
Abstract
Acne is a complex disease with multiple pathogenic factors that act together to produce clinical disease [1]. Dystrophic keratinization is involved in the formation of the plug in the follicle (comedo or microcomedo) that is the central lesion of acne. Hormonal stimulation of the sebaceous gland occurs at puberty which causes production of sebum, a complex mixture of lipids that consists of about 50 % triglycerides. Triglycerides are a rich carbon source for lipase-producing bacteria and are a powerful determinant of the skin microflora through the production of inhibitory fatty acids [2]. Propionibacterium acnes, an anaerobic diphtheroid, dominates the follicular microflora after puberty and is the stimulus for inflammatory acne. The study of acne pathogenesis has been hampered by the lack of a suitable animal model. Although animals can be induced to have keratinous impactions in the follicles, they cannot be induced to have inflammatory lesions since animal sebum lacks triglycerides and P. acnes will not colonize the follicle [3].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
FormalPara Core Messages-
P. acnes can induce inflammatory response by activating innate immune cells, such as monocytes/macrophages.
-
P. acnes may simultaneously produce both high- and low-molecular-weight chemoattractants.
-
TLRs are transmembrane proteins that are activated by common microbial structures such as endotoxin and peptidoglycan to trigger production of pro-inflammatory cytokines such as IL12 and IL18; production of antimicrobial peptides such as defensins; and matrix metalloproteinases.
-
P. acnes contains TLR ligands that have been shown to trigger cytokine production.
1 Introduction
Acne is a complex disease with multiple pathogenic factors that act together to produce clinical disease [1]. Dystrophic keratinization is involved in the formation of the plug in the follicle (comedo or microcomedo) that is the central lesion of acne. Hormonal stimulation of the sebaceous gland occurs at puberty which causes production of sebum, a complex mixture of lipids that consists of about 50 % triglycerides. Triglycerides are a rich carbon source for lipase-producing bacteria and are a powerful determinant of the skin microflora through the production of inhibitory fatty acids [2]. Propionibacterium acnes, an anaerobic diphtheroid, dominates the follicular microflora after puberty and is the stimulus for inflammatory acne. The study of acne pathogenesis has been hampered by the lack of a suitable animal model. Although animals can be induced to have keratinous impactions in the follicles, they cannot be induced to have inflammatory lesions since animal sebum lacks triglycerides and P. acnes will not colonize the follicle [3].
Early attempts to explain inflammation in acne were based on study of sebum composition in acne patients. Free fatty acids were found to be elevated in skin surface lipid from patients with inflammatory acne. These were derived from lipolysis of triglycerides and were suggested to be a trigger for inflammation in acne. Therapy that reduced lesions, e.g., tetracycline, reduced the free fatty acids to normal levels, apparent confirmation that the fatty acids were central to acne inflammation. Later work showed that the lipid fraction of microcomedones was not inflammatory and that only the P. acnes-containing fractions induced inflammation when injected intradermally. Free fatty acids were found to be the result of P. acnes’ metabolism and to be present in proportion to the bacterial population rather than being a facet of aberrant sebaceous gland function; and more attention began to be paid to the role of the organism itself in acne. This was not a new thought, Unna, Sabouraud, and Fleming all speculated on the possibility that the “acne bacillus” was involved in acne. Fleming went so far as to demonstrate increased agglutination of the organism by sera from acne patients, but the idea lapsed until the mid-1970s (reviewed in [4]).
2 Propionibacterium acnes and Acne
Propionibacterium acnes is the predominant organism living on sebaceous regions of the skin. A facultative anaerobe, P. acnes grows in the sebaceous follicle and is carried onto the skin surface by the flow of sebum. P. acnes derives nutrition from the triglyceride fraction of sebum and hence is absent or low in children, but rises rapidly at puberty when androgens stimulate the start of sebum secretion acne [5].
The innate immune system is composed of the various host defense systems that act in the absence of an established immune response and includes the skin barrier, phagocytes, complement, antimicrobial peptides, and pattern recognition receptors such as the Toll-like receptors (TLR). P. acnes interacts with the innate immune system in various ways that are involved in the production of acne lesions.
Puhvel and Sakamoto [6] studied the contents of comedones in vitro and found that comedonal material attracted human neutrophils. The attractant was water soluble and of low molecular weight. A similar factor was detected in the supernatant of P. acnes cultures. Lee et al. [7] found that P. acnes also produced higher-molecular-weight chemotactic factors, one of which was the lipase molecule itself. Subsequent studies found that P. acnes may simultaneously produce both high- and low-molecular-weight chemoattractants and that the majority of neutrophil chemotactic activity in P. acnes culture supernatant was less than 2 kDa. The amount of chemotactic material produced was proportional to the P. acnes population and is of a molecular weight that might conceivably diffuse from an intact follicle [8]. The comedo may also contain other inflammatory factors. Allaker et al. [9] showed that P. acnes produces compounds that have histamine-like activity, and Helgren et al. [10] demonstrated prostaglandin-like activity in P. acnes culture supernatants. Most recently, Ingham et al. [11] found significant levels of interleukin 1 (IL-1)-like activity and tumor necrosis factor (TNF)-like molecules in a majority of open comedones.
Once neutrophils arrive at the comedo, gross rupture of follicular epithelium may be caused by enzymatic digestion of the follicular wall by neutrophil lysosomal hydrolytic enzymes. In vitro studies have shown that neutrophils readily secrete their degradative enzymes extracellularly when exposed to P. acnes that has been opsonized by C3b or immunoglobulin. Release of hydrolases is greatest when the anti-P. acnes antibody titer is elevated [12]. These degradative lysosomal enzymes are capable of digesting tissue and may promote further comedonal rupture. Finally, P. acnes itself also elaborates proteases and other degradative enzymes, which may also play some part in comedonal rupture.
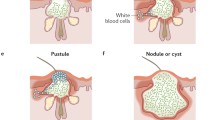
After exposure of comedonal contents to the immune system, a clinically detectable inflammation may result. The magnitude of the response is variable. Small, superficial papulopustules or deep nodules may develop. Complement deposition has been demonstrated in both early and late acne lesions, suggesting at least one means by which inflammation may be promoted. P. acnes is thought to be the cause of this deposition because the organism is a potent activator of both the classic and alternative complement pathways. The alternative pathway is triggered by P. acnes cell wall carbohydrate and the classical pathway by P. acnes–antibody complexes and is activated in proportion to the antibody titer [13, 14]. Crude comedonal material also activates complement by the classical and alternative pathways and this activation is also stimulated by anti-P. acnes antibody [15].
P. acnes can also induce inflammatory response by activating innate immune cells, such as monocytes/macrophages. Vowel et al. demonstrated that P. acnes induces pro-inflammatory cytokine production in monocytes, although the exact mechanism by which this occurs was not known [16]. Recently, with the discovery of the Toll-like receptors, we have a better understanding of how innate immune cells respond to microbes and how this leads to immune response [17].
TLRs are transmembrane proteins that are activated by common microbial structures such as endotoxin and peptidoglycan to trigger production of pro-inflammatory cytokines such as IL12 and IL18; production of antimicrobial peptides such as defensins; and matrix metalloproteinases [18]. P. acnes contains TLR ligands that have been shown to trigger cytokine production [19, 20].
Finally, P. acnes is a persistent inflammatory stimulus, being only degraded by about 10 % per 24 h period, in contrast to other organisms, e.g., Staphylococcus aureus which is degraded to soluble components within hours [15]. Injected radiolabeled P. acnes persists in the skin for many weeks and provided explanation for the inflammation that lingers long after severely inflamed lesions are treated.
3 Explaining the Variation in Acne Severity
After puberty, most individuals have stable P. acnes populations, some degree of microcomedo formation and significant sebum secretion, yet only some have inflammatory acne, and only a proportion of those have severe disease. In fact, when specific factors (e.g., sebum secretion, fatty acid concentration, or P. acnes populations) are compared in patients with and without acne, clear differences may be difficult to detect. In each category the acne population as a group is higher than the normal group, but great overlap exists in the range of values in each disease cohort (e.g., some persons have minimal or no acne, but high sebum production and some with acne have lower values), which suggests that each of these important factors is involved in but is not the determining cause of inflammatory acne (reviewed in [21]).
A second observation to be accounted for is that in severe inflammatory acne almost all lesions arise from microcomedones, whereas larger comedones in patients with noninflammatory acne become clinically inflamed only rarely, yet show histologic evidence of previous subclinical inflammatory episodes [1]. Likewise, clinically uninflamed microcomedones from persons with no apparent acne contain neutrophil markers suggesting earlier, limited inflammatory episodes that were not sufficiently severe to produce a clinical lesion [22].
Finally, the familial association of severe acne must be explained. The observation that severe acne is familial has been made by most clinicians, but studies are few. Acne conglobata and hidradenitis may have autosomal dominant single-gene inheritance and reports have been published of acne of similar severity in monozygotic twins [23–26]. Thus, factors favoring the development of severe acne may be genetically determined.
An explanation that accounts for all these observations centers on differing individual reactivity to P. acnes. There is support for this concept. In vitro studies found P. acnes to generate greater complement activation and lysosomal enzyme release in the presence of anti-P. acnes antibodies [27]. Patients with acne have elevated precipitating, agglutinating, and complement-fixing antibody titers to P. acnes but not to other organisms [28–31]. The antibody titers increase in proportion to the severity of acne inflammation, with little or no overlap between the normal and most severe acne groups.
Some studies have addressed the identity of the P. acnes antigens potentially relevant to acne. One study found that the anti-P. acnes antibody response in a group of patients with severe nodular acne was apparently uniform, directed against a carbohydrate structure in the cell wall [31]. Of all the potential protein and carbohydrate antigens present in the P. acnes to which they were exposed, these patients appeared to hyper-respond to a single antigen. This reactivity was not detected in patients with less severe acne and the mechanism by which it occurs has not been elucidated. Ingham et al. [32] have made a complementary observation.
Cell-mediated immunity to P. acnes is also increased in proportion to acne severity. P. acnes-stimulated lymphocyte transformation is elevated in mononuclear cells from inflammatory acne patients and skin test reactivity to P. acnes is elevated in proportion to the severity of acne inflammation [33, 34]. Wilcox et al. [35] recently studied the cellular responses in acne lesions from patients prone to scarring and those not likely to scar. They found that non-scarring patients had a greater initial influx of lymphocytes than the scarring patients, but the scarring patients had a much greater proportion of memory-effector cells, with the implication that the more severe acne patient has an immunological predisposition to severity.
Thus, there is data in support of the concept that the difference between those with inflammatory acne and those without disease is their immune reactivity to P. acnes and that, in at least one sense, acne may be considered a hypersensitivity disease. In past generations there were attempts to produce a vaccine against P. acnes with an eye to heightening immunity and preventing disease. In light of current data that would have a counterproductive and indeed disease-inducing result, but perhaps a desensitizing regimen might be possible.
References
Kligman AM. An overview of acne. J Invest Dermatol. 1974;62:268–87.
McGinley KJ, Webster GF, Ruggieri MR, Leyden JJ. Regional variations of cutaneous propionibacteria, correlation of Propionibacterium acnes populations with sebaceous secretion. J Clin Microbiol. 1980;12:672–5.
Webster GF, Ruggieri MR, McGinley KJ. Correlation of Propionibacterium acnes populations with the presence of triglycerides on non-human skin. Appl Environ Microbiol. 1981;41:1269–70.
Webster GF. Acne. Curr Prob Dermatol. 1996;8:240–62.
Marples RR, McGinley KJ. Corynebacterium acnes and other anaerobic diphtheroids from human skin. J Med Microbiol. 1974;7:349–61.
Puhvel SM, Sakamoto M. Cytotaxin production by comedonal bacteria. J Invest Dermatol. 1978;71:324–9.
Lee WL, Shalita AR, Sunthralingam K. Neutrophil chemotaxis to P. acnes lipase and its inhibition. Infect Immun. 1982;35:71–8.
Webster GF, Leyden JJ, Tsai CC. Characterization of serum independent polymoprphonuclear leukocyte chemotactic factors produced by Propionibacterium acnes. Inflammation. 1980;4:261–71.
Allaker RP, Greenman J, Osborne RH. The production of inflammatory compounds by Propionibacterium acnes and other skin organisms. Br J Dermatol. 1987;117(2):175–83.
Hellgren L, Vincent J. New group of prostaglandin-like compounds in P. acnes. Gen Pharmacol. 1983;14(1):207–8.
Ingham E, Eady EA, Goodwin CE, Cove JH, Cunliffe WJ. Pro-inflammatory levels of interleukin-1 alpha-like bioactivity are present in the majority of open comedones in acne vulgaris. J Invest Dermatol. 1992;98(6):895–901.
Webster GF, Leyden JJ, Tsai CC, Baehni P, McArthur WP. Polymorphonuclear leukocyte lysosomal release in response to Propionibacterium acnes in vitro and its enhancement by sera from inflammatory acne patients. J Invest Dermatol. 1980;74(6):398–401.
Webster GF, McArthur WR. Activation of components of the alternative pathway of complement by Propionibacterium acnes cell wall carbohydrate. J Invest Dermatol. 1982;79:137–40.
Webster GF, Nilsson UR, McArthur WR. Activation of the alternative pathway of complement by Propionibacterium acnes cell fractions. Inflammation. 1981;5:165–76.
Webster GF, Leyden JJ, Musson RA, Douglas SD. Susceptibility of Propionibacterium acnes to killing and degradation by human monocytes and neutrophils in vitro. Infect Immun. 1985;49:116–21.
Vowels BR, Yang S, Leyden JJ. Induction of proinflammatory cytokines by a soluble factor of Propionibacterium acnes: implications for chronic inflammatory acne. Infect Immun. 1995;63:3158–65.
Suzuki S, Duncan GS, et al. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature. 2002;416:750–6.
Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76.
Kim J, Ochoa MT, Krutzik SR, et al. Activation of toll-like receptor 2 in acne triggers inflammatory cytokine responses. J Immunol. 2002;169:1535–41.
Yoshimura A, Lien E, Ingalls RR, et al. Recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor-2. J Immunol. 1999;163:1–5.
Webster GF. Inflammation in acne vulgaris. J Am Acad Dermatol. 1995;33(2 Pt 1):247–53.
Webster GF, Kligman AM. A method for the assay of inflammatory mediators in follicular casts. J Invest Dermatol. 1979;73(4):266–8.
Fitzsimmins JS, Guilbert PR. A family study of hidradinitis suppurativa. J Med Genet. 1985;22:367–73.
Fitzsimmons JS, Fitzsimmons EM, Gilbert G. Familial hidradenitis suppurativa: evidence in favour of single gene transmission. J Med Genet. 1984;21(4):281–5.
Quintal D, Jackson R. Aggressive squamous cell carcinoma arising in familial acne conglobata. J Am Acad Dermatol. 1986;14:207–14.
Tosti A, Guerra L, Bettoli V, et al. Solid facial edema as a complication of acne in twins. J Am Acad Dermatol. 1987;17:843–4.
Webster GF, Leyden JJ, Norman ME, Nilsson UR. Complement activation in acne vulgaris: in vitro studies with Propionibacterium acnes and Propionibacterium granulosum. Infect Immun. 1978;22(2):523–9.
Ashbee HR, Muir SR, Cunliffe WJ, Ingham E. IgG subclasses specific to Staphylococcus epidermidis and Propionibacterium acnes in patients with acne vulgaris. Br J Dermatol. 1997;136(5):730–3.
Puhvel SM, Barfatani M, Warnick M, Sternberg TH. Study of antibody levels to C. acnes in the serum of acne patients. Arch Dermatol. 1964;90:421–7.
Puhvel SM, Hoffman IK, Sternberg TH. Corynebacterium acnes. Presence of complement fixing antibodies to corynebacterium acnes in the sera of patients with acne vulgaris. Arch Dermatol. 1966;93(3):364–6.
Webster GF, Indrisano JP, Leyden JJ. Antibody titers to Propionibacterium acnes cell wall carbohydrate in nodulocystic acne patients. J Invest Dermatol. 1985;84(6):496–500.
Ingham E, Gowland G, Ward RM, Holland KT, Cunliffe WJ. Antibodies to P. acnes and P. acnes exocellular enzymes in the normal population at various ages and in patients with acne vulgaris. Br J Dermatol. 1987;116(6):805–12.
Puhvel SM, Amirian D, Weintraub J, Reisner RM. Lymphocyte transformation in subjects with nodulo cystic acne. Br J Dermatol. 1977;97(2):205–11.
Puhvel SM, Hoffman IK, Reisner RM, Sternberg TH. Dermal hypersensitivity of patients with acne vulgaris to Corynebacterium acnes. J Invest Dermatol. 1967;49(2):154–8.
Wilcox HE, Farrar MD, Cunliffe WJ, Holland KT, Ingham E. Resolution of inflammatory acne vulgaris may involve regulation of CD4+ T-cell responses to Propionibacterium acnes. Br J Dermatol. 2007;156(3):460–5.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Webster, G.F. (2014). Inflammation in Acne. In: Zouboulis, C., Katsambas, A., Kligman, A. (eds) Pathogenesis and Treatment of Acne and Rosacea. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-540-69375-8_12
Download citation
DOI: https://doi.org/10.1007/978-3-540-69375-8_12
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-540-69374-1
Online ISBN: 978-3-540-69375-8
eBook Packages: MedicineMedicine (R0)