
Abstract
The role of RAS in the development of hypertension and cardiovascular and renal diseases has been extensively studied and is well established. The impact of oxidative stress in vascular homeostasis has also been clearly defined. Many of the cellular effects of Ang II appear to be mediated by ROS generated by NAD(P)H oxidase. Here we provide an overview of ROS physiology in human vessels and in particular the interaction with RAS, as well as a discussion on mechanisms by which therapeutic interventions on RAS affect redox signaling in the vascular wall at a clinical level.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
Introduction
Reactive oxygen species (ROS) are derived from several oxidant systems present within vessel walls, and elimination is achieved through intra- and extracellular antioxidant defense enzymatic systems (Antoniades 2003). While ROS play a fundamental role in the immune system’s defense against mechanism against systemic infection, excess levels of ROS are cytotoxic and contribute to cell death. In addition, the tight control of ROS production and signaling regulated diverse cellular functions in the vasculature. Consequently, redox signaling is essential for the maintenance of vascular homeostasis, and the loss of redox homeostasis is an important aspect of vascular disease pathogenesis (Antoniades 2003).
The endothelium serves as a major source of ROS (Misra et al. 2009). Endothelial dysfunction will inevitably lead to an imbalance in production of vascular protective agents and factors promoting oxidative stress, such as free radicals. It has been postulated that vascular endothelial dysfunction leads to the increased formation of free radicals or ROS such as superoxide (O2 •−), the hydroxyl radical (−OH), and the peroxynitrite anion (ONOO−), which all contribute to oxidative cellular destruction via their damaging actions on nucleic acids, lipids, and cell membranes (Misra et al. 2009). Interestingly ROS may exhibit beneficial activity as a consequence of their cytotoxic properties as a defense mechanism against infection (Hanze 2007).
Here we provide an overview of ROS physiology in human vessels and in particular the interaction with the renin–angiotensin system (RAS) as well as a discussion on mechanisms by which therapeutic interventions on RAS affect redox signaling in the vascular wall at a clinical level.
Physiology of RAS
The activation of RAS resulting in elevated concentrations of the principal effector peptide, angiotensin (Ang) II, has a pivotal role in the pathophysiological development of cardiovascular disease and is implicated in the pathogenesis of a variety of conditions such as heart failure, stroke, and renal failure (Paul 2006).
The “classical” view of RAS (Fig. 53.1) became widely accepted after early studies on the subject, whereby the glycoprotein angiotensinogen (AGT) is cleaved by renin to generate the decapeptide Ang I (McFarlane 2003). Ang I is then cleaved by angiotensin-converting enzyme (ACE, peptidyl dipeptidase A), which is a membrane-bound metalloprotease, to form the octapeptide Ang II. Ang II is the most active peptide of the RAS and acts via selective binding to G protein-coupled receptors, of which four subtypes (AT 1–4) have been identified (Fig. 53.2). The “classical” hemodynamic actions of Ang II in the regulation of blood pressure and blood volume, through vasoconstriction, aldosterone, and vasopressin release, and sodium and water retention are mediated by angiotensin type 1 receptors (AT1R) (Levy 2004). Through its actions on this receptor, Ang II also contributes a key role in cell proliferation, superoxide formation, fibrosis, inflammation, thrombosis, endothelial dysfunction, and nephrosclerosis (Duprez 2006; Ferrario and Strawn 2006) (Fig. 53.3).

The “classical” RAS. When blood volume is low, juxtaglomerular cells in the kidneys secrete renin directly into circulation. Plasma renin then carries out the conversion of angiotensinogen released by the liver to angiotensin I. Angiotensin I is subsequently converted to angiotensin II by the enzyme angiotensin-converting enzyme found in the lungs. Angiotensin II is a potent vasoactive peptide that causes blood vessels to constrict, resulting in increased blood pressure. Angiotensin II also stimulates the secretion of the hormone aldosterone from the adrenal cortex. Aldosterone causes the tubules of the kidneys to increase the reabsorption of sodium and water into the blood. This increases the volume of fluid in the body, which also increases blood pressure

Ang II is the most active peptide of the RAS. Although most of its well-known actions take place through the stimulation of AT 1 receptors, now is well established that more than one G protein-coupled receptors participate in these complex pathways

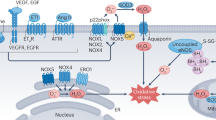
Schema depicting the renin–angiotensin system and its interaction with the kinin system and vascular redox state. Potential differences between the effects of ACEIs and ARBs can readily be appreciated from this figure. Ang II can increase NADPH oxidase activity via AT1 receptor leading to ROS release. ACE angiotensin-converting enzyme, AT1 angiotensin type 1, AT2 angiotensin type 2, ACEIs ACE inhibitors, ARBs AT1 receptor blockers, BP blood pressure, DRIs direct renin inhibitors, ECM extracellular matrix, NADPH nicotinamide adenine dinucleotide phosphate, PAI-1 plasminogen activator inhibitor-1, PG prostaglandin, RBF renal blood flow, ROS reactive oxygen species TGF-β transforming growth factor-β, ROS reactive oxygen species
Conversely, the activation of AT2R has been postulated to counteract both the short- and long-term effects of AT1R, resulting in potentially beneficial vasodilatory and antiproliferative effects (Stoll 1995) (Fig. 53.3). There is also a strong body of evidence supporting the role of AT2R in the regulation of cellular growth, differentiation, apoptosis, and regeneration of neuronal tissue (Steckelings 2005). The function of the AT3R is currently unknown, and the AT4R primarily mediated the release of plasminogen activator inhibitor-1 (PAI-1), the main physiological mediator of fibrinolysis, suggesting a pro-thrombotic role for AT4R.
The complexity of the RAS is far more extensive than initially believed, and the classical view has been expanded in recent years by the discovery of alternative enzyme systems and novel effector peptides. The discovery of several novel components of the RAS such as prorenin receptors (Nguyen and Danser 2008) and the G protein-coupled receptor Mas (Santos 2003) have added to this newly appreciated complexity of the RAS. Renin is synthesized as an inactive preprohormone that is proteolytically cleaved to generate prorenin, which is then released into the circulation (Pimenta and Oparil 2009) and is activated by proteolytic and non-proteolytic mechanisms to generate the active proteolytic enzyme renin. The renin/prorenin receptors appear to activate mitogen-activated protein kinases ERK1 and ERK2, increasing the possibility that renin and prorenin may in fact have direct proliferative and metabolic effects that may be independent of Ang II.
The postural response in blood pressure is brought about by the rapid action of Ang II. This physiological function is achieved by stimulation of smooth muscle cell contraction mediated by the “classical” G protein-dependent pathway. The G protein activates phospholipase C, resulting in the production of inositol-1,4,5-trisphosphate (IP3) and diacylglycerol, initiating Ca2+ release into the cytoplasm. Enhanced by Ca2+ binding to calmodulin and subsequent myosin activation, the interaction between myosin and actin results in smooth muscle cell contraction.
Pathological Effects of RAS: The Role of ROS
The pathological effects of RAS such as increased growth, hypertrophy, and inflammatory processes are the result of higher concentrations of Ang II or the prolonged exposure to Ang II (Touyz 2004). These processes are partially mediated by O2 •−and H2O2. The enzyme responsible for Ang II-induced oxidative stress is the NADPH oxidase (nicotinamide adenine dinucleotide phosphate) (Griendling 1994). Ang II can increase NADPH oxidase activity by inducing a rapid translocation of the small GTPase rac1 to the cell membrane (Wassmann 2001) or by phosphorylation and translocation of the NADPH oxidase subunit p47phox to the membrane (Mehta and Griendling 2007) via protein kinase C (PKC). PKC also leads to activation of the JAK/STAT (Janus kinase (JAK), signal transducer, and activator of transcription) mitogenic pathway, which induces the expression of early growth responsive genes (Berk and Corson 1997). Other components of the Ang II-induced pathways to growth, hypertrophy, and inflammation are the redox-sensitive proteins c-Src and the epidermal growth factor receptor (EGFR) (Ushio-Fukai 2001). Among others, EGFR activates the Ras/Raf/ERK cascade, which upregulates c-Fos, which together with c-Jun, activated by c-Src via JNK, forms the transcription factor activator protein-1 (Mehta 2007).
Further intensification of the Ang II enhances survival of cells and possibly significant DNA damage. Increased amounts of O2 •−and H2O2 activate more redox-sensitive proteins, such as the p38/MAPK that activate the pro-survival factor Akt, inhibiting a series of pro-apoptotic proteins (Kim and Chung 2002).
Aldosterone, another key effector of the RAS, is a major regulator of sodium and potassium balance and thus extracellular volume and has potent profibrotic effects (Johar 2006). Aldosterone release is stimulated by Ang II, and there is increasing evidence that aldosterone may mediate and exacerbate the deleterious effects of Ang II. Acting through mineralocorticoid receptors, aldosterone promotes endothelial dysfunction and thrombosis, reduces vascular compliance and baroreceptor function, and induces myocardial and vascular fibrosis (Struthers and MacDonald 2004). The observed elevation in blood pressure and circulating volume resulting from the effects of Ang II and aldosterone on their target organs establishes a negative feedback loop that inhibits renin release. Maintenance of feedback inhibition of renin release is critically dependent on Ang II-mediated activation of the AT1R.
The Role of ACEI and ARBs in Cardiovascular Disease
The ACE–Ang II–AT1R axis has long been thought to be the critical pathway for the RAS in controlling cardiovascular function. Several classes of antihypertensive/cardiorenal protective agents, including β blockers (BBs), direct renin inhibitors (DRIs), angiotensin-converting enzyme inhibitors (ACEIs), AT1 receptor blockers (ARBs), and mineralocorticoid receptor antagonists (MRAs), act by interrupting signaling in the RAS. ACEIs and ARBs have been the major therapeutic strategies for the treatment of hypertension and other cardiovascular disorders (Eberhardt 1993; Papadopoulos and Votteas 2006). ACEIs and ARBs are accepted as first-line agents in the treatment of hypertension and heart failure. Evidence also supports the role of these classes in both the prevention and treatment of nephropathy.
The cardioprotective and reno-protective effects of ACEIs and ARBs through blood pressure reduction and the improvement in endothelial function in patients with hypercholesterolemia and coronary artery disease, independent of changes in blood pressure, have been well documented (Hornig 2001). Large-scale randomized clinical studies support the hypothesis that their effects are due to actions that go beyond blood pressure reduction per se (Yusuf 2000). For example, the ACEI ramipril and the ARB losartan may favorably alter endothelial and/or vascular structure/function. Large prospective randomized trials have established the role of these drugs administered as monotherapy in patients with hypertension, left ventricular hypertrophy and systolic dysfunction, heart failure, myocardial infarction, and chronic renal disease (Schmieder 1996). Moreover, studies such as the HOPE study, the ONTARGET, and the TRANSCEND trial extended the benefit of these agents to patients at high risk for cardiovascular events who do not have LV dysfunction (Yusuf 2000). ACEI improves endothelial function in patients with coronary artery disease and in hypertensive patients. Furthermore, the Heart Outcomes Prevention Evaluation (HOPE) trial (Yusuf 2000) demonstrated that treatment with ramipril greatly reduced the incidence of death, myocardial infarction, and stroke in high-risk patients without heart failure. Also, the European Trial on Reduction of Cardiac Events (EUROPA) demonstrated a 20 % reduction with perindopril of the relative risk for cardiovascular endpoints in a patient population with stable coronary heart disease (Fox 2003). In contrast, the Prevention of Events with Angiotensin-Converting Enzyme Inhibition (PEACE) trial could not show that patients with stable coronary artery disease and largely intact ventricular function have a therapeutic benefit from the addition of ACEIs to modern conventional therapy. The failure of ACEI to reduce the cardiovascular events in this trial may have been attributable to the low overall event rate of hard endpoints, such as myocardial infarction or death in this patient population.
The Role of ACEI and ARBs on Oxidative Stress and Endothelial Function
The primary stimulus leading to enhanced O2 •− production and endothelial dysfunction in diseased blood vessels is thought to be the increased levels of Ang II. Ang II activates NADPH oxidases via AT1R stimulation (Griendling 2000). In addition, the AT1R is upregulated in vitro by low-density lipoproteins (Warnholtz 1999). Therefore, ACEIs and ARBs may have indirect antioxidant effects by preventing the activation of NADPH oxidase (Warnholtz 1999) and/or increasing the activity of SOD3 (Hornig 2001). Hornig et al. reported that ACEIs, ramipril, and losartan improve endothelial function in patients with coronary artery disease by increasing the bioavailability of NO through reduction of oxidative stress within the arterial wall. Another study suggests that ARBs have vascular protective effects beyond blood pressure reduction. Thus, losartan stimulates endothelial nitric oxide synthase (eNOS) phosphorylation and suppresses TNF-α (tissue necrosis factor)-induced endothelial apoptosis by activating the VEGFR2/PI3K/Akt pathway (Watanabe 2005). A most recent study has demonstrated that ACEIs prevent progression of vascular damage, with an inverse correlation between circulating endothelial progenitor-cell levels and intima media thickness progression (Honjo 2011).
The improvement in eNOS functionality may also be attributed to ARBs as it has been shown that losartan may restore glomerular NO production by increasing GCH1 protein expression and elevating BH4 levels in diabetic rats (Satoh 2008). The ARBs valsartan and irbesartan have anti-atherosclerotic effects by increasing eNOS mRNA stability, enhancing eNOS phosphorylation at Ser1177, decreasing NADPH oxidase expression, augmenting vascular BH4 levels, and restoring eNOS uncoupling (Imanishi 2008; Nussberger 2008). Recent studies have shown that ARBs significantly suppressed the expression of NADPH oxidase p22(phox) in the aortic walls of patients with thoracic aortic aneurysm which according to the authors is caused by the pleiotropic effects of ARBs on vascular metabolism (Honjo 2011).
Some of the beneficial effects of ACEI may be attributable to the effects on an ACE signaling cascade, resulting in an improvement in endothelial function that appears to be independent of effects of ACEI on vasoactive mediators (Fleming 2006). The binding of an ACEI to the ACE present as an ectoenzyme at the cell surface activates casein kinase 2, resulting in phosphorylation of a serine residue at the extreme C-terminal end of the molecule. This in turn results in the activation of ACE-associated c-Jun N-terminal kinase, probably via activation of mitogen-activated protein kinase kinase 7. This leads to accumulation of phosphorylated c-Jun in the nucleus and enhancement of DNA-binding activity of activator protein–1, which increases the expression of both ACE and cyclooxygenase-2. Although it might appear paradoxical, an increase in ACE during continued exposure to an ACEI could have beneficial effects associated with ACE signaling (Fleming 2005). Furthermore, increase in cyclooxygenase-2 expression also has a beneficial effect on endothelial function because the main product of this pathway in endothelial cells is the vasodilator and anti-aggregator prostanoid prostacyclin.
Fleming et al. have identified a novel functional role of ACE involved in outside–in signaling. These investigators showed that binding of an ACEI to ACE led to the activation of signal events that were likely to affect the expression of several proteins. The characterization of the novel signaling pathway via ACE also suggests that some of the beneficial effects of ACEIs can be attributed to the activation of a distinct ACE signaling cascade, rather than to changes in Ang II and bradykinin levels. The beneficial effects of ACEIs go beyond the inhibition of ACE to decrease Ang II or increase kinin levels. ACEI also affects kinin B1 and B2 receptor (B1R and B2R) signaling, which may underlie some of their therapeutic usefulness. Studies indicate that ACEI and some ARBs increase B2R functions as allosteric enhancers by inducing a conformational change in ACE. ACEI acts also as direct allosteric B1R agonists. When ACEIs enhance B2R and B1R signaling, they augment NO production. These actions may contribute to the pleiotropic therapeutic effects of ACEI in various cardiovascular disorders (Erdos 2010). It is also well established that ACEI reduces the levels of CD40L (Tousoulis 2007; Antoniades 2009a), a protein primarily expressed on activated T cell, while recent studies have demonstrated that ACEI increase the levels of adiponectin, a hormone modulating a number of metabolic processes (Antoniades 2009b).
Numerous studies have compared the commonly shared actions of all ACEIs in terms of their antihypertensive and anti-inflammatory actions. One study compared captopril (an ACEI with sulfhydryl group) with enalapril (an ACEI without sulfhydryl group) and finally suggested that ACEIs with sulfhydryl group (such as captopril) can protect the vascular endothelium against the damages induced by L-methionine (Liu 2006). The beneficial effects of captopril were related to attenuating the decrease in paraoxonase-1 activity and NO levels. Another study investigating again the role sulfhydryl group compared zofenopril versus lisinopril in myocardial infarcted heart failure rats (Buikema 2000). The conclusion was similar with the previous study with the sulfhydryl group having a potential advantage in improvement of endothelial dysfunction through increased activity of NO after release from the endothelium into the vessel wall.
The idea of further “downstream” blockade, postulated to result in a more complete inhibition of RAS and to surpass the “ACE escape” phenomenon, leads to the development of the ARBs. ARBs block the deleterious effects of Ang II at AT1R, whereas the beneficial effects of kinins might be diminished. However, increased levels of kinins, initially thought to be an adverse effect of ACEIs, actually promote vasodilation and are beneficial. The ARB therapy can result in the activation of AT2R, resulting in potentially beneficial anti-inflammatory, antithrombotic, and antiproliferative effects. Investigators have reported relative reductions in myocardial infarction with non-ARB antihypertensive treatment compared with ARB-based treatment, giving rise to the so-called ARB–myocardial infarction paradox (Julius 2004; Strauss and Hall 2006). It has been proposed that this results from unopposed activation of AT2R. Although these receptors are generally regarded as having favorable effects, studies in some animal models have shown that they can have hypertrophic and pro-inflammatory effects (Strauss and Hall 2006). Mice lacking AT2R are protected against cardiac hypertrophy resulting from aortic banding, whereas overexpression of the AT2R in isolated human cardiomyocytes is associated with hypertrophy (Senbonmatsu 2000). Furthermore, AT2R activation has been shown to stimulate production of matrix metalloproteinase-1, a collagenase involved in degradation of the fibrous cap of atherosclerotic plaques (D’Amore 2005). Hence, AT2R activation could contribute to plaque instability and thrombus formation (Strauss and Hall 2006).
Furthermore ACEIs and ARBs are involved in the process of atherogenesis and oxidative stress via reducing the levels of ADMA (asymmetric dimethylarginine) through activation of DDAH (dimethylarginine dimethylaminohydrolase) (Trocha 2010; Antoniades 2009c). It is well established that reduced levels of ADMA improve eNOS coupling (Trocha 2010; Antoniades 2009c). However, the exact mechanism through which RAS inhibitors modulate ADMA metabolism is not clear. Ang II increases ROS formation by vascular NADPH oxidase (Kalinowski and Malinski 2004). Due to inactivation of DDAH by ROS, ACEI and ARB might improve ADMA metabolism by ameliorating oxidative stress. Indeed, in some studies serum markers of oxidative stress have been reduced by these drugs (Fu 2005; Ito 2001). Napoli et al. compared the effects of two ACE inhibitors: zofenopril, which contains reduced sulfhydryl groups and thus possesses direct radical-scavenging properties, and enalapril, which does not contain –SH groups and has no antioxidant activity (Napoli 2004). They demonstrated that zofenopril was much more effective in reducing ADMA concentration (Napoli 2004). However, in other studies (Ito 2002), no change in serum lipid peroxidation products was observed in patients treated with ACEI, and, therefore, other mechanisms should be considered. Because shear stress increases ADMA formation by endothelial cells, RAS blockade could decrease ADMA by lowering blood pressure. Simultaneous depression of both ADMA observed by some authors (Chen 2002) would be consistent with this possibility. However, in other studies (Ito 2002) ACEI or ARB decreased ADMA despite having no effect on blood pressure. Also, the observation that only perindopril but not bisoprolol reduces ADMA in hypertensive patients despite similar decrease in blood pressure suggests that effect on blood pressure does not play a pivotal role (Ito 2001). In most studies with ACEI or ARB, no changes in renal function were observed. Thus, it is unlikely that reduction of ADMA concentration resulted from the improvement of renal excretion. Indeed, in patients with type 2 diabetes, treatment with perindopril reduced plasma ADMA but had no effect on urinary ADMA (Ito 2002).
Conclusions
The role of RAS in the development of hypertension and cardiovascular and renal diseases has been well established. However, ROS are generated by a wide range of enzymatic systems located in both vascular endothelium and vascular wall, and they have a key role in vascular homeostasis. Therapeutic interventions to RAS have been proven to modify the clinical outcome in patients with cardiac disease. Classical drugs blocking RAS such as ACEI and ARBs have been studied extensively. However, it is still unclear if all these drugs modify cardiovascular risk through redox mechanisms. Further research is required to identify or develop new therapeutic agents that are able to selectively target redox signaling in human vascular disease states through blockage of RAS.
Abbreviations
- ACE:
-
Angiotensin-converting enzyme
- ACEIs:
-
Angiotensin-converting enzyme inhibitors
- ADMA:
-
Asymmetric dimethylarginine
- AGT:
-
Angiotensinogen
- Ang:
-
Angiotensin
- ARBs:
-
AT1 receptor blockers
- AT1R:
-
Angiotensin type 1 receptors
- AT2R:
-
Angiotensin type 2 receptors
- BB:
-
β-blocker
- BH4 :
-
Tetrahydrobiopterin
- BP:
-
Blood pressure
- DDAH:
-
Dimethylarginine dimethylaminohydrolase
- DRI:
-
Direct renin inhibitors
- EGFR:
-
Epidermal growth factor receptor
- eNOS:
-
Endothelial nitric oxide synthase
- JAK:
-
Janus kinase
- MRAs:
-
Mineralocorticoid receptor antagonists
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- PAI-1:
-
Plasminogen activator inhibitor-1
- PRA:
-
Plasma renin activity
- PKC:
-
Protein kinase C
- RAS:
-
Renin–angiotensin system
- ROS:
-
Reactive oxygen species
- TNF:
-
Tissue necrosis factor
References
Antoniades C, Antonopoulos AS, Tousoulis D, Stefanadis C (2009a) Adiponectin: from obesity to cardiovascular disease. Obes Rev 10(3):269–279
Antoniades C, Bakogiannis C, Tousoulis D, Antonopoulos AS, Stefanadis C (2009b) The CD40/CD40 ligand system: linking inflammation with atherothrombosis. J Am Coll Cardiol 54(8):669–677
Antoniades C, Shirodaria C, Leeson P, Antonopoulos A, Warrick N, Van-Assche T et al (2009c) Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur Heart J 30(9):1142–1150
Antoniades C, Tousoulis D, Tentolouris C, Toutouzas P, Stefanadis C (2003) Oxidative stress, antioxidant vitamins, and atherosclerosis. From basic research to clinical practice. Herz 28(7):628–638
Berk BC, Corson MA (1997) Angiotensin II signal transduction in vascular smooth muscle: role of tyrosine kinases. Circ Res 80(5):607–616
Buikema H, Monnink SH, Tio RA, Crijns HJ, de Zeeuw D, van Gilst WH (2000) Comparison of zofenopril and lisinopril to study the role of the sulfhydryl-group in improvement of endothelial dysfunction with ACE-inhibitors in experimental heart failure. Br J Pharmacol 130(8):1999–2007
Chen JW, Hsu NW, Wu TC, Lin SJ, Chang MS (2002) Long-term angiotensin-converting enzyme inhibition reduces plasma asymmetric dimethylarginine and improves endothelial nitric oxide bioavailability and coronary microvascular function in patients with syndrome X. Am J Cardiol 90(9):974–982
D’Amore A, Black MJ, Thomas WG (2005) The angiotensin II type 2 receptor causes constitutive growth of cardiomyocytes and does not antagonize angiotensin II type 1 receptor-mediated hypertrophy. Hypertension 46(6):1347–1354
Duprez DA (2006) Role of the renin-angiotensin-aldosterone system in vascular remodeling and inflammation: a clinical review. J Hypertens 24(6):983–991
Eberhardt RT, Kevak RM, Kang PM, Frishman WH (1993) Angiotensin II receptor blockade: an innovative approach to cardiovascular pharmacotherapy. J Clin Pharmacol 33(11):1023–1038
Erdos EG, Tan F, Skidgel RA (2010) Angiotensin I-converting enzyme inhibitors are allosteric enhancers of kinin B1 and B2 receptor function. Hypertension 55(2):214–220
Ferrario CM, Strawn WB (2006) Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol 98(1):121–128
Fleming I (2006) Signaling by the angiotensin-converting enzyme. Circ Res 98(7):887–896
Fleming I, Kohlstedt K, Busse R (2005) New fACEs to the renin-angiotensin system. Physiology (Bethesda) 20:91–95
Fox KM (2003) Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomised, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet 362(9386):782–788
Fu YF, Xiong Y, Guo Z (2005) A reduction of endogenous asymmetric dimethylarginine contributes to the effect of captopril on endothelial dysfunction induced by homocysteine in rats. Eur J Pharmacol 508(1–3):167–175
Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW (1994) Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 74(6):1141–1148
Griendling KK, Sorescu D, Ushio-Fukai M (2000) NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 86(5):494–501
Hanze J, Weissmann N, Grimminger F, Seeger W, Rose F (2007) Cellular and molecular mechanisms of hypoxia-inducible factor driven vascular remodeling. Thromb Haemost 97(5):774–787
Honjo T, Yamaoka-Tojo M, Inoue N (2011) Pleiotropic effects of ARB in vascular metabolism–focusing on atherosclerosis-based cardiovascular disease. Curr Vasc Pharmacol 9(2):145–152
Hornig B, Landmesser U, Kohler C, Ahlersmann D, Spiekermann S, Christoph A et al (2001) Comparative effect of ace inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in patients with coronary artery disease: role of superoxide dismutase. Circulation 103(6):799–805
Imanishi T, Tsujioka H, Ikejima H, Kuroi A, Takarada S, Kitabata H et al (2008) Renin inhibitor aliskiren improves impaired nitric oxide bioavailability and protects against atherosclerotic changes. Hypertension 52(3):563–572
Ito A, Egashira K, Narishige T, Muramatsu K, Takeshita A (2001) Renin-angiotensin system is involved in the mechanism of increased serum asymmetric dimethylarginine in essential hypertension. Jpn Circ J 65(9):775–778
Ito A, Egashira K, Narishige T, Muramatsu K, Takeshita A (2002) Angiotensin-converting enzyme activity is involved in the mechanism of increased endogenous nitric oxide synthase inhibitor in patients with type 2 diabetes mellitus. Circ J 66(9):811–815
Johar S, Cave AC, Narayanapanicker A, Grieve DJ, Shah AM (2006) Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J 20(9):1546–1548
Julius S, Kjeldsen SE, Weber M, Brunner HR, Ekman S, Hansson L et al (2004) Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial. Lancet 363(9426):2022–2031
Kalinowski L, Malinski T (2004) Endothelial NADH/NADPH-dependent enzymatic sources of superoxide production: relationship to endothelial dysfunction. Acta Biochim Pol 51(2):459–469
Kim D, Chung J (2002) Akt: versatile mediator of cell survival and beyond. J Biochem Mol Biol 35(1):106–115
Levy BI (2004) Can angiotensin II type 2 receptors have deleterious effects in cardiovascular disease? Implications for therapeutic blockade of the renin-angiotensin system. Circulation 109(1):8–13
Liu YH, Liu LY, Wu JX, Chen SX, Sun YX (2006) Comparison of captopril and enalapril to study the role of the sulfhydryl-group in improvement of endothelial dysfunction with ACE inhibitors in high dieted methionine mice. J Cardiovasc Pharmacol 47(1):82–88
McFarlane SI, Kumar A, Sowers JR (2003) Mechanisms by which angiotensin-converting enzyme inhibitors prevent diabetes and cardiovascular disease. Am J Cardiol 91(12A):30H–37H
Mehta PK, Griendling KK (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292(1):C82–C97
Misra MK, Sarwat M, Bhakuni P, Tuteja R, Tuteja N (2009) Oxidative stress and ischemic myocardial syndromes. Med Sci Monit 15(10):RA209–RA219
Napoli C, Sica V, de Nigris F, Pignalosa O, Condorelli M, Ignarro LJ et al (2004) Sulfhydryl angiotensin-converting enzyme inhibition induces sustained reduction of systemic oxidative stress and improves the nitric oxide pathway in patients with essential hypertension. Am Heart J 148(1):e5
Nguyen G, Danser AH (2008) Prorenin and (pro)renin receptor: a review of available data from in vitro studies and experimental models in rodents. Exp Physiol 93(5):557–563
Nussberger J, Aubert JF, Bouzourene K, Pellegrin M, Hayoz D, Mazzolai L (2008) Renin inhibition by aliskiren prevents atherosclerosis progression: comparison with irbesartan, atenolol, and amlodipine. Hypertension 51(5):1306–1311
Papadopoulos DP, Votteas V (2006) Role of perindopril in the prevention of stroke. Recent Pat Cardiovasc Drug Discov 1(3):283–289
Paul M, Poyan Mehr A, Kreutz R (2006) Physiology of local renin-angiotensin systems. Physiol Rev 86(3):747–803
Pimenta E, Oparil S (2009) Role of aliskiren in cardio-renal protection and use in hypertensives with multiple risk factors. Ther Clin Risk Manag 5(3):459–464
Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I et al (2003) Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA 100(14):8258–8263
Satoh M, Fujimoto S, Arakawa S, Yada T, Namikoshi T, Haruna Y et al (2008) Angiotensin II type 1 receptor blocker ameliorates uncoupled endothelial nitric oxide synthase in rats with experimental diabetic nephropathy. Nephrol Dial Transplant 23(12):3806–3813
Schmieder RE, Martus P, Klingbeil A (1996) Reversal of left ventricular hypertrophy in essential hypertension. A meta-analysis of randomized double-blind studies. JAMA 275(19):1507–1513
Senbonmatsu T, Ichihara S, Price E Jr, Gaffney FA, Inagami T (2000) Evidence for angiotensin II type 2 receptor-mediated cardiac myocyte enlargement during in vivo pressure overload. J Clin Invest 106(2):R1–R5
Steckelings UM, Kaschina E, Unger T (2005) The AT2 receptor—a matter of love and hate. Peptides 26(8):1401–1409
Stoll M, Steckelings UM, Paul M, Bottari SP, Metzger R, Unger T (1995) The angiotensin AT2-receptor mediates inhibition of cell proliferation in coronary endothelial cells. J Clin Invest 95(2):651–657
Strauss MH, Hall AS (2006) Angiotensin receptor blockers may increase risk of myocardial infarction: unraveling the ARB-MI paradox. Circulation 114(8):838–854
Struthers AD, MacDonald TM (2004) Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 61(4):663–670
Tousoulis D, Antoniades C, Nikolopoulou A, Koniari K, Vasiliadou C, Marinou K et al (2007) Interaction between cytokines and sCD40L in patients with stable and unstable coronary syndromes. Eur J Clin Invest 37(8):623–628
Touyz RM (2004) Reactive oxygen species and angiotensin II signaling in vascular cells – implications in cardiovascular disease. Braz J Med Biol Res 37(8):1263–1273
Trocha M, Szuba A, Merwid-Lad A, Sozanski T (2010) Effect of selected drugs on plasma asymmetric dimethylarginine (ADMA) levels. Pharmazie 65(8):562–571
Ushio-Fukai M, Griendling KK, Becker PL, Hilenski L, Halleran S, Alexander RW (2001) Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 21(4):489–495
Warnholtz A, Nickenig G, Schulz E, Macharzina R, Brasen JH, Skatchkov M et al (1999) Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation 99(15):2027–2033
Wassmann S, Laufs U, Baumer AT, Muller K, Konkol C, Sauer H et al (2001) Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol Pharmacol 59(3):646–654
Watanabe T, Suzuki J, Yamawaki H, Sharma VK, Sheu SS, Berk BC (2005) Losartan metabolite EXP3179 activates Akt and endothelial nitric oxide synthase via vascular endothelial growth factor receptor-2 in endothelial cells: angiotensin II type 1 receptor-independent effects of EXP3179. Circulation 112(12):1798–1805
Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G (2000) Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The heart outcomes prevention evaluation study investigators. N Engl J Med 342(3):145–153
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this entry
Cite this entry
Koumallos, N., Sepehripour, A., Dimarakis, I., Paschalis, A., Nasir, A., Yonan, N. (2014). Reactive Oxygen Species Biology and Angiotensin Regulation of Human Vascular Tone – the Role of Angiotensin-Converting Enzyme (ACE) Inhibitors and AT1 Receptor Blockers (ARBs). In: Laher, I. (eds) Systems Biology of Free Radicals and Antioxidants. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-30018-9_67
Download citation
DOI: https://doi.org/10.1007/978-3-642-30018-9_67
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-30017-2
Online ISBN: 978-3-642-30018-9
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences