Kasabov, N. (eds) Springer Handbook of Bio-/Neuroinformatics. Springer Handbooks. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-30574-0_8
Download citation
DOI: https://doi.org/10.1007/978-3-642-30574-0_8
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-30573-3
Online ISBN: 978-3-642-30574-0
eBook Packages: EngineeringEngineering (R0)