
Abstract
The measurement of biological activity is required for therapeutic antibodies at release of a new manufacturing batch and throughout the product life cycle. However, the applicable regulatory guidelines provide relatively little advice with respect to the number and types of bioassays to be employed. For example, 21CFR610.10 states that potency tests “shall consist of either in vitro or in vivo tests, or both, which have been specifically designed for each product so as to indicate its potency in a manner adequate to satisfy the interpretation of potency” where potency is “the specific ability of capacity of the product, as indicated by appropriate laboratory tests or by adequately controlled clinical data obtained through the administration of the product in the manner intended, to effect a given result.” ICH Q6B lists animal-based biological tests, cell culture-based biological tests, biochemical tests and ligand and receptor binding tests as examples of biological tests. However, neither guideline provides any assistance on how to select the most appropriate bioassay format(s) for a given product. Since biological activity is determined based on individual product characteristics and mechanism of action, there is considerable decision-making involved when selecting, developing, switching, and maintaining appropriate bioassays. This chapter discusses the types of biological assays, the current practices and regulatory expectations regarding the potency test format. A key question for antibody therapeutics that acts as agonists or antagonists of a ligand-receptor interaction, is the question of cell-based versus binding assays. Development and validation considerations for biological assays will also be discussed. This chapter will particularly focus on special considerations for biosimilar antibody therapeutics and provide a strategy for the biological testing methods, plan, and statistical assessment for the assessment of functional biosimilarity. Key differences in the application of a battery of in vitro biological activity assays between a biosimilar and a novel biologic, and case studies will be discussed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- Biological activity assays
- Bioassays
- Potency assays
- Receptor binding
- Antibody-dependent cell-mediated cytotoxicity (ADCC)
- Complement-dependent cytotoxicity (CDC)
- Reference standard
- Assaycontrol
- Method development and qualification
- Method validation
- Method transfer
- Assay variability
- Accuracy
- Precision
- Statistical assessment
- Training
- Parallelism
- Data Analysis
Introduction
Monoclonal antibodies are in general complex large molecules, exhibiting many different physicochemical attributes and can exhibit more than one biological activity (both Fab and Fc functions). To ensure their quality and the robustness of process of production requires assessment of several physicochemical attributes including structure, purity, biological potency, stability and consistency batch to batch. In order to establish a risk based approach to assure consistency, characterization of the protein product should be undertaken during product development to establish what are the product attributes and their criticality, to create an overall control strategy.
Physicochemical analyses rely on the unique characteristics of a protein, and use multiple methods to characterize a structural or functional attribute. It is often the case that each test only provides detailed information about a single characteristic, although multi-attribute methods have been developed that can replace several existing types of tests. Even though the data from these assays can help provide the potential for an impact of differences in these attributes on the biological activity (PK, safety and/or efficacy), they are yet unable to predict the biological activity of the vast majority of biological products and therefore bioassays are an essential part of the characterization of the biological activity (Mire-Sluis 2001).
Biosimilar products are intended to be as close in all the areas described above as possible to the reference product, with any differences ‘not having any meaningful clinical impact (i.e. safety and efficacy)’. Therefore, it is essential during biosimilar development that its potency and biological activity be established as such. Most of considerations for the development of bioassays to use both for the biosimilarity exercise as well as for eventual lot release are not significantly different from those for an innovator program but are essential if the program is to be a success. The specific considerations on how to use bioassays to illustrate biosimilarity (such as using the same assay to compare side by side, statistical aspects etc.) are described in this chapter.
Definition of Bioassay and Potency Assays
In the context of this chapter on biosimilar monoclonal antibodies, a bioassay is an analytical procedure utilizing a biological reporter system (resulting in a biological/functional response), the purpose of which is to measure the amount of active analyte or effective constituent in a biological product, i.e. to determine its biological potency.
The term bioassay should not be confused with a potency assay. Potency is the ability of a material to exert its intended activity and may not necessarily have to be measured in a biological system. Bioassays used for quality can illustrate the batch-to-batch consistency of biological potency of a product as well as defining the actual potency of each lot of product. The amount of product required to provide the optimal therapeutic biological activity in humans, as reflected in the therapeutic dose, is determined by clinical trials. This issue is extremely important as the biological potency measured for quality purposes and therapeutic dose are two very different issues. For example, antibody products that are intended to block the binding of one protein to another can have their potency measured in a binding assay. However, it is often the case that binding alone is not the sole biological endpoint of the product and a cell based format may provide a more relevant assay e.g., prevention of ligand binding to its receptor on the cell surface and induction of Fc activity. In this case, both a receptor based binding assay and a prevention of ligand induction of cell activity bioassay could be used. Cell based assays can be replaced as a potency assay with binding assays such as immunoassays and receptor-ligand methods although they do not measure the ability of a protein to induce a ‘functional’ biological response. Therefore, a thorough correlation between a bioassay and binding assays is required to show the latter can replace the former as a suitable potency assay, to be discussed later in the chapter.
Types of Biological Activity Assays for Antibody Therapeutics
Biological activity assays can be carried out in vivo or in vitro. The most appropriate method for assessing biological activity is to compare the biological activity of a sample to that of a well-characterized potency reference standard (Sasardic and Mire-Sluis 2000). Where possible, it is preferable to use an assay with a biological role that correlates with a clinical response. Although, it is not always necessary or possible to mimic therapeutic activity for a potency assay that is used to assess quality or efficacy, a justification is needed to the regulatory authorities. Biological activity assays for a biosimilar assessment may or may not have been conducted by the originator. It is critical that both the reference product and the biosimilar are tested in the same assay.
Binding Assays
Ligand Binding Assay
Ligand binding assay (LBA) is an assay, or an analytic procedure, that relies on the binding of ligand or target protein molecules to receptors, antibodies or other macromolecules. A detection method is used to determine the presence and extent of the ligand-receptor complexes formed, and this is usually determined electrochemically or through a fluorescence detection method. This type of analytic test can be used as potency assay to test the biological activity of therapeutic antibodies.
As further complement to LBA, cell-based binding assays (CBA) acts as an indispensable part in determining the exact mechanism of action (MoA) for antibody products. Regulatory agencies expect that cell-free LBA outcomes should be eventually translated into a cell-based format, so that antibody potency and bioactivity can be better assessed in a more biologically relevant environment. The cell-based system can offer many advantages, such as preservation of the native target form, and higher sensitivity towards physiochemical changes (i.e. glycosylation patterns and conformational alternations).
Competitive Ligand Binding
Competitive ligand binding (CLB) assay refers to how therapeutic antibodies exert their biological function by preventing a ligand from binding to its receptor on the cell surface. For these products, a CLB assay offers direct measurement of the product’s inhibition of ligand binding to its intended target receptor and may be suitable for potency testing.
The most common type of binding assay is the Enzyme Linked Immuno-Sorbent Assay (ELISA), which can be developed relatively quickly and typically offers robust performance. With the advancement of technology, various “homogeneous” immunoassays have been developed and successfully utilized for potency measurement in QC settings. Examples are Time Resolved Homogeneous Fluorescence Resonance Energy Transfer assays, Amplified Luminescence Proximity Homogeneous assays (such as AlphaLISA) and Proximity Based Electrochemiluminescence Immunoassays. These homogeneous immunoassays eliminate the need for wash steps, and the simple “mix and go” procedures result in decreased assay time and potential analyst error. In some cases, superior signal-to-noise ratio and better overall assay performance, as compared to traditional ELISA, may be achieved. However, custom protein conjugation may be required, and assay performance is highly dependent on the quality of these critical reagents (tagged proteins, donor and acceptor beads, etc.). In addition to immunoassays, Surface Plasmon Resonance (SPR) assays have also been utilized to measure product binding to its intended target. In an SPR assay, protein-protein interaction is detected in real time through changes in mass due to adsorption at the chip surface. Data generated can be used to calculate the binding constant; therefore, SPR assays can be particularly useful during product development. Although SPR assays have not been used as widely as QC methods for potency measurement but have been adopted sometimes for product characterization, they have been particularly used for biosimilar development as part of biosimilarity assessment in comparison with the reference products.
Bioassays
The choice of bioassay depends upon the nature of the monoclonal antibody, its intended therapeutic use and whether biological activity is only measurable in whole animals. It is obviously desirable to develop in vitro biological activity assays where possible because they offer distinct advantages over using live animals. One also has to consider which assay to choose during the biosimilar antibody development lifecycle, since biosimilars require earlier and more extensive biological characterizations than that of the innovative antibody development. It must also be stressed that one cannot be assured that the removal and use of cells from tissues or the use of clonal cell lines in in vitro formats represents what is occurring in vivo and thus most, if not all, bioassays are a surrogate marker for biological activity.
In Vivo Bioassays
Earliest attempts to measure biological activity often took the form of an in vivo bioassay, where protein was administered to animals and the response in those animals measured. The use of such assays in monoclonal antibody development has occurred, as an example, to understand the overall consequences blocking of ligands, or to understand if an antibody exhibits Fc functions in its activity, in vivo. However, it is difficult to reduce inter-animal variability in estimates of potency, and in vivo bioassays are expensive and labor intensive. In order for an in vivo assay to provide valid estimates of biological potency, a large number of animals are required to account for this variation. A great deal of care and expense is required to decrease any variation through breeding, housing and feeding of animals. A balance must also be maintained between the large number of animals that could be used to provide several data points for potency estimates and humane, ethical and economic pressures to reduce the use of laboratory animals for assays.
It can be argued that testing in vivo provides biological potency tests more relevant to the clinical use of biologicals because a “whole body” approach takes into account bioavailability, serum half-life, toxicities etc. However, this argument is incorrect because biological assays are not intended to mimic the biological activity of a product in the clinical situation. As described, bioassays are intended to be used for quality control and illustrate the batch-to-batch consistency of biological potency of a product (Thorpe et al. 1997). Bioassays are key to the analytical biosimilarity assessment between the reference product and biosimilar product, as well as for the comparability study during biosimilar development.
However, there may be a case to be made for in vivo testing where a combination of physicochemical and biological tests cannot detect differences known to impact on in vivo activity. Such issues could involve complex glycosylation relevant to biological half-lives or modified monoclonal antibodies.
Attempts to avoid the requirement for live animals testing has led to the production of many different formats for the in-vitro estimation of biological potency for a wide range of monoclonal antibodies.
In Vitro Tissue Based Bioassays
The use of in vivo assays as described, can be useful for characterization of a monoclonal antibody. However, one can use an approach that is more stable, yet retains some of the advantages of in vivo assays by developing in vitro bioassays where cells or tissues from animals are cultured in the laboratory and used as responders to the test protein. Assays for monoclonal antibodies that had an impact on the hematopoietic system used cells from the blood or bone marrow. Monoclonal antibodies that act on solid tissues, such as interfering with growth factors and hormones, require assays that involve the removal of the specific tissue on which they act and its homogenization into single cells that can then be cultured and exposed to protein in vitro (Mire-Sluis and Thorpe 1995). However, donor-to-donor variability still occurs in these systems and pure populations of target cells are difficult to achieve.
In Vitro Cell Line Based Bioassays
Using clonal cell lines that respond to specific ligands is a significant improvement as a source of materials for bioassays. The cellular response of ligand dependent cell lines can take a variety of forms, but is most often proliferation or inhibition of proliferation, expression of cellular markers or enzymes, cytotoxicity, or anti-viral activity. The use of murine cell lines increases specificity in some cases, as they may not respond to proteins that are species restricted in their activity.
Taking advantage of recombinant DNA technology has allowed for the cloning of specific receptors and their expression on previously non-responsive cell lines. This can create a specific, responsive cell line for almost any protein with a cellular receptor, without the need to screen a wide range of existing cell lines or tumor cells for responsiveness.
Reporter Gene Based Bioassays
While transfected receptor cell lines can offer selective responsiveness, such lines are still prone to the variability that occurs during the extended periods required for some induced biological function to appear (e.g., cell division, maturation or celldeath). Therefore, the development of bioassays that identify the activation of the genes involved in that function can be much more rapid and robust. The format of these assays is to introduce a plasmid containing a promoter (or rather a relevant region) known to be involved in the expression of genes induced by a test ligand. The promoter region is linked to a reporter gene that subsequently is expressed on ligand binding to its receptor.
The earliest forms of such assays, termed reporter gene assays, used luciferase expression as a marker for gene activation induced by test ligands. This enzyme catalyzes a reaction that results in light formation detectable by luminometers. In recent years, even more sensitive reporter gene systems have been devised, including green fluorescent protein and beta-galactosidase. Due to the shorter time required for significant expression of reporter genes, 2 h as opposed to days for standard bioassays, the assays appear less affected by extraneous influences and are therefore less variable and more precise.
Biological Assay Selection Based on Their Therapeutic Mechanism of Actions (MoA)
Agonistic MoA
Agonistic biotherapeutics, e.g. cytokines, growth hormones, agonistic mAbs, exert pharmaceutical activity by directly binding and activating a cellular receptor. Cell-based assays should be used whenever possible for this class of biotherapeutics. For agonistic mAbs biotherapeutics, if efforts to identify and/or develop a cell-based assay are unsuccessful during the early stages of drug development, regulatory input should be sought for using an alternative non-cell-based potency assay in the interim while a reliable cell-based potency assay will continue to be developed to support later studies.
Antagonistic MoA
Monoclonal Antibodies Binding to Soluble Ligands
A mAb biotherapeutic belonging to this class exerts its pharmacological effect by binding directly to a humoral target (a ligand presents in body fluid), which prevents the interaction of the ligand with the target receptor and obstructs its biological function. Since drug binds to the ligand in circulation and does not involve direct interaction with cells, a non cell-based binding assay suitably reflects the therapeutic MoA and may be deemed acceptable for potency assay.
Anti-receptor MAbs
An antagonistic mAb targeting a receptor directly blocks the biological function of cellular receptor. For example, the therapeutic antibody prevents the receptor specific ligand from binding to it and inhibits the downstream ligand mediated activation of signaling pathway. Since this category of biotherapeutics functions through an inhibitory mechanism and the drug-target interaction involves cells, either cell-based or binding assays may be suitable for potency assay. However, it is important to emphasize that for this class of products, the selection of a proper potency assay format requires a thorough understanding of the structural attributes of the cell surface receptor. If the antagonist targets a hetero-oligomeric receptor, a cell-based assay may be the most appropriate format for potency assessment. In a competitive ligand binding (CLB) assay, the multiple subunits of the purified recombinant receptor may not retain their native structure, function and integrity and would thereby fail to represent the drug-target interaction as it occurs in vivo. Similarly, if the target receptor requires a co-receptor to exert its biological function, a cell-based assay would be a better choice compared to the CLB assay. In a CLB assay, it is not feasible to couple the receptor and the co-receptor in a conformation that resembles their native orientation and structure when associated with the cellular membrane (Hu et al. 2015).
Soluble Receptors/Receptor Fusion Proteins
Soluble receptors or receptor fusion proteins, such as etanercept (Peppel et al. 1991) and abatacept (Korhonen and Moilanen 2009), function as an antagonist by binding the ligand and preventing its interaction with the cognate cellular receptor. When drug binds to the ligand in circulation and does not involve direct interaction with cells, a binding assay suitably reflects the therapeutic MoA may be deemed acceptable for potency assay. However, a cell-based potency assay may be needed when the target ligand is cell membrane-associated.
Multiple or Novel MoA
Besides the above-mentioned classes, “novel” biotherapeutics with diverse “modalities” and more complicated therapeutic MoAs have emerged recently for drug development. These include biotherapeutics with multiple functional domains such as antibody-drug conjugates (ADCs), antibodies with effector function, multi-specific molecules (a biotherapeutic binding to multiple drug targets), oligonucleotides, gene therapies and cell-based therapies, etc. This chapter will focus on defining the potency assay selection for biotherapeutics with MoAs mediated by Fab and Fc domains, such as ADCs and antibodies with effector function. Oligonucleotides, gene therapies, and cell-based therapies are not covered here, although the basic principle of designing assays based on the MoAs would still apply for these treatment remedies. For multi-specific biotherapeutics, separate potency assays may be needed for measuring biological responses specific to each functional domain. There are exceptions when only one potency assay is required for development if the potency of the respective domain function can be measured in one multiplex assay. Compared to the potency assay specific to each functional domain in separate assays, the multiplex approach monitors the simultaneous interaction of drug with its respective targets in one single assay and thereby closely mimics the therapeutic MoAs. This would allow more precise potency assessment since additional steric hindrance is observed when drug binds to multiple targets instead of one target.
Antibody Drug Conjugates (ADCs)
ADCs are unique immunoconjugates that couple a cytotoxic drug to a monoclonal antibody through a peptide or small molecule linker for targeted cancer therapy (Sievers and Senter 2013). An ADC binds to a specific tumor marker on cancer cells via mAb to deliver the cytotoxic payload or drug through cell membrane internalization for therapeutic intervention. For ADC molecules, a cell-based potency assay using read-out associated with cell death or proliferation that reflect the therapeutic MoA is a scientifically justified potency platform. In addition, a binding potency assay is also required that can measure either ADC binds to a specific tumor marker or the binding to both naked Ab portion and cytotoxic payload portion of the ADC.
Effector Function of MAbs
Therapeutic antibodies may manifest their clinical efficacy through effector functions such as antibody-dependent cell-mediated cytotoxicity (ADCC), antibody dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC) (Jiang et al. 2011; Wu et al. 2015). MAbs with ADCC and CDC function induce target cell lysis following specific binding of antibody variable region to the target antigen on cell surface and the interaction of antibody Fc with either FcγRs on effector cells or complement (Jiang et al. 2011). Since target cell lysis is the end-point resulting from the drug’s pharmaceutical activity, cell-based effector function assays which directly measure target cell lysis would be reflective of the relevant MoA for this category of antibody biotherapeutics. However, such effector function assays often suffer from relatively higher variability compared to regular cell-based assays due to the employment of two cell types (i.e. target and effector cells) (Schnueriger et al. 2011). If a cell-based effector assay is not feasible to serve as potency assay, other assay formats such as a cell-based binding assay or a non cell-based CLB assay may be acceptable to serve as the surrogate assay. However, cell-based effector assay should be used for biological characterization to bridge with the binding assay.
Biological Assays in Biosimilar Product Life-Cycle Management
Biological assays of various types can be used at the very early stages for establishing the quality target product profile (QTPP) of the reference products as well as developing biosimilar products to match the QTPP of reference products. These can provide information of the types of activities shown by the reference product. During product development, bioassays are invaluable for investigations aimed at characterizing the biological activities of the product and for stability, dosing and formulation studies. After product development, the assays are used to show batch-to-batch consistency and product (final form) stability under the proposed storage conditions as well as defining the biological potency of the product (Mire-Sluis et al. 1996).
Different bioassays may be needed for different purposes at different stages of biosimilar monoclonal antibody product development. Biological assays used early in product identification and characterization may need to be designed to maximize biological relevance and information content and could be less precise, accurate and rugged as well as being different from assays used later in product development to determine potency, stability and batch-to-batch consistency.
For example, the impact of glycosylation on a biosimilar monoclonal antibody either increased or decreased in comparison to the reference product can be studied by exploring the impact on product potency using in vivo, binding, antibody dependent cell-mediated cytotoxicity (ADCC), or complement dependent cytotoxicity (CDC) assays. The function of other structural characteristics of a product (e.g., size) should also be understood and included in the design of comparability studies.
For the development of both biosimilar and innovative molecules, biological assays, whether binding or cell-based functional assays, should be run concurrently so that a developer can select the appropriate potency assay in later development and have a body of data to support that choice. For example, if a bioassay is deemed too variable or not optimal for quality control testing the manufacturer should consider switching back to the binding assays. However, when replacing a bioassay with a binding or other surrogate assay, data must be gathered to demonstrate a strong correlation between the assays. Therefore, you should develop a cell-based assay as soon as possible because of the time required to do so and to gain experience with the cell line. This also allows ample time to gather correlation data between the assays, which should mitigate the regulatory risk associated with a poorly justified method. A parallel path can be built between the bioassay and a binding assay that justifies the selection of the final method.
The final release potency method should be “locked down” and in place prior to the pivotal trial. This would include the final internal controls of the assay (e.g. assay suitability) as well as the ranges in which the assay can be performed appropriately. This presents some advantages for either an innovator or biosimilar product. This provides a good deal of experience with the final potency method before submission, which offers a true estimate of method performance and success rate. In addition, fewer validations and bridging studies can be performed if a small number of methods are used throughout the clinical phases. However, it may be necessary to use multiple potency assays until a clear understanding of the product attributes and MoA has been achieved. Ultimately, the most well characterized, precise bioassay reflective of the mechanism of action is generally selected as the lot release potency assay to support commercialization of the product. Regardless of the final assay format, appropriate design, validation, and analysis are necessary if an assay is to provide reproducible and meaningful data.
Cell Based Assay Optimization and Method Remediation
For a cell-based bioassay, one of the most important factors is to choose a cell line that responds well to the drug and that response should be durable. The cell line should thus be stable, meaning that cell growth and response to the drug are consistent over time. This requires an understanding of the cellular growth patterns and receptor expression kinetics. A developer should determine how cell responsiveness and receptor expression are affected by passage number, cell density, feeding schedules and days in culture. Establishing these cell traits during development can help ensure a consistent and robust cellular response to the drug. The output used to measure cellular activity (e.g., fluorescence, luminescence) should be quantitative and indicative of a robust cellular response. Therefore, a primary goal in cell line and output selection should be to maximize the signal-to-noise ratio of the response.
Carefully controlled bioassays are technically demanding, relying heavily on the competence of staff carrying out the assays to accurately and reproducibly dilute and pipette solutions. However, automation of both bioassays and immunoassays have been particularly successful, but the capital investment is large. Therefore, the design of any bioassay must take into account factors that introduce variability and the analysis of bioassays must test for variability if results are to be statistically valid. A titration of the test material has to be made and compared to a titration of a reference material, with particular attention paid to comparisons of the linear portion of the dose-response curve. At least three points on the linear portion of the dose response curve are required to compare sample and reference curves.
Proper assay design also integrates multiple strategies to minimize variability and bias. There should be as few handling steps and reagents as possible to minimize dilution or technical errors. Most bioassays and immunoassay use microtiter plate that are particularly prone to position effects that can result in variability of data. To reduce the effects of position within microtiter plate assays, randomization of the position of sample titration curves within plates is recommended, as is the inclusion of a standard reference preparation on each plate; again, preferably in different positions. The use of coded duplicates in the assessment of variability and bias is particularly valuable.
A Reference Standard and Assay Control should be established as early as possible for continuous trending of assay performance. When performing a bioassay for biosimilar, an in-house Reference Standard, often from one of the GMP drug substance batch, needs to be qualified and established against pre-defined acceptance criteria. Bioassay always uses the Reference Standard to establish system suitability and assay acceptance criteria. Concurrently, bioassay also needs to use an “Assay Control”, which can be a lot of reference product. Thus, one always runs both biosimilar and reference product, side-by-side, and use the same passage of cells. The system suitability, or acceptance criteria, of a bioassay should be sufficient to ensure that the assay remains in control between runs. System suitability criteria often include requirements for cell viability, cell count, passage number, the signal-to-noise ratio, internal control potency, and parallelism, but may include any parameter that is determined to be important in minimizing inter-assay variability. Additionally, several statistical tools can be used to improve assay robustness.
These studies are similar to validation testing, but they are not as protocol driven and are performed at earlier phases in development to demonstrate that they are suitable for use. Proper robustness studies are also key to method transfer and performance trending because they establish the method variability that may exist between runs without detriment to the results. An experienced biostatistician can aid in experimental designs to determine the component variance analysis, or the factors in the assay that contribute most to variability. Assay performance can be improved significantly by understanding and controlling for these factors.
Proper analyst training is also of great importance in delivering consistent and reliable assay results. Because an analyst is generally one of the most significant sources of bioassay variability, the focus of training should be to limit this source of variability to whatever extent possible. Implementation of these practices will yield a bioassay that is well controlled and usable as a quality control release assay.
When designing an immunoassay, one has to consider whether proteins on a plastic surface or in solution have the same affinity as those on a cell or in solution. In addition, are the receptors/ligand oriented in the same way? This can be an issue when considering the hydrophobicity of full length receptors. Reproducibility of plate coating is also important to assess.
During immunoassay development, it is also important to judge if binding of a monoclonal antibody to a receptor/ligand on a plate reflects functionality. Binding to a protein on a plastic plate does not always mean the product is functional (there could be sticky degradants, charged variants etc.)—the same applies to a monoclonal antibody binding to its ligand on a plate that might not always assure the ligand is then neutralized in vivo.
Method Validation
The appropriate validation of any assay used for the characterization and release of biosimilar monoclonal antibodies is critical. Even though there are general regulatory guidelines for assessment of the validity of an assay, details contained in the US Pharmacopoeia are specific to bioassays. However, it is up to the assay developer to use these guidelines and develop in house protocols based on sound scientific principles and the nature of the assay. Assay characteristics with associated acceptance criteria such as reproducibility, robustness, signal to noise ratio etc. should be contained in a predefined validation protocol. When more than one biosimilar monoclonal antibody is produced in a facility, specificity should be part of the validation criteria.
The fundamental condition for any assay validity however is the condition of biosimilarity of sample and reference standard; that is, the dose–response relations (i.e. slope, asymptotes etc.) for the sample and the reference standard should be identical. During assay development, substantial information about dose–response curves should be collected in order to select an optimal dose range for potency estimation assays. After such data are available, analysis in terms of log doses is often found preferable to analysis in terms of absolute units.
The optimal assay range is often chosen in a linear (or linear under suitable transformation) part of the log dose–response relation. In such a situation, the condition of biosimilarity becomes a condition that the log dose–response line for the sample should be parallel to that for the reference standard, i.e. a parallel line assay. Provided at least three or ideally more doses of each preparation are included in the assay, the conditions of linearity and parallelism of the log dose–response lines can be tested in the individual assay. Moreover, the slope of the line or other characteristics of the responses may also provide information about conformity or otherwise with the previously determined complete dose–response relation.
Various assumptions about the statistical nature of the assay response data must be satisfied if estimates derived from such analyses, and the tests for the conditions of linearity and parallelism given by such analysis, are to be valid.
One must always assume that the “experimental units” providing the response represent a random selection from a defined population of such units. For example, results obtained for cells in a microtiters plate applied earlier may be different from those applied later. Units may differ because of a temperature, oxygen or humidity gradient across the plate. If an assay extends over several microtiters plates such differences between wells become even greater so these and other factors must become part of the definition of the “experimental unit”.
During assay validation, one should also assess the precision of an assay. Precision is a measure of assay variability as it illustrates how similar the results of an assay are when several estimates of potency are provided. This should not be confused with accuracy, which is a measure of how close an assay result is to the ‘correct’ result. An assay can be very precise, but give the wrong answer i.e. is inaccurate.
Assay repeatability is the precision of the assay internally (intra assay variability)–how repeated estimates within a single assay compare with each other. Intermediate precision is the precision of the assay when performed on different occasions or by different analysts, but within the same laboratory (inter-assay variability); i.e. How the results of independent bioassays compare to each other. Lastly, reproducibility is the precision of the assay when performed by different laboratories. This is measured by collaborative study between laboratories.
Critical Reagents
Reagents that are deemed critical through assay qualification studies should be well characterized and tightly controlled, monitored, and thus ‘qualified’. Whenever feasible, critical reagents should not be single-sourced, meaning that they should be available from more than a single vendor. This precaution will prevent an inability to perform assays if one source is suddenly unable to provide the quantity or quality of the reagent required. The stability of critical reagents has to be assessed so a shelf life can be applied that ensures new material is made before the original material becomes ineffective.
With all cell line based assays, careful evaluation of the stability of the cell line should be carried out. Cell lines can often lose their biological responsiveness over time, so it is important to have a well-characterized cell bank and some idea of how long a line can be passaged before its response becomes compromised. Therefore, cell lines should normally be cultured for a pre-specified period or number of cell doublings and then replaced with an early passage of cells.
Different batches of fetal calf serum used to maintain cell cultures can greatly affect the performance of bioassays and should be carefully screened prior to use. Some batches of sera can provide excellent maintenance of cell lines (i.e. cells grow rapidly), but result in poor bioassays with high backgrounds or low stimulation indices. Screening of sera should include both performance in cell maintenance and in bioassays.
For both immunoassays and cell based assays, the type of microtiter plate and its materials of construction cannot be underestimated. Changing from one manufacturer to another can impact the ability of adherent cells to stick to the plate, the same for antigen or antibody coatings in immunoassays.
For immunoassays, receptor or ligands used are usually critical reagents, as are any conjugated antibodies used in the assay. Each should be well characterized before use.
Whenever critical reagents are identified during assay development, it is necessary to ensure enough time is provided between replacing the reagents to assure the assay still performs as expected. A suitable critical reagent replacement protocol should be in place to follow when the time comes, with associated predefined acceptance criteria.
Legacy Potency Assay and Assay Replacement/Comparability
Although it might not appear relevant for a biosimilar product for its initial approval, one must always consider that the original bioassay could be replaced. For example, a cell based assay with an immunoassay, or to a more robust cell line, as one gains more experience with the product—even if the innovator was using a cell based assay at the time of the biosimilar approval.
Regardless of product type or history, the replacement of a bioassay in potency testing is not possible without a strong body of data that strongly correlates product activity between assays. It is advisable to start putting that together early in development, devising a parallel path that provides extensive experience with all assays and includes testing of multiple lots and product variants. The data must be combined with a strong knowledge of the MoA to demonstrate that product potency is well represented by the surrogate assay(s).
An interesting aspect of the replacement of a bioassay in potency testing is the fact that a robust and precise bioassay is required for the effort to end in success. It is unrealistic to expect you could replace a poor bioassay with a quality surrogate assay. That is because the results of a surrogate assay will not be accurate and reliable unless they were correlated to a bioassay that possessed those features in the first place. Given the value of a quality bioassay, this could represent an interesting dilemma to any company considering the use of a non cell-based assay for potency testing of biosimilar monoclonal antibodies.
When replacing one assay with another, one should test a set of samples side by side to justify that the assay is able to detect changes in potency if the product differs in some way. These samples can include: several lots of product (bulk and FDF, SKU’s), existing product variants (aggregates, oxidized etc.), temperature degradation over time, freeze/thaw, light exposure degradation over time, proteolytic degradants, pH exposure, glycosylation variants and any relevant in process materials.
In addition to lot release, stability testing has to be considered when changes to a bioassay are being proposed. For stability testing, a surrogate assay may not need to be the most sensitive assay for product change, but it must be able to detect all aspects of change that are important for potency. Of course, extensive characterization studies must be performed to determine what changes occur to a product over time and the impact of each on its potency. It may then be possible to combine this knowledge with risk assessments to ensure that the surrogate assay provides necessary coverage for accurate and reliable potency testing.
To assure regulatory acceptance of any assay comparability plan, a comparability protocol should be written outlining:
-
Detailed description of both assays
-
Statistical plan
-
Sample plan
-
Testing plan
-
Data to be presented
-
Acceptance criteria
Discussing any bioassay replacement plan with regulators ahead of time is advisable.
Method Tech Transfer
Transferring a method between sites often occurs as one moves from the clinical manufacturing site to the commercial scale site. Whilst there is an increasing movement in the rapid development of biosimilar monoclonal antibodies to do both clinical and commercial manufacturing at the same site (thus alleviating the need to transfer assays) for approval, method transfer would still have to occur if import testing was to be required as products are approved globally.
Therefore, a company should have specific protocols for how to transfer an assay to ensure it performs in the expected way between sites and continues to do reproducibly over time. Having drift between assay sites can cause considerable issues if not monitored for and addressed in a timely manner.
A suitable protocol should be created with the necessary contents similar to assessing comparability between to assays as described above (e.g. sampling and analysis plan, acceptance criteria etc.). It is highly recommended that both the sending and receiving site execute the protocol on the same samples and results examined carefully for any bias, even if acceptance criteria are met. A drive to simply use existing sending site data as the comparator to the receiving site can cause issues if the same samples are not executed around the same timeframe (i.e. sample stability can shift results).
The type and number of samples should be based on the known inherent variability of the assay and should be derived in consultation with a statistician.
Special Considerations of Biological Assays Comparing Innovative Biologics and Biosimilar
Regulatory Expectations and Current Practices on Potency Test
The regulatory pathway for biosimilar medicines is a unique and thoughtful process. It is designed to help ensure the development and approval of high-quality biosimilar medicines. Approved biosimilar medicines should have no clinically meaningful differences in terms of safety and efficacy from the relevant reference product, based on the totality of evidence from analytical, nonclinical, pharmacokinetic, and clinical studies. The totality of evidence represents a new approach by the FDA to the development of a new biologic product.
Robust analytical testing, including comparative structural and functional characterization, should be employed to establish high biosimilarity of the biosimilar and the reference product. Nonclinical testing will be used to evaluate the toxicity and safety profiles of the biosimilar. Comparative human pharmacokinetic and pharmacodynamic studies and clinical immunogenicity assessment will also need to be established. If residual uncertainty exists, comparative trials may be required (based on recent approvals, the FDA has required studies).
Functional assays can serve multiple purposes in the characterization of protein products. These assays act to complement physicochemical analyses and are a qualitative measure of the function of the protein product. Depending on the structural complexity of the protein and available analytical technology, the physicochemical analysis may not be able to confirm the integrity of the higher order structures. Instead, the integrity of such structures can usually be inferred from the product’s biological activity. If the clinically relevant MoAs are known for the reference product, the functional assays should reflect these MoAs. Multiple functional assays should, in general, be performed as part of the analytical biosimilarity assessments. The assessment of functional activity is also useful in providing an estimate of the specific activity of a product as an indicator of manufacturing process consistency, as well as product purity, potency, and stability.
If a reference product exhibits multiple functional activities, a set of appropriate assays designed to evaluate the range of relevant activities for that product should be performed. For example, with proteins that possess multiple functional domains expressing enzymatic and receptor-mediated activities, one should evaluate both activities. For products where functional activity can be measured by more than one parameter (e.g., enzyme kinetics or interactions with blood clotting factors), the comparative characterization of each parameter between products should be assessed.
It is recognizable that some types of biological assays have potential limitations, such as high variability, that might preclude detection of small but significant differences between the proposed biosimilar product and the reference product. Because a highly variable assay may not provide a meaningful assessment as to whether the proposed product is highly similar to the reference product, efforts should be made to develop bioassays that are less variable, more sensitive to changes in the functional activities of the product. In addition, in vitro bioactivity assays may not fully reflect the clinical activity of the protein. For example, these assays generally do not predict the bioavailability (pharmacokinetics and biodistribution) of the product, which can affect pharmacodynamics and clinical performance. Also, bioavailability can be dramatically altered by subtle differences in glycoform distribution or other posttranslational modifications. Thus, these limitations should be taken into account when assessing the robustness of the quality of data supporting biosimilarity and the need for additional information that may address residual uncertainties.
How to Select a Potency Assay
Potency assays play a pivotal role in determination of potency of protein products. As required by U.S. regulation, an assessment of potency is required for the licensure of the biopharmaceuticals defined in 21 CFR 601.2. Ideally, a potency assay should reflect the product’s MoA, be sensitive to changes in product critical quality attributes, and stability indicating. The potency test should be validated as per ICH Q2 (R1). Developing a robust, sensitive, and relevant potency assay represents a substantive challenge both in planning and execution. Selecting the best potency assay format (i.e., in vivo or in vitro) should be based on scientific knowledge of the product-target interactions, therapeutic effect elicited through the product-target interaction, and the assay performance itself based on the status of assay’s validation and qualification. System suitability and assay specification acceptance criteria are usually set as a numerical range and should be adjusted throughout the product development to reflect the manufacturing and clinical experience.
Several regulatory and guidance documents are published by the Food and Drug Administration (FDA), the International Committee on Harmonization (ICH), the United States Pharmacopeia (USP) (2012a, b), and the European Pharmacopeia (Ph. Eur.) (European Directorate for the Quality of Medicines 2004) to cover different aspects of bioassay validation. Despite the availability of these documents, often there are questions related to implementation and interpretation of these guidelines. Assay validation demonstrates that the assay, when performed per the SOP, is adequately precise and accurate for use in product release and stability studies.
The legacy potency assay employed by the originator product can always serve as a starting point for potency assay selection for biosimilar development. Nonetheless, it is possible that fast, homogeneous and precise bioassays reflective of product’s MoA can be used to replace the variable legacy potency assay. In this case, a method “bridging” or “comparative” study may be needed to demonstrate the equivalent performance of two methods in detecting changes impacting bioactivity and demonstrating similar stability indicating properties.
Reference Standards
The design of these assays and calculation of relative potency for a product rely heavily on Reference Standards. This is not to be confused with ‘reference product’ to which the biosimilar is compared to the innovator (reference) product. Selecting and establishing the right material to serve as the Reference Standard is important. The biological response of a test sample is directly compared against the Reference Standard in a potency assay. Thus, the Reference Standard is ideally generated from a similar manufacturing process as the test sample and with known stability data under intended storage conditions. Moreover, the Reference Standard should be evaluated thoroughly through multiple runs in the potency assay (n > 10) to establish a “normal” range for EC50, hill slope, and upper and lower asymptotes when the assay uses a 4-PL data-fitting model commonly used for potency assay evaluation. When the Reference Standard is deemed appropriate for a given assay, allocate sufficient quantities of material for future assays. It is likely that the material will be used not only for assay development and validation, but also for sample testing when its shelf life allows. When the current lot is close to depletion, retain some samples for use in a bridging study to compare with the new Reference Standard.
If there is a suitable, publicly available, and well-established Reference Standard for the protein product, a physicochemical and/or functional comparison of the proposed product with this standard may also provide useful information. Although studies with such a Reference Standard may be useful, they do not satisfy the BPCI Act’s requirement to demonstrate the biosimilarity of the proposed product to the U.S.-licensed reference product. For example, if an International Standard for calibration of potency is available, a comparison of the relative potency of the proposed product with this potency standard should be performed. As recommended in ICH Q6B, an in-house Reference Standard(s) should always be established, qualified and used for control of the manufacturing process and product.
An International Reference Standard, when applicable, can be obtained from a nationally or internationally recognized source. Alternative material, including material generated in-house, may be qualified and designated as a Primary or Working In-House Reference Standard. In-house primary Reference Standard material must be prepared from lot(s) representative of production and clinical batches, and qualified following established procedures that include characterization testing requirements and specifications/assay acceptance criteria, as well as stability testing procedures. A process is established for succession planning of Reference Standards.
In summary, analytical studies carried out to support the approval of a proposed product should not focus solely on the characterization of the proposed product in isolation. Rather, these studies should be part of a broad comparison that includes, but is not limited to, the proposed product, the reference product, applicable Reference Standards, and consideration of relevant publicly available information.
Number of Lots Required for Physiocochemical and Functional Biosimilarity Studies
Extensive and robust comparative physicochemical and functional studies should be performed to evaluate whether the proposed product and the reference product are highly similar. A meaningful assessment as to whether the proposed product is highly similar to the reference product depends on, among other things, the capabilities of available state-of-the-art analytical assays to assess, for example, the molecular weight of the protein, complexity of the protein (higher order structure and posttranslational modifications), degree of heterogeneity, functional properties, impurity profiles, and degradation profiles denoting stability. Physicochemical and functional characterization studies should be sufficient to establish relevant quality attributes including those that define a product’s identity, quantity, safety, purity, and potency. The product-related impurities, product-related substances, and process-related impurities should be identified, characterized as appropriate, quantified, and compared with multiple lots of the proposed product to multiple lots of the reference product, to the extent feasible and relevant, as part of an assessment of the potential impact on the safety, purity, and potency of the product (Food and Drug Administration (FDA) 2012).
In general, at least 15 lots of reference product and 10 Drug Substance lots of the proposed biosimilar product are required to be used for analytical biosimilarity assessment—although this is highly dependent on the methods use and their variability (Tsong et al. 2017). Therefore, it is strongly recommended that a statistician be consulted to select the appropriate, statistically valid, number of lots to show biosimilarity.
Addressing Assay Variability
A successful bioassay suitable for validation and final-product lot release may take multistage development and fine-tuning to reach a final design. Although many roadblocks can present on the way to a robust bioassay, controlling variables at early stage assay development and careful quality control in assay performance are key to a meaningful potency test to ensure product quality.
Here we focus on potential aspects to consider when building a consistent potency assay that is suitable as a release test.
Cell Type/Cell Line Selection
To develop a cell-based potency assay, there are many factors that need to be considered. Firstly, determine which cells are appropriate. If possible, select a type that is relevant to a product’s MoA and is known to respond well to the product. For instance, when developing a mAb that binds to a cancer cell marker and subsequently leads to growth inhibition of target cells, screen several malignant cell lines that express that marker. The most responsive cell line should be selected, although one must consider stability of the cell line to the response above sensitivity. There is no benefit in having a cell line that exhibits a strong response if it is either highly variable or loses reactivity too quickly.
Primary cells in general should not be used because of their potential for lot-to-lot, donor-to-donor variability. However, in some cases where primary cells must be used, consider appropriate approaches to minimize cell heterogeneity. That can be done by securing a large lot of cells or isolating a subpopulation when feasible. Ready-to-use frozen cells can be helpful in reducing assay variability.
Peripheral blood mononucleated cells (PBMCs) are commonly used in bioassays for product characterization. But PBMCs lack consistency in potency tests in general, primarily because only a subset of cells generates the response of interest. Furthermore, the percentage and activity of different subpopulations of PBMCs vary from run to run and between lots. Instead, there should be attempts to isolate a desired cell population and use the “purer” cells in a potency assay.
Selected cell lines need to be extensively characterized. Information about cloning history, genetic stability, gene copy number, growth characteristics, and passage limits all should be established. At a minimum, evaluate passage limits and vial-to-vial consistency in the potency assay. In addition, create and store phase- appropriate cell banks. It is not unusual to use a research-grade cell bank for early phase potency assay development. However, when a product progresses to phase 2–3, it is critical that you make and fully characterize a cell bank generated under a more controlled laboratory environment. Whenever a new bank is generated—in addition to the standard purity and identity testing—test cells from that new bank in the assay to ensure that the assay parameters are comparable with the current bank.
Lastly, ensure that cells are in the necessary physiological state and behave in the potency assay as expected. For suspension cells, establish the minimum and maximum cell density for culture maintenance. Spent media should not be used as it may impact cell growth and metabolism, and cause unwanted cell selection. It is also important to not under or over trypsinize adherent cells as it can potentially damage the cell membrane. Cells should not be allowed to grow over confluent to prevent potential cell transformation.
Procedural Accuracy
Because of the inherent, non-robust nature of potency assays, a robust potency assay requires the use of well-defined and accurate procedures. From a stock solution, both a Reference Standard and test sample are diluted over multiple steps to the final working dilution (concentration) range tested in the assay. In addition, a potency assay involves pipetting cell suspension onto 96-well microplates and mixing with other reagents. Without accurate pipetting, there is no solid foundation for a robust potency assay.
We do not discuss pipetting techniques at length here, but rather offer a few quick points to consider. First, work with a volume that is close to each pipette’s calibration volume. Second, use prewet tips to increase consistency. Third, except for cell suspension, all reagents should be at room temperature for accurate pipetting. Last, use reverse pipetting when dealing with viscous liquids.
Incubation temperature and time should be well controlled. By contrast with an assay performed in an R&D environment, a potency assay must have a well-defined range for acceptable incubation temperature and time. Many good laboratory practice (GLP) or GMP laboratories have incubation chambers (incubator, refrigerator, or freezer) for 37 °C, refrigerated, or frozen conditions but no chambers for room temperature. As a result, plates are placed on the bench top for room-temperature incubation. This “room temperature” can range from 20 to 35 °C, even 15–40 °C. Fluctuations across the range of temperatures can significantly affect assay outcomes. For incubation steps that are performed at room temperature, using an incubator set at 20–25 °C can reduce assay variability. As for incubation time, do not use a wide range of times for critical incubation steps, if possible. For example, a 60 ± 10 min time window is much better than 1–2 h.
Consistent washing steps are essential for controlling assay background and precision between replicate wells. Whether using manual washing or an automated plate washer, be consistent and allow only one washing step method in the procedure. When an assay requires manual washing, ensure that all analysts wash plates in a similar way—working through the plate at the same orientation, adding wash buffer at similar speed, and washing adjacent rows at similar intervals. When using a plate washer, make sure the same setting is used every time.
Proper and timely calibration and maintenance of equipment also can contribute to procedural accuracy. All equipment used in GMP assays should be validated for their intended use.
Assay Training
For an assay that is not completely automated, the analyst is the largest source of assay variability. This is especially true for a bioassay that involves multiple dilution steps and manipulation of test sample, cells, and reagents. Onsite training can be conducted when transferring an assay to a different laboratory. This training provides the personnel from the sending and the receiving laboratories an opportunity to observe each other. Cross-training allows analysts to identify steps that might not be documented in an assay’s standard operating procedures but are important to assay performance. On many occasions, the sending lab SME can provide information about equipment or reagents that differ between the sending and receiving laboratories.
When an assay is performed infrequently, a periodic requalification program can familiarize analysts with assays and prevent potential assay failure due to long gaps between assay performance. The frequency of requalification depends on the complexity of the assay and the proficiency of the analyst. Generally, if an analyst has not run a given assay for 6 months, a requalification run should be performed before performing a GMP release test.
Data Analysis
The design of a bioassay that reports a relative potency value for a test sample against the Reference Standard takes into account run-to-run variability to some degree. Some assays are still highly variable despite thorough evaluations of the sources of variability. That is possibly attributed to the wide and unpredictable biological response being measured. For such assays, averaging final potency results from two or three independent setups or runs can be a useful approach to reduce the risk of the assay results being influenced by random factors. This strategy has been adopted by many scientists developing potency assays, especially for effector assays such as an antibody-dependent cell-mediated cytotoxicity (ADCC) assay or a complement-dependent cytotoxicity (CDC) assay. In such cases, the assay is qualified or validated based on two or three runs, the same as described in governing documents.
Assay Troubleshooting
There are several approaches for troubleshooting a non-robust bioassay. Dissecting a complex bioassay to individual steps is sometimes very helpful. When the response from cells plated in a 96-well microplate is measured after incubation with a number of reagents, evaluate the response after each step, if possible, to identify the problematic step in the procedure. Starting from a base plate with cells only often provides some clues such as position/edge effect or uneven cell seeding or growth.
A design of experiments (DoE) study is a useful tool to evaluate multiple variables systemically. You can perform DoE at the assay development stage to identify optimal assay conditions or for assay troubleshooting. For example, ligand concentrations, incubation time, and cell density all can be incorporated into one DoE, rather than be part of separate evaluations. DoE enables assessment of the impact from related experimental conditions that cannot be achieved by changing variables one at a time.
Data trending should be implemented to monitor performance of a potency assay. Key factors that could potentially affect assay outcomes, such as operator, cell seeding and harvest density, passage number, material lots, and equipment identification should be recorded. Other assay parameters such as EC50 values, hill slopes, and upper to lower asymptote ratios can also be trended. Those data often can answer questions such as (1) What has changed from when the assay was running well? (2) Is there a trend? And (3) What is the most likely root cause for the assay failure?
Data trending also helps detect data shift or drift before a system suitability failure or out-of-specification or out-of-trending event. Once a trend has been identified, preventative actions should be taken to prevent assay failure.
Setting limits goes hand-in-hand with data trending. For example, when a trend shows that an assay does not work well once cells have been cultured for more than 20 passages, then set a cell passage limit in that assay protocol. Knowing method limits such as cell passages, specific reagent lots expiry, and analyst-specific parameters is valuable and helps exclude potential factors that could introduce variability.
Critical Quality Attributes and Their Relationship to Potency
The critical quality attributes of a reference product, how each impacts safety, efficacy, pharmacokinetics, and overall quality, is fundamentally important to producing a high-quality biosimilar. A biologic drug is extremely complex and typically has more than 1 hundred features or “attributes.” Some of these attributes are important to the different ways the body can recognize proteins and are therefore critical to the safety, efficacy, and pharmacokinetics of the drug. These are known as “critical quality attributes.”
An understanding of which attributes are important to each function for each product is important to obtain the best possible match. Some attributes work individually to drive a biological function, and some work in a composite manner. A biosimilar will not be exactly like its reference product and some features will not match, but the critical quality attributes need to match so that the biosimilar medicine and the original biologic work in the same way, that is, have the same biological function for every patient. Similarly, a structural match is also desirable, but it is feasible to not have a precise structural match while still preserving function. However, an understanding of how physicochemical attributes especially ones such as glycoforms and charged variants impact potency, should be part of biosimilar product characterization. For each biological therapeutic, the differences between the reference product and the biosimilar in the key critical attributes should be the primary focus as they are expected to drive the “potential for biological differences”. Any bioassays that can help resolve the residual uncertainty would help define the clinical development of the proposed biosimilar.
Biological Dose-Response Modeling, Parallelism and Data Analysis
The potency of a biological therapeutic is often determined relative to a Reference Standard, such as via parallel line analysis. Measurement of relative potency is only meaningful if the test sample behaves as a dilution or concentration of the Reference Standard, and exhibits a parallel relationship to the Reference Standard. Such similarity is called parallelism. Graphically, parallelism is observed where the dose-response curve of the sample is a horizontal shift of that of the Reference Standard on the logarithmic dose axis. The amount of shift represents the logarithm of relative potency (USP 2012b). As a necessary sample acceptance criterion for bioassay, there is a need to assess parallelism before the results of a bioassay are interpreted. The requirement for the evaluation of parallelism appears in both the United States Pharmacopeia (USP) (USP 2012a, b) and European Pharmacopeia (EP) (European Directorate for the Quality of Medicines 2004).
Parallelism is a necessary condition for the relative potency of a bioassay to be meaningful. Difference and equivalence tests are two major statistical methods used for parallelism testing, with the latter being recommended for use in the revision of USP Chapters <1032> and <1034>. The recommendation is largely motivated by the criticism that the difference test may reject parallelism even for insignificant differences when the sample size is large or the assay is too precise, and that it fails to reject parallelism of non-parallel curves when sample size is small or the assay is imprecise. Therefore, the method rewards assays of small sample sizes and large variability, and thus does not offer adequate protection to consumer’s risk. From a compliance perspective, the equivalence test may be the preferred method because it makes the control of consumer’s risk possible, and encourages the manufacturer to improve its assay so as to provide better protection to the producer’s risk. However, implementation of this method can be challenging for laboratories that lack experience in statistical analysis and software development. Development of such a parallelism testing enabling tool is important for a laboratory to be compliant.
A customized assay analysis template that is incorporated into a fully GMP compliant software package (Yang et al. 2012). The template automates USP-recommended parallelism testing method based on a 4PL model, and it is simple to use. It makes the implementation of the USP guidance both practical and feasible. A case study demonstrates that the equivalence test can fail non-parallel samples and pass parallel samples. The tool can easily be generalized to bioassays with other types of non-linear response data such as 5-parameter logistic function. Overall, we show that an equivalence approach for parallelism testing, as recommended by USP, can be implemented in a simple, QC-friendly, compliant, and validatable manner.
Analytical Biosimilarity Assessment
FDA currently recommends the use of a statistical approach to evaluate quality attributes of proposed biosimilar products that is consistent with the risk assessment principles set forth in the International Conference on Harmonization Quality Guidelines Q8, Q9, Q10, and Q11. Consistent with these principles, FDA recommends an analytical biosimilarity assessment that is based on a tiered system in which approaches of varying statistical rigor are used (Tsong et al. 2017; Christl 2015).
One approach to determining the tier to which a particular quality attribute would be assigned will depend upon a criticality risk ranking of quality attributes with respect to their potential impact on activity, PK/PD, safety, and immunogenicity with quality attributes being assigned to tiers commensurate with their risk.
-
For quality attributes with the highest risk ranking (Tier 1), equivalency testing would be recommended and generally would include assay(s) that evaluate clinically relevant mechanism(s) of action of the product for each indication for which approval is sought.
-
For assessing quality attributes with lower risk ranking (Tier 2), FDA recommends the use of quality ranges (mean ± X σ, where X should be appropriately justified.
-
For the lowest risk ranking (Tier 3), FDA recommends an approach that uses raw data/graphical comparisons
In addition to criticality, other factors should be considered in assigning quality attributes and assays to a particular tier using this approach. This could include, but is not limited to, the levels of the attribute in both the reference product and proposed biosimilar product (as determined by the biosimilar sponsor’s testing), the sensitivity of an assay to detect differences between products, if any, and an understanding of the limitations in the type of statistical analysis that can be performed due to the nature of a quality attribute. Therefore, while many attributes may be considered high risk, not all would need to be included in Tier 1 testing. FDA recommends that sponsors submit their proposal for ranking attributes that will be assessed in each tier to gain agreement from the Agency prior to performing the statistical assessment.
FDA also recommends that sponsors carefully assess their analytical biosimilarity plan to identify and address any other factors that could potentially impact the ability to demonstrate that a biosimilar product is highly similar to the reference product. For example, considering the ages of the biosimilar and reference product lots tested, optimizing assays, and pre-specifying the criteria under which wider biosimilarity acceptance criteria for a particular assay would be considered appropriate.
However, it should be noted that while a statistical approach to evaluate quality attributes of a biosimilar product may be considered in support of the demonstration that the biosimilar product is highly similar to the reference product, the determination if a biosimilar product is highly similar to the reference product will also be based upon the totality of the evidence relevant to the assessment.
Case Studies
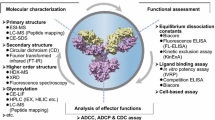
Therapeutic antibodies rely on two types of functionalities to achieve clinical efficacy: target-specific binding by the Fab (antigen-binding fragment) domain and immune-mediated effector functions—such as ADCC and CDC—via interaction of the Fc domain with receptors on various cell types. The Fc portion of a therapeutic antibody may therefore have an important role in its mechanism of action through its influence on either ADCC or CDC. Based on their putative mechanism of action, therapeutic antibodies can generally be classified into three categories, from which their potential for Fc functionality can be ranked (Jiang et al. 2011).
The reference product is a Class I antibody that recognizes and binds to cell-bound antigen and the Fc effector functions, ADCC and CDC, are part of the MoAs. The analytical biosimilarity assessment is accomplished through extensively characterizing the physicochemical and biological properties of reference products and the proposed biosimilar product. The case study here focuses only on biological characterization with the application of a battery of in vitro biological activity assays.
Tier 1 biological assays measure quality attributes with the highest risk ranking and evaluate clinically relevant MoAs of the product. Equivalency testing will be employed for these assays that include the following:
-
ADCC
-
CDC
Tier 2 biological assays assess quality attributes with lower risk ranking, and quality ranges (mean ± 3SD) will be examined. These Tier 2 biological assays include:
-
Apoptosis
-
Cell surface antigen binding
-
C1q binding
-
FcγRI binding
-
FcγRIIa binding
-
FcγRIIb binding
-
FcγRIIIa (F/V) binding
-
Neonatal Fc receptor (FcRn) binding
ADCC Assay
An ADCC assay measures the biological activity of an antibody against tumor target cells. In order to induce cell lysis of the tumor target cells, Fab and Fc regions of the antibody need to bind to cell surface antigen on tumor target cells and CD16 on NK effector cells, respectively.
An engineered CD16-expressing human NK cell line (NK92-CD16) was used as effector cells while antigen-expressing cell line was used as target cells. CytoTox-Glo™ cytotoxicity assay kit (Promega) was applied and ADCC activity was quantified by measuring the ratio of EC50 Ref Std/EC50 Test Sample × 100% in luminescence (Molecular Devices, SpectraMax®L). The relative potency was calculated via software SoftMax® Pro 5.4.5 (Molecular Devices). Figure 16.1 is representative dose response curves of ADCC for the proposed biosimilar product vs. reference product.

Representative ADCC dose response curves for the proposed Biosimilar Product, Reference Product (EU) and Reference Product (US)
An equivalence test was performed applying ± 1.5SD. Results show that the reference products and biosimilar product were statistically equivalent, indicating that the ADCC activity of the proposed biosimilar product is equivalent to that of the reference products.
CDC Assay
CDC measures complement mediated cellular cytotoxicity. The reference product can mediate CDC by binding to cell surface antigen on tumor target cells and recruiting complement complex via C1q binding from serum to target cells. For CDC assay, human serum was used for C1q source and antigen-expressing tumor cells were sued as target cells. CytoTox-Glo™ cytotoxicity assay kit (Promega) was applied and CDC activity was quantified by measuring the ratio of EC50 Ref Std/EC50 Test Sample × 100% in luminescence (Molecular Devices, SpectraMax®L). The relative potency was calculated via software SoftMax® Pro 5.4.5 (Molecular Devices). Figure 16.2 is representative dose response curves of CDC for the proposed biosimilar product vs. reference product.

Representative CDC dose response curves for the proposed Biosimilar Product, Reference Product (EU) and Reference Product (US)
Equivalence test was performed applying ± 1.5SD. Results show that the reference products and biosimilar product were statistically equivalent, indicating that the CDC activity of the proposed biosimilar product is equivalent to that of the reference products.
Apoptosis Assay
Antibody induced apoptosis was determined using antigen-expressing tumor cell line and Caspase 3/7-Glo™ assay kit (Promega). Apoptosis activity was quantified by measuring the ratio of EC50 Ref Std/EC50 Test Sample × 100% in luminescence (Molecular Devices, SpectraMax®L). The relative potency was calculated via software SoftMax® Pro 5.4.5 (Molecular Devices). Figure 16.3 is representative dose response curves of Apoptosis for the proposed biosimilar product vs. reference product.

Representative Apoptosis dose response curves for the proposed Biosimilar Product, Reference Product (EU) and Reference Product (US)
The biosimilarity range for apoptosis assay was set to be 94.9–105.3% which represents the mean ± 3SD of data from 28 different batches of reference products. All results (94.9–99.2%) of proposed biosimilar batches were within the biosimilarity range of reference products.
Cell Surface Antigen Binding Assay
Binding activities of the proposed biosimilar product and reference products to cell surface antigen were determined by a flow cytometric method. The relative binding activity was calculated by comparing to a Reference Standard by measuring the ratio of EC50 Ref Std/EC50 Test Sample × 100% in fluorescence (Beckman Dickinson). The relative potency was calculated via software SoftMax® Pro 5.4.5 (Molecular Devices). Figure 16.4 is representative dose response curves of cell surface antigen binding for the proposed biosimilar product vs. reference product.

Representative cell surface antigen binding dose response curves for the proposed Biosimilar Product, Reference Product (EU) and Reference Product (US)
The biosimilarity range for antigen binding assay was set to be 94.2–111.9% which represents the mean ± 3SD of data from 25 different batches of reference products. The range of the antigen binding activities of the proposed biosimilar were 92.6–100.6% whereas reference products (EU) and (US) were 97.9–109.4%.
From method qualification of the cell surface antigen binding assay, intermediate precision was 8.1%. Thus, 1.7% difference between lower limit of biosimilarity range and the lowest value of proposed biosimilar (94.2% and 92.6% each) might be due to assay variability. In addition, ADCC, CDC and apoptosis activities of proposed biosimilar product, which represent the MoAs of the reference products, were equivalent or similar to the reference products (see above). Furthermore, cell surface antigen binding activity of all GMP batches of the proposed biosimilar were close to 100% (100.0–100.6%).
C1q Binding Assay
C1q binding plays an important role in CDC function. The binding activity was measured by ELISA using a microplate reader (Molecular Devices). The relative binding activity was calculated by comparing to a Reference Standard. Figure 16.5 is representative dose response curves of C1q binding for the proposed biosimilar product vs. reference product. The biosimilarity range of reference products was 86.1–105.4% and all data of the proposed biosimilar product were within the biosimilarity range.

Representative C1q binding dose response curves for the proposed Biosimilar Product, Reference Product (EU) and Reference Product (US)
FcγR I Binding Assay
As the antibody binds to the immobilized FcγR I on the chip surface, the accumulation of protein results in a change of the refractive index. To generate binding curves, each test sample was serially diluted from 4000 to 125 nM. The binding affinity is evaluated using BIA evaluation software (GE Healthcare). The relative binding activity was calculated by comparing to a Reference Standard.
The biosimilarity range for the reference products was 80.3–118.9%. The proposed biosimilar product showed a range of 95.7–113.9%, well within the biosimilarity range of the reference products.
FcγR IIa Binding Assay
The FcγR IIa binding activity was determined using Biacore™ T200 (GE Healthcare). The binding affinity is evaluated using BIA evaluation software (GE Healthcare). The relative binding activity was calculated by comparing to a Reference Standard.
The biosimilarity range for the reference products was 90.2–107.9%. The proposed biosimilar product showed a range of 94.7–107.0%, well within the biosimilarity range of the reference products.
FcγR IIb Binding Assay
The FcγR IIb binding activity was determined using Biacore™ T200 (GE Healthcare). To generate a binding curve, each test sample was serially diluted from 20,000 to 625 nM. The binding affinity is evaluated using BIA evaluation software (GE Healthcare). The relative binding activity was calculated by comparing to a Reference Standard.
The biosimilarity range for the reference products was 90.7–104.6%. The proposed biosimilar product showed a range of 96.1–101.3%, well within the biosimilarity range of the reference products.
FcγR IIIa-Phe Binding Assay
The FcγRIIIa-Phe binding activity was determined using Biacore™ T200 (GE Healthcare). To generate a binding curve, each test sample was serially diluted from 4000 to 125 nM. The binding affinity is evaluated using BIA evaluation software (GE Healthcare). The relative binding activity was calculated by comparing to a Reference Standard.
The biosimilarity range for the reference products was 79.3–114.7%. The proposed biosimilar product showed a range of 94.9–123.3%. Three batches of the proposed biosimilar product had a slightly higher binding affinity than that of reference products. However overall range of the proposed biosimilar product was overlapped with that of reference products, and the observed difference is unlikely to affect the biological functions of the product, as ADCC, CDC and apoptosis activities of the proposed biosimilar product are equivalent or similar to that of reference products.
FcγR IIIa-Val Binding Assay
The FcγR IIIa-Val binding activity was determined using Biacore™ T200 (GE Healthcare). To generate a binding curve, each test sample was serially diluted from 2000 to 62.5 nM. The binding affinity is evaluated using BIA evaluation software (GE Healthcare). The relative binding activity was calculated by comparing to a Reference Standard.
The biosimilarity range for the reference products was 77.5–111.6%. The proposed biosimilar product showed a range of 88.7–114.0%. Two batches of the proposed biosimilar product had a slightly higher binding affinity than that of reference products. However overall range of the proposed biosimilar product was overlapped with that of reference products, and the observed difference is unlikely to affect the biological functions of the product, as ADCC, CDC and apoptosis activities of the proposed biosimilar product are equivalent or similar to that of reference products.
FcRn Binding Assay
The FcRn binding activity was determined using Biacore™ T200 (GE Healthcare). To generate a binding curve, each test sample was serially diluted from 4000 to 125 nM. The binding affinity is evaluated using BIA evaluation software (GE Healthcare). The relative binding activity was calculated by comparing to a Reference Standard.
The biosimilarity range for the reference products was 78.6–113.2%. The proposed biosimilar product showed a range of 99.4–132.3%. However, FcRn binding activity was considered for Tier 3 analysis. In addition, clinical studies have already demonstrated PK equivalence between proposed biosimilar product and the reference products. Therefore, considering overlapped range and method intrinsic variation, it is concluded that the proposed biosimilar product was similar to the reference products in FcRn binding.
Summary of the Case Studies
A series of Fab-related biological assays (Apoptosis and antigen binding assays), as well as Fc-related biological assays (ADCC, CDC, C1q binding, Fcγ RI, Fcγ RIIa, Fcγ RIIb, FcγR IIIa-Phe and FcγR IIIa-Val binding) and the FcRn binding assays were all performed in order to assess the biosimilarity of functional properties between the proposed biosimilar product and the reference products. All results except antigen binding were within the equivalence margin or biosimilarity range of the reference products. Even though the antigen binding activity of the biosimilar product was slightly lower than that of reference products, major functional bioactivities such as ADCC, CDC and apoptosis of the biosimilar product did not show any significant difference from that of the reference products. Overall, the biosimilar product is highly similar to the reference products in biological functions.
Concluding Remarks
The development of monoclonal antibodies has been an ongoing process for many years. The rapid rate at which they are produced to clinical grade requires reassurance obtained through the rigorous testing applied to such biological products. Bioassays and assays based on physicochemical principles address different aspects of the characteristics of biologicals. The data produced by these different types of procedures complement each other to provide a spectrum of information on the substance and different batches of product. Although some of this may overlap, the different assay types provide data that relate to different properties of the molecule in question. Advances in physicochemical and biological analytical sciences enable protein products to be characterized extensively in their physicochemical and biological properties. These analytical procedures have improved the ability to identify and characterize not only the desired product but also product-related substances and product- and process-related impurities. Advances in manufacturing science and production methods, as well as advances in analytical sciences, may enhance the likelihood that a proposed product can be demonstrated to be highly similar to a reference product by better targeting the reference product’s physiochemical and functional properties.
A relatively common misconception is that bioassays are so variable and imprecise that the results obtained are not usable for quantitative purposes and thus it is argued that they serve limited purpose. Although this might apply if a bad choice of assay is made, it can be avoided by careful selection of bioassay methodology, format and analysis. Therefore, carefully designed, validated and correctly analyzed bioassays can provide suitably quantitative information. In fact, bioassays have been able to detect differences in activity of biosimilar monoclonal antibodies that could not be readily predicted by physicochemical testing alone, especially as it relates to Fc functionality. Multiple functional assays should, in general, be performed as part of the analytical biosimilarity assessments. The assessment of functional activity is also useful in providing an estimate of the specific activity of a product as an indicator of manufacturing process consistency, as well as product purity, potency, and stability.
References
Christl L. Overview of regulatory pathway and FDA’s guidance for development and approval of biosimilar products in US. Presented at FDA ODAC meeting, 7 Jan 2015, Silver Spring, Maryland. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM436387.pdf.
European Directorate for the Quality of Medicines. Chapter 5.3. Statistical analysis. European pharmacopoeia. Strasbourg: European Directorate for the Quality of Medicines; 2004. p. 473–507.
Food and Drug Administration (FDA). Scientific considerations in demonstrating biosimilarity to a reference product, 2012.
Hu J, Wala I, Han H, Nagatani J, Barger T, Civoli F, Kaliyaperumal A, Zhuang Y, Gupta S. Comparison of cell-based and non-cell-based assay platforms for the detection of clinically relevant anti-drug neutralizing antibodies for immunogenicity assessment of therapeutic proteins. J Immunol Methods. 2015;419:1–8.
Jiang XR, Song A, Bergelson S, Arroll T, Parekh B, May K, Chung S, Strouse R, Mire-Sluis A, Schenerman M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nature Rev Drug Discov. 2011;10(2):101–11.
Korhonen R, Moilanen E. Abatacept. A novel CD80/86-CD28 T cell co-stimulation modulator, in the treatment of rheumatoid arthritis. Basic Clin Pharmacol Toxicol. 2009;104:276–84.
Mire-Sluis AR. Progress in the use of biological assays during the development of biotechnology products. Pharm Res. 2001;18(9):1239–46.
Mire-Sluis AR, Thorpe R. Quantitative cell line based bioassays for cytokines. J Immunol Methods. 1995;187:191–9.
Mire-Sluis AR, Gaines-Das R, Gerrard T, Padilla A, Thorpe R. Biological assays: their role in the development and quality control of biological medicinal products. Biologicals. 1996; 24:351–61.
Peppel K, Crawford D, Beutler B. A tumor necrosis factor (TNF) receptor-IgG heavy chain chimeric protein as a bivalent antagonist of TNF activity. J Exp Med. 1991;174:1483–9.
Sasardic D, Mire-Sluis AR. Biological standardization and control at the World Health Organization: successes, current issues and future challenges. Biologicals. 2000;28:47–53.
Schnueriger A, Grau R, Sondermann P, Schreitmueller T, Marti S, Zocher M. Development of a quantitative, cell-line based assay to measure ADCC activity mediated by therapeutic antibodies. Mol Immunol. 2011;48(12–13):1512–7.
Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64: 15–29.
Thorpe R, Wadhwa M, Mire-Sluis AR. The use of bioassays for the characterisation and control of biological therapeutic products produced by biotechnology. Dev Biol Stand. 1997;91:79–88.
Tsong Y, Dong X, Shen M. Development of statistical methods for analytical similarity assessment. J Biopharm Stat. 2017;27(2):197–205.
USP. Chapter <1032>. United States Pharmacopeia (USP): the national formulary. Rockville: USP Pharmacopeia Convention, Inc.; 2012a.
USP. Chapter <1034>. United States Pharmacopeia (USP): the national formulary. Rockville: USP Pharmacopeia Convention, Inc.; 2012b.
Wu YL, Li J, Kim HJ, Liu X, Liu WY, Akhgar A, Bowen MA, Spitz S, Jiang XR, Roskos L, White W. A novel antibody dependent cellular cytotoxicity (ADCC)-based reporter neutralizing antibody (NAb) assay in support of benralizumab clinical development. AAPS J. 2015;17(6):1417–26.
Yang H, Kim HJ, Zhang L, Schenerman M, Strouse R, Jiang XR. Implementation of parallelism testing for four-parameter logistic model in bioassays. PDA J Pharm Sci Tech. 2012;66:262–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 American Association of Pharmaceutical Scientists
About this chapter

Cite this chapter
Jiang, XR., Mire-Sluis, A. (2018). Biological Activity Assays for Antibody Therapeutics. In: Gutka, H., Yang, H., Kakar, S. (eds) Biosimilars. AAPS Advances in the Pharmaceutical Sciences Series, vol 34. Springer, Cham. https://doi.org/10.1007/978-3-319-99680-6_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-99680-6_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-99679-0
Online ISBN: 978-3-319-99680-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)