
Abstract
EMAP is a recently described late-onset and rapidly progressive geographic macular atrophy starting in the fifth decade, associated with pseudodrusen or reticular drusen in the posterior pole and paving stone degeneration in the retinal periphery.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
FormalPara SynonymsMacular atrophy, Drusen and pseudodrusen, EMAP
1 Introduction
EMAP is a recently described late-onset and rapidly progressive geographic macular atrophy starting in the fifth decade, associated with pseudodrusen or reticular drusen in the posterior pole and paving stone degeneration in the retinal periphery.
2 Clinical Features
2.1 Presentation
2.1.1 Presentation
The first symptoms appear at the end of the fourth decade [4]. The patient complains of loss of distant visual acuity and a paracentral scotoma. There are often problems of photophobia, of dark adaptation and of colour vision. The paracentral scotoma enlarges in a few years, resulting in loss of central vision. When the disease has progressed to the stage of areolar atrophy, the patients may complain of nyctalopia.
2.1.2 Fundus Appearance
The disease may be diagnosed at a routine examination even before functional signs appear. The macula may already present some changes although the areolar atrophy is not yet seen. Some pigmented areas are seen at the macular borders as well as minuscule white dots, very similar to small hard drusen (Fig. 34.1). Around the macula multiple larger greyish flecks, which are more or less confluent and which can extend outside the vessels of the posterior pole towards the periphery, correspond to reticular drusen [1, 2, 5, 8]. These flecks, which are especially visible in red-free light and in fluorescein angiography, give a reticular aspect to the affected area (Fig. 34.2).


The two top pictures show the initial changes (patient of Fig. 34.1). The mixture of more or less fused micro-drusen and of pseudodrusen provokes a reticular aspect to the fundus. The chorioretinal atrophy starts above the macula. The two pictures in the bottom correspond to a 70-year-old patient, where the areolar atrophy was already present 15 years earlier. The areolar atrophy is limited to the macular region. Pseudodrusen are seen on fluorescein angiography and there are paving stone lesions in the periphery
The atrophy will extend horizontally over four to five disc diameters, whereas the vertical diameter is often slightly larger (Fig. 34.2, 34.3, 34.4, and 34.5). Its borders are polycyclic. Paving stone degeneration is present.

A 61-year-old woman with bilateral geographic macular atrophy, reticular drusen and paving stone degeneration in the retinal periphery

This woman was 45 years old in 1994. She complains of progressive loss of vision, photophobia, hemeralopia and dyschromatopsia. In 2002 the patient’s vision was 1/20 RE and 1/40 LE. The amplitudes of both the photopic and scotopic ERG responses were diminished. The rod ERG further deteriorated. Note the progression of the lesions between 1994 and 2008. In 2008 in both eyes a rupture of Bruch’s membrane crosses the macula, not seen in the previous pictures

Same patient as Fig. 34.4 in 2008. Colour pictures, fluorescein angiography and autofluorescence
The disease is often accompanied by a moderate myopia (−0.5 to −5.00).
2.1.3 Investigations
Visual Fields. The macular lesion rapidly provokes a dense central scotoma, whereas the peripheral field remains normal.

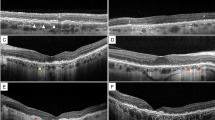
Early stage in a 48-year-old female patient. There is already a thinning of the retina with less marked foveal depression and irregular thickness of the photoreceptor-pigment epithelium layers
Colour Vision . Most commonly an acquired dyschromatopsia in the blue-yellow axis is found, sometimes the axis is undetermined, and rarely an alteration in the red-green axis is noticed.
Fluorescein Angiography . In the early stages of the disease, fluorescein angiography shows diffuse pigment epithelial changes in the macular region and sometimes beginning chorioretinal atrophy in the parafoveal region (Fig. 34.2b); the chorioretinal atrophy further extends to an areolar aspect (Fig. 34.2d). The pseudodrusen around the macula remain dark [2]. A reticular aspect persists with staining of the cluster of small hard drusen (Fig. 34.2).
The ERG is progressively affected; the scotopic responses are more reduced than the photopic ones.
The EOG is initially normal but is progressively affected with progressing chorioretinal atrophy in the posterior pole.
OCT . OCT shows the thinning of the retina, but the pseudodrusen at the limit of the macular lesion are not visible with this technique. When Bruch’s membrane has disappeared, the choroidal signal is enhanced (Fig. 34.6).
Dark Adaptation . The dark adaptation curve is elevated with a disruption of the alpha point [4].
2.1.4 Classic and Atypical Forms
The disease may appear more or less early and its severity may be variable. The four major symptoms are the presence of areolar macular atrophy, drusen, pseudodrusen and paving stone peripheral degeneration. The pseudodrusen coupled with fused cluster of small hard drusen may be localized to the posterior pole or extend up to the peripheral paving stone degeneration.
2.1.5 Evolution
The central areolar atrophy progresses in a few years and will ultimately lead to legal blindness.
2.1.6 Diagnosis and Differential Diagnosis
The diagnosis is based on the four main criteria and the relatively rapid evolution. EMAP is easily distinguished from cone-rod dystrophies by the absence of osteoblastic pigment and on the basis of the ERG [7], from Sorsby pseudo-inflammatory dystrophy by the absence of the typical retinal deposit from North Carolina dystrophy which is already present early in life and shows little progression [9], from Sorsby central areolar atrophy which after complete disappearance of the retinal pigment epithelium in the posterior pole gives rise to a spherical atrophy without polycyclic borders and which usually progresses over several decades [3, 10, 11] and from late-onset macular dystrophy with well-demarcated atrophic areas [6, 12], as is the case in gyrate atrophy and age-related geographic atrophy which usually appears after the age of 50 years (Figs. 34.7, 34.8, and 34.9).

Choroidal atrophy and possible RPE tear

Left eye of the patient of Fig. 34.7. Subfoveal neovascular membrane after PDT. Rupture of Bruch’s membrane and on OCT displacement of the various retinal layers

Right eye of the patient of Fig. 34.1. Marked areolar atrophy and linear rupture of Bruch’s membrane
3 Genetics
Because of the bilaterality and the symmetrical aspect of the lesions, a hereditary aetiology is considered but as the disease only appears late in life, an autosomal dominant inheritance cannot be proved, although family history often reveals that one of the parents possibly already deceased and suffered from retinal blindness.
4 Epidemiology
EMAP is rare but not exceptional. The pseudodrusen or the reticular drusen are not necessarily part of the syndrome as they can appear at any age, are more frequent in women than in men and increase with age. In the older age group, the prevalence can reach up to 2.4 % [5].
Summary for the Clinician
-
EMAP is a newly described macular dystrophy.
-
It is probably inherited in an autosomal dominant way.
-
It is quite similar and is often confused with age-related macular degeneration.
-
It starts at the end of the fourth decade.
-
The evolution is related to macular atrophy and ruptures of Bruch’s membrane.
-
The disease leads to legal blindness.
-
The locus of the affected gene has not yet been localized.
References
Arnold JJ, Quaranta M, Soubrane G, Sarks SH, Coscas G. Indocyanine green angiography of drusen. Am J Ophthalmol. 1997;124:344–56.
Arnold JJ, Sarks SH, Killingsworth MC, Sarks JP. Reticular pseudodrusen. A risk factor in age-related maculopathy. Retina. 1995;15:183–91.
Ashton N. Central areolar choroidal sclerosis: a histo-pathological study. Br J Ophthalmol. 1953;37:140.
Hamel C, Meunier I, Arndt C, Ben Salah S, Lopez S, Bazalgette C, Bazalgette C, Zanlonghi X, Arnaud B, Defoort-Dhellemmes S, Puech B. Extensive macular atrophy with pseudodrusen-like appearance: a new clinical entity. Am J Ophthalmol. 2009;147:609–20.
Klein R, Meuer SM, Knudtson MD, Iyengar SK, Kobrin Klein BE. The epidemiology of retinal reticular drusen. Am J Ophthalmol. 2008;145:317–26.
Kuntz CA, Jacobson SG, Cideciyan AV, Li ZY, Stone EM, Possin D, Milam AH. Sub-retinal pigment epithelial deposits in a dominant late-onset retinal degeneration. Invest Ophthalmol Vis Sci. 1996;37:1772–82.
Michaelides M, Hardcastle AJ, Hunt DM, Moore AT. Progressive cone and cone-rod dystrophies: phenotypes and underlying molecular genetic basis. Surv Ophthalmol. 2006;51:232–58.
Mimoun G, Soubrane G, Coscas G. Macular drusen. J Fr Ophtalmol. 1990;13:511–30.
Small KW, Puech B, Mullen L, Yelchits S. North Carolina macular dystrophy phenotype in France maps to the MCDR1 locus. Mol Vis. 1997;3:1.
Sorsby A. The dystrophies of the macula. Br J Ophthalmol. 1940;24:469–533.
Sorsby A, Mason MEJ, Gardner N. A fundus dystrophy with unusual features (late onset and dominant inheritance of a central retinal lesion showing oedema, haemorrhage and exudates developing into generalized choroidal atrophy with massive pigment proliferation). Br J Ophthalmol. 1949;33:67–97.
Styles CJ, Dhillon B, Wright AF. The diagnosis of autosomal dominant late-onset retinal degeneration in two sisters. Eye. 2003;17:530–2.51:232–258.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Puech, B., De Laey, JJ. (2014). Extensive Macular Atrophy with Pseudodrusen-Like Appearance. In: Puech, B., De Laey, JJ., Holder, G. (eds) Inherited Chorioretinal Dystrophies. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-540-69466-3_34
Download citation
DOI: https://doi.org/10.1007/978-3-540-69466-3_34
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-540-69464-9
Online ISBN: 978-3-540-69466-3
eBook Packages: MedicineMedicine (R0)