
Abstract
The differential impact of distinct antiestrogens (AEs) is the result of varying structural perturbations they confer to estrogen receptors (ERs) when these small-molecule synthetic compounds compete with endogenous hormones, such as 17β-estradiol. These structural changes translate to altered ability of ERs to conscript cofactors and consequently alter the transcription of their target genes. AEs, depending on the mechanism of action, are classified as either selective estrogen receptor modulators (SERMs), which display tamoxifen-like partial agonism, or as selective estrogen receptor downregulators (SERDs) that confer structurally induced posttranslational modifications (PTMs) that destine these receptors for proteosomal degradation. The conformational plasticity of the ER helix 12 (H12) and how its dynamics and conformational sampling is altered by different AEs are crucial to cofactor recruitment and selectivity, translating to varying degrees of receptor modulation and downstream functional effects. Dissecting these conformational state fluctuations within the context of variable cofactor profiles in different tissues, PTM induction, and emergence of hormonal treatment-related resistance mutations in ERs could lead to improved design of novel therapeutic molecules for breast cancer.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
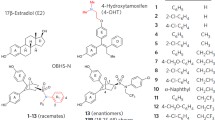
Estrogen receptors (ERs) are ligand-dependent transcription factors regulated by the main circulating estrogen hormone, 17β-estradiol (E2), which is normally produced by the ovaries or via alternative metabolic pathway starting with precursor hormones, such as testosterone [1]. Associated metabolites of E2, estriol and estrone, are also estrogen agonists but generally weaker than E2, and some have been found to have tissue-specific roles (Fig. 1a) [2]. These receptors regulate the function of the female reproductive system, control bone density maintenance, and have protective roles on the central nervous and cardiovascular systems. The effect of E2 on target tissues and organs is mediated by two distinct receptors, ERα (NR3A1) and ERβ (NR3A2), which are encoded by distinct genes [3, 4]. ERs have been implicated in pathological conditions ranging from breast and uterine cancers to cardiovascular and bone disease [5, 6]. Small synthetic molecules, such as antiestrogen steroid or steroid mimics that are designed to block ERs, are used to treat breast cancer (Fig. 1b–e). Several antiestrogens demonstrate tissue-specific activity, such as selective estrogen receptor modulators (SERMs ), tamoxifen and raloxifene (Fig. 1b, c).

Estrogen receptor agonists and antagonists. (a) Most abundant circulating estrogens, estrone, 17β-estradiol, and estriol. (b) Tamoxifen, its active metabolite, 4-hydroxytamoxifen, and SERMs derived from tamoxifen. (c) SERM antiestrogens with a steroid-like backbone and a tertiary amine side chain. (d) Pure antiestrogens with long side chains attached to a steroid-like scaffold. (e) SERDs with steroid-like scaffolds and an acrylic acid functional group
Structural studies have illuminated ligand-binding induced conformational reorganization of ER ligand-binding domain (LBD ) that leads to stabilization of the ER dimer, thereby promoting interaction with coregulator proteins [7, 8]. Since coregulator proteins have cell-specific expression, estrogens have distinct cellular effects. In general, ERα is the principal receptor mediating E2 signaling in the mammary gland, skeletal muscle, uterus, adipose tissue, and pituitary gland, while ERβ plays a less dominant role in these tissues. In contrast, ERβ is found to be dominant in the central nervous and cardiovascular systems, as well as in the lung, ovary, and prostate gland [9,10,11]. Understanding the structural properties of ER and the molecular mechanisms underlying ligand-dependent conformational changes has led to the development of more selective ER ligands for more effective antiestrogen therapy. Antiestrogens, in a simplistic binary conformational description (i.e., active vs. inactive receptor), are thought to occupy the ligand-binding pocket, thereby blocking E2 access and locking ER into inactive conformations not conducive to coactivator recruitment. However, current evidence suggests an antagonist-specific continuum of conformational states in ERs that allow exposure of unique surfaces for coregulator recruitment [12, 13]. Furthermore the cell type-specific profiles of coregulators also dictate the transcriptional activities of ERs bound to antagonists [14].
Detailed ER domain structural analysis has proven critical to our understanding of receptor function [15]. X-ray crystallography studies provide structural snapshots that have revealed mechanisms of E2-ER interaction thereby providing invaluable clues to future drug design targeting the estrogen receptor. This chapter will focus on how the structural perturbations in estrogen receptors induced as a result of interaction with agonists and antagonists, posttranslational modifications (PTMs ), and endocrine treatment-induced resistance mutations affect cofactor recruitment, transcription of target genes, and antiestrogenicity.
2 Structural Organization of ERs
2.1 Architecture and Sequence Homology of ER Subtypes
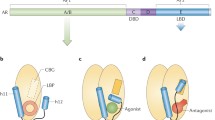
Similar to other transcription factors in the nuclear hormone receptor (NHR) family, ERs have distinct domains with structural and functional roles (Fig. 2) [16]. ERα and ERβ are encoded by distinct genes with varying expression levels in different tissues. Full-length ERα is a 66-kDa protein containing 595 amino acids [17], whereas ERβ is slightly smaller at 60 kDa, spanning 530 amino acids [18].

Schematic of the ER architecture and details of the structural regions. A/B domain at the N-terminus contains activation function-1 (AF-1), the central C region is the DNA-binding domain (DBD ), the hinge D contains the nuclear localization signal (NLS), region E is the ligand-binding domain and overlaps with the AF-2, and finally, domain F is the C-terminus of the AF-2 and contains the conformationally flexible helix 12 (H12). Percent homology of ERβ with respect to ERα are reported
Typically, there are six functional regions [A–E (and in some receptors such as ERα, an F region as well)] in NHRs with significant level of sequence homology. Transcriptional activation is facilitated by two activation functions (AF) within the ERs, namely, the constitutively active AF-1 at the NH2 terminus (A/B region, the least conserved between the ERs) and the ligand-dependent AF-2 at the COOH-terminal region (E region) that overlaps with the ligand-binding domain (LBD ). The two ER subtypes share an overall sequence identity of 47%, which is primarily driven by high sequence identity in the central DNA-binding domain (DBD ; C domain) and ligand-binding domain (LBD; E domain) with 94% and 59% identity between the receptor isotypes, respectively. Region D is considered a flexible hinge that also contains the nuclear localization signal (36% identity) [19]. The highly conserved DBD is responsible for DNA binding and recognition, while the LBD located at the COOH-terminal region is the site for small-molecule ligand binding. Both subtypes show high affinity for E2, which consequently stimulate transcription of an ER responsive gene containing an estrogen-responsive element (ERE) [20].
2.2 ER Ligand-Binding Domain Structure and the Helix 12 Conformational Switch
Like the LBDs of all NHRs, the ER LBDs form three-layered antiparallel α-helical folds [21]. ERα LBD has 12 helices, where the central core is formed by helices H5/H6, H9, and H10 sandwiched between helical layers, L1 (H1–4 and H7) and L2 (H8 and H11), which creates a ligand-binding pocket associated with helices H3, H6, H8, H11, H12, and the hairpin S1/S2 [22]. The dynamically mobile H12 and small two-stranded antiparallel β-sheet flank the major triple layer scaffold [21, 22]. ERβ closely resembles ERα as both have an unstructured F region. However, the ERβ COOH terminus has only an extremely short extended F region. While the F domain of ERα appears to have a role on transcriptional activity modulation, dimerization, receptor stabilization, and coactivator recruitment, the analogous role in ERβ remains unclear [23,24,25].
The LBDs of ERα and ERβ display considerable structural similarities, with the ligand-binding pocket of ERβ differing only at two residue positions with respect to ERα. Amino acid residues outside the binding cleft influence the size and shape of the respective ER pockets, which explains the subtype-selective binding of certain ligands, as exemplified by the ERβ-specific agonist, diarylpropionitrile [26]. Subtype-selective agonists and antagonists are invaluable for dissecting the biological effects specific to ERα and ERβ, which could corroborate findings from ER-knockout animal models.
Dimerization is crucial to ER function as amino acid substitutions that interfere with dimer formation abrogate receptor transcriptional activity [27]. The dimerization domain of ERα is created predominantly by helix 11, with some contribution from the DBD , amino terminal ends, and residues from H8 and loop H9/H10 of each monomer [28]. The ERα dimer binds ligands via hydrogen bond interactions and hydrophobic contacts with nonpolar ligands in a hydrophobic groove formed by helices, H3, H4, H5, and H12 [29]. Charged residues, namely, E353, R394, H524 and E260, R301, H430 in human ERα and Rat ERβ, respectively, stabilize the binding of agonist and antagonist by interacting with the hydroxyl groups of the estrogenic steroidal backbone.
The ligand-dependent transcriptional activation function-2 (AF-2) of ER is a conformationally dynamic region of the LBD that contains the conformational switch, helix 12 (H12). Depending on the class of ligand bound to ER, H12 is oriented differently with respect to the rest of the LBD (Fig. 3). Binding of an agonist, 17β-estradiol (E2), positions H12 over the ligand-binding cavity to generate a competent AF-2 interaction surface for coactivator docking, which is essential for transcriptional activation (Fig. 3a, b). Specifically, this opens up a new surface consisting of D538, L539, E542, and M543 to facilitate the interaction with a coactivator [22, 30, 31]. In this H12 conformation, the AF2 surface is conducive to recruitment of helical segments with LXXLL motif, where L and X are leucine and any residue, respectively—a motif found in many coactivators including the p160 steroid receptor coactivators (SRC) [30, 32]. In addition, the 17-OH group in E2 interacts with H524, which is forced to form an H-bond with the peptidic carbonyl group of E419 in loop 6–7. This facilitates the salt bridge formation between E339 from H3 and E419 from H7 to form a salt bridge network with K531 from H11 that favors the agonist orientation of H12 (Fig. 4a) [22].

ERα bound to agonists or antagonists with or without coactivator peptide. (a) ERα LBD bound to estradiol (E2) (PDB ID 1ERE). (b) ERα LBD bound to E2 and SRC-2 NR box 3 (PDB ID 1GWR). (c) ERα LBD bound to 4-hydroxytamoxifen (4-OHT ) antagonist (PDB ID 3ERT). (d) ERα LBD bound to SERM antagonist, raloxifene (PDB ID 1ERR)

(a) ERα LBD bound to E2 and SRC-2 NR box 3 reveals two salt bridges: K531-E419 and K531-E339 (PDB ID 1GWR). (b) When bound to the 4-OHT antagonist (PDB ID 3ERT), the salt bridges are disrupted
In contrast, binding of an antagonist, such as 4-hydroxytamoxifen (4-OHT ), disrupts the formation of the salt bridge network (Fig. 4b), thereby conferring increased conformational plasticity to H12 and ability to adopt a conformation that occludes the AF-2 groove, which physically blocks coactivator binding (Fig. 3c, d). This physical occlusion is made possible by an internal sequence in H12 that mimics an LXXLL motif, which enables a part of H12 to bind the coactivator groove [30]. A more compelling alternative explanation is that H12 contains an extended corepressor box sequence that binds the AF-1 surface, thereby preventing or hampering corepressor interaction [33, 34]. The latter explains why deletion of H12 confers strong enhancement of ER interaction with corepressors, such as NCoR and SMRT [35, 36]. Even though the importance of NR corepressors to ER signaling remains unclear, studies show that both agonist- and antagonist-bound ERs are capable of recruiting other proteins that repress ER activity [37]. Molecular dynamics (MD) simulation of 4-hydroxytamoxifen (4-OHT )-bound ERα demonstrates structural flexibility of H12, which fluctuates from an initial antagonist position to structurally distinct continuum of H12 positions between an agonist and antagonist conformation, explaining the mixed agonist-antagonist effects of 4-OHT [38].
2.3 ER DNA-Binding Domain Structure and Response Element Recognition
The centrally positioned DNA-binding domains of the ERs are highly conserved and interact with identical DNA sequences. Crystal structures of ERα DBD with or without DNA reveal a topology characterized by two zinc finger-like motifs consisting of four cysteine residues that each coordinate with Zn2+ in a tetrahedral geometry (Fig. 5) [39,40,41,42]. Amino acid residues in the D box contribute to ER dimerization and discriminate half site spacing, while residues in the P box are involved in estrogen response element (ERE) recognition. Specifically, P box residues E203, G204, and A207 determine DNA-binding specificity and sequence discrimination and are critical to ERE binding. EREs, which are located at various positions from the transcription start site and/or within a gene locus, are variations of the palindromic sequence, 5′-GGTCAnnnTGACC-3′, where n is any nucleotide acting as a spacer [43, 44].

The DNA-binding domain (DBD ) of estrogen receptor α (ERα) dimerbound to the consensus sequence of the estrogen response element (ERE), GGTCAnnnTGACC, where n are nonspecific bases acting as spacer (PDB ID 1HCQ). The first zinc-finger module called P box determine DNA-binding specificity, while the second zinc module termed D box is involved in half-site spacing discrimination
Gene expression modulated by the binding of E2-complexed ER (E2-ER) to EREs relies on a signaling pathway described as “ERE-dependent” [45,46,47,48,49]. Meanwhile, regulation of target gene expression that is mediated by transcription factor interaction with E2-ER, such as activation protein (AP) 1 and stimulatory protein (SP) 1 bound to associated regulatory elements on DNA, is classified as “ERE-independent” and employs a signaling pathway mechanism that remains unclear [45, 46, 49, 50]. ER could interact with transcription factors directly or indirectly through coregulatory proteins using interaction surfaces that include the DBD , while transcription control is conferred by combined effects transmitted through the NH2 and COOH termini.
2.4 The Intrinsically Disordered ER NH2 Terminus
The amino terminus (domain A/B in Fig. 2) encompasses the AF-1 region and is highly variable among members of the nuclear hormone receptor family [51]. In yeast and chicken cells, ERα AF-1 functions independently of the AF-2 in a ligand- and promoter-independent manner, but decoupling of AF-1 from AF-2 in mammalian cells resulted in a dysfunctional AF-1 in terms of influencing transcription [52,53,54,55]. Therefore, ERα AF-1 function depends on three factors, namely, cell type, ligand agonism, and structural integrity of the LBD . This is supported by further studies that demonstrate full activity of ER only when AF-1 and AF-2 are functionally integrated [54, 56,57,58].
Due to the intrinsically disordered nature of the ER AF-1, the underlying biochemical and structural mechanism of AF-1 action remains elusive. Interestingly, this disordered nature leads to the formation of a large ensemble of rapidly and reversibly interconverting conformational states [51, 59,60,61]. Inter-domain allosteric cooperativity, protein interaction, and posttranslational modifications (i.e., phosphorylation) control these conformational changes. For instance, interaction of the TATA box-binding protein upon interacting with the NH2 terminus of ERα induces formation of an ordered structure [61]. Meanwhile, the S118 phosphorylation of ERα complexed with either E2 or tamoxifen via growth factor signaling led to Pin1 recruitment, which isomerizes the S118-P199 bond from cis to trans and promotes a conformational change that favors ligand-independent and agonist-inducible ERα activity [62]. Protein interactions with ERα are important for inducing conformational changes that stabilize interaction with coregulatory proteins , which translates to effective transcription [51]. In contrast, ERβ NH2 terminus is devoid of AF-1 [48, 58, 63,64,65], does not interact with the COOH terminus [58], and impairs the ERβ-ERE interactions [66].
3 Structure-Activity Relationships in Antiestrogen Therapy
3.1 Selective ER Modulators and Selective ER Downregulators
Selective ER modulators (SERMs ) such as tamoxifen , raloxifene, and analogues (Fig. 1b, c) are ER ligands that display gene- and/or tissue-specific agonist/antagonist activity. The first clinically approved SERM, tamoxifen, is the standard therapeutic regimen for all stages of breast cancer and has benefitted 70% of women with ERα-positive breast cancer [67, 68]. Aside from the antagonist effects of tamoxifen that inhibit breast cancer proliferation, the drug has desirable agonist effects on bone and lipid profiles [69,70,71,72]. However, tamoxifen and its active metabolite, 4-hydroxytamoxifen (OHT ) has an estrogenic effect in the uterus of mice and rat models, which translates to higher risk of endometrial cancer development during the course of treatment [73, 74]. Moreover, the emergence of tamoxifen resistance without loss of ERα expression has been observed in primary tumors in majority of metastatic cancer patients [75, 76], although remissions are observed after tamoxifen withdrawal or altered treatment regimen, suggesting ongoing ER signaling activity in some tamoxifen-resistant tumors [77, 78].
To hamper the unwanted side effects while concomitantly improving the efficacy of tamoxifen, synthetic analogues (Fig. 1b) were designed, such as the halogenic/pyrrolidino derivatives, toremifene and idoxifene, and the secondary amine variant of 4-OHT , endoxifen [67]. Unfortunately, these tamoxifen analogues did not demonstrate improved efficacy or prevent drug resistance [67, 77, 79]. The benzothiophene SERM derivative, raloxifene (Fig. 1c), retained 76% of tamoxifen efficacy while reducing endometrial cancer incidence but is ineffective against tamoxifen-resistant breast cancer cells [67, 77, 80]. Meanwhile, the raloxifene analogue, arzoxifene, failed to be on par with tamoxifen efficacy for metastatic breast cancer in a phase III clinical trial [81] despite being more potent than tamoxifen and 4-OHT on inhibiting human mammary carcinoma cell proliferation [82, 83]. Relative to tamoxifen and raloxifene, a structural analogue bazedoxifene is more effective than other SERMs at inhibiting gene expression in MCF-7 cells and hampering the growth of tamoxifen-resistant xenograft [84].
Steroidal compounds with long side chains, such as ICI 164,384, ICI 182,780 (fulvestrant), and RU 58668 (Fig. 1d), were developed to minimize partial agonist activity. These drugs were initially referred to as pure antiestrogens (AEs) due to their lack of partial agonist effects in breast and endometrial cell lines [85,86,87,88] but were later designated as selective ER downregulators (SERDs ) as they promote ERα degradation via the ubiquitin-proteasome pathway in ERα-positive breast cancer cells [89,90,91,92]. The pure AE character of fulvestrant did not confer an advantage over tamoxifen against advanced or metastatic breast cancer [79, 93], which could be attributed to its poor pharmacokinetic properties. This limitation is circumvented by doubling the intramuscular injection dosage to 500 mg, which increased patient survival rate [94,95,96]. In comparison, certain SERM derivatives of tamoxifen, such as GW7604, GDC-0810, and AZD9496, demonstrate SERD ability to induce ERα degradation with similar efficacy as fulvestrant but with improved oral bioavailability [78, 97,98,99].
In aggregate, antiestrogens demonstrate varying SERD activity, ranging from drugs that lack ERα downregulating capacity, such as tamoxifen, to SERM analogues with disparate levels of ERα reduction ability (e.g., raloxifene, bazedoxifene, GDC-0810, and GW7604). The strongest SERD activity is associated with pure AEs with long side chains, such as ICI 164,384, fulvestrant, and RU 58668.
3.2 Molecular Rationale of Antiestrogen Effects
AEs bind to ERα LBD akin to estradiol—with bulky side chains attached at steroid core positions 7α or 11β conferring antiestrogenicity by positioning these substituents between H3 and H11 of the binding cavity. Presence of bulky functional groups of different length and size results in structural rearrangements that can cause varying levels of steric hindrance to the positioning of H12 over the ligand-binding pocket.
Tamoxifen, raloxifene, and synthetic SERM analogues (Fig. 1b, c) contain alkylaminoethoxy side chains with varying tertiary amine groups. The dimethylamino group of tamoxifen or the piperidyl group of raloxifene sterically hinders the positioning of H12 to the coactivator-binding site. In an unobstructed state, H12 will dock to the coactivator groove via its hydrophobic residues, L540, M543, and L544, in a manner similar to the LXXLL motif in coactivators [22, 30, 100]. Synthetic analogues and ER mutants were made to validate this structural observation. For instance, a raloxifene derivative with a nitrogen-to-carbon replacement of a crucial compound side chain abrogated the drug’s antagonist activity [101]. Furthermore, substitution of the crucial D351 residue to glutamic acid altered the effect of raloxifene from a pure antagonist to a tamoxifen-like antagonist in HepG2 cells. A hydrogen bond binds the tertiary amine of raloxifene to D351 in ERα LBD ; the mutation to E351 altered this crucial interaction [22, 102]. In transfected MDA-MB-231 cells, a similar mutation D351G abolished tamoxifen-induced expression of TGFA—an estrogen target gene [103]. In addition, a D351A substitution rendered ERα inactive on a reporter gene in tamoxifen-induced HepG2 cells [102], corroborating the importance of D351 in mediating the partial agonist activity of SERMs .
Relative to SERMs , pure AEs such as fulvestrant and ICI 164,384 (Fig. 1d) have longer side chains. A crystal structure of the latter with rat ERβ shows position 7α protruding out of the ligand-binding cleft reminiscent of SERM side chains but bends at carbon 5 by 90°, thereby positioning the rest of the chain onto the coactivator-binding surface [104]. Side chains L261, M264, I265, and L286 in the coactivator-binding surface of rat ERβ form hydrophobic contacts with the terminal n-butyl group of ICI 164,384, which displaces H12 from the same position in crystal structures of 4-OHT and raloxifene-bound ERα (Fig. 3c, d). The long side chain sterically clashes with H12 residues L540 and M543 in the agonist conformation and with L536 and L540 when H12 is positioned in the coactivator-binding surface. Substitution of the aforementioned residues to alanine increased the pure AE-induced ERα transcriptional activity [105,106,107,108].
In contrast to ICI 164,384, fulvestrant antiestrogenicity is not affected by D351 mutations, but introduction of tertiary amine functional group in analogues ZK-253 and ZK-703 improved growth inhibition ability toward mouse xenografts from tamoxifen-resistant and estrogen-sensitive breast cancer cell lines [109]. However, these studies did not address whether direct interaction with pure AEs is crucial for enhanced activity.
Aside from amine functional groups, the effect of chain length was also investigated. To reveal optimal chain length for pure antiestrogenicity, derivatives of ICI 164,384 with variable side chain lengths were synthesized. Side chain lengths consisting of 15–19 atoms display optimal antiestrogenicity, while shorter side chains (13–14 carbon atoms) show agonist or SERM-like activity in reporter assays in HepG2 cells transiently transfected with ERα. These results suggest that longer chains are necessary for pure AEs to reach the coactivator-binding surface. Moreover, hydrophobicity and presence of terminal pentafluoropentyl group are crucial factors for pure AE activity, as supported by a study that shows higher potency and efficacy of fulvestrant relative to ICI 164,384 for growth inhibition in in cellula and in vivo human breast cancer models [88].
Partial or full SERD activity has been observed upon changing the shorter side chains of SERM analogues. For instance, the derivative bazedoxifene differs from raloxifene by having a larger heterocyclic amine ring (Fig. 1c) conferring enhanced steric clash with H12. Furthermore, GW5638 (Etacstil), the prodrug of the active metabolite GW7604 (Fig. 1e), is a tamoxifen derivative where the dimethylaminoethyl group is substituted with an acrylic acid side chain. In its protonated state, the carboxylate group in GW5638 form hydrogen bonds with the peptidic backbone of H12 and E351, inducing a conformation of H12 where the side chain of hydrophobic residues L536, L539, L540, and M543 are pointing toward the aqueous environment, which effectively increases the hydrophobic surface area of H12 relative to 4-OHT -bound ERα while concomitantly maintaining interaction in the coactivator-binding surface [110]. Therefore, pure antiestrogenicity positively correlates with hydrophobic surface area of H12 and is independent of H12 positioning in crystal structures. However, the effect of these structural perturbations on protein-protein interactions and overall ERα stability is not yet clear.
3.3 Effect of AE Binding on Cofactor Recruitment and Gene Transcription
The AF1 and AF2 activation functions at the N- and C-termini, respectively, are utilized by ERs to conscript a large number of cofactors in the presence of agonists. Such cofactors include chromatin remodeling complexes, histone acetyltransferases (HAT ), methyltransferases (HMTs ) and deacetylases (HDACs ), and transcriptional machinery components [111]. The altered recruitment of cofactors to ERα is conformationally induced by AEs that modify the protein surface available for interaction [84]. HATs such as SRC1–3 (NCOA1–3) and CMP/p300 and HMTs including CARM1 and PRMT1 are some of the coactivators that interact directly with the AF2 of E2-ERα or perturb it allosterically [112, 113]. Tamoxifen, but not raloxifene, selectively recruit SRC-1 to promoter genes in Ishikawa and ECC-1 cell lines. Repression of SRC-1 in Ishikawa cells abrogates the partial agonist activity of tamoxifen on target genes [14]. In contrast, when SRC-1 is overexpressed in MCF-7 cells, the behavior of tamoxifen switches from agonist to antagonist suggesting that difference of SRC-1 expression level in breast and uterine cells explains the tissue-specific tamoxifen effects on transcription [14]. Analogously, SRC-2 and p300 overexpression in HeLa cells transfected with ERα amplified the partial agonist activity of tamoxifen but only have moderate and negligible effects in the presence of raloxifene and fulvestrant, respectively [36]. These results suggest that presence of coactivators may contribute to cell- and gene-specific partial agonist activity of SERMs . In addition, 11% of breast tumors show increased SRC-3 expression and is associated with unfavorable prognosis and tumor phenotype, which is explained by the impact of SRC-3 on the cell cycle regulation of both ERα+ and ERα− tumors [114].
The activity and ligand-independent AF-1 function of ERα is linked to the partial agonist activity of tamoxifen and, to some degree, of raloxifene in a cell- and promoter-specific manner [56, 115, 116]. Case in point is the agonist effect of tamoxifen in HEC1 cells that is dependent on the AF1 of ERα [65]. In addition, Zwart and coworkers swapped the AF1 domain of ERα with that of ERβ and consequently abolished tamoxifen-induced transcriptional activity in U2OS cells, showing that the AF1 region is crucial to the partial agonist activity of tamoxifen . This result is corroborated by studies that show the ability of ERα, but not of ERβ, to conscript SRC-1 via the AF1 region [117, 118].
ERα recruits corepressors NCOR1 and NCOR2 (SMRT) in the presence of tamoxifen in MCF-7 cells resulting in the repression of estrogen target genes. Increase of ER target gene expression is observed in the same cells in the presence of tamoxifen after siRNA knockdown of the aforementioned corepressors. Analogously, recruitment of corepressors is absent on genes upregulated by tamoxifen in Ishikawa cells [14, 119, 120]. Moreover, SMRT2 overexpression in HepG2 cells inhibits partial agonist activity of tamoxifen [121]. In comparison to SERMs , ERα bound to fulvestrant is more efficient than raloxifene or tamoxifen at recruiting NCOR1 C-terminal fragment in ChIP experiments in HeLa cells [36]. However, the difference in corepressor recruitment mechanism between SERMs and SERDs remains elusive. It only became possible for raloxifene-bound ERα to co-crystallize with a corepressor NR sequence after H12 deletion, where the peptide occupies the AF2 surface subtended by H3 and H5, and with the raloxifene side chain packed against the peptide N-terminus. Further studies are needed to confirm whether the differential H12 conformation between SERM- and SERD-bound ERα increases corepressor recruitment in the presence of SERDs.
3.4 Impact of ERα Posttranslational Modifications to Pure Antiestrogenicity
Cofactor recruitment is likely modulated by ER posttranslational modifications (PTMs ). Mass spectrometry has become an invaluable tool for the identification of acetylation, methylation, and phosphorylation sites in ERα. A study shows that phosphorylation of S104, S106, and S118 in the AF1 region and of S305 in the AF2 may be linked with tamoxifen resistance [122]. Presence of pure AEs induce the phosphorylation of the same serine residues on the AF-1 region, but the link of these PTMs to SERD transcriptional downregulation remains unclear [123, 124]. MCF-7 breast cancer cells with dephosphorylated Y537 displayed increased sensitization to SERMs and fulvestrant [125]. Sensitization of breast cancer cells to AEs may also be induced by other PTMs on ERα, such as acetylation, SUMOylation, ubiquitination, and methylation [122, 126].
SERDs induce the degradation of ERα in breast and uterine cancer cell lines. Presence of 4-OHT increases ERα expression [92, 106, 127], with some level of decrease in the presence of endoxifen, raloxifene, and bazedoxifene and substantial decrease in the presence of pure AE, such as fulvestrant [84]. Moreover, ERα ubiquitination is doubled in the presence of fulvestrant [92].
ERα turnover differs in the presence of AEs and E2. For instance, α-amanitin transcriptional inhibition prevents E2-induced degradation of ERα, but not by fulvestrant [128]. Similarly, partial inhibition of ERα degradation by E2 but not by pure AEs is afforded by cycloheximide or kinase inhibitor treatment [129, 130]. In spite of the aforementioned differences in degradation mechanisms between E2 and SERDs , the Neddylation pathway seems to be important for both E2- and pure AE-induced turnover [131].
Overexpression of ERα to saturate the degradation process has no effect on the capacity of SERDs to act as AEs in MCF-7 cells [132]. Moreover, the steady-state level of ERα unexpectedly increased in the presence of fulvestrant in HepG2 cells, but this ligand still functioned as an inverse agonist while tamoxifen has partial agonist activity [106, 127] suggesting an alternative mechanism to afford enhanced efficacy of pure AEs for inhibiting ERα activity in HepG2 cells. SUMOylation of ERα is strongly induced by pure AEs in MCF-7 breast cancer cells, HEK293 and HepG2 cells, and abolishing SUMOylation attenuates transcription in the presence of pure AEs, with no effect on the corresponding activity induced by E2 or tamoxifen, suggesting that SUMOylation contributes to pure antiestrogenicity [127]. Interestingly, SUMOylation activity peaked at 15–19 carbon atom chain length and decreased when chain length is >22, which correlates with inverse agonist activity in HepG2 cells and with the ability of the AE side chain to dock at the coactivator-binding cleft. Furthermore, the SERM raloxifene also induce SUMOylation to a lesser extent, which positively correlates with its ability to suppress basal transcription activity in HepG2 cells [127]. Possibly, differential SUMOylation could explain the varying SERM effects in different tissues.
3.5 Effect of ER Mutations on AE Action
Emergence of endocrine treatment resistance remains a challenging issue in treatment of patients with ER+ breast cancers. After developing resistance, majority of tumors still express ERα, suggesting the role of ERα in tumor growth. Coactivator overexpression inducing estrogen-dependent transcription is a potential mechanism of desensitization, as is the signaling pathway activation that controls the activity of ERα and/or its associated coactivators [133]. A recent review has highlighted the role of ERα mutations as an additional hormonal treatment resistance mechanism [134], as initially hinted by a constitutively active ERα Y537N mutant isolated from metastatic breast cancer cells [135]. Majority of hormone therapy-resistant tumors contain gain-of-function mutations, such as E380Q, L536Q/R, Y537S/C/N, and D538G that result in ERα activity that is ligand-independent [136,137,138]. These mutants show higher levels of S118 phosphorylation, enhanced recruitment of SRC1–3, increased ligand-independent tumor growth, and/or S118 phosphorylation [138,139,140].
Crystal structures of ERα mutants Y537S and D438G in the apo state adopt an agonist-like conformation [138,139,140,141]. As a result, affinity of binding of E2 and 4-OHT to said mutants is tenfold weaker relative to wild-type ERα, and higher doses of 4-OHT and fulvestrant are required to affect levels of activity inhibition in mutant ERα similar to wild type. This could lead to clinical resistance to AE therapy when required concentrations for activity suppression of ER mutants are not reached [136, 138, 140]. Furthermore, the structural changes relative to wild type in 4-OHT-bound ERα mutant LBDs may result in different effects on ER target genes at saturation [139].
4 Epilogue
ERα and ERβ have similar structures but display distinct as well as overlapping regulatory potentials in cells in a tissue-specific manner. Antiestrogens have diverse conformations and structures that modulate AF-1 and/or AF-2 activity that translate to varying levels of antiestrogenicity in breast cancer cells. The conformational dynamics of AE binding to ERs has several downstream consequences on posttranslational modifications and ER degradation mechanisms and needs to be explored further. Hormone therapy resistance is caused by the emergence of ER mutants that need to be characterized for their individual responses to various clinically available AEs, which will guide the design of future drugs for breast cancer.
References
Gruber CJ, Tschugguel W, Schneeberger C, Huber JC (2002) Production and actions of estrogens. N Engl J Med 346(5):340–352. https://doi.org/10.1056/NEJMra000471
Gruber DM, Huber JC (1999) Conjugated estrogens--the natural SERMs. Gynecol Endocrinol 13(Suppl 6):9–12
Green S, Walter P, Kumar V, Krust A, Bornert JM, Argos P, Chambon P (1986) Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature 320(6058):134–139. https://doi.org/10.1038/320134a0
Leygue E, Dotzlaw H, Lu B, Glor C, Watson PH, Murphy LC (1998) Estrogen receptor beta: mine is longer than yours? J Clin Endocrinol Metab 83(10):3754–3755. https://doi.org/10.1210/jcem.83.10.5187-1
Deroo BJ, Korach KS (2006) Estrogen receptors and human disease. J Clin Invest 116(3):561–570. https://doi.org/10.1172/JCI27987
Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA (2001) Mechanisms of estrogen action. Physiol Rev 81(4):1535–1565. https://doi.org/10.1152/physrev.2001.81.4.1535
Bai Y, Giguere V (2003) Isoform-selective interactions between estrogen receptors and steroid receptor coactivators promoted by estradiol and ErbB-2 signaling in living cells. Mol Endocrinol 17(4):589–599. https://doi.org/10.1210/me.2002-0351
Mak HY, Hoare S, Henttu PM, Parker MG (1999) Molecular determinants of the estrogen receptor-coactivator interface. Mol Cell Biol 19(5):3895–3903
Harris HA (2007) Estrogen receptor-beta: recent lessons from in vivo studies. Mol Endocrinol 21(1):1–13. https://doi.org/10.1210/me.2005-0459
Hewitt SC, Winuthayanon W, Korach KS (2016) What’s new in estrogen receptor action in the female reproductive tract. J Mol Endocrinol 56(2):R55–R71. https://doi.org/10.1530/JME-15-0254
Hamilton KJ, Arao Y, Korach KS (2014) Estrogen hormone physiology: reproductive findings from estrogen receptor mutant mice. Reprod Biol 14(1):3–8. https://doi.org/10.1016/j.repbio.2013.12.002
Heldring N, Nilsson M, Buehrer B, Treuter E, Gustafsson JA (2004) Identification of tamoxifen-induced coregulator interaction surfaces within the ligand-binding domain of estrogen receptors. Mol Cell Biol 24(8):3445–3459
Paige LA, Christensen DJ, Gron H, Norris JD, Gottlin EB, Padilla KM, Chang CY, Ballas LM, Hamilton PT, McDonnell DP, Fowlkes DM (1999) Estrogen receptor (ER) modulators each induce distinct conformational changes in ER alpha and ER beta. Proc Natl Acad Sci U S A 96(7):3999–4004
Shang Y, Brown M (2002) Molecular determinants for the tissue specificity of SERMs. Science 295(5564):2465–2468. https://doi.org/10.1126/science.1068537
Pike AC (2006) Lessons learnt from structural studies of the oestrogen receptor. Best Pract Res Clin Endocrinol Metab 20(1):1–14. https://doi.org/10.1016/j.beem.2005.09.002
Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P (1987) Functional domains of the human estrogen receptor. Cell 51(6):941–951
Ponglikitmongkol M, Green S, Chambon P (1988) Genomic organization of the human oestrogen receptor gene. EMBO J 7(11):3385–3388
Ogawa S, Inoue S, Watanabe T, Hiroi H, Orimo A, Hosoi T, Ouchi Y, Muramatsu M (1998) The complete primary structure of human estrogen receptor beta (hER beta) and its heterodimerization with ER alpha in vivo and in vitro. Biochem Biophys Res Commun 243(1):122–126. https://doi.org/10.1006/bbrc.1997.7893
Muramatsu M, Inoue S (2000) Estrogen receptors: how do they control reproductive and nonreproductive functions? Biochem Biophys Res Commun 270(1):1–10. https://doi.org/10.1006/bbrc.2000.2214
Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA (1997) Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138(3):863–870. https://doi.org/10.1210/endo.138.3.4979
Pike AC, Brzozowski AM, Hubbard RE (2000) A structural biologist's view of the oestrogen receptor. J Steroid Biochem Mol Biol 74(5):261–268
Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M (1997) Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 389(6652):753–758. https://doi.org/10.1038/39645
Arao Y, Hamilton KJ, Coons LA, Korach KS (2013) Estrogen receptor alpha L543A,L544A mutation changes antagonists to agonists, correlating with the ligand binding domain dimerization associated with DNA binding activity. J Biol Chem 288(29):21105–21116. https://doi.org/10.1074/jbc.M113.463455
Koide A, Zhao C, Naganuma M, Abrams J, Deighton-Collins S, Skafar DF, Koide S (2007) Identification of regions within the F domain of the human estrogen receptor alpha that are important for modulating transactivation and protein-protein interactions. Mol Endocrinol 21(4):829–842. https://doi.org/10.1210/me.2006-0203
Montano MM, Muller V, Trobaugh A, Katzenellenbogen BS (1995) The carboxy-terminal F domain of the human estrogen receptor: role in the transcriptional activity of the receptor and the effectiveness of antiestrogens as estrogen antagonists. Mol Endocrinol 9(7):814–825. https://doi.org/10.1210/mend.9.7.7476965
Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA (2001) Estrogen receptor-beta potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem 44(24):4230–4251
Lees JA, Fawell SE, White R, Parker MG (1990) A 22-amino-acid peptide restores DNA-binding activity to dimerization-defective mutants of the estrogen receptor. Mol Cell Biol 10(10):5529–5531
Tamrazi A, Carlson KE, Daniels JR, Hurth KM, Katzenellenbogen JA (2002) Estrogen receptor dimerization: ligand binding regulates dimer affinity and dimer dissociation rate. Mol Endocrinol 16(12):2706–2719. https://doi.org/10.1210/me.2002-0250
Vajdos FF, Hoth LR, Geoghegan KF, Simons SP, LeMotte PK, Danley DE, Ammirati MJ, Pandit J (2007) The 2.0 A crystal structure of the ERalpha ligand-binding domain complexed with lasofoxifene. Protein Sci 16(5):897–905. https://doi.org/10.1110/ps.062729207
Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL (1998) The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95(7):927–937
Warnmark A, Treuter E, Gustafsson JA, Hubbard RE, Brzozowski AM, Pike AC (2002) Interaction of transcriptional intermediary factor 2 nuclear receptor box peptides with the coactivator binding site of estrogen receptor alpha. J Biol Chem 277(24):21862–21868. https://doi.org/10.1074/jbc.M200764200
Tetel MJ (2009) Nuclear receptor coactivators: essential players for steroid hormone action in the brain and in behaviour. J Neuroendocrinol 21(4):229–237. https://doi.org/10.1111/j.1365-2826.2009.01827.x
Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, Gustafsson JA (2007) Estrogen receptors: how do they signal and what are their targets. Physiol Rev 87(3):905–931. https://doi.org/10.1152/physrev.00026.2006
Heldring N, Pawson T, McDonnell D, Treuter E, Gustafsson JA, Pike AC (2007) Structural insights into corepressor recognition by antagonist-bound estrogen receptors. J Biol Chem 282(14):10449–10455. https://doi.org/10.1074/jbc.M611424200
Huang HJ, Norris JD, McDonnell DP (2002) Identification of a negative regulatory surface within estrogen receptor alpha provides evidence in support of a role for corepressors in regulating cellular responses to agonists and antagonists. Mol Endocrinol 16(8):1778–1792. https://doi.org/10.1210/me.2002-0089
Webb P, Nguyen P, Kushner PJ (2003) Differential SERM effects on corepressor binding dictate ERalpha activity in vivo. J Biol Chem 278(9):6912–6920. https://doi.org/10.1074/jbc.M208501200
Dobrzycka KM, Townson SM, Jiang S, Oesterreich S (2003) Estrogen receptor corepressors -- a role in human breast cancer? Endocr Relat Cancer 10(4):517–536
Chakraborty S, Levenson AS, Biswas PK (2013) Structural insights into Resveratrol’s antagonist and partial agonist actions on estrogen receptor alpha. BMC Struct Biol 13:27. https://doi.org/10.1186/1472-6807-13-27
Schwabe JW, Chapman L, Finch JT, Rhodes D (1993) The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell 75(3):567–578
Schwabe JW, Chapman L, Finch JT, Rhodes D, Neuhaus D (1993) DNA recognition by the oestrogen receptor: from solution to the crystal. Structure 1(3):187–204
Schwabe JW, Chapman L, Rhodes D (1995) The oestrogen receptor recognizes an imperfectly palindromic response element through an alternative side-chain conformation. Structure 3(2):201–213
Schwabe JW, Neuhaus D, Rhodes D (1990) Solution structure of the DNA-binding domain of the oestrogen receptor. Nature 348(6300):458–461. https://doi.org/10.1038/348458a0
Klinge CM (2001) Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res 29(14):2905–2919
Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, Chiu KP, Lipovich L, Barnett DH, Stossi F, Yeo A, George J, Kuznetsov VA, Lee YK, Charn TH, Palanisamy N, Miller LD, Cheung E, Katzenellenbogen BS, Ruan Y, Bourque G, Wei CL, Liu ET (2007) Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet 3(6):e87. https://doi.org/10.1371/journal.pgen.0030087
Bjornstrom L, Sjoberg M (2002) Mutations in the estrogen receptor DNA-binding domain discriminate between the classical mechanism of action and cross-talk with Stat5b and activating protein 1 (AP-1). J Biol Chem 277(50):48479–48483. https://doi.org/10.1074/jbc.C200570200
Cheung E, Acevedo ML, Cole PA, Kraus WL (2005) Altered pharmacology and distinct coactivator usage for estrogen receptor-dependent transcription through activating protein-1. Proc Natl Acad Sci U S A 102(3):559–564. https://doi.org/10.1073/pnas.0407113102
Hall JM, Couse JF, Korach KS (2001) The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem 276(40):36869–36872. https://doi.org/10.1074/jbc.R100029200
Huang J, Li X, Hilf R, Bambara RA, Muyan M (2005) Molecular basis of therapeutic strategies for breast cancer. Curr Drug Targets Immune Endocr Metabol Disord 5(4):379–396
Safe S (2001) Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm 62:231–252
Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P (2000) Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol 74(5):311–317
Kumar R, Thompson EB (2003) Transactivation functions of the N-terminal domains of nuclear hormone receptors: protein folding and coactivator interactions. Mol Endocrinol 17(1):1–10. https://doi.org/10.1210/me.2002-0258
Bocquel MT, Kumar V, Stricker C, Chambon P, Gronemeyer H (1989) The contribution of the N- and C-terminal regions of steroid receptors to activation of transcription is both receptor and cell-specific. Nucleic Acids Res 17(7):2581–2595
Metzger D, Ali S, Bornert JM, Chambon P (1995) Characterization of the amino-terminal transcriptional activation function of the human estrogen receptor in animal and yeast cells. J Biol Chem 270(16):9535–9542
Tasset D, Tora L, Fromental C, Scheer E, Chambon P (1990) Distinct classes of transcriptional activating domains function by different mechanisms. Cell 62(6):1177–1187
Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, Chambon P (1989) The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell 59(3):477–487
Benecke A, Chambon P, Gronemeyer H (2000) Synergy between estrogen receptor alpha activation functions AF1 and AF2 mediated by transcription intermediary factor TIF2. EMBO Rep 1(2):151–157. https://doi.org/10.1038/sj.embor.embor609
Kraus WL, McInerney EM, Katzenellenbogen BS (1995) Ligand-dependent, transcriptionally productive association of the amino- and carboxyl-terminal regions of a steroid hormone nuclear receptor. Proc Natl Acad Sci U S A 92(26):12314–12318
Yi P, Bhagat S, Hilf R, Bambara RA, Muyan M (2002) Differences in the abilities of estrogen receptors to integrate activation functions are critical for subtype-specific transcriptional responses. Mol Endocrinol 16(8):1810–1827. https://doi.org/10.1210/me.2001-0323
Kumar R, Litwack G (2009) Structural and functional relationships of the steroid hormone receptors’ N-terminal transactivation domain. Steroids 74(12):877–883. https://doi.org/10.1016/j.steroids.2009.07.012
Kumar R, Zakharov MN, Khan SH, Miki R, Jang H, Toraldo G, Singh R, Bhasin S, Jasuja R (2011) The dynamic structure of the estrogen receptor. J Amino Acids 2011:812540. https://doi.org/10.4061/2011/812540
Warnmark A, Wikstrom A, Wright AP, Gustafsson JA, Hard T (2001) The N-terminal regions of estrogen receptor alpha and beta are unstructured in vitro and show different TBP binding properties. J Biol Chem 276(49):45939–45944. https://doi.org/10.1074/jbc.M107875200
Rajbhandari P, Finn G, Solodin NM, Singarapu KK, Sahu SC, Markley JL, Kadunc KJ, Ellison-Zelski SJ, Kariagina A, Haslam SZ, Lu KP, Alarid ET (2012) Regulation of estrogen receptor alpha N-terminus conformation and function by peptidyl prolyl isomerase Pin1. Mol Cell Biol 32(2):445–457. https://doi.org/10.1128/MCB.06073-11
Cowley SM, Parker MG (1999) A comparison of transcriptional activation by ER alpha and ER beta. J Steroid Biochem Mol Biol 69(1–6):165–175
Delaunay F, Pettersson K, Tujague M, Gustafsson JA (2000) Functional differences between the amino-terminal domains of estrogen receptors alpha and beta. Mol Pharmacol 58(3):584–590
McInerney EM, Weis KE, Sun J, Mosselman S, Katzenellenbogen BS (1998) Transcription activation by the human estrogen receptor subtype beta (ER beta) studied with ER beta and ER alpha receptor chimeras. Endocrinology 139(11):4513–4522. https://doi.org/10.1210/endo.139.11.6298
Huang J, Li X, Maguire CA, Hilf R, Bambara RA, Muyan M (2005) Binding of estrogen receptor beta to estrogen response element in situ is independent of estradiol and impaired by its amino terminus. Mol Endocrinol 19(11):2696–2712. https://doi.org/10.1210/me.2005-0120
Martinkovich S, Shah D, Planey SL, Arnott JA (2014) Selective estrogen receptor modulators: tissue specificity and clinical utility. Clin Interv Aging 9:1437–1452. https://doi.org/10.2147/CIA.S66690
Jordan C (2002) Historical perspective on hormonal therapy of advanced breast cancer. Clin Ther 24(Suppl A):A3–A16
Love RR, Barden HS, Mazess RB, Epstein S, Chappell RJ (1994) Effect of tamoxifen on lumbar spine bone-mineral density in postmenopausal women after 5 years. Arch Intern Med 154(22):2585–2588. https://doi.org/10.1001/archinte.154.22.2585
Love RR, Wiebe DA, Feyzi JM, Newcomb PA, Chappell RJ (1994) Effects of tamoxifen on cardiovascular risk factors in postmenopausal women after 5 years of treatment. J Natl Cancer Inst 86(20):1534–1539
Turner RT, Wakley GK, Hannon KS, Bell NH (1988) Tamoxifen inhibits osteoclast-mediated resorption of trabecular bone in ovarian hormone-deficient rats. Endocrinology 122(3):1146–1150. https://doi.org/10.1210/endo-122-3-1146
Ward RL, Morgan G, Dalley D, Kelly PJ (1993) Tamoxifen reduces bone turnover and prevents lumbar spine and proximal femoral bone loss in early postmenopausal women. Bone Miner 22(2):87–94
Davies P, Syne JS, Nicholson RI (1979) Effects of estradiol and the antiestrogen tamoxifen on steroid hormone receptor concentration and nuclear ribonucleic acid polymerase activities in rat uteri. Endocrinology 105(6):1336–1342. https://doi.org/10.1210/endo-105-6-1336
Martin L, Middleton E (1978) Prolonged oestrogenic and mitogenic activity of tamoxifen in the ovariectomized mouse. J Endocrinol 78(1):125–129
Jordan VC (2004) Selective estrogen receptor modulation: concept and consequences in cancer. Cancer Cell 5(3):207–213
Musgrove EA, Sutherland RL (2009) Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer 9(9):631–643. https://doi.org/10.1038/nrc2713
Ali S, Buluwela L, Coombes RC (2011) Antiestrogens and their therapeutic applications in breast cancer and other diseases. Annu Rev Med 62:217–232. https://doi.org/10.1146/annurev-med-052209-100305
McDonnell DP, Wardell SE, Norris JD (2015) Oral selective estrogen receptor downregulators (SERDs), a breakthrough endocrine therapy for breast cancer. J Med Chem 58(12):4883–4887. https://doi.org/10.1021/acs.jmedchem.5b00760
Howell A (2006) Pure oestrogen antagonists for the treatment of advanced breast cancer. Endocr Relat Cancer 13(3):689–706. https://doi.org/10.1677/erc.1.00846
Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, Bevers TB, Fehrenbacher L, Pajon ER, Wade JL 3rd, Robidoux A, Margolese RG, James J, Runowicz CD, Ganz PA, Reis SE, McCaskill-Stevens W, Ford LG, Jordan VC, Wolmark N, National Surgical Adjuvant B, Bowel P (2010) Update of the National Surgical Adjuvant Breast and Bowel Project study of tamoxifen and raloxifene (STAR) P-2 trial: preventing breast cancer. Cancer Prev Res (Phila) 3(6):696–706. https://doi.org/10.1158/1940-6207.CAPR-10-0076
Deshmane V, Krishnamurthy S, Melemed AS, Peterson P, Buzdar AU (2007) Phase III double-blind trial of arzoxifene compared with tamoxifen for locally advanced or metastatic breast cancer. J Clin Oncol 25(31):4967–4973. https://doi.org/10.1200/JCO.2006.09.5992
Palkowitz AD, Glasebrook AL, Thrasher KJ, Hauser KL, Short LL, Phillips DL, Muehl BS, Sato M, Shetler PK, Cullinan GJ, Pell TR, Bryant HU (1997) Discovery and synthesis of [6-hydroxy-3-[4-[2-(1-piperidinyl)ethoxy]phenoxy]-2-(4-hydroxyphenyl)]b enzo[b]thiophene: a novel, highly potent, selective estrogen receptor modulator. J Med Chem 40(10):1407–1416. https://doi.org/10.1021/jm970167b
Suh N, Glasebrook AL, Palkowitz AD, Bryant HU, Burris LL, Starling JJ, Pearce HL, Williams C, Peer C, Wang Y, Sporn MB (2001) Arzoxifene, a new selective estrogen receptor modulator for chemoprevention of experimental breast cancer. Cancer Res 61(23):8412–8415
Wardell SE, Nelson ER, Chao CA, McDonnell DP (2013) Bazedoxifene exhibits antiestrogenic activity in animal models of tamoxifen-resistant breast cancer: implications for treatment of advanced disease. Clin Cancer Res 19(9):2420–2431. https://doi.org/10.1158/1078-0432.CCR-12-3771
Barsalou A, Gao W, Anghel SI, Carriere J, Mader S (1998) Estrogen response elements can mediate agonist activity of anti-estrogens in human endometrial Ishikawa cells. J Biol Chem 273(27):17138–17146
Bowler J, Lilley TJ, Pittam JD, Wakeling AE (1989) Novel steroidal pure antiestrogens. Steroids 54(1):71–99
Van de Velde P, Nique F, Bouchoux F, Bremaud J, Hameau MC, Lucas D, Moratille C, Viet S, Philibert D, Teutsch G (1994) RU 58,668, a new pure antiestrogen inducing a regression of human mammary carcinoma implanted in nude mice. J Steroid Biochem Mol Biol 48(2–3):187–196
Wakeling AE, Dukes M, Bowler J (1991) A potent specific pure antiestrogen with clinical potential. Cancer Res 51(15):3867–3873
Dauvois S, Danielian PS, White R, Parker MG (1992) Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc Natl Acad Sci U S A 89(9):4037–4041
El Khissiin A, Leclercq G (1999) Implication of proteasome in estrogen receptor degradation. FEBS Lett 448(1):160–166
Gibson MK, Nemmers LA, Beckman WC Jr, Davis VL, Curtis SW, Korach KS (1991) The mechanism of ICI 164,384 antiestrogenicity involves rapid loss of estrogen receptor in uterine tissue. Endocrinology 129(4):2000–2010. https://doi.org/10.1210/endo-129-4-2000
Wijayaratne AL, McDonnell DP (2001) The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J Biol Chem 276(38):35684–35692. https://doi.org/10.1074/jbc.M101097200
Howell A, Robertson JF, Abram P, Lichinitser MR, Elledge R, Bajetta E, Watanabe T, Morris C, Webster A, Dimery I, Osborne CK (2004) Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: a multinational, double-blind, randomized trial. J Clin Oncol 22(9):1605–1613. https://doi.org/10.1200/JCO.2004.02.112
Di Leo A, Jerusalem G, Petruzelka L, Torres R, Bondarenko IN, Khasanov R, Verhoeven D, Pedrini JL, Smirnova I, Lichinitser MR, Pendergrass K, Garnett S, Lindemann JP, Sapunar F, Martin M (2010) Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor-positive advanced breast cancer. J Clin Oncol 28(30):4594–4600. https://doi.org/10.1200/JCO.2010.28.8415
Di Leo A, Jerusalem G, Petruzelka L, Torres R, Bondarenko IN, Khasanov R, Verhoeven D, Pedrini JL, Smirnova I, Lichinitser MR, Pendergrass K, Malorni L, Garnett S, Rukazenkov Y, Martin M (2014) Final overall survival: fulvestrant 500 mg vs 250 mg in the randomized CONFIRM trial. J Natl Cancer Inst 106(1):djt337. https://doi.org/10.1093/jnci/djt337
Robertson JF, Lindemann J, Garnett S, Anderson E, Nicholson RI, Kuter I, Gee JM (2014) A good drug made better: the fulvestrant dose-response story. Clin Breast Cancer 14(6):381–389. https://doi.org/10.1016/j.clbc.2014.06.005
Bentrem D, Dardes R, Liu H, MacGregor-Schafer J, Zapf J, Jordan V (2001) Molecular mechanism of action at estrogen receptor alpha of a new clinically relevant antiestrogen (GW7604) related to tamoxifen. Endocrinology 142(2):838–846. https://doi.org/10.1210/endo.142.2.7932
Lai A, Kahraman M, Govek S, Nagasawa J, Bonnefous C, Julien J, Douglas K, Sensintaffar J, Lu N, Lee KJ, Aparicio A, Kaufman J, Qian J, Shao G, Prudente R, Moon MJ, Joseph JD, Darimont B, Brigham D, Grillot K, Heyman R, Rix PJ, Hager JH, Smith ND (2015) Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J Med Chem 58(12):4888–4904. https://doi.org/10.1021/acs.jmedchem.5b00054
Wijayaratne AL, Nagel SC, Paige LA, Christensen DJ, Norris JD, Fowlkes DM, McDonnell DP (1999) Comparative analyses of mechanistic differences among antiestrogens. Endocrinology 140(12):5828–5840. https://doi.org/10.1210/endo.140.12.7164
Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson JA, Carlquist M (1999) Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J 18(17):4608–4618. https://doi.org/10.1093/emboj/18.17.4608
Grese TA, Cho S, Finley DR, Godfrey AG, Jones CD, Lugar CW 3rd, Martin MJ, Matsumoto K, Pennington LD, Winter MA, Adrian MD, Cole HW, Magee DE, Phillips DL, Rowley ER, Short LL, Glasebrook AL, Bryant HU (1997) Structure-activity relationships of selective estrogen receptor modulators: modifications to the 2-arylbenzothiophene core of raloxifene. J Med Chem 40(2):146–167. https://doi.org/10.1021/jm9606352
Dayan G, Lupien M, Auger A, Anghel SI, Rocha W, Croisetiere S, Katzenellenbogen JA, Mader S (2006) Tamoxifen and raloxifene differ in their functional interactions with aspartate 351 of estrogen receptor alpha. Mol Pharmacol 70(2):579–588. https://doi.org/10.1124/mol.105.021931
MacGregor Schafer J, Liu H, Bentrem DJ, Zapf JW, Jordan VC (2000) Allosteric silencing of activating function 1 in the 4-hydroxytamoxifen estrogen receptor complex is induced by substituting glycine for aspartate at amino acid 351. Cancer Res 60(18):5097–5105
Pike AC, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, Li YL, Gustafsson JA, Carlquist M (2001) Structural insights into the mode of action of a pure antiestrogen. Structure 9(2):145–153
Arao Y, Hamilton KJ, Ray MK, Scott G, Mishina Y, Korach KS (2011) Estrogen receptor alpha AF-2 mutation results in antagonist reversal and reveals tissue selective function of estrogen receptor modulators. Proc Natl Acad Sci U S A 108(36):14986–14991. https://doi.org/10.1073/pnas.1109180108
Lupien M, Jeyakumar M, Hebert E, Hilmi K, Cotnoir-White D, Loch C, Auger A, Dayan G, Pinard GA, Wurtz JM, Moras D, Katzenellenbogen J, Mader S (2007) Raloxifene and ICI182,780 increase estrogen receptor-alpha association with a nuclear compartment via overlapping sets of hydrophobic amino acids in activation function 2 helix 12. Mol Endocrinol 21(4):797–816. https://doi.org/10.1210/me.2006-0074
Mahfoudi A, Roulet E, Dauvois S, Parker MG, Wahli W (1995) Specific mutations in the estrogen receptor change the properties of antiestrogens to full agonists. Proc Natl Acad Sci U S A 92(10):4206–4210
Norris JD, Fan D, Stallcup MR, McDonnell DP (1998) Enhancement of estrogen receptor transcriptional activity by the coactivator GRIP-1 highlights the role of activation function 2 in determining estrogen receptor pharmacology. J Biol Chem 273(12):6679–6688
Hoffmann J, Bohlmann R, Heinrich N, Hofmeister H, Kroll J, Kunzer H, Lichtner RB, Nishino Y, Parczyk K, Sauer G, Gieschen H, Ulbrich HF, Schneider MR (2004) Characterization of new estrogen receptor destabilizing compounds: effects on estrogen-sensitive and tamoxifen-resistant breast cancer. J Natl Cancer Inst 96(3):210–218
Wu YL, Yang X, Ren Z, McDonnell DP, Norris JD, Willson TM, Greene GL (2005) Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol Cell 18(4):413–424. https://doi.org/10.1016/j.molcel.2005.04.014
Hall JM, McDonnell DP (2005) Coregulators in nuclear estrogen receptor action: from concept to therapeutic targeting. Mol Interv 5(6):343–357. https://doi.org/10.1124/mi.5.6.7
Johnson AB, O’Malley BW (2012) Steroid receptor coactivators 1, 2, and 3: critical regulators of nuclear receptor activity and steroid receptor modulator (SRM)-based cancer therapy. Mol Cell Endocrinol 348(2):430–439. https://doi.org/10.1016/j.mce.2011.04.021
Smith CL, O’Malley BW (2004) Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev 25(1):45–71. https://doi.org/10.1210/er.2003-0023
Burandt E, Jens G, Holst F, Janicke F, Muller V, Quaas A, Choschzick M, Wilczak W, Terracciano L, Simon R, Sauter G, Lebeau A (2013) Prognostic relevance of AIB1 (NCoA3) amplification and overexpression in breast cancer. Breast Cancer Res Treat 137(3):745–753. https://doi.org/10.1007/s10549-013-2406-4
Metivier R, Penot G, Flouriot G, Pakdel F (2001) Synergism between ERalpha transactivation function 1 (AF-1) and AF-2 mediated by steroid receptor coactivator protein-1: requirement for the AF-1 alpha-helical core and for a direct interaction between the N- and C-terminal domains. Mol Endocrinol 15(11):1953–1970. https://doi.org/10.1210/mend.15.11.0727
Tzukerman MT, Esty A, Santiso-Mere D, Danielian P, Parker MG, Stein RB, Pike JW, McDonnell DP (1994) Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. Mol Endocrinol 8(1):21–30. https://doi.org/10.1210/mend.8.1.8152428
Merot Y, Metivier R, Penot G, Manu D, Saligaut C, Gannon F, Pakdel F, Kah O, Flouriot G (2004) The relative contribution exerted by AF-1 and AF-2 transactivation functions in estrogen receptor alpha transcriptional activity depends upon the differentiation stage of the cell. J Biol Chem 279(25):26184–26191. https://doi.org/10.1074/jbc.M402148200
Webb P, Nguyen P, Shinsako J, Anderson C, Feng W, Nguyen MP, Chen D, Huang SM, Subramanian S, McKinerney E, Katzenellenbogen BS, Stallcup MR, Kushner PJ (1998) Estrogen receptor activation function 1 works by binding p160 coactivator proteins. Mol Endocrinol 12(10):1605–1618. https://doi.org/10.1210/mend.12.10.0185
Keeton EK, Brown M (2005) Cell cycle progression stimulated by tamoxifen-bound estrogen receptor-alpha and promoter-specific effects in breast cancer cells deficient in N-CoR and SMRT. Mol Endocrinol 19(6):1543–1554. https://doi.org/10.1210/me.2004-0395
Lavinsky RM, Jepsen K, Heinzel T, Torchia J, Mullen TM, Schiff R, Del-Rio AL, Ricote M, Ngo S, Gemsch J, Hilsenbeck SG, Osborne CK, Glass CK, Rosenfeld MG, Rose DW (1998) Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc Natl Acad Sci U S A 95(6):2920–2925
Smith CL, Nawaz Z, O’Malley BW (1997) Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol 11(6):657–666. https://doi.org/10.1210/mend.11.6.0009
Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L (2011) Cracking the estrogen receptor’s posttranslational code in breast tumors. Endocr Rev 32(5):597–622. https://doi.org/10.1210/er.2010-0016
Ali S, Metzger D, Bornert JM, Chambon P (1993) Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. EMBO J 12(3):1153–1160
Thomas RS, Sarwar N, Phoenix F, Coombes RC, Ali S (2008) Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is important for estrogen receptor-alpha activity. J Mol Endocrinol 40(4):173–184. https://doi.org/10.1677/JME-07-0165
Suresh PS, Ma S, Migliaccio A, Chen G (2014) Protein-tyrosine phosphatase H1 increases breast cancer sensitivity to antiestrogens by dephosphorylating estrogen receptor at Tyr537. Mol Cancer Ther 13(1):230–238. https://doi.org/10.1158/1535-7163.MCT-13-0610
Ascenzi P, Bocedi A, Marino M (2006) Structure-function relationship of estrogen receptor alpha and beta: impact on human health. Mol Asp Med 27(4):299–402. https://doi.org/10.1016/j.mam.2006.07.001
Hilmi K, Hussein N, Mendoza-Sanchez R, El-Ezzy M, Ismail H, Durette C, Bail M, Rozendaal MJ, Bouvier M, Thibault P, Gleason JL, Mader S (2012) Role of SUMOylation in full antiestrogenicity. Mol Cell Biol 32(19):3823–3837. https://doi.org/10.1128/MCB.00290-12
Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F (2003) Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol Cell 11(3):695–707
Borras M, Laios I, el Khissiin A, Seo HS, Lempereur F, Legros N, Leclercq G (1996) Estrogenic and antiestrogenic regulation of the half-life of covalently labeled estrogen receptor in MCF-7 breast cancer cells. J Steroid Biochem Mol Biol 57(3–4):203–213
Marsaud V, Gougelet A, Maillard S, Renoir JM (2003) Various phosphorylation pathways, depending on agonist and antagonist binding to endogenous estrogen receptor alpha (ERalpha), differentially affect ERalpha extractability, proteasome-mediated stability, and transcriptional activity in human breast cancer cells. Mol Endocrinol 17(10):2013–2027. https://doi.org/10.1210/me.2002-0269
Fan M, Bigsby RM, Nephew KP (2003) The NEDD8 pathway is required for proteasome-mediated degradation of human estrogen receptor (ER)-alpha and essential for the antiproliferative activity of ICI 182,780 in ERalpha-positive breast cancer cells. Mol Endocrinol 17(3):356–365. https://doi.org/10.1210/me.2002-0323
Wardell SE, Marks JR, McDonnell DP (2011) The turnover of estrogen receptor alpha by the selective estrogen receptor degrader (SERD) fulvestrant is a saturable process that is not required for antagonist efficacy. Biochem Pharmacol 82(2):122–130. https://doi.org/10.1016/j.bcp.2011.03.031
Nardone A, De Angelis C, Trivedi MV, Osborne CK, Schiff R (2015) The changing role of ER in endocrine resistance. Breast 24(Suppl 2):S60–S66. https://doi.org/10.1016/j.breast.2015.07.015
Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R (2015) ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol 12(10):573–583. https://doi.org/10.1038/nrclinonc.2015.117
Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA (1997) An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res 57(7):1244–1249
Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, Ferrer-Lozano J, Perez-Fidalgo JA, Cristofanilli M, Gomez H, Arteaga CL, Giltnane J, Balko JM, Cronin MT, Jarosz M, Sun J, Hawryluk M, Lipson D, Otto G, Ross JS, Dvir A, Soussan-Gutman L, Wolf I, Rubinek T, Gilmore L, Schnitt S, Come SE, Pusztai L, Stephens P, Brown M, Miller VA (2014) Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res 20(7):1757–1767. https://doi.org/10.1158/1078-0432.CCR-13-2332
Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L, Gursky A, Siddiqui J, Tomlins SA, Roychowdhury S, Pienta KJ, Kim SY, Roberts JS, Rae JM, Van Poznak CH, Hayes DF, Chugh R, Kunju LP, Talpaz M, Schott AF, Chinnaiyan AM (2013) Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 45(12):1446–1451. https://doi.org/10.1038/ng.2823
Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, Hudis C, Chen D, Taran T, Hortobagyi G, Greene G, Berger M, Baselga J, Chandarlapaty S (2013) ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 45(12):1439–1445. https://doi.org/10.1038/ng.2822
Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, Toy W, Green B, Panchamukhi S, Katzenellenbogen BS, Tajkhorshid E, Griffin PR, Shen Y, Chandarlapaty S, Katzenellenbogen JA, Greene GL (2016) Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. elife 5. https://doi.org/10.7554/eLife.12792
Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, Yelensky R, Brown M, Miller VA, Sarid D, Rizel S, Klein B, Rubinek T, Wolf I (2013) D538G mutation in estrogen receptor-alpha: a novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res 73(23):6856–6864. https://doi.org/10.1158/0008-5472.CAN-13-1197
Nettles KW, Bruning JB, Gil G, Nowak J, Sharma SK, Hahm JB, Kulp K, Hochberg RB, Zhou H, Katzenellenbogen JA, Katzenellenbogen BS, Kim Y, Joachmiak A, Greene GL (2008) NFkappaB selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nat Chem Biol 4(4):241–247. https://doi.org/10.1038/nchembio.76
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
de Vera, I.M.S., S. Wanninayake, U., Burris, T.P. (2019). Structural Insights into Estrogen Receptors and Antiestrogen Therapies. In: Zhang, X. (eds) Estrogen Receptor and Breast Cancer. Cancer Drug Discovery and Development. Humana Press, Cham. https://doi.org/10.1007/978-3-319-99350-8_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-99350-8_10
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-99349-2
Online ISBN: 978-3-319-99350-8
eBook Packages: MedicineMedicine (R0)