
Abstract
Dysentery is bloody diarrhea caused by infection with certain bacteria or parasites. The most common bacterial causes are members of the Genus Shigella. Shigella are Gram-negative intracellular bacterial pathogens that cause diarrheal disease by infecting intestinal epithelial cells. Following invasion of intestinal cells, Shigella induce host cell cytoskeletal rearrangements and interfere with host cell signal transduction cascades. These effects are mediated by multiple different effector proteins that are translocated from the bacterial cell into the host cell through a type three secretion system. Translocated Shigella effector proteins modulate the host immune response, which contributes to inflammation during infection and to clearance of the organism. Antibiotics are available and effective against Shigella infection; however, isolates resistant to routine antibiotics are increasingly frequent in many areas of the world. Vaccine development is an ongoing area of research.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Dysentery is bloody diarrhea caused by infection with certain bacteria or parasites. The most common bacterial causes are members of the genus Shigella, the biology of which is discussed in this chapter. The most common parasitic causes of dysentery are the amoebae.
Shigella spp. are nonmotile Gram-negative, nonspore forming, non-lactose fermenting, facultative anaerobic bacillus-shaped bacterium that are very closely related to Escherichia coli. Within the genus Shigella are four species (S. dysenteriae, S. flexneri, S. boydii, and S. sonnei) and multiple serotypes of each species. S. dysenteriae is the most virulent pathogen within this genus; it was first discovered and isolated by the Japanese microbiologist Kiyoshi Shiga in 1898. The epidemiology and pathology of this microbe is of particular clinical significance as Shigella is associated with severe diarrheal disease and dysentery in humans. The organism is spread from person to person through contact with contaminated food and water products.
The complete genome of Shigella includes a single circular chromosome, one large virulence plasmid, and a variable number of small plasmids, which collectively encode genes for a type three secretion system and multiple virulence factors that enable this pathogen to invade epithelial cells, manipulate the host cytoskeleton, spread through tissue, and modulate the innate immune response.
Taxonomy
The genus Shigella is very closely related to the genus Escherichia; both belong to the family Enterobacteriaceae. In the current classification, within the genus Shigella are four species: S. dysenteriae (serogroup A), S. flexneri (serogroup B), S. boydii (serogroup C), and S. sonnei (serogroup D). The most virulent of the Shigella species, S. dysenteriae, was discovered by the Japanese microbiologist Kiyoshi Shiga during a severe outbreak in 1898. Serogroups A and C (S. flexneri and S. sonnei) are most commonly associated with clinical disease. Within each serogroup are multiple serotypes: serogroup A (S. dysenteriae), 12 serotypes; serogroup B (S. flexneri), 6 serotypes; serogroup C (S. boydii), 23 serotypes; and serogroup D (S. sonnei), 1 serotype (Table 14.1 ). The four species share several key features, including lack of motility, inability to form spores, and inability to ferment lactose, and all four species are facultative anaerobes. Shigella species are differentiated from one another using a method of serotyping, which is based on antigen type.
Habitat
Humans are the only natural host for Shigella. Monkeys and certain small animals can be infected in the laboratory, but are not natural hosts. In most cases, spread of disease from one individual to another occurs via the fecal-oral route, typically via contamination of the hand. However, with increasing frequency, spread involves ingestion of contaminated foodstuffs or contaminated water.
Epidemiology
Humans are the only natural reservoir of Shigella. Annually in the United States, Shigella are estimated to cause approximately 450,000 cases (Mead et al. 1999), with about four cases per 100,000 population (2009). Annually worldwide, they are estimated to cause 165 million infections (Kotloff et al. 1999). The species most commonly associated with sporadic infections and outbreaks are S. flexneri and S. sonnei. S. flexneri is overall the most common serogroup isolated from clinical infections worldwide. It is most prevalent in the developing world, whereas S. sonnei is the most prevalent serogroup in Europe and the United States. S. flexneri and S. sonnei are associated with endemic forms of the disease, while S. dysenteriae serotype 1 is responsible for most epidemics. S. dysenteriae infections in North America are most commonly due to serotype 1, whereas in other areas of the world, other serotypes of S. dysenteriae have largely replaced serotype 1. Infections due to Shigella typically occur in situations of overcrowding or poor hygiene and sanitation, such as day care centers, institutions for the mentally disabled, and cruise ships.
Evolution and Genomics
The genome of Shigella consists of a single circular chromosome, a virulence plasmid, and multiple smaller “cryptic” plasmids. The sequence of the entire Shigella genome, including the chromosome, the virulence plasmid, and the cryptic plasmids, was completed in the early 2000s by several independent groups of researchers (Jin et al. 2002; Nie et al. 2006; Wei et al. 2003; Yang et al. 2005, Venkatesan et al. 2001, Buchrieser et al. 2000). The genomes of five different strains, representing all four species of Shigella, S. dysenteriae, S. flexneri, S. boydii, and S. sonnei, are currently available (Jin et al. 2002; Nie et al. 2006; Wei et al. 2003; Yang et al. 2005). The circular chromosome of S. flexneri is 4.6 Mbp (4,599,354) with a G + C content of 50.9 % and 4,084 predicted genes (Wei et al. 2003). The large virulence plasmid, which is present in all isolates of Shigella and is required for virulence, is 0.220 Mbp (220 kbp), while the size and number of additional plasmids vary depending on the isolate.
Several studies have shown high sequence similarity between the genomes of Shigella spp. and Escherichia coli. Early studies using DNA hybridization revealed that these two microbes are taxonomically indistinguishable (Brenner et al. 1972). More recent studies using multilocus enzyme electrophoresis, comparative genomic hybridization, and multilocus sequence typing have confirmed early predictions of the high sequence similarity between Shigella spp. and E. coli (Lan and Reeves 2002; Pupo et al. 1997, 2000).
Sequence analysis of eight housekeeping genes in four different regions of the chromosome of multiple species of Shigella, S. boydii, S. dysenteriae, S. flexneri, and S. sonnei, revealed that Shigella spp. evolved from E. coli at least 35,000–270,000 years ago. Sequence variation of these eight genes and more extensive sequence analysis of housekeeping genes suggest multiple independent lines of evolution (Pupo et al. 2000; Yang et al. 2007). Based on these studies, it is now well accepted that Shigella belongs to the species E. coli, instead of belonging to its own separate genus (Schroeder and Hilbi 2008) and that pathogenic strains that are commonly known as Shigella spp. emerged from E. coli at least seven times during evolution (Fig. 14.1 ).


Sequence of evolutionary events that result in the classification of Shigella as a distinct genus. Through a series of gene acquisition and gene loss events, Shigella acquired a virulence plasmid and multiple pathogenicity islands and lost genes required for flagella and fimbriae synthesis, genes encoding proteins whose activity inhibits virulence (e.g., CadA and OmpT), and genes for catabolic pathways (Adapted from Peng et al. (2009), Schroeder and Hilbi (2008)).
Comparative genomics studies further substantiate the genetic and taxonomic relationship between Shigella and E. coli. The genomes of Shigella and E. coli are only 1.5 % divergent (Fukushima et al. 2002; Lan and Reeves 2002; Pupo et al. 1997, 2000). The chromosome of S. flexneri is slightly smaller (4,599,354 bp) than that of enterohemorrhagic E. coli (4,639,221 bp) (Perna et al. 2001; Wei et al. 2003). The overall organization of the two chromosomes is similar, consisting of large regions of backbone with islands. S. flexneri has a slighter larger amount of backbone (82 %) than enterohemorrhagic and uropathogenic E. coli (75 %) and 200 more pseudogenes than these E. coli (Wei et al. 2003).
Shigella spp. are most closely related to enteroinvasive E. coli (EIEC) as opposed to other strains of E. coli (Lan et al. 2004; Yang et al. 2007). Shigella spp. and EIEC evolved from other E. coli via convergent evolution (Lan et al. 2004; Pupo et al. 2000) involving multiple events of gene acquisitions, horizontal gene transfer, and genetic loss, through gene deletion (Yang et al. 2007). Shigella spp. acquired the large virulence plasmid and five chromosomal pathogenicity islands (SHI-1, SHI-2, SHI-3, SHI-O, and SRL) (Ingersoll et al. 2002; Luck et al. 2001; Ochman et al. 2000; Peng et al. 2009; Purdy and Payne 2001; Rajakumar et al. 1997; Schroeder and Hilbi 2008; Vokes et al. 1999). Shigella spp. lost genes for flagella synthesis, rendering the organism nonmotile, and for fimbriae synthesis (Al Mamun et al. 1996; Hacker et al. 1990; Tominaga et al. 2005). Shigella spp. also lost the gene encoding the outer membrane protein OmpT, a protease that can cleave the outer membrane protein IcsA (VirG) at the bacterial surface, and acquired on the virulence plasmid the gene encoding a similar yet more highly regulated protease (IcsP, SopA). IcsA, described below, is required for actin polymerization and intracellular spread (Bernardini et al. 1989; Lett et al. 1989). The genes involved in the biosynthesis of cadaverine, the small polyamine product of lysine decarboxylation, were lost during the evolution of both Shigella spp. and EIEC from E. coli (Casalino et al. 2003; Maurelli et al. 1998). The presence of cadaverine during Shigella infection leads to delayed lysis of the phagocytic vacuole by intracellular bacteria, decreased transmigration of polymorphonuclear leukocytes across the infected epithelium, and consequent attenuation of the infection (Fernandez et al. 2001; Maurelli et al. 1998; McCormick et al. 1999). Shigella spp. also lost genes for the L-aspartate-dihydroxyacetone and lactose fermentation pathways (Ito et al. 1991; Prunier et al. 2007a, b; Yang et al. 2005).
The chromosomal loci that have been acquired during evolution are designated chromosomal pathogenicity islands SHI-1, SHI-2, SHI-3, SHI-O, and the Shigella resistance locus (SRL) (Ingersoll et al. 2002). Pathogenicity islands are large genomic regions that encode virulence factors and are typically characterized by a G + C content and codon usage that are distinct from the chromosome. Acquired by horizontal gene transfer events, pathogenicity islands are often associated with mobile genetic elements and insertion sequences (Dobrindt et al. 2004). SHI-1 encodes the immunoglobulin A-like protease SigA (Al-Hasani et al. 2000), the serine protease Pic (Henderson et al. 1999), and the enterotoxin ShET1 (Fasano et al. 1995, 1997). SHI-2 encodes ShiD and ShiA, which has been shown to interfere with the T-cell immune response during infection (Ingersoll et al. 2003; Ingersoll and Zychlinsky 2006). SHI-2 and SHI-3 encode factors involved in iron acquisition, including the siderophore aerobactin and enterochelin receptors (Luck et al. 2001; Nassif et al. 1987; Purdy and Payne 2001; Vokes et al. 1999). SHI-O, which is present in a subset of strains, contains genes that modify the O-antigen of lipopolysaccharide in ways that contribute to virulence (Huan et al. 1997; Lindberg et al. 1991; Zhong 1999). SRL encodes genes for antibiotic resistance, including tetracycline, chloramphenicol, ampicillin, and streptomycin (Luck et al. 2001; Turner et al. 2001, 2003). Collectively, these genetic acquisition and loss events have lead to the evolution of Shigella spp. from E. coli as a discrete pathogen adapted to a distinct, predominantly intracellular, lifestyle.
Pathogenesis and Virulence Factors
A key distinguishing feature of Shigella is its ability to invade host intestinal epithelial cells. The factors required for invasion are encoded on a large plasmid, known as the “virulence plasmid” or “invasion plasmid,” which is present in all virulent strains (Parsot 2009; Sansonetti et al. 1982). Two adjacent loci on the virulence plasmid confer invasion capabilities: the mxi-spa locus, which encodes the structural components of the type three secretion system (T3SS), and the ipa (invasion-related plasmid-encoded antigens) locus, which encodes multiple different factors, including those required for delivery of effector proteins into host cells, transcriptional regulators, chaperones, and effector proteins (Schroeder and Hilbi 2008). The T3SS is essential for Shigella invasion, as plasmid-cured strains and strains carrying disruptions or deletions of any of the T3SS structural genes are unable to invade (Sansonetti et al. 1982). Expression of the genes encoding the structural proteins and of many of the effectors is regulated by two virulence plasmid-encoded transcription activators, VirB and VirF (Le Gall et al. 2005; Schroeder and Hilbi 2008). VirF, a member of the AraC family of transcription activators, activates transcription of virB and icsA (virG) in response to increase in temperature to 37 °C (Hale 1991; Tobe et al. 1993). VirB activates transcription of the T3SS structural proteins and the type three secreted invasion proteins (Porter and Dorman 1997).
The Shigella spp. T3SS is a multi-protein apparatus in the bacterial cell envelope that allows for the transport of effector proteins from the bacterial cell cytoplasm across both the bacterial cell envelope and the host epithelial cell plasma membrane into the host epithelial cell cytoplasm (Blocker et al. 2001). The apparatus consists of a gated channel that traverses the inner membrane, the periplasm, and the outer membrane and extends in the form of a long needle into the extracellular space. Activation of secretion is initiated upon contact with host epithelial cells (Enninga et al. 2005). Upon activation, three translocators and 25 or more effector proteins are delivered through the T3SS apparatus into the host epithelial cells (Table 14.2 ) (Enninga et al. 2005; Parsot 2009).
Delivery of proteins through the T3SS occurs in an orderly fashion, with the proteins involved in the formation of a pore in the host cell membrane being delivered first, followed by the effector proteins involved in the entry process, and lastly by the effector proteins that modulate later stages of infection, including those that participate in bacterial intercellular spread and those that manipulate the innate immune response. The genes encoding the proteins that are secreted early, including IpaB, IpaC, and IpaD, which are involved in pore formation, IpaA and IpgB1, which participate in entry, and IcsB, which functions in avoidance of autophagy, along with their chaperones, are transcribed independent of MxiE (see below) (Le Gall et al. 2005; Parsot 2009). Consequently, they are preformed in the bacterial cell and, upon contact with the host cell, are ready to be secreted.
A second group of proteins secreted by the T3SS are those that modulate later stages of infection. As a rule, transcription of these effectors is dependent on MxiE, an AraC family transcription activator encoded within the T3SS locus, whose transcription is regulated by VirB. Transcription by MxiE is intricately co-regulated by IpgC, the chaperone for the translocases IpaB and IpaC. Prior to host cell contact, IpgC is bound to IpaB and IpaC, preventing their premature association (Menard et al. 1994). Upon contact, IpaB and IpaC are dissociated from IpgC in the bacterial cytoplasm and are secreted, whereupon they interact with each other to form a pore in the host plasma membrane (Menard et al. 1994). Concurrently, IpgC becomes available to serve as co-activator of MxiE-mediated transcription (Mavris et al. 2002a, b). Prior to MxiE-IpgC assembly, MxiE is in complex with OspD1 and the chaperone Spa15, which effectively inhibit the activity of MxiE (Page et al. 2002; Parsot et al. 2005). Four effectors secreted by the T3SS, VirA, OspB, OspC1, and OspF, are partially regulated by MxiE-mediated transcriptional activation (Parsot 2009).
In addition to the transcriptional regulation described above, Spa32 negatively regulates the length of the T3SS apparatus needle that extends from the bacterial surface and controls the selection of substrates for secretion, whereas Spa33 regulates Ipa protein secretion (Magdalena et al. 2002; Schuch and Maurelli 2001). Under anaerobic conditions, such as those present in the lumen of the human intestine, transcription of spa32 and spa33 are repressed by FNR, a conserved regulator of anaerobic metabolism, and the needles become long (Marteyn et al. 2010). Close to the epithelial surface, however, oxygen tension increases sufficiently to de-repress transcription of spa32 and spa33, triggering molecular events necessary for cellular entry (Marteyn et al. 2010).
The best-described functions of IpaB, IpaC, and IpaD are as translocators that form a pore in the host plasma membrane (Menard et al. 1994). In addition, they are thought to possibly gate the pore and anchor the T3SS needle to the plasma membrane. IpaB and IpaC are positioned at the tip of the T3SS apparatus from where they integrate into cholesterol-rich domains of the plasma membrane (De Geyter et al. 1997; Harrington et al. 2006; Lafont et al. 2002), creating a pore and a conduit between the bacterial and host cells that allows for the subsequent delivery of effector proteins (Blocker et al. 1999; Espina et al. 2006; Menard et al. 1993, 1994; Veenendaal et al. 2007). IpgD is also positioned at the tip of the T3SS apparatus, where in addition to providing scaffolding, it also regulates secretion (Picking et al. 2005; Schroeder and Hilbi 2008). IpaD is anchored directly to MxiH, the protein that forms the needle of the T3SS apparatus (Zhang et al. 2007). The precise signals responsible for activating secretion and the molecular mechanisms by which these signals trigger the assembly of the T3SS apparatus tip upon epithelial cell contact are unknown and yet are active areas of research. In addition to their roles as translocators, IpaB and IpaC have also been shown to have effector-like activities.
The early effector IpaA causes localized actin depolymerization at the sites of bacterial invasion, leading to enhanced bacterial uptake (Bourdet-Sicard et al. 1999). The effects of IpaA activity are mediated by its direct interaction with the host actin cytoskeletal protein vinculin. The C-terminal domain of IpaA binds to the amino-terminal head domain of vinculin (Bourdet-Sicard et al. 1999; Demali et al. 2006; Ramarao et al. 2007; Tran Van Nhieu et al. 1997). Independent of its interaction with vinculin, IpaA induces weakening of cellular adhesion to the extracellular matrix (Demali et al. 2006). Spa15 serves as a chaperone for IpaA (Page et al. 2002).
The early effector protein IpgB1 activates a cellular pathway that induces formation of membrane ruffles, likely by serving as a GTP exchange factor (GEF) for the Rho GTPase RhoG. Like RhoG, IpgB1 interacts with the cellular protein complex ELMO-Dock180, which results in activation of the actin nucleation-promoting factors Rac1 and Cdc42 and actin-mediated formation of membrane ruffles (Handa et al. 2007; Ohya et al. 2005). IpgB2, a homologue of IpgB1, serves as a GEF for the Rho GTPase RhoA (Klink et al. 2010) and activates the immune modulator NF-κB (Fukazawa et al. 2008), but its molecular function in Shigella pathogenesis remains uncertain. OspB also activates NF-κB by a mechanism that is unclear (Fukazawa et al. 2008).
IpgD, an early type three secreted effector, is an inositol phosphatase that promotes membrane ruffling during bacterial entry and alters cellular survival and lysosomal degradation pathways (Niebuhr et al. 2000; Pendaries et al. 2006; Ramel et al. 2011). IpgD specifically mediates the dephosphorylation of phosphatidylinositol 4,5-bisphosphate (PI-(4,5)P2) to yield phosphatidylinositol 5-monophosphate (PI-(5)P) (Niebuhr et al. 2002). IpgD-induced formation of PI-(5)P results in activation of the PI3-kinase signaling pathway that leads to Akt phosphorylation in a manner that depends on the epidermal growth factor receptor (EGFR), which modulates endosomal trafficking (Pendaries et al. 2006; Ramel et al. 2011). Increased levels of PI-(5)P lead to decreased lysosomal degradation and increased host cell survival (Pendaries et al. 2006; Ramel et al. 2011).
Following Shigella uptake into host epithelial cells, a second wave of effectors is secreted via the T3SS into the Shigella-containing vacuole and, after vacuolar lysis, into the cell cytoplasm. Some of these effectors are important for pathogen survival, others are essential for Shigella dissemination through the epithelial cell layer, and others modulate the host immune response. Finally, the functions of other effectors are less well defined.
Shigella effectors that mediate lysis of the Shigella-containing vacuole are unknown. Early work suggested that IpaB is involved in this process (High et al. 1992), but given what has been learned since about the role of IpaB in secretion of other effectors, the mechanism of its involvement in vacuolar lysis is unclear. Lysis of the vacuole releases the bacterium into the cell cytoplasm, where it utilizes the cellular actin polymerization machinery to move. The bacterium polymerizes actin into a tail at one end of the bacterial body (Fig. 14.2 ). Recruitment of the actin polymerization machinery to the bacterium depends on the Shigella outer membrane protein IcsA (VirG) (Bernardini et al. 1989; Lett et al. 1989), which is a member of the autotransporter family of proteins and is not secreted by the T3SS. IcsA binds the cellular actin nucleation-promoting factor N-WASP, and N-WASP is activated by the cellular protein Toca-1, whereupon it recruits and activates the actin polymerizing complex Arp2/3 (Leung et al. 2008; Lommel et al. 2001; Snapper et al. 2001; Suzuki et al. 1998). Polymerization of the tail propels the bacterium to the cell periphery, whereupon through processes that are incompletely understood, it utilizes diaphanous formins to generate protrusions of the plasma membrane that enclose the bacterium (Heindl et al. 2010). Bacterium-containing protrusions are engulfed by adjacent cells, leading to spread of the bacterium into these cells.


Actin tail assembly by Shigella during infection. (a) Polymerized actin; (b) bacterial and cellular DNA; (c) overlay of polymerized actin (red) and DNA staining (blue). Arrows, bacteria at the tip of actin tails
In addition to recruiting actin polymerization machinery, IcsA can be recognized by the cellular autophagy protein Atg5. Atg5 recognition activates the autophagosome formation pathway. Autophagy is a cellular pathway that engulfs foreign objects present in the cytoplasm, such as intracellular bacteria, and kills and degrades them. The type three secreted effector IcsB shares the same binding region and has higher affinity than Atg5 for IcsA, such that IcsB binding to IcsA masks Atg5 recognition and allows the bacterium to escape detection and destruction via autophagy (Ogawa and Sasakawa 2006; Ogawa et al. 2005).
Shigella spp. encode two copies of the type three secreted effectors OspE, OspE1, and OspE2, which have nearly identical protein sequences. OspE proteins interact with integrin-linked kinase (ILK) within sites of cellular attachment to the extracellular matrix, causing stabilization of these attachment sites and preventing cell release from the substratum during infection (Kim et al. 2009).
The type three secreted effector VirA is homologous and structurally similar and can partially functionally complement EspG, a type three secreted effector of enterohemorrhagic Escherichia coli (EHEC) and enteropathogenic E. coli (EPEC) (Davis et al. 2008; Elliott et al. 2001; Germane and Spiller 2011; Selyunin et al. 2011). EspG regulates endomembrane trafficking through interactions with ADP-ribosylation factor GTPases and p21-activated kinases (Germane and Spiller 2011; Selyunin et al. 2011). However, the specific function of VirA remains uncertain, as a possible role in endomembrane trafficking has not been examined and data indicating a role in microtubule destabilization and protease activity are conflicting (Germane et al. 2008; Yoshida et al. 2006).
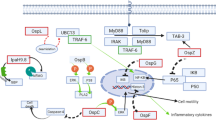
Among the type three secreted effectors of Shigella spp. is a family of 5–7 effector proteins designated IpaH, including some encoded on the Shigella chromosome (Ashida et al. 2007). IpaH proteins contain a conserved C-terminal domain and a variable N-terminal domain. The C-terminal domains function as E3 ligases (Rhode et al. 2007; Singer et al. 2008; Zhu et al. 2008), proteins that target specific substrates for degradation via the cellular ubiquitination pathway. The N-terminal domain is the site of a leucine-rich repeat domain, which is a classical pathogen-associated molecular pattern recognition site involved in the host epithelial cell immune response during pathogen infection (Bell et al. 2003; Hartman et al. 1990; Okuda et al. 2005; Parsot 2009; Venkatesan et al. 1991). The substrate specificity of the IpaH proteins is determined by the N-terminal domain and has been identified for only one, IpaH9.8, which targets NEMO/IKKγ, a host inflammatory response modulator, for degradation, thereby dampening the NF-κB regulated inflammatory response (Ashida et al. 2010). In the same pathway, OspG, a type three effector protein that is not a member of the IpaH family, binds ubiquitinated ubiquitin-conjugating enzymes, thereby preventing the degradation of the NF-kB inhibitor IκBα (Kim et al. 2005).
Two other type three secreted effector proteins involved in modulation of the host inflammatory response are OspF and OspZ. OspF possesses phosphothreonine lyase activity, an unusual enzymatic activity. It irreversibly dephosphorylates components of the mitogen-activated protein kinase (MAPK) signaling pathway, leading to inhibition of this pathway (Arbibe et al. 2007; Kramer et al. 2007). OspZ inhibits the nuclear translocation of NF-kB (Newton et al. 2010). As for the activity of IpaH9.8 and OspG, the activities of OspF and OspZ attenuate the host inflammatory response. The functions of several type three effectors, including OspC1, OspC2, OspC3, OspD2, and OspD3, are currently unknown (Table 14.2 ).
Shigella and the Immune Response
Shigella infection is generally restricted to the mucosal layer of the large intestine. The organism is able to survive the environment of the stomach due to acid resistance mechanisms (Gorden and Small 1993). Once at the epithelial lining of the large intestine, Shigella may be taken up by M-cells, which are specialized in gut-lumen sampling; uptake by M-cells leads to transcytosis of the bacteria across the epithelial layer (Sansonetti et al. 1996; Wassef et al. 1989). Transcytosis enables Shigella to enter the epithelial cell lining at the basolateral surface, instead of at the apical surface, and it also enables bacterial interactions with macrophages and dendritic cells within the mucosa (Mounier et al. 1992; Sansonetti et al. 1999). Shigella may also enter cells by disrupting epithelial intercellular junctions (Perdomo et al. 1994a; Sakaguchi et al. 2002). Whether organisms also enter cells from the apical side of the epithelium in vivo is uncertain.
When phagocytosed by macrophages, Shigella evades killing by triggering apoptosis, which is accompanied by the release of massive amounts of the pro-inflammatory cytokines interleukin (IL)-1β and IL-18 (Islam et al. 1997; Sansonetti et al. 2000; Zychlinsky et al. 1992; Zychlinsky et al. 1996). Release of IL-1β triggers intestinal inflammation (Sansonetti et al. 1995). Release of IL-18 is associated with an antimicrobial response that involves NK (natural killer) cell activation and the production of interferon (IFN)-γ, which is critical for mounting an innate immune response against microbial infection (Hilbi et al. 1997; Le-Barillec et al. 2005; Sansonetti et al. 2000; Way et al. 1998).
Shigella enters the intestinal epithelium by the basolateral surface of cells (Sansonetti et al. 1986). Following entry, internalized bacteria escape the uptake vacuole, replicate within the cytoplasm, and utilize the host cytoskeleton to move to the cell periphery and into adjacent cells. All eukaryotic cells possess mechanisms for eliminating intracellular foreign bodies, including autophagy and activation of the innate immune response. Shigella has evolved mechanisms to evade each of these host responses.
Eukaryotic cells possess a lysosomal degradation pathway called autophagy that serves both to recover nutrients during periods of starvation and to rid the cell of undesirable particles, including invading pathogens. Upon entry into cells, Shigella is surrounded by a vacuolar membrane, which it rapidly lyses. The remnants of the vacuolar membrane are degraded by the autophagy pathway (Dupont et al. 2009). Then, a tug-of-war is staged between the intracytoplasmic bacteria and the innate immune response, in which some of the intracytoplasmic bacteria succumb to autophagy and some escape. Those that are destined to succumb are ubiquitinated and may be surrounded by a scaffold of the cytoskeletal protein septin, before being engulfed in cellular membranes and degraded (Mostowy et al. 2010; Ogawa et al. 2005). Escape from autophagy is mediated at least in part by the type three secreted effector protein IcsB, which blocks binding of the autophagy protein Atg5 to the surface of Shigella (Ogawa et al. 2005).
Peptidoglycan fragments from intracellular bacteria are sensed by the pattern recognition receptor Nod1, the activation of which results in NF-kB activation and subsequent release of the pro-inflammatory cytokine IL-8 (Girardin et al. 2003; Pedron et al. 2003; Philpott et al. 2000; Sansonetti et al. 1999). IL-8 is responsible for the recruitment of polymorphonuclear leukocytes (PMNs) to the sites of Shigella infection (Sansonetti et al. 2000; Singer and Sansonetti 2004). Infiltrating PMNs entrap and kill invading bacteria but also contribute to the destruction of the epithelial cell lining, which further enables the entry and invasion of more Shigella into intestinal epithelial cells (Perdomo et al. 1994a; Perdomo et al. 1994b). The ability of PMNs to destroy invading bacteria contributes to the resolution of infection (Brinkmann et al. 2004; Mandic-Mulec et al. 1997; Zhang et al. 2001).
The adaptive immune response to Shigella provides partial protection against subsequent infection (Taylor et al. 1989). Particularly surprising is the observation that although Shigella are intracellular pathogens, individuals who have been infected are protected in a serotype-specific manner (Ferreccio et al. 1991; Lerman et al. 1994; Mel et al. 1965, 1968, 1971), suggesting that protection is mediated by the humoral immune response and not the cellular immune response. In animal models, serotype-specific IgA can provide protection (Phalipon et al. 1995), yet is not required for protective immunity (Way et al. 1999), suggesting that IgG is the protective isotype. Whereas it is known that Shigella blocks aspects of the adaptive cellular immune response (Jehl et al. 2011), how it does so is unclear.
Clinical Disease Due to Shigella spp
Shigella causes diarrhea and dysentery, a diarrheal syndrome characterized by blood and white blood cells in the stool. Shigella is a human pathogen, with no reservoir in other animals. In the majority of cases, the organism is acquired from an infected individual by direct human-to-human spread. In other cases, the organism is acquired from food or water that has been contaminated by an infected individual. The incubation period averages 3 days, with a range of 1–7 days.
The infectious inoculum is as few as 10–100 bacteria (Dupont et al. 1989), in large part because the organism is relatively resistant to stomach acid, such that even when only a small number of organisms are ingested, a sufficient number gain access to the intestine, where they replicate and cause disease. As a consequence, outbreaks are common in day care centers, mental institutions, and other settings where housing is crowded or hygiene suboptimal, and secondary infection rates among family members are reported as high as 20 %.
Shigella infect intestinal epithelial cells of the sigmoid colon and rectum, the distal most segments of the colon. The organism is thought to enter the epithelium largely by transcytosis of microfold (M) cells, whose normal function is to sample antigens from the intestinal lumen. Following transcytosis to the subepithelium, Shigella enter into the epithelial cells using a type three secretion system apparatus (T3SS). Once within the cells, the organism spreads into adjacent cells. The release of pro-inflammatory cytokines leads to an acute inflammatory cell infiltrate. The combination of bacterial spread through the epithelium and the inflammatory response leads to local destruction of the epithelium with ulceration and abscess formation and, in many cases, blood and white blood cells in the stool. Symptoms characteristically include severe abdominal cramping, rectal urgency (tenesmus), frequent small loose stools, general malaise, and fever. In the absence of antibiotic therapy, the diarrhea is typically self-limited and resolves in 7 or fewer days.
Significant complications are uncommon. Approximately 4 % of infected individuals will have transient seeding of the bloodstream (bacteremia), 2.5 % will experience obstruction of the intestine, and a small percentage of children will develop rectal prolapse or seizures. In infection due to S. dysenteriae 1, 3 % of individuals will develop a severe dilatation of the colon, called toxic megacolon, which is treated with surgery.
Two uncommon, yet important, complications of Shigella infection are post-infectious arthritis (formerly Reiter syndrome) and hemolytic-uremic syndrome. Occurring in a small percentage of cases, post-infectious arthritis develops 1–2 weeks after the diarrhea and may be accompanied by conjunctivitis and painful urination (urethritis). Seventy percent of patients who develop this syndrome have the haplotype HLA-B27. Post-infectious arthritis can occur following infection with any of several enteric and urethral bacterial pathogens, including Campylobacter, Salmonella, and Yersinia spp.
Hemolytic-uremic syndrome is a potentially life-threatening complication that is characterized by the combination of anemia due to hemolysis of red blood cells, decreased platelets, and kidney failure due to injury of the renal glomeruli. Most commonly affected are children under the age of 5, with 5–25 % suffering from some degree of permanent kidney dysfunction. The damage is mediated by Stx toxin (formerly Shiga toxin), which among Shigella spp. is encoded only by S. dysenteriae 1.
Treatment with antibiotics is recommended for all individuals infected with Shigella. Prognosis is excellent, with nearly all individuals recovering fully. No Shigella vaccines are currently approved for use, although both live attenuated vaccines and subunit vaccines that combine purified protein and lipopolysaccharide (LPS) are under development. Prevention of spread of Shigella depends on meticulous hand hygiene.
Laboratory Identification, Isolation, and Clinical Diagnosis of Shigella Infection
The diagnosis of Shigella infection is made by culture of the organism from stool samples. Shigella can be isolated from stool of infected individuals. In approximately 4 % of infections, Shigella can also be isolated from the bloodstream. Organisms cannot be isolated from other body sites. Stool is plated both on nonselective indicator media and on selective media. The nonselective media is typically MacConkey agar, on which Shigella spp. grow as white colonies and E. coli grow as red colonies (Fig. 14.3a ). Selective media include Salmonella Shigella agar, which, as the name suggests, is selective for growth of Salmonella and Shigella spp., and Hektoen enteric agar, which is both selective for growth of Salmonella and Shigella spp. and differentiates between the two on the basis of the appearance of the colonies. On Hektoen, Salmonella spp. grow as black colonies because they produce hydrogen sulfite, whereas Shigella spp. grow as green colonies because they do not (Fig. 14.3b ). To maximize the likelihood of recovering the organism from stool, it is generally advised to initially plate it on MacConkey agar (nonselective media) and then re-streak lactose negative colonies onto selective media. Colonies are convex, with smooth edges, and translucent, with a typical diameter of 0.5–2.0 mm, and on MacConkey are white in color.


Growth of enteric pathogens on selective agar. (a) Enteric pathogens were grown on MacConkey agar. Lactose non-fermentors, including Shigella, form white colonies, and lactose fermentors, such as E. coli, form pink colonies. EHEC, enterohemorrhagic E. coli. (b) On Hektoen enteric agar, Salmonella form black colonies due to production of hydrogen sulfite (H2S), Shigella form green colonies, and lactose-fermenting bacteria, such as EHEC, form orange colonies
Confirmation of the genus as Shigella is performed using biochemical tests. The organism is oxidase negative, catalase positive, Voges-Proskauer and Simmons citrate negative, lysine decarboxylase negative, arginine dihydrolase negative, and variable for indole production and ornithine reaction. It does not produce hydrogen sulfite, does not hydrolyze urea, does not utilize malonate, and does not grow on potassium cyanide (KCN) agar. Shigella spp. ferment glucose, but do not ferment lactose. S. dysenteriae can be discriminated from the other species of Shigella by its inability to ferment mannitol, and S. sonnei can be discriminated from the others by its ability to produce ornithine decarboxylase. Most clinical microbiology laboratories determine the species of a Shigella isolate by O-antigen typing (serotyping) using O-antigen specific antisera.
Treatment and Vaccine Development
All individuals infected with Shigella should receive a course of antibiotics. Agents that are recommended include the fluoroquinolones (e.g., ciprofloxacin), azithromycin, or trimethoprim-sulfamethoxazole. Resistance to ciprofloxacin, ampicillin, and trimethoprim-sulfamethoxazole is increasing worldwide, so whenever possible, the selection of an antibiotic should be based on laboratory susceptibility data. If left untreated, Shigella infection will resolve over 5–7 days. Antibiotic treatment has been shown to shorten the duration of illness by a couple of days (Christopher et al. 2010).
At present, no vaccine for Shigella is approved for use in the United States. Several distinct types of vaccines are being developed, including subunit vaccines, live attenuated vaccines, and outer membrane vesicle vaccines. The subunit vaccines under development consist of various combinations of purified IpaB and IpaC, translocases of the type three secretion system, purified IpaD, the type three secretion system needle tip, and purified lipopolysaccharide (LPS) (Martinez-Becerra et al. 2011; Riddle et al. 2011). The live attenuated vaccines under development carry deletions in genes involved in intercellular motility (icsA), the toxins ShET2-1 and ShET2-2 (senA, senB), acetylation of LPS (msbB1, msbB2), and guanine biosynthesis (guaBA) (Barnoy et al. 2010, 2011; Ranallo et al. 2010; Wu et al. 2011). Outer membrane vesicle vaccines consist of outer membrane vesicles purified from virulence strains (Camacho et al. 2011).
Conclusion
Shigella is a Gram-negative intracellular bacterial pathogen that causes diarrheal disease by infecting intestinal epithelial cells. Following invasion of intestinal cells, Shigella induces host cell cytoskeletal rearrangements and interferes with host cell signal transduction cascades. These effects are mediated by multiple different effector proteins that are translocated from the bacterial cell into the host cell through a type three secretion system. Translocated Shigella effector proteins modulate the host immune response, which contributes to inflammation during infection and to clearance of the organism. Antibiotics are available and effective against Shigella infection; however, isolates resistant to routine antibiotics are increasingly frequent in many areas of the world. Vaccine development is an ongoing area of research.
References
Al Mamun AA, Tominaga A, Enomoto M (1996) Detection and characterization of the flagellar master operon in the four Shigella subgroups. J Bacteriol 178:3722–3726
Al-Hasani K, Henderson IR, Sakellaris H, Rajakumar K, Grant T, Nataro JP, Robins-Browne R, Adler B (2000) The sigA gene which is borne on the she pathogenicity island of Shigella flexneri 2a encodes an exported cytopathic protease involved in intestinal fluid accumulation. Infect Immun 68:2457–2463
Arbibe L, Kim DW, Batsche E, Pedron T, Mateescu B, Muchardt C, Parsot C, Sansonetti PJ (2007) An injected bacterial effector targets chromatin access for transcription factor NF-kappaB to alter transcription of host genes involved in immune responses. Nat Immunol 8:47–56
Ashida H, Toyotome T, Nagai T, Sasakawa C (2007) Shigella chromosomal IpaH proteins are secreted via the type III secretion system and act as effectors. Mol Microbiol 63:680–693
Ashida H, Kim M, Schmidt-Supprian M, Ma A, Ogawa M, Sasakawa C (2010) A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKgamma to dampen the host NF-kappaB-mediated inflammatory response. Nat Cell Biol 12:66–73, sup pp 1–9
Barnoy S, Jeong KI, Helm RF, Suvarnapunya AE, Ranallo RT, Tzipori S, Venkatesan MM (2010) Characterization of WRSs2 and WRSs3, new second-generation virG(icsA)-based Shigella sonnei vaccine candidates with the potential for reduced reactogenicity. Vaccine 28:1642–1654
Barnoy S, Baqar S, Kaminski RW, Collins T, Nemelka K, Hale TL, Ranallo RT, Venkatesan MM (2011) Shigella sonnei vaccine candidates WRSs2 and WRSs3 are as immunogenic as WRSS1, a clinically tested vaccine candidate, in a primate model of infection. Vaccine 29:6371–6378
Bell JK, Mullen GE, Leifer CA, Mazzoni A, Davies DR, Segal DM (2003) Leucine-rich repeats and pathogen recognition in toll-like receptors. Trends Immunol 24:528–533
Bernardini ML, Mounier J, d’Hauteville H, Coquis-Rondon M, Sansonetti PJ (1989) Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci USA 86:3867–3871
Blocker A, Gounon P, Larquet E, Niebuhr K, Cabiaux V, Parsot C, Sansonetti P (1999) The tripartite type III secreton of Shigella flexneri inserts IpaB and IpaC into host membranes. J Cell Biol 147:683–693
Blocker A, Jouihri N, Larquet E, Gounon P, Ebel F, Parsot C, Sansonetti P, Allaoui A (2001) Structure and composition of the Shigella flexneri “needle complex”, a part of its type III secreton. Mol Microbiol 39:652–663
Bourdet-Sicard R, Rudiger M, Jockusch BM, Gounon P, Sansonetti PJ, Nhieu GT (1999) Binding of the Shigella protein IpaA to vinculin induces F-actin depolymerization. EMBO J 18:5853–5862
Brenner DJ, Fanning GR, Skerman FJ, Falkow S (1972) Polynucleotide sequence divergence among strains of Escherichia coli and closely related organisms. J Bacteriol 109:953–965
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535
Buchrieser C, Glaser P, Rusniok C, Nedjari H, D’Hauteville H, Kunst F, Sansonetti P, Parsot C (2000) The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol Microbiol 38(4):760–771
Camacho AI, de Souza J, Sanchez-Gomez S, Pardo-Ros M, Irache JM, Gamazo C (2011) Mucosal immunization with Shigella flexneri outer membrane vesicles induced protection in mice. Vaccine 29:8222–8229
Casalino M, Latella MC, Prosseda G, Colonna B (2003) CadC is the preferential target of a convergent evolution driving enteroinvasive Escherichia coli toward a lysine decarboxylase-defective phenotype. Infect Immun 71:5472–5479
Centers for Disease Control and Prevention Publication (2009) Preliminary FoodNet Data on the incidence of infection with pathogens transmitted commonly through food – 10 States, 2008. MMWR Morb Mortal Wkly Rep 58:333–7
Christopher PR, David KV, John SM, Sankarapandian V (2010) Antibiotic therapy for Shigella dysentery. Cochrane Database Syst Rev 8: CD006784
Davis J, Wang J, Tropea JE, Zhang D, Dauter Z, Waugh DS, Wlodawer A (2008) Novel fold of VirA, a type III secretion system effector protein from Shigella flexneri. Protein Sci 17:2167–2173
De Geyter C, Vogt B, Benjelloun-Touimi Z, Sansonetti PJ, Ruysschaert JM, Parsot C, Cabiaux V (1997) Purification of IpaC, a protein involved in entry of Shigella flexneri into epithelial cells and characterization of its interaction with lipid membranes. FEBS Lett 400:149–154
Demali KA, Jue AL, Burridge K (2006) IpaA targets beta1 integrins and rho to promote actin cytoskeleton rearrangements necessary for Shigella entry. J Biol Chem 281:39534–39541
Dobrindt U, Hochhut B, Hentschel U, Hacker J (2004) Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol 2:414–424
Dupont HL, Levine MM, Hornick RB, Formal SB (1989) Inoculum size in shigellosis and implications for expected mode of transmission. J Infect Dis 159:1126–1128
Dupont N, Lacas-Gervais S, Bertout J, Paz I, Freche B, Van Nhieu GT, van der Goot FG, Sansonetti PJ, Lafont F (2009) Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe 6:137–149
Elliott SJ, Krejany EO, Mellies JL, Robins-Browne RM, Sasakawa C, Kaper JB (2001) EspG, a novel type III system-secreted protein from enteropathogenic Escherichia coli with similarities to VirA of Shigella flexneri. Infect Immun 69:4027–4033
Enninga J, Mounier J, Sansonetti P, Tran Van Nhieu G (2005) Secretion of type III effectors into host cells in real time. Nat Methods 2:959–965
Espina M, Olive AJ, Kenjale R, Moore DS, Ausar SF, Kaminski RW, Oaks EV, Middaugh CR, Picking WD, Picking WL (2006) IpaD localizes to the tip of the type III secretion system needle of Shigella flexneri. Infect Immun 74:4391–4400
Fasano A, Noriega FR, Maneval DR Jr, Chanasongcram S, Russell R, Guandalini S, Levine MM (1995) Shigella enterotoxin 1: an enterotoxin of Shigella flexneri 2a active in rabbit small intestine in vivo and in vitro. J Clin Invest 95:2853–2861
Fasano A, Noriega FR, Liao FM, Wang W, Levine MM (1997) Effect of Shigella enterotoxin 1 (ShET1) on rabbit intestine in vitro and in vivo. Gut 40:505–511
Fernandez IM, Silva M, Schuch R, Walker WA, Siber AM, Maurelli AT, McCormick BA (2001) Cadaverine prevents the escape of Shigella flexneri from the phagolysosome: a connection between bacterial dissemination and neutrophil transepithelial signaling. J Infect Dis 184:743–753
Ferreccio C, Prado V, Ojeda A, Cayyazo M, Abrego P, Guers L, Levine MM (1991) Epidemiologic patterns of acute diarrhea and endemic Shigella infections in children in a poor periurban setting in Santiago, Chile. Am J Epidemiol 134:614–627
Fukazawa A, Alonso C, Kurachi K, Gupta S, Lesser CF, McCormick BA, Reinecker HC (2008) GEF-H1 mediated control of NOD1 dependent NF-kappaB activation by Shigella effectors. PLoS Pathog 4:e1000228
Fukushima M, Kakinuma K, Kawaguchi R (2002) Phylogenetic analysis of Salmonella. Shigella, and Escherichia coli strains on the basis of the gyrB gene sequence. J Clin Microbiol 40:2779–2785
Germane KL, Spiller BW (2011) Structural and functional studies indicate that the EPEC effector, EspG, directly binds p21-activated kinase. Biochemistry 50:917–919
Germane KL, Ohi R, Goldberg MB, Spiller BW (2008) Structural and functional studies indicate that Shigella VirA is not a protease and does not directly destabilize microtubules. Biochemistry 47:10241–10243
Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, Tedin K, Taha MK, Labigne A, Zahringer U, Coyle AJ, DiStefano PS, Bertin J, Sansonetti PJ, Philpott DJ (2003) Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300:1584–1587
Gorden J, Small PL (1993) Acid resistance in enteric bacteria. Infect Immun 61:364–367
Hacker J, Bender L, Ott M, Wingender J, Lund B, Marre R, Goebel W (1990) Deletions of chromosomal regions coding for fimbriae and hemolysins occur in vitro and in vivo in various extraintestinal Escherichia coli isolates. Microb Pathog 8:213–225
Hale TL (1991) Genetic basis of virulence in Shigella species. Microbiol Rev 55:206–224
Handa Y, Suzuki M, Ohya K, Iwai H, Ishijima N, Koleske AJ, Fukui Y, Sasakawa C (2007) Shigella IpgB1 promotes bacterial entry through the ELMO-Dock180 machinery. Nat Cell Biol 9:121–128
Harrington A, Darboe N, Kenjale R, Picking WL, Middaugh CR, Birket S, Picking WD (2006) Characterization of the interaction of single tryptophan containing mutants of IpaC from Shigella flexneri with phospholipid membranes. Biochemistry 45:626–636
Hartman AB, Venkatesan M, Oaks EV, Buysse JM (1990) Sequence and molecular characterization of a multicopy invasion plasmid antigen gene, ipaH, of Shigella flexneri. J Bacteriol 172:1905–1915
Heindl JE, Saran I, Yi CR, Lesser CF, Goldberg MB (2010) Requirement for formin-induced actin polymerization during spread of Shigella flexneri. Infect Immun 78:193–203
Henderson IR, Czeczulin J, Eslava C, Noriega F, Nataro JP (1999) Characterization of pic, a secreted protease of Shigella flexneri and enteroaggregative Escherichia coli. Infect Immun 67:5587–5596
High N, Mounier J, Prevost MC, Sansonetti PJ (1992) IpaB of Shigella flexneri causes entry into epithelial cells and escape from the phagocytic vacuole. EMBO J 11:1991–1999
Hilbi H, Chen Y, Thirumalai K, Zychlinsky A (1997) The interleukin 1beta-converting enzyme, caspase 1, is activated during Shigella flexneri-induced apoptosis in human monocyte-derived macrophages. Infect Immun 65:5165–5170
Huan PT, Bastin DA, Whittle BL, Lindberg AA, Verma NK (1997) Molecular characterization of the genes involved in O-antigen modification, attachment, integration and excision in Shigella flexneri bacteriophage SfV. Gene 195:217–227
Ingersoll MA, Zychlinsky A (2006) ShiA abrogates the innate T-cell response to Shigella flexneri infection. Infect Immun 74:2317–2327
Ingersoll M, Groisman EA, Zychlinsky A (2002) Pathogenicity islands of Shigella. Curr Top Microbiol Immunol 264:49–65
Ingersoll MA, Moss JE, Weinrauch Y, Fisher PE, Groisman EA, Zychlinsky A (2003) The ShiA protein encoded by the Shigella flexneri SHI-2 pathogenicity island attenuates inflammation. Cell Microbiol 5:797–807
Islam D, Veress B, Bardhan PK, Lindberg AA, Christensson B (1997) In situ characterization of inflammatory responses in the rectal mucosae of patients with shigellosis. Infect Immun 65:739–749
Ito H, Kido N, Arakawa Y, Ohta M, Sugiyama T, Kato N (1991) Possible mechanisms underlying the slow lactose fermentation phenotype in Shigella spp. Appl Environ Microbiol 57:2912–2917
Jehl SP, Doling AM, Giddings KS, Phalipon A, Sansonetti PJ, Goldberg MB, Starnbach MN (2011) Antigen-specific CD8(+) T cells fail to respond to Shigella flexneri. Infect Immun 79:2021–2030
Jin Q, Yuan Z, Xu J, Wang Y, Shen Y, Lu W, Wang J, Liu H, Yang J, Yang F, Zhang X, Zhang J, Yang G, Wu H, Qu D, Dong J, Sun L, Xue Y, Zhao A, Gao Y, Zhu J, Kan B, Ding K, Chen S, Cheng H, Yao Z, He B, Chen R, Ma D, Qiang B, Wen Y, Hou Y, Yu J (2002) Genome sequence of Shigella flexneri 2a: insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res 30:4432–4441
Kim DW, Lenzen G, Page AL, Legrain P, Sansonetti PJ, Parsot C (2005) The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc Natl Acad Sci USA 102:14046–14051
Kim M, Ogawa M, Fujita Y, Yoshikawa Y, Nagai T, Koyama T, Nagai S, Lange A, Fassler R, Sasakawa C (2009) Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nature 459:578–582
Klink BU, Barden S, Heidler TV, Borchers C, Ladwein M, Stradal TE, Rottner K, Heinz DW (2010) Structure of Shigella IpgB2 in complex with human RhoA: implications for the mechanism of bacterial guanine nucleotide exchange factor mimicry. J Biol Chem 285:17197–17208
Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, Adak GK, Levine MM (1999) Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull World Health Organ 77:651–666
Kramer RW, Slagowski NL, Eze NA, Giddings KS, Morrison MF, Siggers KA, Starnbach MN, Lesser CF (2007) Yeast functional genomic screens lead to identification of a role for a bacterial effector in innate immunity regulation. PLoS Pathog 3:e21
Lafont F, Tran Van Nhieu G, Hanada K, Sansonetti P, van der Goot FG (2002) Initial steps of Shigella infection depend on the cholesterol/sphingolipid raft-mediated CD44-IpaB interaction. EMBO J 21:4449–4457
Lan R, Reeves PR (2002) Escherichia coli in disguise: molecular origins of Shigella. Microbes Infect 4:1125–1132
Lan R, Alles MC, Donohoe K, Martinez MB, Reeves PR (2004) Molecular evolutionary relationships of enteroinvasive Escherichia coli and Shigella spp. Infect Immun 72:5080–5088
Le Gall T, Mavris M, Martino MC, Bernardini ML, Denamur E, Parsot C (2005) Analysis of virulence plasmid gene expression defines three classes of effectors in the type III secretion system of Shigella flexneri. Microbiology 151:951–962
Le-Barillec K, Magalhaes JG, Corcuff E, Thuizat A, Sansonetti PJ, Phalipon A, Di Santo JP (2005) Roles for T and NK cells in the innate immune response to Shigella flexneri. J Immunol 175:1735–1740
Lerman Y, Yavzori M, Ambar R, Sechter I, Wiener M, Cohen D (1994) Epidemic spread of Shigella sonnei shigellosis and evidence for development of immunity among children attending day-care centers in a communal settlement (Kibbutz). J Clin Microbiol 32:1092–1094
Lett MC, Sasakawa C, Okada N, Sakai T, Makino S, Yamada M, Komatsu K, Yoshikawa M (1989) virG, a plasmid-coded virulence gene of Shigella flexneri: identification of the virG protein and determination of the complete coding sequence. J Bacteriol 171:353–359
Leung Y, Ally S, Goldberg MB (2008) Bacterial actin assembly requires toca-1 to relieve N-wasp autoinhibition. Cell Host Microbe 3:39–47
Lindberg AA, Karnell A, Weintraub A (1991) The lipopolysaccharide of Shigella bacteria as a virulence factor. Rev Infect Dis 13(Suppl 4):S279–S284
Lommel S, Benesch S, Rottner K, Franz T, Wehland J, Kuhn R (2001) Actin pedestal formation by enteropathogenic Escherichia coli and intracellular motility of Shigella flexneri are abolished in N-WASP-defective cells. EMBO Rep 2:850–857
Luck SN, Turner SA, Rajakumar K, Sakellaris H, Adler B (2001) Ferric dicitrate transport system (Fec) of Shigella flexneri 2a YSH6000 is encoded on a novel pathogenicity island carrying multiple antibiotic resistance genes. Infect Immun 69:6012–6021
Magdalena J, Hachani A, Chamekh M, Jouihri N, Gounon P, Blocker A, Allaoui A (2002) Spa32 regulates a switch in substrate specificity of the type III secreton of Shigella flexneri from needle components to Ipa proteins. J Bacteriol 184:3433–3441
Mandic-Mulec I, Weiss J, Zychlinsky A (1997) Shigella flexneri is trapped in polymorphonuclear leukocyte vacuoles and efficiently killed. Infect Immun 65:110–115
Marteyn B, West NP, Browning DF, Cole JA, Shaw JG, Palm F, Mounier J, Prevost MC, Sansonetti P, Tang CM (2010) Modulation of Shigella virulence in response to available oxygen in vivo. Nature 465:355–358
Martinez-Becerra FJ, Kissmann JM, Diaz-McNair J, Choudhari SP, Quick AM, Mellado-Sanchez G, Clements JD, Pasetti MF, Picking WL (2011) A broadly protective Shigella vaccine based on Type III secretion apparatus proteins. Infect Immun 80(3):1222–1231
Maurelli AT, Fernandez RE, Bloch CA, Rode CK, Fasano A (1998) “Black holes” and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc Natl Acad Sci USA 95:3943–3948
Mavris M, Page AL, Tournebize R, Demers B, Sansonetti P, Parsot C (2002a) Regulation of transcription by the activity of the Shigella flexneri type III secretion apparatus. Mol Microbiol 43:1543–1553
Mavris M, Sansonetti PJ, Parsot C (2002b) Identification of the cis-acting site involved in activation of promoters regulated by activity of the type III secretion apparatus in Shigella flexneri. J Bacteriol 184:6751–6759
McCormick BA, Fernandez MI, Siber AM, Maurelli AT (1999) Inhibition of Shigella flexneri-induced transepithelial migration of polymorphonuclear leucocytes by cadaverine. Cell Microbiol 1:143–155
Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C, Griffin PM, Tauxe RV (1999) Food-related illness and death in the United States. Emerg Infect Dis 5:607–625
Mel DM, Terzin AL, Vuksic L (1965) Studies on vaccination against bacillary dysentery. 1. Immunization of mice against experimental Shigella infection. Bull World Health Organ 32:633–636
Mel DM, Arsic BL, Nikolic BD, Radovanic ML (1968) Studies on vaccination against bacillary dysentery. 4. Oral immunization with live monotypic and combined vaccines. Bull World Health Organ 39:375–380
Mel D, Gangarosa EJ, Radovanovic ML, Arsic BL, Litvinjenko S (1971) Studies on vaccination against bacillary dysentery. 6. Protection of children by oral immunization with streptomycin-dependent Shigella strains. Bull World Health Organ 45:457–464
Menard R, Sansonetti PJ, Parsot C (1993) Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J Bacteriol 175:5899–5906
Menard R, Sansonetti P, Parsot C (1994) The secretion of the Shigella flexneri Ipa invasins is activated by epithelial cells and controlled by IpaB and IpaD. EMBO J 13:5293–5302
Mostowy S, Bonazzi M, Hamon MA, Tham TN, Mallet A, Lelek M, Gouin E, Demangel C, Brosch R, Zimmer C, Sartori A, Kinoshita M, Lecuit M, Cossart P (2010) Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe 8:433–444
Mounier J, Vasselon T, Hellio R, Lesourd M, Sansonetti PJ (1992) Shigella flexneri enters human colonic Caco-2 epithelial cells through the basolateral pole. Infect Immun 60:237–248
Nassif X, Mazert MC, Mounier J, Sansonetti PJ (1987) Evaluation with an iuc::Tn10 mutant of the role of aerobactin production in the virulence of Shigella flexneri. Infect Immun 55:1963–1969
Newton HJ, Pearson JS, Badea L, Kelly M, Lucas M, Holloway G, Wagstaff KM, Dunstone MA, Sloan J, Whisstock JC, Kaper JB, Robins-Browne RM, Jans DA, Frankel G, Phillips AD, Coulson BS, Hartland EL (2010) The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kappaB p65. PLoS Pathog 6:e1000898
Nie H, Yang F, Zhang X, Yang J, Chen L, Wang J, Xiong Z, Peng J, Sun L, Dong J, Xue Y, Xu X, Chen S, Yao Z, Shen Y, Jin Q (2006) Complete genome sequence of Shigella flexneri 5b and comparison with Shigella flexneri 2a. BMC Genomics 7:173
Niebuhr K, Jouihri N, Allaoui A, Gounon P, Sansonetti PJ, Parsot C (2000) IpgD, a protein secreted by the type III secretion machinery of Shigella flexneri, is chaperoned by IpgE and implicated in entry focus formation. Mol Microbiol 38:8–19
Niebuhr K, Giuriato S, Pedron T, Philpott DJ, Gaits F, Sable J, Sheetz MP, Parsot C, Sansonetti PJ, Payrastre B (2002) Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S. flexneri effector IpgD reorganizes host cell morphology. EMBO J 21:5069–5078
Ochman H, Lawrence JG, Groisman EA (2000) Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304
Ogawa M, Sasakawa C (2006) Intracellular survival of Shigella. Cell Microbiol 8:177–184
Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C (2005) Escape of intracellular Shigella from autophagy. Science 307:727–731
Ohya K, Handa Y, Ogawa M, Suzuki M, Sasakawa C (2005) IpgB1 is a novel Shigella effector protein involved in bacterial invasion of host cells. Its activity to promote membrane ruffling via Rac1 and Cdc42 activation. J Biol Chem 280:24022–24034
Okuda J, Toyotome T, Kataoka N, Ohno M, Abe H, Shimura Y, Seyedarabi A, Pickersgill R, Sasakawa C (2005) Shigella effector IpaH9.8 binds to a splicing factor U2AF(35) to modulate host immune responses. Biochem Biophys Res Commun 333:531–539
Page AL, Sansonetti P, Parsot C (2002) Spa15 of Shigella flexneri, a third type of chaperone in the type III secretion pathway. Mol Microbiol 43:1533–1542
Parsot C (2009) Shigella type III secretion effectors: how, where, when, for what purposes? Curr Opin Microbiol 12:110–116
Parsot C, Ageron E, Penno C, Mavris M, Jamoussi K, d’Hauteville H, Sansonetti P, Demers B (2005) A secreted anti-activator, OspD1, and its chaperone, Spa15, are involved in the control of transcription by the type III secretion apparatus activity in Shigella flexneri. Mol Microbiol 56:1627–1635
Pedron T, Thibault C, Sansonetti PJ (2003) The invasive phenotype of Shigella flexneri directs a distinct gene expression pattern in the human intestinal epithelial cell line Caco-2. J Biol Chem 278:33878–33886
Pendaries C, Tronchere H, Arbibe L, Mounier J, Gozani O, Cantley L, Fry MJ, Gaits-Iacovoni F, Sansonetti PJ, Payrastre B (2006) PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J 25:1024–1034
Peng J, Yang J, Jin Q (2009) The molecular evolutionary history of Shigella spp. and enteroinvasive Escherichia coli. Infect Genet Evol 9:147–152
Perdomo JJ, Gounon P, Sansonetti PJ (1994a) Polymorphonuclear leukocyte transmigration promotes invasion of colonic epithelial monolayer by Shigella flexneri. J Clin Invest 93:633–643
Perdomo OJ, Cavaillon JM, Huerre M, Ohayon H, Gounon P, Sansonetti PJ (1994b) Acute inflammation causes epithelial invasion and mucosal destruction in experimental shigellosis. J Exp Med 180:1307–1319
Perna NT, Plunkett G 3rd, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, Posfai G, Hackett J, Klink S, Boutin A, Shao Y, Miller L, Grotbeck EJ, Davis NW, Lim A, Dimalanta ET, Potamousis KD, Apodaca J, Anantharaman TS, Lin J, Yen G, Schwartz DC, Welch RA, Blattner FR (2001) Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409:529–533
Phalipon A, Kaufmann M, Michetti P, Cavaillon JM, Huerre M, Sansonetti P, Kraehenbuhl JP (1995) Monoclonal immunoglobulin A antibody directed against serotype-specific epitope of Shigella flexneri lipopolysaccharide protects against murine experimental shigellosis. J Exp Med 182:769–778
Philpott DJ, Yamaoka S, Israel A, Sansonetti PJ (2000) Invasive Shigella flexneri activates NF-kappa B through a lipopolysaccharide-dependent innate intracellular response and leads to IL-8 expression in epithelial cells. J Immunol 165:903–914
Picking WL, Nishioka H, Hearn PD, Baxter MA, Harrington AT, Blocker A, Picking WD (2005) IpaD of Shigella flexneri is independently required for regulation of Ipa protein secretion and efficient insertion of IpaB and IpaC into host membranes. Infect Immun 73:1432–1440
Porter ME, Dorman CJ (1997) Differential regulation of the plasmid-encoded genes in the Shigella flexneri virulence regulon. Mol Gen Genet 256:93–103
Prunier AL, Schuch R, Fernandez RE, Maurelli AT (2007a) Genetic structure of the nadA and nadB antivirulence loci in Shigella spp. J Bacteriol 189:6482–6486
Prunier AL, Schuch R, Fernandez RE, Mumy KL, Kohler H, McCormick BA, Maurelli AT (2007b) nadA and nadB of Shigella flexneri 5a are antivirulence loci responsible for the synthesis of quinolinate, a small molecule inhibitor of Shigella pathogenicity. Microbiology 153:2363–2372
Pupo GM, Karaolis DK, Lan R, Reeves PR (1997) Evolutionary relationships among pathogenic and nonpathogenic Escherichia coli strains inferred from multilocus enzyme electrophoresis and mdh sequence studies. Infect Immun 65:2685–2692
Pupo GM, Lan R, Reeves PR (2000) Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. Proc Natl Acad Sci USA 97:10567–10572
Purdy GE, Payne SM (2001) The SHI-3 iron transport island of Shigella boydii 0–1392 carries the genes for aerobactin synthesis and transport. J Bacteriol 183:4176–4182
Rajakumar K, Sasakawa C, Adler B (1997) Use of a novel approach, termed island probing, identifies the Shigella flexneri she pathogenicity island which encodes a homolog of the immunoglobulin A protease-like family of proteins. Infect Immun 65:4606–4614
Ramarao N, Le Clainche C, Izard T, Bourdet-Sicard R, Ageron E, Sansonetti PJ, Carlier MF, Tran Van Nhieu G (2007) Capping of actin filaments by vinculin activated by the Shigella IpaA carboxyl-terminal domain. FEBS Lett 581:853–857
Ramel D, Lagarrigue F, Pons V, Mounier J, Dupuis-Coronas S, Chicanne G, Sansonetti PJ, Gaits-Iacovoni F, Tronchere H, Payrastre B (2011) Shigella flexneri infection generates the lipid PI5P to alter endocytosis and prevent termination of EGFR signaling. Sci Signal 4:61
Ranallo RT, Kaminski RW, George T, Kordis AA, Chen Q, Szabo K, Venkatesan MM (2010) Virulence, inflammatory potential, and adaptive immunity induced by Shigella flexneri msbB mutants. Infect Immun 78:400–412
Rhode J, Fogoros S, Zick S, Wahl H, Griffith KA, Huang J, Liu JR (2007) Ginger inhibits cell growth and modulates angiogenic factors in ovarian cancer cells. BMC Complement Altern Med 7:44
Riddle MS, Kaminski RW, Williams C, Porter C, Baqar S, Kordis A, Gilliland T, Lapa J, Coughlin M, Soltis C, Jones E, Saunders J, Keiser PB, Ranallo RT, Gormley R, Nelson M, Turbyfill KR, Tribble D, Oaks EV (2011) Safety and immunogenicity of an intranasal Shigella flexneri 2a Invaplex 50 vaccine. Vaccine 29:7009–7019
Sakaguchi T, Kohler H, Gu X, McCormick BA, Reinecker HC (2002) Shigella flexneri regulates tight junction-associated proteins in human intestinal epithelial cells. Cell Microbiol 4:367–381
Sansonetti PJ, Kopecko DJ, Formal SB (1982) Involvement of a plasmid in the invasive ability of Shigella flexneri. Infect Immun 35:852–860
Sansonetti PJ, Ryter A, Clerc P, Maurelli AT, Mounier J (1986) Multiplication of Shigella flexneri within HeLa cells: lysis of the phagocytic vacuole and plasmid-mediated contact hemolysis. Infect Immun 51:461–469
Sansonetti PJ, Arondel J, Cavaillon JM, Huerre M (1995) Role of interleukin-1 in the pathogenesis of experimental shigellosis. J Clin Invest 96:884–892
Sansonetti PJ, Arondel J, Cantey JR, Prevost MC, Huerre M (1996) Infection of rabbit Peyer’s patches by Shigella flexneri: effect of adhesive or invasive bacterial phenotypes on follicle-associated epithelium. Infect Immun 64:2752–2764
Sansonetti PJ, Arondel J, Huerre M, Harada A, Matsushima K (1999) Interleukin-8 controls bacterial transepithelial translocation at the cost of epithelial destruction in experimental shigellosis. Infect Immun 67:1471–1480
Sansonetti PJ, Phalipon A, Arondel J, Thirumalai K, Banerjee S, Akira S, Takeda K, Zychlinsky A (2000) Caspase-1 activation of IL-1beta and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity 12:581–590
Schroeder GN, Hilbi H (2008) Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin Microbiol Rev 21:134–156
Schuch R, Maurelli AT (2001) Spa33, a cell surface-associated subunit of the Mxi-Spa type III secretory pathway of Shigella flexneri, regulates Ipa protein traffic. Infect Immun 69:2180–2189
Selyunin AS, Sutton SE, Weigele BA, Reddick LE, Orchard RC, Bresson SM, Tomchick DR, Alto NM (2011) The assembly of a GTPase-kinase signalling complex by a bacterial catalytic scaffold. Nature 469:107–111
Singer M, Sansonetti PJ (2004) IL-8 is a key chemokine regulating neutrophil recruitment in a new mouse model of Shigella-induced colitis. J Immunol 173:4197–4206
Singer AU, Rohde JR, Lam R, Skarina T, Kagan O, Dileo R, Chirgadze NY, Cuff ME, Joachimiak A, Tyers M, Sansonetti PJ, Parsot C, Savchenko A (2008) Structure of the Shigella T3SS effector IpaH defines a new class of E3 ubiquitin ligases. Nat Struct Mol Biol 15:1293–1301
Snapper SB, Takeshima F, Anton I, Liu CH, Thomas SM, Nguyen D, Dudley D, Fraser H, Purich D, Lopez-Ilasaca M, Klein C, Davidson L, Bronson R, Mulligan RC, Southwick F, Geha R, Goldberg MB, Rosen FS, Hartwig JH, Alt FW (2001) N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat Cell Biol 3:897–904
Suzuki T, Miki H, Takenawa T, Sasakawa C (1998) Neural Wiskott-Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J 17:2767–2776
Taylor DN, Bodhidatta L, Brown JE, Echeverria P, Kunanusont C, Naigowit P, Hanchalay S, Chatkaeomorakot A, Lindberg AA (1989) Introduction and spread of multi-resistant Shigella dysenteriae I in Thailand. Am J Trop Med Hyg 40:77–85
Tobe T, Yoshikawa M, Mizuno T, Sasakawa C (1993) Transcriptional control of the invasion regulatory gene virB of Shigella flexneri: activation by virF and repression by H-NS. J Bacteriol 175:6142–6149
Tominaga A, Lan R, Reeves PR (2005) Evolutionary changes of the flhDC flagellar master operon in Shigella strains. J Bacteriol 187:4295–4302
Tran Van Nhieu G, Ben-Ze’ev A, Sansonetti PJ (1997) Modulation of bacterial entry into epithelial cells by association between vinculin and the Shigella IpaA invasin. EMBO J 16:2717–2729
Turner SA, Luck SN, Sakellaris H, Rajakumar K, Adler B (2001) Nested deletions of the SRL pathogenicity island of Shigella flexneri 2a. J Bacteriol 183:5535–5543
Turner SA, Luck SN, Sakellaris H, Rajakumar K, Adler B (2003) Molecular epidemiology of the SRL pathogenicity island. Antimicrob Agents Chemother 47:727–734
Veenendaal AK, Hodgkinson JL, Schwarzer L, Stabat D, Zenk SF, Blocker AJ (2007) The type III secretion system needle tip complex mediates host cell sensing and translocon insertion. Mol Microbiol 63:1719–1730
Venkatesan MM, Buysse JM, Hartman AB (1991) Sequence variation in two ipaH genes of Shigella flexneri 5 and homology to the LRG-like family of proteins. Mol Microbiol 5:2435–2445
Venkatesan MM, Goldberg MB, Rose DJ, Grotbeck EJ, Burland V, Blattner FR (2001) Complete DNA sequence and analysis of the large virulence plasmid of Shigella flexneri. Infect Immun. 69(5):3271–3285
Vokes SA, Reeves SA, Torres AG, Payne SM (1999) The aerobactin iron transport system genes in Shigella flexneri are present within a pathogenicity island. Mol Microbiol 33:63–73
Wassef JS, Keren DF, Mailloux JL (1989) Role of M cells in initial antigen uptake and in ulcer formation in the rabbit intestinal loop model of shigellosis. Infect Immun 57:858–863
Way SS, Borczuk AC, Dominitz R, Goldberg MB (1998) An essential role for gamma interferon in innate resistance to Shigella flexneri infection. Infect Immun 66:1342–1348
Way SS, Borczuk AC, Goldberg MB (1999) Adaptive immune response to Shigella flexneri 2a cydC in immunocompetent mice and mice lacking immunoglobulin A. Infect Immun 67:2001–2004
Wei J, Goldberg MB, Burland V, Venkatesan MM, Deng W, Fournier G, Mayhew GF, Plunkett G 3rd, Rose DJ, Darling A, Mau B, Perna NT, Payne SM, Runyen-Janecky LJ, Zhou S, Schwartz DC, Blattner FR (2003) Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457 T. Infect Immun 71:2775–2786
Wu T, Grassel C, Levine MM, Barry EM (2011) Live attenuated Shigella dysenteriae type 1 vaccine strains overexpressing shiga toxin B subunit. Infect Immun 79:4912–4922
Yang F, Yang J, Zhang X, Chen L, Jiang Y, Yan Y, Tang X, Wang J, Xiong Z, Dong J, Xue Y, Zhu Y, Xu X, Sun L, Chen S, Nie H, Peng J, Xu J, Wang Y, Yuan Z, Wen Y, Yao Z, Shen Y, Qiang B, Hou Y, Yu J, Jin Q (2005) Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res 33:6445–6458
Yang J, Nie H, Chen L, Zhang X, Yang F, Xu X, Zhu Y, Yu J, Jin Q (2007) Revisiting the molecular evolutionary history of Shigella spp. J Mol Evol 64:71–79
Yoshida S, Handa Y, Suzuki T, Ogawa M, Suzuki M, Tamai A, Abe A, Katayama E, Sasakawa C (2006) Microtubule-severing activity of Shigella is pivotal for intercellular spreading. Science 314:985–989
Zhang Z, Jin L, Champion G, Seydel KB, Stanley SL Jr (2001) Shigella infection in a SCID mouse-human intestinal xenograft model: role for neutrophils in containing bacterial dissemination in human intestine. Infect Immun 69:3240–3247
Zhang L, Wang Y, Olive AJ, Smith ND, Picking WD, De Guzman RN, Picking WL (2007) Identification of the MxiH needle protein residues responsible for anchoring invasion plasmid antigen D to the type III secretion needle tip. J Biol Chem 282:32144–32151
Zhong QP (1999) Pathogenic effects of Opolysaccharide from Shigella flexneri strain. World J Gastroenterol 5:245–248
Zhu Y, Li H, Hu L, Wang J, Zhou Y, Pang Z, Liu L, Shao F (2008) Structure of a Shigella effector reveals a new class of ubiquitin ligases. Nat Struct Mol Biol 15:1302–1308
Zychlinsky A, Prevost MC, Sansonetti PJ (1992) Shigella flexneri induces apoptosis in infected macrophages. Nature 358:167–169
Zychlinsky A, Thirumalai K, Arondel J, Cantey JR, Aliprantis AO, Sansonetti PJ (1996) In vivo apoptosis in Shigella flexneri infections. Infect Immun 64:5357–5365
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this entry
Cite this entry
Jandu, N., Goldberg, M.B. (2013). Dysentery. In: Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-30144-5_100
Download citation
DOI: https://doi.org/10.1007/978-3-642-30144-5_100
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-30143-8
Online ISBN: 978-3-642-30144-5
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences