
Abstract
This chapter describes the process of bringing radiopharmaceuticals from preclinical investigations to first-in-man studies, specifically the first application of a new radiopharmaceutical in an imaging study in humans aiming to demonstrate the potential of the radiotracer to image a specific molecular target. This translational trajectory – often called moving “from bench to bedside” – includes several steps that require attention to specific regulations. As these regulations differ throughout the world, we will focus on the current situation in Europe. This chapter will focus on the philosophy developed in the European Union by European agencies and associations – for example, the European Medicines Agency (EMA) and the European Pharmacopoeia (Ph. Eur.) – as well as societies such as the European Association of Nuclear Medicine (EANM).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Clinical translation
- Investigational Medicinal Product Dossier (IMPD)
- Good manufacturing practice (GMP)
- Good clinical practice (GCP)
- Radiopharmaceutical
- Toxicity
- Legislation
- Clinical trial
Introduction
This chapter describes the process of bringing radiopharmaceuticals from preclinical to first-in-man studies, specifically the first application of a new radiopharmaceutical in an imaging study in humans aiming to demonstrate the potential of the radiotracer to image a specific molecular target. This translational trajectory – often called moving “from bench to bedside” – includes several steps that require attention to specific regulations. As these regulations differ throughout the world, we will focus on the current situation in Europe. This chapter will focus on the philosophy developed in the European Union by European agencies and associations – e.g. the European Medicines Agency (EMA) and the European Pharmacopoeia (Ph. Eur.) – as well as societies such as the European Association of Nuclear Medicine (EANM).
As the first-in-man administration of a new radiopharmaceutical may hold safety risks for the volunteer, information derived from preclinical data regarding the toxicity, radiation dosimetry, product quality, and imaging potential of the radiopharmaceutical need to be available before human administration. All of this information is collected in an Investigational Medicinal Product Dossier (IMPD), and each of these topics will be discussed in more detail in this chapter. See Table 1 for references to legal binding documents, guidelines, and recommendations; see Table 2
for definitions of pertinent terms.
Investigational Medicinal Product Dossier (IMPD)
The European Union (EU) has produced a specific legislative framework for the use of radiopharmaceuticals in clinical trials. The preparation of an Investigational Medicinal Product Dossier (or IMPD) as part of the clinical trial application process is an essential step and is required by Regulation 536/2014 (“The Clinical Trials Regulation”). However, there are situations in which a simplified IMPD will be sufficient. A simplified IMPD may be submitted if information has been assessed previously as part of a marketing authorization in any Member State or a clinical trial under a competent authority (http://www.imp-dossier.eu/). The IMPD should include all the necessary information related to the chemical and pharmaceutical quality of the drug and product substances, as well as non-clinical data related to pharmacology, pharmacokinetics, radiation dosimetry, and toxicology. Of course, both a description of the clinical trial and a risk assessment must be included as well.
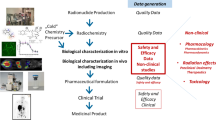
The format of the IMPD is described by the European Medicines Agency (EMA) “Guideline on the requirements to the chemical and pharmaceutical quality documentation concerning Investigational Medicinal Product in clinical trials” (Fig. 1). IMPD1 contains two main sections related to the production of the radiopharmaceutical: the “Drug Substance” (the active pharmaceutical ingredient, or API, the S-section) and the “Drug Product” (or finished product, described in the P-section). These parts are further divided into subsections that address more detailed topics, such as chemical information on the new entity, batch production and analysis, analytical methods, release criteria, etc. With respect to the documentation required during a marketing authorization application (MAA), information included in the IMPD should particularly focus on the risk aspects of the radiopharmaceutical (such as a justification of its use as well as data on toxicity and radiation dosimetry).

Schematic template displaying the main sections of an Investigational Medicinal Product Dossier (IMPD)
In the case of most PET radiotracers, the drug substance is not isolated and characterized during the preparation of the radiopharmaceutical, especially when the process is continuous and automated. Therefore, in the proposed guidelines, information in various 2.2.1.S subsections is not necessary, and the required details can instead be provided in the corresponding 2.2.1.P subsections [EANM guideline for the preparation of an Investigational Medicinal Product Dossier (IMPD)].
Toxicity Issues/Dosimetry
Information on toxicity as an indicator of the safety of the IMP should be included in the IMPD as part of the non-clinical pharmacology section. As the requirements for toxicity are addressed in a variable way within Europe, a position paper has been published by the Radiopharmacy Committee of the EANM addressing toxicology studies for new diagnostic and therapeutic radiopharmaceuticals [1]. This paper excludes endogenous and ubiquitous substances in human such as radiolabeled amino acids, as they are present in the body anyway and no toxicity studies would therefore be required.
To better understand how to address different points of view regarding toxicity, two distinct scenarios are recognized with respect to the reaction of a radionuclide with a non-radioactive precursor:
Scenario #1
The radiolabeling reaction of the radionuclide with a chemical precursor proceeds quantitatively. Therefore, no purification is required to separate the product and the unreacted radionuclide. In these cases, the precursor is typically used in a large molar excess over the radionuclide (e.g. the complexation of a radiometal by a chelator-bearing biomolecule). As a result, all components – including the precursor (or precursor hydrolysis product) and the resulting radiopharmaceutical active ingredient – are injected into the patient. In this case, the precursor or precursor hydrolysis product should be subjected to preclinical toxicity studies.
Scenario #2
The radiolabeling reaction of the radionuclide with a chemical precursor does not proceed quantitatively. In this scenario, purification is required to separate the desired radioactive compound from the reaction mixture, including the unreacted radionuclide and the precursor. In this case, the molecule containing a stable isotope of the intended radioactive nuclide should be used (e.g. 19F instead of 18F) for toxicity testing.
Based on the above scenarios and taking into consideration the generally accepted toxicity guidelines, the EANM has described a new approach for the assessment of toxicology based upon three distinct toxicological limits: (1) <1.5 μg, (2) <100 μg, and (3) >100 μg.
Less Than 1.5 μg
The <1.5 μg limit is based on the Threshold of Toxicological Concern (TTC) concept. A TTC value of 1.5 μg/day intake of a genotoxic impurity is considered to be associated with an acceptable risk – excess cancer risk of <1 in 100,000 over a lifetime – for most pharmaceuticals. Based on case-by-case judgments for radiopharmaceuticals applied in amounts of <1.5 μg per dose, it can be considered that no toxicology tests are needed. However, a risk assessment on potential toxicity should be included. This risk assessment of potential toxicity may be performed by in silico screening and (quantitative) structure-activity relationship (Q)SAR. For radiopharmaceuticals, doses of <1.5 μg can be achieved when the radiotracer is produced with high molar activity. For example, in the case of a 250 MBq dose of a radiopharmaceutical with a molecular weight of 300 and a molar activity of 50,000 GBq/mmol, only 1.5 μg of tracer is actually injected. In light of the fact that next-generation cameras are much more sensitive – and therefore require fewer MBq of activity – this dosage of <1.5 μg will be much easier to achieve in the future.
Less Than 100 μg
In this case, we are dealing with the so-called microdosing concept, and the “Note for guidance on non-clinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals” (CPMP/ICH/286/95) can be applied. Typically, a 100-fold larger dose than the clinical dose is tested in 30 rodents. More specifically, ten animals/sex are examined on the day following the injection, and five animals/sex are examined after 14 days (via hematology, clinical chemistry, necropsy, and histopathology). Subsequently, allometric scaling should be applied to translate from animal to human doses. It is important to note that at present, in vivo toxicology tests must be performed in compliance with GLP standards. Alternatively, the 1000-fold scaling is mentioned in the same guideline and may be followed if allometric scaling is not used. Both approaches can be used and may be subject to negotiation with the appropriate authorities.
One major limitation of this microdosing approach is that it does not take into account that pharmacological and toxicological effects are usually not determined by the mass but the molar amount administered. As a result, the toxicological effects of larger molecules such as proteins or peptides can be underestimated. In light of this, in the case of larger molecules such as proteins, the FDA’s “Guidance for Industry, Investigators, and Reviewers: Exploratory IND Studies” sets the limit to <30 nmoles.
To reduce time-consuming and costly toxicity studies, biodistribution data (often including imaging) from preclinical studies can be used to assess toxicity as well. These studies give detailed quantitative data on the accumulation of the drug in tissues and its elimination via excretion pathways. Based on these in vivo data, extended single dose toxicity studies can be focused primarily on risk organs and tissues. Such arguments must be made on a case-by-case basis, and the rationale for this approach must be described in detail in the application process.
More Than 100 μg
Dosages of more than 100 μg of a substance may be required for imaging with radiolabeled peptides, proteins, or antibodies or for therapeutic applications. Under these circumstances, masses in excess of 100 μg are used because cold peptide/protein has been added to the formulated radiopharmaceutical to modify the biodistribution (e.g. to uptake in organs such as the liver). In this case, the “Note for guidance on non-clinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals” (CPMP/ICH/286/95) can also be applied. An extended single dose toxicity study should be done in both a rodent and non-rodent species, as well as a test for genotoxicity (usually an Ames test). Apart from following the current guidelines, it is also necessary to perform a risk assessment for each compound in order to evaluate which toxicological studies are needed and/or useful. Therefore, a scientific advice meeting with the appropriate authorities can be very helpful before starting expensive toxicity studies.
Radiation Dosimetry
Before conducting a study in humans, an estimate of the radiation dosimetry of the radiotracer is required. The intended radiation dose to a patient should always be justified and is a requirement by the local/national authorities in the application for the clinical trial. Generally speaking, the radiation dose can be estimated from animal biodistribution data using OLINDA software and then later confirmed in humans. This can be performed using imaging in nonhuman primates or via multi-timepoint biodistribution studies in rodents in conjunction with a Medical Internal Radiation Dose (MIRD) system for calculations. A dose equation consisting of biological and physical parameters is proposed by MIRD. The biological parameters are determined by the time that the radioactivity spends in each organ and the physiological effects of the decay of the radiopharmaceutical. Therefore, knowledge of the distribution of the radioactivity within the body is required, data which can be obtained by extrapolation from preclinical experiments, external measurements with a PET or a SPECT camera, and estimations using compartmental models or measurements of excretory fluids. The physical parameters depend on the nature of the radiation, the absorption characteristics, and the anatomical model. The nuclear characteristics of any radionuclide can be found in MIRD radionuclide data as well as decay schemes published by the Society of Nuclear Medicine in 1989. In addition, source/target organ configurations, absorbed fractions, and S-values can be found in MIRD pamphlets.
The guidelines of the International Commission on Radiological Protection (ICRP 62) are adapted to estimate the risks and consequences of radiation doses received by patients. Several risk categories are defined. The radiation dose of all radiopharmaceuticals falls within categories IIb and III. The ICRP has described these categories as follows:
Category IIb: Effective Dose Range 1–10 mSv (Adults)
This category involves risks to the irradiated individual of the order of 1 in 10,000. The degree of benefit to society from studies in this category should be “moderate”; the benefit would be expected to be “aimed directly at the diagnosis, cure, or prevention of disease.”
Category III: Effective Doses Greater Than 10 mSv (Adults)
Here, the risks to the irradiated individual are estimated at greater than one in a thousand. This is a moderate risk for a single exposure but might be considered as verging on the unacceptable for continued or repeated exposures. To justify investigations in this category, the benefit would have to be “substantial and usually directly related to the saving of life or the prevention or mitigation of serious disease.” Doses should be kept below the threshold for deterministic effects unless these are necessary for the therapeutic effect.
In practice, the radiation dose to a patient should be kept as low as reasonable achievable (the ALARA principle). The next-generation PET and SPECT cameras are significantly more sensitive than the currently used instruments, so in the future, lower amounts of radioactivity can be administered to humans.
Preclinical Requirements
Data from preclinical studies should be collected in order to assess whether the new radiopharmaceutical performs according to the expectations. The results from these studies need to be summarized in the IMPD. The following preclinical data illustrate that the interaction of the radiopharmaceutical with the intended target is the major driver of uptake and thus contributes to the justification for using a new radiopharmaceutical in humans:
-
Plasma and metabolic stability
-
Affinity for the target
-
Ex vivo biodistribution data obtained in appropriate animal models
-
Calculations of non-specific and non-saturable binding
-
Imaging data obtained in appropriate animal models
-
Pharmacokinetic data obtained in appropriate animal models
-
Non-clinical pharmacology
-
Toxicity (discussed above)
-
Radiation dosimetry (discussed above)
With respect to the first item on the list, the stability of a radiopharmaceutical is important because the overly rapid breakdown of the radiotracer can prevent its interaction with the intended target. Furthermore, a fundamental understanding of the metabolic fate of a radiopharmaceutical can be extraordinarily helpful in assessing its in vivo performance. For example, it is crucial to know whether the radiolabeled metabolites of the parent tracer have binding affinity for the same target as the intact radiopharmaceutical. In addition, the non-specific uptake of metabolites in the tissue of interest can cause confounding results. Knowledge of the identity of metabolites can also help determine the optimal radiolabeling position in the molecule. Ideally, the radiolabel should be excreted rapidly upon metabolic breakdown. Along these lines, it is advisable to perform metabolite studies with cold reference material in human liver microsomes to assess the metabolic stability of the radiopharmaceutical and the chemical identity of potential metabolites. Information on metabolic stability can be obtained from biodistribution studies.
The affinity of a radiopharmaceutical for its target is also a critical parameter. A radiotracer is often chemically modified after radiolabeling, especially in the case of labeling with 18F or radiometals. As a result, it is important to test the affinity of the new molecular entity for its target. Affinities are usually determined by competition assays for interaction with the target (e.g. receptors, transporters, enzymes). Of course, the radiopharmaceutical must have sufficient affinity to ensure contrast with its surrounding tissue. As uptake and contrast are also determined by the expression levels of the target, the ratio between the density of the target and the affinity of the radiotracer should be assessed. In the case of receptor-targeted radiopharmaceuticals, the ratio B max/K D can be used and should be larger than 4 and preferably >10.
Tissue uptake data obtained either from imaging or biodistribution experiments can provide information on several of the in vivo characteristics of the radiopharmaceutical, including its uptake in target-rich tissues (specific binding), uptake in target-negative tissues (non-specific binding), excretion pathways, and pharmacokinetics. Critically, animal welfare legislation should also be followed when obtaining preclinical biodistribution and/or imaging data. This legislation is based on the 3R approach – replacement, reduction, and refinement – and should therefore include a justification for the selected animal model, a justification of the required number of animals for the study, and an evaluation of alternative approaches which could yield comparable information. The European legislation describing the protection of animals with respect to scientific research is Directive 2010/63.
Generally speaking, it is recommended to first use healthy mice or rats to investigate the pharmacokinetic profile, excretion profile, and non-specific uptake of a radiopharmaceutical. To further test the specificity and selectivity of a radiotracer, the following experiments could be employed:
-
The use of animal models with increased expression of the target, most commonly disease models such as tumor-bearing animals (though many animal models are available for cardiovascular, brain, and inflammation research as well)
-
The use of knockout mice
-
Blockade experiments based on the co-administration of a known competitive ligand or substrate
-
Displacement studies based on the administration of a competitive ligand or substrate in the equilibration phase
Relevant Clinical Data
All available clinical information on the radiopharmaceutical and/or its non-radioactive reference counterpart should be collected in the IMPD as it might be useful to assess its safety. This includes information on absorption, distribution, metabolism, and excretion (ADME). Available clinical data and information on the investigated patient groups, related drugs/radiopharmaceuticals, adverse events, and radiation dosimetry in combination with other exposures in the study (i.e. CT or other radiopharmaceuticals) should be included as well.
Regulations for the Production of a Radiopharmaceutical
When bringing a novel radiopharmaceutical into the clinic, specific requirements for its production must be considered. According to European regulations, a radiopharmaceutical is “Any medicinal product which, when ready for use, contains one or more radionuclides (radioactive isotopes) included for a medicinal purpose” (Art. 1.6 Directive 2001/83/EC). Since it is a medicinal product, a radiopharmaceutical must comply with all the requirements for such products, though specific considerations exist in relation to radiopharmaceuticals due to their unique traits. Namely, they are radioactive products, and the radiation dose to the patient must hence always be considered. Furthermore, their radioactive nature means that their composition is not constant (due to radioactive decay) and that their preparation process has some peculiarities that we shall discuss in more detail below.
The manufacturing or importation of medicinal products – including investigational medicinal products – is subject to a manufacturing or import authorization. The holder of such an authorization is obliged to comply with the principles and guidelines of good manufacturing practice (GMP) for medicinal products and to use as starting materials only active substances (active pharmaceutical ingredients) that have been manufactured in accordance with GMP. The principles and guidelines of GMP concerning medicinal products for human use and investigational medicinal products are in Commission Directive 2003/94/EC, the so-called GMP Directive. In addition, many detailed GMP guidelines from the European Medicines Agency (EMA) exist as well. Nonetheless, the special nature of radiopharmaceuticals has necessitated special provisions. One such special provision stems from the radioactive nature of radiopharmaceuticals, resulting in the fact that they are subject to both radiation protection legislation (designed for the protection of personnel) and GMP legislation (designed for the protection of the patient).
Good Manufacturing Practices
Good manufacturing practices (GMP) are the basis for ensuring that medicinal products are produced in such a way that it can be guaranteed that they are fit for their intended use, comply with the requirements of the marketing or clinical trial authorizations, and do not place patients at risk due to inadequate safety, quality, or efficacy. To achieve this quality objective reliably, there must be a comprehensively designed and correctly implemented pharmaceutical quality system that incorporates GMP and quality risk management (QRM). The pharmaceutical quality system involves quality management, good manufacturing practice, quality control, product quality review, and quality risk management. Quality management – in which good manufacturing practice is included – is a wide-ranging concept that covers all matters that individually or collectively influence the quality of a product. GMP is concerned with both production and quality control. In a GMP-based system, all processes are defined, systematically reviewed, and shown to be capable of consistently providing medicinal products of the required quality and complying with their specifications. Validation is a crucial part of GMP, meaning that all critical steps of manufacturing processes as well as significant changes to these processes are validated.
The scope of GMP includes the following aspects of the production of a medicinal product: the pharmaceutical quality system, personnel, premises and equipment, documentation, production, quality control, self-inspection, and outsourced activities. Complaints and product recalls must also be taken into account. Each of the aforementioned topics is addressed in a specific chapter of the GMP guidelines (EudraLex Chap. 4). In addition, these guidelines also include several annexes that deal with specific topics related to GMP production. Of these, there are several that are especially important in the context of the production of radiopharmaceuticals for investigational purposes in humans: manufacture of radiopharmaceuticals (Annex 3), manufacture of investigational medicinal products (Annex 13), manufacture of sterile products (Annex 1), computerized systems (Annex 11), qualification and validation (Annex 15), and parametric release (Annex 17). These annexes are especially relevant because they address some of the unique aspects of the production of radiopharmaceuticals. For example, radiotracers are most often produced using computerized systems to ensure robustness and reproducibility and to provide radiation protection for the operator. Furthermore, in many cases, due to the extremely short life of radiopharmaceuticals, not all of the quality controls of the final MP (i.e. sterility) can be finished before the radiopharmaceutical is released for human use. The application of GMP in the production of radiopharmaceuticals is intended to ensure not only that the subjects to whom these radiopharmaceuticals are administered are not placed at risk but also that the results of the clinical trials are not compromised by inadequate safety, quality, or efficacy due to unsatisfactory manufacture. Notwithstanding the production of radiopharmaceuticals under GMP, in many cases – especially in hospitals and academia-based radiopharmacies –radiopharmaceuticals are produced in accordance with an individual prescription for an individual patient or in accordance with a pharmacopoeia monograph. The preparation of such radiopharmaceuticals can be done under the provisions stated in Article 3.1 or 3.2 of Directive 2001/83, that is, magistral or officinal preparations. Such preparations are considered out of the scope of the directive and regulated at the national level. This has led to substantial variations with respect to whether (or not) such an approach can be used for radiopharmaceuticals in different EU countries. In principle, such a radiopharmaceutical could not be used in clinical trials according to the Clinical Trial Directive. However, the new Clinical Trial Regulation has an exception – Article 63.2 Regulation 536/2014 – for diagnostic radiopharmaceuticals used in clinical trials under some circumstances: diagnostic radiopharmaceuticals used as IMPs when the production process is carried out in hospitals and when they are intended to be used exclusively in hospitals. In this case, there is no need for GMP production. This decision follows the spirit of “proportionate risk” in the new regulation and allows that in some specific cases, it should be possible to allow deviations from those rules in order to facilitate the conduct of a clinical trial. Therefore, the applicable rules should allow for some flexibility, provided that subject safety as well as the reliability and robustness of the data in the clinical trial are not compromised.
We have recently seen two new relevant documents related with GMP, albeit both of them will only become applicable once the Clinical Trial regulation is applicable. Such documents are the new Regulation (EU) 2017/1569 specifying principles of and guidelines for good manufacturing practice for investigational medicinal products for human use and the new Directive (EU) 2017/1572 supplementing Directive 2001/83/EC of the European Parliament and of the Council as regards the principles and guidelines of good manufacturing practice for medicinal products for human use.
Needless to say, GMP is not the only way to ensure the adequate quality of a medicinal product: rather, it is just one of the most widely used ways to do it. We should not forget that the implementation of strict GMP for the production of radiopharmaceuticals could (in many cases) introduce so many hurdles that the availability of critical radiotracers for trials is reduced. Yet still, GMP is intended to ensure that there is consistency between batches of the same investigational medicinal product used in the same or different clinical trials and that changes during the development of an investigational medicinal product are adequately documented and justified.
Validation
Validation is the act of proving that any procedure, process, equipment, material, activity, or system actually leads to the expected results, while qualification indicates the actions and operations aimed to demonstrate that a system or piece of equipment is properly installed, works correctly, and leads to the expected results. In any case, qualification may be considered a part of validation. General Principles on Validation and Qualification are outlined in Annex 15 of GMP, while the validation of analytical methods are outlined in the Note for Guidance on validation of analytical procedures: text and methodology [ICH Q(2) guideline]. In any case, both documents are very general, though radiopharmaceuticals require specific validation protocols because they are radioactive and their shelf life is often extremely short. When dealing with the production of radiopharmaceuticals, the proper qualification of all equipment involved in production or QC is of the utmost importance. This would be the first step in the overall validation of the processes in which this equipment is used. When qualifying equipment used for the measurement of radioactivity, issues including the range of activity utilized, the energy and type of radiation used, and the efficiency of the detectors under each of these conditions must be considered.
The overall validation activities should be described in a general document – the validation master plan, VMP – that should not only detail a general validation policy with a description of the intended working methodology but also all of the issues related to the overall validation process. All validation activities must be extensively documented. Further information on the overall process can be found in an article by Todde et al. [2].
In addition, good laboratory practices (GLP) should be followed whenever possible, especially with respect to non-clinical pharmacology and toxicology data. The principles of GLP promote the quality and validity of data in the testing of chemicals and prevent fraudulent practices. In this way, requirements including organization, personnel, the integrity and traceability of quality management system data, inspections, archiving, the cross-contamination of data and materials, the qualification and validation of equipment and experimental methods, and the storage of materials must be considered.
EU Regulation Related to Clinical Trials
The regulation of clinical trials in the EU has been ruled by Directive 2001/20/EC (the “Clinical Trial Directive”) and was concretized further by Directive 2005/28/EC (the “GCP Directive”), both of which lay down principles and detailed guidelines for good clinical practice (GCP). In addition, the preparation of medicinal products for clinical trials had to follow the principles established in Directive 2003/94/EC (the “GMP Directive”), as we have previously explained. However, because a directive needs transposition to the national legislation corpus of the different states in the EU, substantial differences in its practical implementation have emerged across Europe. Soon after its implementation in the practice of the Clinical Trial Directive, the negative effects that this regulation had on clinical research in Europe became evident. Patients and researchers from academia, foundations, hospitals, research networks, and industry alike criticized the directive mainly for its disproportionately stringent regulatory requirements, the high costs associated with satisfying these regulations, and the lack of harmonization of the applicable rules necessary for multinational clinical trials.
The principal negative attributes of the CT Directive were (1) the legislative differences between different nations; (2) the obstacles to the conduct of clinical trials; (3) the significant expense of the highly demanding regulatory requirements, irrespective of the level of risk of the trial; (4) the sluggish pace of the trial implementation process; and (5) the theoretically similar but practically different ethical and regulatory requirements between countries. Not surprisingly, this has led to a decrease in investigator-driven studies since its implementation.
Regulation 536/2014 (“The Clinical Trials Regulation”) was approved in April 2014 and replaced Directive 2001/20. However, the new regulation is not applicable yet. The main characteristics of the new regulation are (1) it repeals the Clinical Trial Directive; (2) as it needs no transposition and is enforceable “as is,” it ensures that the rules for conducting clinical trials are identical throughout Europe; (3) the new regulation is focused on patient safety and reasonable and proportionate risk assessment; (4) it facilitates multicenter transnational clinical trials; and (5) it established a streamlined application procedure that greatly simplifies the overall authorization procedures. In summary, the new procedures will ensure patient safety and public health, promote strict scientific and ethical reviews, avoid administrative delays, and encourage prompt answers for applicants.
For the specific case of radiopharmaceuticals, Regulation 536/2014 introduces two very relevant changes that are exceptions to the general rules. First, it establishes that there is no need to hold an authorization for the preparation of radiopharmaceuticals used as diagnostic (not therapeutic) IMPs under specific circumstances. And second, it establishes there is no need for the GMP production of these diagnostic radiopharmaceuticals, as the regulation itself allows for some flexibility provided that subject safety – as well as the reliability and robustness of the data generated in the clinical trial – is not compromised. As previously stated, while GMP are the most common way to ensure the quality of the products, we must emphasize that it is not the only method that can be used to ensure the quality and safety of radiopharmaceuticals provided a sufficiently robust pharmaceutical quality control system is implemented.
As a whole, the new CT Regulation establishes a new framework for clinical research in the EU. It tries to correct all of the problems and drawbacks that the old CT Directive generated and focuses on the protection of subjects involved in CTs using reasonable and proportionate risk assessment as well as the overall simplification of procedures. Regarding radiopharmaceuticals, very relevant changes have been introduced for diagnostic radiotracers that will hopefully make clinical research easier. Hopefully, all the changes introduced by the regulation will help increase and facilitate clinical research in the EU, not only for sponsored CTs but also for investigations promoted in the academia environment.
Specifics for the Preparation and Use of Radiopharmaceuticals for Research Applications in Humans in Different EU Countries
Numerous differences exist among the different EU countries with respect to the use of novel radiopharmaceuticals in humans, mainly due to the fact that the currently available pan-European regulation for clinical trials is the CT Directive until the new CT Regulation becomes applicable (probably by 2018). In addition, the in-house preparation of radiopharmaceuticals can be considered under the umbrella of “pharmacy practice” in some countries, while this is not the case in others. This has led to significant heterogeneity and means that procedures that can be done in some countries cannot be done in the same way in others [3, 4].
Guidelines and Guidance Documents
Apart from the aforementioned legislation, there are a good number of guidelines and guidance documents published by groups such as the European Medicines Agency (EMA) and the European Association of Nuclear Medicine. Guidelines are not mandatory but rather are recommendations for the effective implementation of legislation; guidances are also recommendations, but in a more specific and detailed form.
Very recently, the Safety Working Party of the CHMP of EMA has recommended the issuing of a guidance on principles for the non-clinical development of radiopharmaceuticals. As a preliminary step, a concept paper – Concept paper on the development of guidance on the non-clinical evaluation of radiopharmaceuticals – has been open for public consultation from August till October 2017. In principle, the Safety Working Party suggests that the paper should be based on current guidelines and the scientific review of the different intended uses of both diagnostic and therapeutic radiopharmaceuticals.
The main documents of interest in this respect are:
-
CHMP/SWP/28367/07: “Guideline on strategies to identify and mitigate risks for first-in-human clinical trials with investigational medicinal products” that covers non-clinical issues for consideration prior to the first administration in humans as well as the design and conduct of trials in the initial phase of single and ascending doses during clinical development.
-
CHMP/QWP/185401/2004: Guideline on the requirements for the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials. This guideline addresses the documentation of the chemical and pharmaceutical quality of IMPs to be submitted to the competent authority for approval prior to beginning a clinical trial in humans.
-
EMA/CHMP/QWP/834816/2015 (draft): Guideline on the requirements for the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials. This guideline replaces the “Guideline on the requirements for the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials” (CHMP/QWP/185401/2004 final). This guideline addresses the documentation on the chemical and pharmaceutical quality of IMPs and AxMPs containing chemically defined drug substances, synthetic peptides, synthetic oligonucleotides, herbal substances, herbal preparations, and chemically defined radioactive/radiolabeled substances to be submitted to the competent authority for approval prior to beginning a clinical trial in humans.
-
CHMP/BWP/534898/2008: Guideline on the requirements for quality documentation concerning biological investigational medicinal products in clinical trials – this guideline addresses the specific documentation requirements on the biological, chemical, and pharmaceutical quality of IMPs containing biological/biotechnology-derived substances. The guidance outlined in this document applies to proteins and polypeptides, their derivatives, and products of which they are components (e.g. conjugates) and thus includes radiolabeled bioconjugates, although they are not even mentioned as such.
-
EANM guideline for the preparation of an Investigational Medicinal Product Dossier (IMPD): This guideline aims to take radiopharmaceutical scientists through the practicalities of preparing an IMPD, in particular giving advice where the standard format is not suitable. Examples of generic IMPDs for three classes of radiopharmaceuticals are given: a small molecule, a kit-based diagnostic test, and a therapeutic radiopharmaceutical.
-
EANM guideline to regulations for radiopharmaceuticals in early phase clinical trials in the EU. The purpose of this guideline is to help investigators by giving an overview of relevant current EU requirements concerning the quality of starting materials and final drug products (the radiopharmaceuticals) as well as the non-clinical safety studies and dosimetry considerations for designing a human clinical trial that includes the use of radiopharmaceuticals.
-
EANM guidance on current good radiopharmacy practice (cGRPP) for the small-scale preparation of radiopharmaceuticals. This guidance is meant as a guidance to Part B of the EANM “Guidelines on Good Radiopharmacy Practice (GRPP)” issued by the Radiopharmacy Committee of the EANM (see www.eanm.org) and covers the small-scale, “in-house” preparation of radiopharmaceuticals which are not kit procedures. The aim is to provide more detailed and practice-oriented guidance to those who are involved in the small-scale preparation of PET, therapeutic, or other radiopharmaceuticals which are not intended for commercial purposes or distribution.
-
EANM guidelines on current good radiopharmacy practice (cGRPP) in the preparation of radiopharmaceuticals. The preparation of radiopharmaceuticals for injection involves adherence to regulations on radiation protection as well as to appropriate rules of working under aseptic conditions, which are covered by these guidelines on good radiopharmacy practice (GRPP)
In addition – and to clarify and facilitate the implementation of Regulation (EU) No. 536/2014 – several recommendation documents have recently been published in EudraLex Vol 10:
-
Auxiliary medicinal products (AxMP) in clinical trials (June 2017). This is the previously named guidance on investigational medicinal products (IMPs) and “noninvestigational medicinal products” (NIMPs). This document includes as AxMP those PET radiopharmaceuticals administered to assess the effect of a new drug whose effects are the primary end point of a clinical trial.
-
Risk proportionate approaches in clinical trials (April 2017). This document provides further information on how a risk proportionate approach can be implemented and also highlights the areas identified in the regulation that allow such adaptation. The aim of risk control is to determine whether the risk is acceptable and, if not, to reduce the risk to an acceptable level. For this purpose, predefined quality tolerance limits should be established. The main components of risk control are risk mitigation, adaptation, and risk acceptance actions (including accountability).
To perform clinical trials, authorizations are required from the medicine agency, which can be the EMA, as well as national/local agencies. These agencies will ask for the IMPD and the Study Protocol (Fig. 2). In addition, authorization is needed from the Ethics Committee requiring investigator brochures, and investigators will need to follow GCP. GCP is an international ethical and scientific quality standard for the design, conduct, recording, and reporting of clinical trials involving humans. The clinical trial should comply to provide public assurance that the rights, safety, and well-being of trial subjects are protected and that the quality and reliability of the data are secured. The key elements of the quality system include:
-
The development, implementation, and maintenance of documented procedures
-
The training of sponsor personnel as well as the personnel in affiliates, at partners, and at trial sites
-
The validation of computerized systems
-
The monitoring of trial sites and technical facilities on-site or by using centralized monitoring techniques
-
The establishment of appropriate data management and quality control procedures
-
The performance of internal and external audits by independent auditors

The authorizations required and parties involved in setting up a clinical trial
Serious adverse events (SAEs) and suspected unexpected serious adverse reactions (SUSARs) must be documented and reported, as they can be the result of the administered radiopharmaceutical. Depending on the local situation, a pharmaco-vigilance document should be kept.
The Future
We foresee the following developments affecting the production of radiopharmaceuticals:
Increased Use of Radiotherapeutic Agents
We believe that in the next few years, we will see an increase in the use of radiotherapeutic agents, mainly radiopharmaceuticals labeled with alpha emitters. The use of these agents poses tremendous challenges during the development process due to their intended toxicity and the difficulties they present with respect to the evaluation of their safety. The new guideline on the non-clinical evaluation of radiopharmaceuticals – which will hopefully be published soon by the EMA – will help clarify the complex world that researchers are currently trying to navigate.
More Sensitive Cameras
Technological advances in both PET and SPECT cameras will produce increased resolution and augmented sensitivity. This could provide more precise data for in vivo pharmacokinetics and biodistribution studies in phase 0 trials.
Trends in Legislation
The implementation of the new Clinical Trial Regulation (hopefully during 2018) will likely boost academic research in the field of diagnostic radiopharmaceuticals given that the regulation, for the very first time, includes specific exemptions for diagnostic radiopharmaceuticals (prepared and used under very specific circumstances).
Risk-Based Approaches to Mitigate Potential Dangers
There is a growing trend of applying risk-based approaches rather than strict rules. This has been prompted by several factors, most notably (1) the fact that one-size-fits-all rules are detrimental to advancing the development of radiopharmaceuticals and decrease the number of clinical trials and (2) the increased awareness by authorities of the specifics of the development of radiopharmaceuticals.
The Bottom Line
-
For investigational medicinal products (IMPs) used under the new Clinical Trial Regulation, GMP is no longer required.
-
Risk assessment must be applied for the evaluation of the toxicity of new radiopharmaceuticals.
-
More sensitive PET and SPECT cameras will result in reduced radiation burdens for patients as well as reductions in the amount of compound injected.
-
The validation of production and analytical methods is a critical component of the synthesis of radiopharmaceuticals.
References
Koziorowski J, Behe M, Decristoforo C, Ballinger J, Elsinga PH, Ferrari V, et al. Position paper on requirements for toxicological studies in the specific case of radiopharmaceuticals. EJNMMI Radiopharm Chem. 2016;1:1.
Todde S, Peitl P, Elsinga PH, Koziorowski J, Ferrari V, Ocak EM, et al. Guidance on validation and qualification of processes and operations involving radiopharmaceuticals. EJNMMI Radiopharm Chem. 2017;2:8.
Decristoforo C, Penuelas I, Patt M, Todde S. New tracers to the clinic: translational studies. QJNM. 2017;61(2):135–44.
Bormans G, Buck A, Chiti A, Cooper M, Croasdale J, Desruet M, et al. Position statement on radiopharmaceutical production for clinical trials. EJNMMI Radiopharm Chem. 2017;2:12.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Peñuelas, I., Elsinga, P.H. (2019). The Clinical Translation Process in Europe. In: Lewis, J., Windhorst, A., Zeglis, B. (eds) Radiopharmaceutical Chemistry. Springer, Cham. https://doi.org/10.1007/978-3-319-98947-1_35
Download citation
DOI: https://doi.org/10.1007/978-3-319-98947-1_35
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-98946-4
Online ISBN: 978-3-319-98947-1
eBook Packages: MedicineMedicine (R0)