
Abstract
Tuberculosis (TB) is a deadly infectious disease of humans caused by Mycobacterium tuberculosis (Mtb). In 2016, there were about 10 million new cases and 1.8 million deaths attributed to TB worldwide (WHO Report 2017). In addition, about a third of the world population has asymptomatic latent Mtb infection (LTBI), of which about 10% can reactivate to symptomatic active TB in their lifetime dependent upon their immune status. However, in a population, Mtb infection results in a heterogeneous outcome, ranging from complete bacterial clearance to the establishment of LTBI or development of a full-blown disease. A key feature of TB pathogenesis is granuloma formation, which has been a subject of intense research for several decades. Although granulomas are thought to protect the host by containing Mtb in a confined area and preventing bacterial dissemination to other parts of the body, it is also likely to act as a safe-harbor for the infecting bacteria to thrive and persist in a niche with a compromised immunity. This chapter summarizes various cellular events underlying TB pathogenesis and the role of different types of immune cells involved in granuloma formation.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Introduction
Tuberculosis (TB), a deadly infectious disease of humans caused by Mycobacterium tuberculosis (Mtb), continues to be a major health threat worldwide. In 2016, there were about 10 million new cases and 1.8 million deaths attributed to TB (WHO Report 2017). The World Health Organization has estimated that about a third of the world population has asymptomatic latent Mtb infection (LTBI) and about 5–10% of individuals develop symptomatic, active primary TB following initial Mtb-infection. However, in a population, Mtb infection results in a heterogeneous outcome, ranging from complete bacterial clearance to the establishment of LTBI or development of a full-blown disease. Although individuals with LTBI are considered to be asymptomatic and noncontagious, about 10% of these individuals can reactivate to symptomatic active TB in their lifetime dependent upon their immune status. The outcome of Mtb infection in humans is dependent on both the host and pathogen-derived factors, including the infectious dose inhaled, the nature of Mtb, and the status of host immunity at the time of infection (Alcais et al. 2005; Zumla et al. 2013). However, a population that remains asymptomatic or do not display disease-like conditions after exposure to Mtb have a chance of 2–23% to remain uninfected for their lifetime. Thus, an individual exposed to Mtb may prevent the establishment of infection. This supports the notion that host immunity is capable of restricting the onset of infection and/or establishing a strong protective response very early after exposure to Mtb. In most of the individuals with LTBI, the bacteria are thought to be maintained inside the granuloma in a dormant form for many years (Lin and Flynn 2010). Active TB can develop either following initial Mtb infection (primary-progressive TB) or by reactivation of LTBI (post-primary TB); 30–50% of active TB patients develop cavities in their lungs (Benator et al. 2002). Lung cavity is an immunologically weak host environment and is permissive for profound bacterial replication. Cavity formation is considered as one of the detrimental host processes and is indicative of final stages of TB. Patients with pulmonary cavitary TB are highly contagious and are the major source of disease transmission in the community (Rodrigo et al. 1997).
TB is an ancient disease of humans and over the years, Mtb has evolved with its host; bacterial persistence that contributes to reactivation and reinfection are two major difficulties associated with eradication of this deadly disease, as they ultimately provide space for drug tolerance/resistance and emergence of new drug-resistant strains of Mtb. A key feature of TB pathogenesis is granuloma formation, which has been a subject of intense research for several decades. Although granulomas are thought to protect the host by containing Mtb in a confined area and preventing bacterial dissemination to other parts of the body, it is also likely to act as a safe-harbor for the infecting bacteria to thrive and persist in a niche with a compromised immunity.
This chapter summarizes various cellular events underlying TB pathogenesis and the role of different types of immune cells involved in granuloma formation.
Tuberculosis Pathogenesis and Granuloma Formation
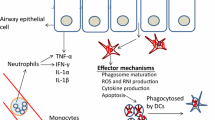
Through sneezing, coughing, talking, singing, and related activities, individuals with active pulmonary TB generate and spread aerosol droplets containing pathogenic Mtb that is capable of infecting new hosts. The inhaled bacilli are primarily engulfed by innate phagocytic cells such as alveolar macrophages, neutrophils, and dendritic cells (DCs) in the lung, where the primary inflammatory response is generated with the release of cytokines and chemokines by the infected phagocytes (Huynh et al. 2011) (Fig. 1). At this level, macrophages engulfing Mtb may be classified into two groups, one which is well-activated and capable of killing the bacteria and the other that is suboptimally or poorly activated and providing a growth niche to this intracellular pathogen.

Architecture and function of granuloma. Following Mtb infection, phagocytes (e.g., macrophage and dendritic cell/DC) uptake the bacteria. The pattern recognition receptors (PRR) present in these antigen presenting cells recognize Mtb antigen that leads to the production of various cytokines and chemokines, which recruit different types of immune cells (neutrophils, T and B cells) to the site of infection to form granuloma. A granuloma is a well-organized cellular structure characterized by the presence of infected macrophages (alveolar, interstitial, foamy, and epithelioid) and DCs that are surrounded by multinucleated giant cells and cuffs of lymphocytes (B and T cells). During disease progression, the granulomas undergo several changes in their structure, immune cell composition and function. Thus, granulomas can be broadly stratified into: (a) fibro-calcific granulomatous nodules, (b) non-necrotic granulomas, (c) caseous necrotic granulomas, and (d) suppurative and cavitary granulomas
The pathogen recognition receptors (PRRs) present on innate immune cells, such as macrophages, recognize the pathogen-associated molecular patterns (PAMP) present in Mtb, and play a central role in the initiation of host innate immune response (Akira et al. 2006; Pahari et al. 2017). PRRs are either membrane-bound, such as toll-like receptors (TLRs) and C-type lectins, or cytoplasmic, such as NOD-like receptors (NLRs) and RNA helicase retinoic acid-inducible gene I (RIG-I) receptors (Hossain and Norazmi 2013). Among the various TLRs identified, TLR2, TLR4, and TLR9 have key roles in sensing different mycobacterial antigens. The TLR2 interacts with mycobacterial lipoproteins such as 19-kDa secreted lipoprotein (LpqH), LprA and LprG and phosphatidyl-myo-inositol mannoside and induces a strong pro-inflammatory response by activating myeloid differentiation factor 88 (MyD88) and TIR domain-containing adaptor protein (TIRAP) signaling pathways (Quesniaux et al. 2004). Similarly, TLR4 interacts with lipopolysaccharide (LPS) of bacteria and activates MyD88-mediated nuclear factor-κB (NF-κB) signaling pathways, thus inducing the expression of pro-inflammatory cytokines and chemokines (Reiling et al. 2002; Bulut et al. 2005). While TLR9 recognizes CpG DNA of bacteria and promotes pro-inflammatory cytokines production (Hemmi et al. 2000), this receptor can also function cooperatively with TLR2 to induce interferon-gamma (IFN-γ) and IL12p40 production, thus playing a critical role in the development of host-protective immunity to infection (Bafica et al. 2005). Recognition of Mtb antigens by various TLRs also induces maturation of DCs, which migrate from the site of infection to draining lymph nodes and present the antigen to naϊve T-cells, thus initiating the adaptive immune response to Mtb infection (Bhatt and Salgame 2007). Moreover, some strains of Mtb activate TLR2, while few others interact with TLR4; this differential stimulation of TLR also leads to differential regulation and/or expression pattern of cytokines and chemokines by the phagocytes and subsequent cellular immune response to the pathogen (Carmona et al. 2013).
There are several types of C-type lectin receptors, such as mannose receptors (MR or CD206) and dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN/CD209), present on phagocytes that interact with Mtb and mediate phagocytosis. In human macrophages, MR recognizes mannose-capped lipoarabinomannan (ManLAM) of Mtb. It has been proposed that MR-mediated phagocytosis delays the fusion of Mtb-containing phagosome with lysosome, by suppressing the production of phosphatidylinositol-3-phosphate in the phagosomes (Rajaram et al. 2017). Moreover, in Mtb-infected DCs, MR-mediated phagocytosis augments anti-inflammatory response, by interfering with pro-inflammatory IL-12 production (Nigou et al. 2001). This also facilitates intracellular survival of Mtb. The DC-SIGN recognizes different types of PAMPs including LAM, ManLAM, and phosphatidylinositol mannosides (PIMs). Similarly to MR, the interaction of DC-SIGN with ManLAM also modulates pro-inflammatory responses and causes immune suppression (Geijtenbeek et al. 2003). The CD14 receptor plays a crucial role in the monocytic cell differentiation and phagocytosis of Mtb (Lingnau et al. 2007). This receptor recognizes mycobacterial LPS and induces a host-protective innate immune response to infection (Bowdish et al. 2009). Moreover, a decreased pulmonary inflammat ory response was observed in Mtb-infected CD14 knockout mice, which suggests that CD14 has a role in regulating inflammation during chronic pulmonary infection (Wieland et al. 2008). In addition to these cell surface receptors, cytoplasmic receptors can also sense mycobacterial antigens. The NLRs, such as NOD2, can interact with mycobacterial muramyl dipeptide (MDP) and induce pro-inflammatory cytokine production (Divangahi et al. 2008). The NOD2 receptor plays a protective role by sensing the pathogenic Mtb that enters the host cell cytoplasm after rupturing phagosome membrane (Pandey et al. 2009). Thus, the interaction of mycobacterial antigens with specific receptor can activate unique downstream signaling pathway, and their cumulative effect on immune cell activation determines the fate of intracellular Mtb as well as the course of infection towards progressive disease or containment and clearance.
Architecture and Function of Granuloma
Granuloma is a well-organized cellular structure, comprised of infected macrophages (alveolar, interstitial, foamy, and epithelioid), dendritic cells, and neutrophils that are surrounded by multinucleated giant cells, blood-derived macrophages, and cuffs of lymphocytes (T and B cells) (Fig. 1 and Table 1). In a well-organized granuloma, freshly recruited phagocytes and lymphocytes surround the Mtb-infected macrophages. At this stage, the granuloma can act as a physiological barrier and prevent the initial dissemination of infection within and between hosts (Flynn et al. 2011). When a granuloma loses its integrity during early stages and becomes permissive for bacterial growth, then the initial infection progresses into primary active TB. During early stages of infection, an elevated level of pro-inflammatory cytokines, such as TNF-α and IFN-γ produced by immune cells, polarizes the macrophages to a predominantly M1 phenotype that is capable of controlling Mtb growth and replication, through their antimicrobial effector functions. In addition, M1 macrophages can efficiently present Mtb antigens to T cells, which mount a host-protective Th1 type immunity by recruiting and activating more innate and adaptive immune cells with a pro-inflammatory phenotype. These cells surround the infected macrophages at the center of granuloma and limit the spread of Mtb (Chakravarty et al. 2008). Thus, the quality and quantity of immune response elicited by M1 macrophages, followed by immune cell recruitment and actvation of CD4+ and CD8+ T cells, mark the type of immune response generated in a granuloma (Fig. 2). However, as the disease progresses, the immune environment changes and the macrophages are more permissive to Mtb growth and polarized toward an anti-inflammatory M2 phenotype, marked with elevated IL-4 and IL-10 production. This facilitates establishment of a Th2 type immune environment with increased IL-2, IL-4, and I-10 production by immune cells; all these events are also associated with elevated lipid metabolism that contributes to formation of foamy macrophages (Kim et al. 2010).

Host immune response during Mtb infection. Th1 immune response plays a critical role during granuloma formation and restricts bacterial growth. Optimal onset of Th1 response along with protracted neutrophil infiltration enables containment of bacteria in the granulomas (left panel), while increased infiltration of neutrophils to the site of infection and impaired Th1 response increase inflammation and tissue destruction that contribute to elevated bacterial load (right panel)
During active disease, the Mtb-infected macrophages at the center of granulomas undergo necrosis that results in the accumulation of caseum, a creamy and acellular region, in which Mtb can survive extracellularly. Uncontrolled necrosis of infected host cells contributes to granuloma rupture and dissemination of pathogen to neighboring tissues facilitating secondary granuloma formation; these are seen in the lungs of about 20% of active TB cases, while reactivation of Mtb from quiescent granulomas contributes to active lung disease in about 80% of cases (Frieden et al. 2003). Even though granulomas are thought to restrict Mtb growth and dissemination, it is also suggested to promote bacterial persistence, which is a potential reservoir for future disease reactivation (Davis and Ramakrishnan 2009). Mycobacterial components are known to stimulate IL-10 secretion thereby suppressing Th1 response (Davis and Ramakrishnan 2009) and promoting M2 macrophage polarization with the development of lipid-rich, foamy macrophages and giant cells (Lugo-Villarino et al. 2012). This phenomenon suggests that granulomas can serve as a niche to support Mtb growth and/or survival by providing host lipids as a carbon source (Davis and Ramakrishnan 2009).
Structural Transformation of Granuloma
The granuloma physically maintains the bulk of Mtb in the organs of infected individuals. However, during progression of infection into active disease, the granulomas undergo several changes, mediated through the immune cells that constitute the granulomas, including structural transformation and metabolic shift (Russell et al. 2009a, b; Cadena et al. 2017). There is a strong positive correlation between the structural organization of granuloma and the extent of disease progression (Marino et al. 2011). While solid granulomas, marked with well-activated immune cells and no necrosis can effectively control Mtb growth and infection, cavitary granulomas with necrotic center are permissive for bacterial growth and disease progression. This suggests that any host debilitating conditions, such as immune deficiency (e.g., HIV infection, immune suppressive treatment) or metabolic disorder (e.g., diabetics) can alter the granuloma’s integrity and significantly affect the disease status. According to their structural organization, granulomas can be broadly stratified into: (a) fibro-calcific nodules, (b) non-necrotic, (c) caseous necrotic, (d) suppurative, and (e) cavitary granulomas (Mattila et al. 2013; Cadena et al. 2017). The fibro-calcific nodules are usually found in individuals with LTBI and believed to have a host protective role in controlling Mtb infection from transmission. These are usually smaller, healing-type granulomas (a.k.a. Ghon’s focus), characterized by mineralized fibrotic tissue and calcification at the center; both these processes can reduce bacterial survival and the probability of disease reactivation (Lin et al. 2009). In these granulomas, the fibrotic center is surrounded by large numbers of lymphocytes, macrophages (predominantly CD163+), and a restricted number of neutrophils (Lin et al. 2012).
The non-necrotic granulomas are highly cellular and characterized by the absence of cellular necrosis at the center, which is surrounded by both CD68+ and CD163+ macrophages. Limited number of neutrophils, abundant HAM56+ foamy macrophages, elevated proportion of epithelioid macrophages that strongly express iNOS and eNOS, and a relatively low frequency of Arg-1 expressing monocytes/macrophages are also observed in these granulomas. The caseous necrotic and suppurative granulomas have almost related features with a necrotic center. In caseous necrotic granuloma, the necrotic region is acellular with moderate number of neutrophils, whereas large number of neutrophils are present in the vicinity of necrotic center in suppurative granulomas (Mattila et al. 2013). In these granulomas, both CD68+ and CD163+ macrophages are distributed in and around the lymphocyte cuff. While the CD163+ macrophages are predominantly present in the peripheral region, HAM56+ foamy macrophages are abundant mainly at the rim of necrotic center. Both iNOS and Arg-1 are expressed abundantly throughout the necrotic granuloma, including lymphocytic cuff and the periphery of necrotic region. It is suggested that the ratio between iNOS and Arg-1 expression levels might play an important role in the structural transformation of granuloma and subsequent disease progression (Mattila et al. 2013; Kramnik and Beamer 2016).
Cavitation of granulomas in the lungs is a prelude to transmission of infection/Mtb within and between host(s). The luminal wall of cavitary lesions have copious number of Mtb actively proliferating extracellularly; since cavitary lesions in pulmonary tuberculosis open into airways and enable aerosol generation, they facilitate disease transmission (Kaplan et al. 2003). These lesions are thought to be manifestations of secondary (post-primary) tuberculosis, marked by elevated levels of regulatory T cells (Tregs) and DEC-205+ DCs (Welsh et al. 2011). Histologically, cavitary granulomas are characterized by severe tissue inflammation, suboptimal activation of innate and adaptive immune cells, and accumulation of B cells in the granuloma (Subbian et al. 2011). The cellular and immunological responses in cavitary lesions are also significantly different from non-necrotic and fibro-capsular lung lesions (Ulrichs et al. 2005). It has been suggested that differential regulation of local immune response at the site of infection determines the evolution of granulomas. Therefore, in patients with active pulmonary TB and in nonhuman primates and rabbit models of TB, remarkable heterogeneity in granulomas can be seen within the same lung that correlates with diseas progression or control of infection (Subbian et al. 2011; Ulrichs et al. 2005; Cadena et al. 2017).
Role of Cytokines in Granuloma Formation
The interaction of Mtb with PRRs of innate immune cells induces production of both pro- and anti-inflammatory cytokines and chemokines. While a pro-inflammatory response further activates the innate and adaptive immune cells and mediates granuloma formation, an anti-inflammatory response dampens inflammation and counteracts the effects of pro-inflammatory responses to avoid tissue damage. Although the former response is thought to be protective against Mtb infection, a delicate balance between pro- and anti-inflammatory responses is crucial for effective control of Mtb infection and tissue destruction (Domingo-Gonzalez et al. 2016). TNF-α is a key pro-inflammatory cytokine, produced by the cells of innate and adaptive immunity following Mtb infection, and plays key roles during granuloma formation and maintenance. It induces the production of chemokines such as CCL5 (RANTES), CXCL9 (MIG), and CXCL10 (IP-10) from various types of immune cells (Akira et al. 2006). These chemokines bind to respective receptors expressed on the surface of activated T cells, B cells, macrophages, and neutrophils and mediate extravasation of leukocytes to the primary site of infection to form the granuloma. TNF-α also maintains granuloma integrity that prevents reactivation and dissemination of Mtb (Chakravarty et al. 2008). In LTBI individuals, neutralization of TNF-α resulted in reactivation of symptomatic, active TB (Keane et al. 2001). This observation has also been confirmed in mouse and rabbit models of Mtb infection, in which treatment with anti-TNF-α antibodies suppressed host immunity, exacerbated disease severity, and bacillary load (Koo et al. 2011; Tsenova et al. 2014). These studies underline the protective role of TNF-α and the importance of granuloma integrity in TB.
Mycobacterial components are presented through infected macrophages and DC to the cells of the adaptive immunity, such as CD4+ and CD8+ T cells, Treg, and natural killer (NK) cells. DCs with engulfed Mtb can migrate from the lungs to regional lymph nodes to prime naive CD4+ and CD8+ T cells that ultimately become effector and memory subsets and play distinct roles in TB pathogenesis (O’Garra et al. 2013). In the granuloma, CD4+ T cells at the periphery of infected macrophages predominantly secrete interferon-γ (IFN-γ), a key pro-inflammatory cytokine, which activates the antimicrobial activities of phagocytes and facilitates antigen-specific T cell response (Cooper 2009). In addition, IFN-γ regulates systemic and local (lung) inflammation by inhibiting IL-17 production and neutrophil infiltration at the site of infection (Nandi and Behar 2011). In contrast, type I IFNs, such as IFN-α and IFN-β, are thought to inhibit the IFN-γ-mediated antimicrobial processes as well as the recruitment of CD4+ and CD8+ cells to the granuloma while promoting neutrophils infiltration to the site of infection (Ordway et al. 2007; Berry et al. 2010).
Cytokines that belong to the IL-1 family, such as IL-1α, IL-1β, and IL-18, can also initiate a pro-inflammatory response during Mtb infection. IL-1α has been shown to induce the production of IL-6 in human lung fibroblast, which activates host-protective effects on Mtb-infected macrophages; whereas IL-1β restricts intracellular Mtb growth in macrophages by enhancing phagosome maturation, autophagy, and production of antimicrobial peptides such as β-defensin 4 (Suwara et al. 2014; Master et al. 2008; Liu et al. 2009; Verway et al. 2013). IL-18 is known to induce pro-inflammatory IFN-γ production from T cells and thus plays a protective role against Mtb infection (Kinjo et al. 2002). Similarly, IL-6 produced by phagocytes upon Mtb infection plays a critical role in the onset of early inflammatory response (Law et al. 1996; Hoheisel et al. 1998). This cytokine regulates neutrophil infiltration (Fielding et al. 2008) and mediates differentiation and maintenance of Th17 cells, which contribute to establishment of an acute inflammatory response during Mtb infection (Jones and Vignali 2011). IL-12 is an important host-protective cytokine against Mtb infection and has vital roles in both innate and adaptive immune responses. IL-12 (IL-12p70) is composed of two subunits, IL-12p35 and IL-12/23p40, and is mainly secreted by Mtb-activated DCs. This cytokine is also a potent inducer of IFN-γ producing T cells, which are important to control Mtb infection (Cooper et al. 2011). It has been shown that infection of DCs by Mtb can trigger IL-12p40 secretion through TLR2 or TLR9 signaling pathways (Bafica et al. 2005). In addition, IL-12p40 regulates adaptive immune response and is required for DC migration and T cell priming in the lymph nodes during Mtb infection (Khader et al. 2006). The importance of IL-12 receptors is highlighted in recent studies, which show that mutations in the IL-12 receptor β1 (IL-12Rβ1) abrogate cellular immune response to IL-12 and compromise protective immunity to Mtb infection. Similarly, autosomal recessive IL-12Rβ1 deficiency in adults and children has been shown to result in severe disseminated forms of TB (Boisson-Dupuis et al. 2011; Tabarsi et al. 2011).
During Mtb infection, a subset of T cells, namely the Th17 cells, mainly produces pro-inflammatory cytokine IL-17, which promotes granuloma formation and plays a significant role in restricting Mtb growth (Okamoto Yoshida et al. 2010). IL-17 has been shown to induce IFN-γ and IL-12 secretion by macrophages and DCs that enhance host-protective Th1 immune responses (Lin et al. 2009). In addition, IL-17 induces the expression of chemokine CXCL13 and promotes the accumulation of CXCR5+ T cells at the site of infection (Gopal et al. 2013). These T cells are long-living and contribute to improved protection offered by BCG vaccination against Mtb infection (Desel et al. 2011; Lindenstrom et al. 2012). However, studies have also shown that IL-17 mediates excessive neutrophil recruitment, which contributes to elevated tissue inflammation and disease pathology during Mtb infection (Cruz et al. 2010). Therefore, it appears that fine-tuning of Th17 cell activation and IL-17 production is important for optimal host protection to Mtb infection. Another cytokine that plays key roles in the differentiation and maintenance of Th17 cells and development of B cell follicles during Mtb infection is IL-23 (Khader et al. 2011). This cytokine also induces the development of IFN-γ producing Th1 cells and proliferation of memory T cells in response to Mtb infection (Wozniak et al. 2006). Following Mtb infection, CD4+ cells, NK cells, and other lymphoid cells also express IL-22, a cytokine that stimulates production of antimicrobial peptides, such as β-defensin and lipocalin (McAleer and Kolls 2014). A higher level of IL-22 is reported in pulmonary TB granulomas (Matthews et al. 2011), where it activates macrophages and restricts intracellular Mtb growth by promoting phagolysosome fusion (Dhiman et al. 2014). In addition, IL-22 has been shown to mediate antigen-specific T cell responses, while suppressing the expansion of infection-induced Tregs, thus contributing positively to vaccine-mediated protective immunity to TB (Dhiman et al. 2009).
In contrast to the Th1 type cells, the Th2 and Tregs cells express key immune-regulatory cytokines, such as IL-4, IL-5, IL-10, IL-13, and TGF-β. These cytokines inhibit Th1 responses and regulate inflammatory response during Mtb infection. IL-4 downregulates the expression of inducible nitric oxide synthase (iNOS) and TLR2, thus contributing to dampening of macrophage activation and disease progression (Gordon 2003; Krutzik et al. 2003). However, increased expression of IL-4 also promotes necrosis of immune cells in the granuloma that facilitate extracellular bacterial replication, disease progression, and lung cavitation (Mazzarella et al. 2003; Bezuidenhout et al. 2009). IL-10 is another important immune-regulatory cytokine induced during Mtb infection. This cytokine exerts anti-inflammatory effect via STAT3-dependent pathway that inhibits macrophage activation (Cassatella et al. 1999; Moore et al. 2001; O’Leary et al. 2011). IL-10 also suppresses T cell proliferation and inhibits pro-inflammatory Th1 and Th17 responses during Mtb infection (Kumar et al. 2013). Similarly, IL-13 promotes Mtb survival by downregulating IFN-γ-induced autophagy in infected phagocytes (Harris et al. 2007). Increased expression of IL-13 reduces the frequency of IFN-γ and IL-17-expressing CD4+ T cells and augments necrotic cell death in TB granulomas (Heitmann et al. 2014). Mycobacterial membrane components, such as lipoarabinomannan (LAM), can induce the production of TGF-β by monocytes and DCs at the sites of infection (Toossi et al. 1995; Condos et al. 1998). This anti-inflammatory cytokine can suppress Th1 immune response by dampening IFN-γ production, antigen presentation, and pro-inflammatory cytokine production in Mtb-infected macrophages (Toossi and Ellner 1998). It has been shown that the combined induction of IL-10, TGF-β, TGF-β receptor 1 (RI) and RII expression can down-modulate the host immune response to Mtb infection and favors subsequent disease progression and bacterial growth (Bonecini-Almeida et al. 2004).
Role of Chemokines in Granuloma Formation
Several chemokines have been reported to mediate the early migration of immune cells to facilitate granuloma formation at the site of Mtb infection (Flynn and Chan 2005). These chemokines are classified as CXC or alpha, CC or beta, C or gamma, and CX3C or delta, based on the location of cysteine residues. Chemokines exert their function through interaction with respective receptors, which are members of the G1 protein coupled receptors (GPCR). The CC-type chemokines, such as monocyte chemoattractant protein-1 (MCP-1/CCL2), macrophage inflammatory protein-1α (MIP-1α/CCL3) and MIP-1β (CCL4) regulated upon activation, normal T cell expressed and secreted (RANTES/CCL5), as well as CXC-type chemokines, such as IL-8, monokine induced by gamma interferon (MIG/CXCL9), interferon gamma inducible protein-10 (IP-10/CXCL10), and stromal cell derived factor-1 (SDF-1/CXCL12), have been demonstrated to play key roles during Mtb infection and granuloma formation (Domingo-Gonzalez et al. 2016).
The CC-chemokine MCP-1 is one of the most potent chemoattractants of immune cells and activator of monocytes and plays an important role in regulating the host-pathogen interactions during Mtb infection. This chemokine is primarily secreted by monocytes, macrophages, and DCs, and it attracts both Th1 and Th2 cells to the site of infection (Siveke and Hamann 1998). However, it has been reported that MCP-1 enhances polarization of naïve T cells to Th2 cells, which can lead to an inefficient control of Mtb infection by the immune cells (Hussain et al. 2011). MIP-1α and MIP-1β are potent chemoattractants/activators of pro-inflammatory cells in the granuloma (Collins and Kaufmann 2001; Algood et al. 2003). These chemokines can also activate granulocytes, such as neutrophils and eosinophils, which lead to acute inflammation (Hsieh et al. 2008). While MIP-1α can induce the production of cytokines such as TNF-α, IL-1β, and IL-6, MIP-1β has been shown to modulate CCL3-induced TNF-α production in macrophages (Fahey et al. 1992). It has been shown that Mtb-infected human alveolar macrophages induce the production of RANTES, which helps to reduce intracellular Mtb growth (Saukkonen et al. 2002). This chemokine can attract immune cells to the site of infection and promote granuloma formation. CCL5 is another host-protective chemokine, which mediates recruitment of IFN-γ-producing, antigen-specific T cells expressing CCR5, a receptor for CCL5. Thus, CCL5 limits the intracellular survival of Mtb and contributes to host protection (Vesosky et al. 2010). In addition, coordinated expression/function of CCL5, perforin, and granulysin in CD8+ T cells facilitates killing of Mtb within infected macrophages (Stegelmann et al. 2005).
The CXC-chemokine, IL-8, is produced by epithelial cells, monocytes, macrophages, and fibroblasts during Mtb infection. This chemokine is an important neutrophil chemoattractant and reported to have a significant role in the inflammatory response and control of Mtb infection (O’Kane et al. 2007). Elevated serum levels of IL-8 were noted in patients with active pulmonary TB, which decreased to basal levels following successful anti-TB chemotherapy treatment (Almeida Cde et al. 2009). Thus, IL-8 has been suggested as a potent biomarker of TB to measure disease severity and treatment efficacy. The IFN-γ inducible chemokines, MIG and IP-10 (CXCL10), are chemotactic for NK cells and activated T lymphocytes. It has been shown that the expression of MIG enhances the recruitment of IFN-γ producing CD4+ T cells to the granulomas, which helps to restrict bacterial growth (Khader et al. 2007). During Mtb infection, IP-10 acts as an immune-inflammatory mediator and plays an important role in the granuloma formation by recruiting activated CXCR3+ T cells to the infection site (Agostini et al. 1998; Zhang et al. 2004). In addition, most of the peripheral CXCR3+ T cells express CD45RO (memory T cells), which is implicated in binding of lymphocytes to endothelial cells. Elevated levels of IP-10 has been detected in the plasma/sera of patients with active TB as well as subclinical and LTBI cases; although this chemokine has been suggested as a promising diagnostic marker for active TB (Strzelak et al. 2012). The stromal cell derived factor-1 (SDF-1) is a homeostatic CXC-type chemokine that is involved in myelopoiesis, B-lymphopoiesis, and localization and retention of progenitor cells in bone marrow as well as in organ development (Moser and Loetscher 2001). Increased levels of SDF-1 have been reported in the plasma of patients with active pulmonary TB (Shalekoff and Tiemessen 2003). A recent study has also shown that during Mtb infection, SDF-1 attracts circulating CXCR4+ B cells to the pleural space, thus playing an important role in B cell trafficking to the site of infection (Feng et al. 2011). Moreover, SDF-1 has been suggested as a diagnostic marker for differentiating TB pleurisy from other forms of TB (Kohmo et al. 2012). Thus, chemokine-mediated recruitment of immune cells to the site infection plays central role in the granuloma formation and disease pathogenesis in TB.
B Cell Response in Tuberculosis
Adaptive immune response in TB, elicited by lymphocytes upon Mtb infection of the host, can be broadly divided into T cell-mediated cellular immunity and B cell-mediated humoral immunity. Although T cell-mediated cellular immune response is crucial in controlling Mtb infection and regulating granuloma formation/maturation, several studies have shown that B cells and their antibody-mediated humoral immune response can also impact the host responses against Mtb infection (Jacobs et al. 2016). A recent study reported that B cell responses are dysregulated in patients with active and latent TB, and this impairment was not observed in individuals after anti-TB treatment (Joosten et al. 2016). This study also shows that B cells augment the effector functions of cellular immune response mediated by T cells. The B cells present in TB granulomas mediate several immune processes such as antigen presentation to T cells, Mtb-specific antibody production, and promoting the development of Th1 response by inducing IL-12 and IFN-γ production (Kozakiewicz et al. 2013; Chan et al. 2014; Bao et al. 2014). Antibodies to Mtb, secreted by the B cells, are thought to be involved in the neutralization of toxin, opsonization, and modulation of complement-mediated lysis. Moreover, IgG antibody specific to mycobacterial antigen has been found in the plasma of TB patients (Daniel et al. 1981). The antibody-mediated immune response is more prominent in the granulomas during disease progression, where bacteria replicate extracellularly in the necrotic material. It is proposed that anti-Mtb antibodies can prevent the establishment or dissemination of Mtb infection by hampering the bacterial adhesion to phagocytes (Schlesinger et al. 1994). For example, in macrophages, the FcR-mediated phagocytosis of Mtb was shown to enhance phagolysosomal fusion and mycopeptide presentation to T cells, thus augmenting the Th1 response (Maglione et al. 2008; Guilliams et al. 2014). In addition, the pre-coating of bacilli with anti-LAM antibodies has been shown to enhance phagolysosomal fusion and to increase the abundance of IFN-γ-expressing CD4+ and CD8+ T cells (de Valliere et al. 2005; Kumar et al. 2015). In addition, higher levels of anti-Mtb isotype IgG3 antibodies have been shown to be associated with preventio of LTBI reactivation in high-risk individuals (Encinales et al. 2010). Similarly, passive administration of Mtb-specific monoclonal antibodies as well as human gamma globulin has been shown to provide a protective effect against Mtb infection (Hamasur et al. 2004; Olivares et al. 2009; Balu et al. 2011). Furthermore, B cells can help to regulate inflammation in the infected tissue by secreting anti-inflammatory cytokines, such as TGF-b, IL-4, and IL-33, and by regulating Th1 and Th17 response (Chan et al. 2014). Moreover, B cells regulate neutrophil infiltration at the site of infection by modulating IL-17 response and thus help to prevent exacerbated inflammatory response during Mtb infection (Kozakiewicz et al. 2013).
Metabolic Shift in Granulomas
Infections by pathogens and/or environmental stresses, such as limited nutrient availability, hypoxia, and low pH, are capable of modulating the central metabolism of immune cells that can alter the protective host response and contribute to disease progression. A metabolic shift toward enhanced glycolysis with diminished oxygen consumption is a key feature of immune cells in TB granulomas (Shi et al. 2016). Mtb infection has been shown to modulate the host metabolome by increasing glucose oxidation and lipid peroxidation that ultimately lead to accumulation of several metabolites such as gluconic acid, lactone, glutaric acid, butanal, and ethane. Importantly, presence of these metabolic intermediates has been confirmed in the sputum samples of patients with active pulmonary TB (du Preez and Loots 2013). Upon Mtb infection, the metabolic reprogramming is mainly found in classically activated, pro-inflammatory M1 macrophages, immunogenic DCs, and activated T cells but not in anti-inflammatory M2 macrophages and tolerogenic DCs. The metabolic shift in TB granuloma has similar characteristics of the Warburg effect reported in cancer cells, which contributes to the increased production of nitric oxide and pro-inflammatory cytokines (Shi et al. 2016; Kiran et al. 2016). During Mtb infection, several genes associated with the Warburg effect (e.g., H+-ATPase, glucose transporters, and glycolytic enzymes such as hexokinase, phosphofructokinase, phosphoglycerate kinase, enolase, ADP-dependent glucose kinase, and lactate dehydrogenase) were shown to be upregulated (Shi et al. 2016). In addition, induction of Warburg effect in the host immune cells is also associated with corresponding increase in the expression of hypoxia-inducible factor-1 alpha (HIF-1 α), which plays a key regulatory role during infection and inflammation (Semenza 2010). In addition, HIF-1α regulates the expression of pro-inflammatory cytokines and differentiation of Th17 cells (Dang et al. 2011). In TB granulomas, HIF-1α-induced Warburg effect is associated with activation of pyruvate kinase M2 (PKM2), a key regulator of glycolysis. Translocation of PKM2 to the nucleus and its interaction with HIF-1α activates expression of pro-inflammatory cytokines, such as IL-1β and the glycolytic enzymes (Palsson-McDermott et al. 2015).
In addition to the shift in glucose metabolism, altered lipid metabolism has also been reported in the immune cells of TB granulomas. Mtb upregulates expression of several host genes involved in lipid sequestration and metabolism such as adipophilin (ADFP), acyl Co-A synthase long-chain family member 1 (ACSL1), and prosaposin (PSAP) in caseous granulomas (Kim et al. 2010). Similarly, an increased production of cholesterol (CHO), cholesterol ester (CE), triacylglycerol (TAG), and lactosylceramide (LacCer) has been reported in TB granulomas (Chatterjee et al. 1997; Garner et al. 2002). This Mtb-induced lipid sequestration can lead to the accumulation of lipid droplets in the cytoplasm and other organelles of macrophages and subsequent foamy macrophage formation (Daniel et al. 2011). Mtb can persist and replicate in foamy macrophages and their presence is an indication of degenerative granuloma and caseous necrosis (Tan and Russell 2015). Furthermore, Mtb can utilize host-derived lipid intermediates, such as TAG as its carbon source for growth and replication (Pandey and Sassetti 2008). Thus, altered immune cell metabolism during Mtb infection can augment bacterial outgrowth and necrosis of granuloma and a delicate balance in host immunometabolism is crucial for an effective host-protective response.
Targeting Granulomas as a Therapeutic Approach for TB
Although granuloma plays a significant role in the prevention of initial stage of Mtb infection towards progressive disease, presence of multiple types of granulomas, ranging from cavitary to necrotic and non-necrotic lesions, within the lungs of active TB patients questions its protective role. Moreover, granulomas, by virtue of their tight-knit cellular structure with fibrotic rims, can be refractory to antibiotics penetration into their necrotic center with caseum, where Mtb thrives; this contributes to prolonged duration of anti-TB treatment (at least 6 months for drug-sensitive pulmonary TB). This also suggests that additional host-directed therapeutic strategy that can modulate the structure (e.g., integrity, vascularization) and function (inflammation, metabolism) of granulomas can be used to enhance the efficacy of existing anti-TB drugs (Kolloli and Subbian 2017). Previous findings show promising trend in this approach. For example, TNF-α plays a significant role in the formation of granuloma and maintenance of its integrity; neutralization of TNF-α with antibody (e.g., Enbrel) treatment leads to disruption of granuloma integrity and enhances antibiotic penetration that augments bacterial clearance and reduces lung pathology (Chakravarty et al. 2008; Bourigault et al. 2013). Similarly, blocking of TNF-α using etanercept during initial stages of TB treatment was shown to accelerate sputum culture conversion and elevate the number of CD4+ T cells by 25% in patients with HIV-associated TB (Wallis et al. 2004). These findings suggest that granuloma integrity is a critical factor for bacterial survival and proliferation, and therapeutic agents that can perturb granuloma structure have the potential to be used as adjunct drug with standard antibiotics to promote efficient bacterial clearance, improve treatment efficacy, and shorten the duration of therapy.
Recently, pharmacological agents that either promote or inhibit angiogenesis have been reported to augment TB treatment. In this strategy, the host vascular endothelial growth factor (VEGF) and angiopoietins (Ang) were targeted. These molecules are potent angiogenic factors and are essential for the growth and proliferation of endothelial cells. Moreover, VEGF and Ang are abundantly expressed in pulmonary TB granulomas (Datta et al. 2015) and elevated levels of these molecules have been reported in patients with active pulmonary TB, compared to healthy controls (Kumar et al. 2016). The increased expression of VEGF and Ang also promotes abnormal, leaky, and dysfunctional blood vessel formation around the granulomatous area. This creates a hypoxic microenvironment in the granuloma that impairs normal immune response and favors Mtb persistence (Osherov and Ben-Ami 2016). Recently, the ability of an anti-VEGF antibody (Bevacizumab) or treatment with SU5416 (tyrosine kinase receptor), and pazopanib (VEGFR inhibitor) in normalizing the vascular structure of granulomas was investigated (Oehlers et al. 2015; Datta et al. 2015). Results from these studies show successful neutralization of VEGR function that reduced hypoxia and facilitated anti-TB drug penetration and killing of Mtb in the granuloma (Oehlers et al. 2015; Datta et al. 2015).
In addition to controlling angiogenesis, recent research also focused on an alternative strategy, which promotes normal angiogenesis in the granulomas to augment anti-TB treatment with antibiotics. Since granulomas are limited in their blood supply and circulation, restoring normal angiogenesis might improve the immune response and delivery of anti-TB drugs. The lack of VEGF in the sera of patients with active, cavitary pulmonary TB supports this approach (Abe et al. 2001). Therefore, any therapeutic approach that promotes expression of angiogenic growth factors such as VEGF, epidermal growth factors, transforming growth factor, platelet-derived growth factor, fibroblast growth factor, and hypoxia-inducible factor can be used to improve the clinical outcome of current anti-TB treatment. Since hormones involved in glucose homeostasis promote angiogenesis, targeting these hormones can be another promising approach to improve TB treatment (Cun et al. 2015). For example, exenatide, an agonist of the glucagon-like peptide-1 receptor that enhances angiogenesis in endothelial cells, has been reported to promote angiogenesis in TB granuloma (Aronis et al. 2013). Although clinical intervention to restore and normalize vascular structure might enhance the efficacy of anti-TB chemotherapy, more preclinical studies are required to assess the beneficial role of targeting granuloma integrity and restoring tissue angiogenesis for better delivery of anti-TB drugs.
Summary and Conclusion
To summarize, following successful entry into the lungs, pathogenic Mtb interacts with host cell receptors present on phagocytic cells in the lungs. This leads to engulfment of the pathogen and secretion of several pro-inflammatory molecules, which aid in the extravasation of various immune cells from the circulation to the site of infection and initiate granuloma formation. With time, the granuloma is packed with various leukocytes and confined to a defined area in the lungs. These host cells produce a plethora of cytokines/chemokines that shapes the quality of immune response prevailing in the granuloma. In general, a pro-inflammatory Th1 response is considered as host protective while an anti-inflammatory Th2 response is considered as counter-protective against Mtb infection; although a delicate balance between Th1 and Th2 response is crucial for optimal control of infection. Within the granuloma, the bacteria are either contained or proliferate and disseminate, depending on the nature of the bacteria and the immune environment prevailing in the granuloma. In general, during early stages of infection, granulomas wall off replication/dissemination of bacteria/disease and play a host-protective role. During progression of infection to active disease, the lung granulomas undergo structural changes and deteriorate to develop central caseous necrosis. At this stage, Mtb actively multiplies extracellularly within the granuloma that can undergo structural changes leading to lung cavitation. In addition, immune cells present in the granuloma can adopt a metabolic shift with enhanced glycolysis and altered lipid metabolism. Considering the limitations associated with current antibiotic-based therapy, adjunct host-directed therapeutic modalities, such as those that target granuloma, is a novel and new treatment approach for TB. In this approach, the structure and/or immune cell function of granulomas can be modulated to increase better anti-TB drug delivery and/or accessibility for efficient bacteriall killing and control of infection. This strategy can improve the efficacy of existing anti-TB drugs, help to reduce the duration therapy, and elevate the quality of clinical outcome following treatment of patients with TB.
References
Abe, Y., Nakamura, M., Oshika, Y., Hatanaka, H., Tokunaga, T., Ohkubo, Y., et al. (2001). Serum levels of vascular endothelial growth factor and cavity formation in active pulmonary tuberculosis. Respiration, 68(5), 496–500.
Agostini, C., Cassatella, M., Zambello, R., Trentin, L., Gasperini, S., Perin, A., et al. (1998). Involvement of the IP-10 chemokine in sarcoid granulomatous reactions. Journal of Immunology, 161(11), 6413–6420.
Akira, S., Uematsu, S., & Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell, 124(4), 783–801.
Alcais, A., Fieschi, C., Abel, L., & Casanova, J. L. (2005). Tuberculosis in children and adults: Two distinct genetic diseases. The Journal of Experimental Medicine, 202(12), 1617–1621.
Algood, H. M., Chan, J., & Flynn, J. L. (2003). Chemokines and tuberculosis. Cytokine & Growth Factor Reviews, 14(6), 467–477.
Almeida Cde, S., Abramo, C., Alves, C. C., Mazzoccoli, L., Ferreira, A. P., & Teixeira, H. C. (2009). Anti-mycobacterial treatment reduces high plasma levels of CXC-chemokines detected in active tuberculosis by cytometric bead array. Memórias do Instituto Oswaldo Cruz, 104(7), 1039–1041.
Aronis, K. N., Chamberland, J. P., & Mantzoros, C. S. (2013). GLP-1 promotes angiogenesis in human endothelial cells in a dose-dependent manner, through the Akt, Src and PKC pathways. Metabolism, 62(9), 1279–1286.
Bafica, A., Scanga, C. A., Feng, C. G., Leifer, C., Cheever, A., & Sher, A. (2005). TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. The Journal of Experimental Medicine, 202(12), 1715–1724.
Balu, S., Reljic, R., Lewis, M. J., Pleass, R. J., McIntosh, R., van Kooten, C., et al. (2011). A novel human IgA monoclonal antibody protects against tuberculosis. Journal of Immunology, 186(5), 3113–3119.
Bao, Y., Liu, X., Han, C., Xu, S., Xie, B., Zhang, Q., et al. (2014). Identification of IFN-gamma- producing innate B cells. Cell Research, 24(2), 161–176.
Benator, D., Bhattacharya, M., Bozeman, L., Burman, W., Cantazaro, A., Chaisson, R., et al. (2002). Rifapentine and isoniazid once a week versus rifampicin and isoniazid twice a week for treatment of drug-susceptible pulmonary tuberculosis in HIV-negative patients: A randomised clinical trial. Lancet, 360(9332), 528–534.
Berry, M. P., Graham, C. M., McNab, F. W., Xu, Z., Bloch, S. A., Oni, T., et al. (2010). An interferon- inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature, 466(7309), 973–977.
Bezuidenhout, J., Roberts, T., Muller, L., van Helden, P., & Walzl, G. (2009). Pleural tuberculosis in patients with early HIV infection is associated with increased TNF-alpha expression and necrosis in granulomas. PLoS One, 4(1), e4228.
Bhatt, K., & Salgame, P. (2007). Host innate immune response to Mycobacterium tuberculosis. Journal of Clinical Immunology, 27(4), 347–362.
Bodnar, K. A., Serbina, N. V., & Flynn, J. L. (2001). Fate of Mycobacterium tuberculosis within murine dendritic cells. Infection and Immunity, 69(2), 800–809.
Boisson-Dupuis, S., El Baghdadi, J., Parvaneh, N., Bousfiha, A., Bustamante, J., Feinberg, J., et al. (2011). IL-12Rbeta1 deficiency in two of fifty children with severe tuberculosis from Iran, Morocco, and Turkey. PLoS One, 6(4), e18524.
Bonecini-Almeida, M. G., Ho, J. L., Boechat, N., Huard, R. C., Chitale, S., Doo, H., et al. (2004). Down- modulation of lung immune responses by interleukin-10 and transforming growth factor beta (TGF-beta) and analysis of TGF-beta receptors I and II in active tuberculosis. Infection and Immunity, 72(5), 2628–2634.
Boselli, D., Losana, G., Bernabei, P., Bosisio, D., Drysdale, P., Kiessling, R., et al. (2007). IFN- gamma regulates Fas ligand expression in human CD4+ T lymphocytes and controls their anti-mycobacterial cytotoxic functions. European Journal of Immunology, 37(8), 2196–2204.
Bourigault, M. L., Vacher, R., Rose, S., Olleros, M. L., Janssens, J. P., Quesniaux, V. F., et al. (2013). Tumor necrosis factor neutralization combined with chemotherapy enhances Mycobacterium tuberculosis clearance and reduces lung pathology. American Journal of Clinical and Experimental Immunology, 2(1), 124–134.
Bowdish, D. M., Sakamoto, K., Kim, M. J., Kroos, M., Mukhopadhyay, S., Leifer, C. A., et al. (2009). MARCO, TLR2, and CD14 are required for macrophage cytokine responses to mycobacterial trehalose dimycolate and Mycobacterium tuberculosis. PLoS Pathogens, 5(6), e1000474.
Bulut, Y., Michelsen, K. S., Hayrapetian, L., Naiki, Y., Spallek, R., Singh, M., et al. (2005). Mycobacterium tuberculosis heat shock proteins use diverse toll-like receptor pathways to activate pro-inflammatory signals. The Journal of Biological Chemistry, 280(22), 20961–20967.
Cadena, A. M., Fortune, S. M., & Flynn, J. L. (2017). Heterogeneity in tuberculosis. Nature Reviews. Immunology, 17(11), 691–702.
Carmona, J., Cruz, A., Moreira-Teixeira, L., Sousa, C., Sousa, J., Osorio, N. S., et al. (2013). Mycobacterium tuberculosis strains are differentially recognized by TLRs with an impact on the immune response. PLoS One, 8(6), e67277.
Cassatella, M. A., Gasperini, S., Bovolenta, C., Calzetti, F., Vollebregt, M., Scapini, P., et al. (1999). Interleukin-10 (IL-10) selectively enhances CIS3/SOCS3 mRNA expression in human neutrophils: Evidence for an IL-10-induced pathway that is independent of STAT protein activation. Blood, 94(8), 2880–2889.
Chakravarty, S. D., Zhu, G., Tsai, M. C., Mohan, V. P., Marino, S., Kirschner, D. E., et al. (2008). Tumor necrosis factor blockade in chronic murine tuberculosis enhances granulomatous inflammation and disorganizes granulomas in the lungs. Infection and Immunity, 76(3), 916–926.
Chan, J., Mehta, S., Bharrhan, S., Chen, Y., Achkar, J. M., Casadevall, A., et al. (2014). The role of B cells and humoral immunity in Mycobacterium tuberculosis infection. Seminars in Immunology, 26(6), 588–600.
Chatterjee, S. B., Dey, S., Shi, W. Y., Thomas, K., & Hutchins, G. M. (1997). Accumulation of glycosphingolipids in human atherosclerotic plaque and unaffected aorta tissues. Glycobiology, 7(1), 57–65.
Collins, H. L., & Kaufmann, S. H. (2001). The many faces of host responses to tuberculosis. Immunology, 103(1), 1–9.
Condos, R., Rom, W. N., Liu, Y. M., & Schluger, N. W. (1998). Local immune responses correlate with presentation and outcome in tuberculosis. American Journal of Respiratory and Critical Care Medicine, 157(3 Pt 1), 729–735.
Cooper, A. M. (2009). Cell-mediated immune responses in tuberculosis. Annual Review of Immunology, 27, 393–422.
Cooper, A. M., Mayer-Barber, K. D., & Sher, A. (2011). Role of innate cytokines in mycobacterial infection. Mucosal Immunology, 4(3), 252–260.
Cruz, A., Fraga, A. G., Fountain, J. J., Rangel-Moreno, J., Torrado, E., Saraiva, M., et al. (2010). Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. The Journal of Experimental Medicine, 207(8), 1609–1616.
Cun, X., Xie, J., Lin, S., Fu, N., Deng, S., Xie, Q., et al. (2015). Gene profile of soluble growth factors involved in angiogenesis, in an adipose-derived stromal cell/endothelial cell co-culture, 3D gel model. Cell Proliferation, 48(4), 405–412.
Dang, E. V., Barbi, J., Yang, H. Y., Jinasena, D., Yu, H., Zheng, Y., et al. (2011). Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell, 146(5), 772–784.
Daniel, T. M., Oxtoby, M. J., Pinto, E., & Moreno, E. (1981). The immune spectrum in patients with pulmonary tuberculosis. The American Review of Respiratory Disease, 123(5), 556–559.
Daniel, J., Maamar, H., Deb, C., Sirakova, T. D., & Kolattukudy, P. E. (2011). Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathogens, 7(6), e1002093.
Datta, M., Via, L. E., Kamoun, W. S., Liu, C., Chen, W., Seano, G., et al. (2015). Anti-vascular endothelial growth factor treatment normalizes tuberculosis granuloma vasculature and improves small molecule delivery. Proceedings of the National Academy of Sciences of the United States of America, 112(6), 1827–1832.
Davis, J. M., & Ramakrishnan, L. (2009). The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell, 136(1), 37–49.
de Valliere, S., Abate, G., Blazevic, A., Heuertz, R. M., & Hoft, D. F. (2005). Enhancement of innate and cell-mediated immunity by antimycobacterial antibodies. Infection and Immunity, 73(10), 6711–6720.
Desel, C., Dorhoi, A., Bandermann, S., Grode, L., Eisele, B., & Kaufmann, S. H. (2011). Recombinant BCG DeltaureC hly+ induces superior protection over parental BCG by stimulating a balanced combination of type 1 and type 17 cytokine responses. The Journal of Infectious Diseases, 204(10), 1573–1584.
Dhiman, R., Indramohan, M., Barnes, P. F., Nayak, R. C., Paidipally, P., Rao, L. V., et al. (2009). IL-22 produced by human NK cells inhibits growth of Mycobacterium tuberculosis by enhancing phagolysosomal fusion. Journal of Immunology, 183(10), 6639–6645.
Dhiman, R., Venkatasubramanian, S., Paidipally, P., Barnes, P. F., Tvinnereim, A., & Vankayalapati, R. (2014). Interleukin 22 inhibits intracellular growth of Mycobacterium tuberculosis by enhancing calgranulin a expression. The Journal of Infectious Diseases, 209(4), 578–587.
Divangahi, M., Mostowy, S., Coulombe, F., Kozak, R., Guillot, L., Veyrier, F., et al. (2008). NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. Journal of Immunology, 181(10), 7157–7165.
Domingo-Gonzalez, R., Prince, O., Cooper, A., & Khader, S. A. (2016). Cytokines and chemokines in Mycobacterium tuberculosis infection. Microbiology Spectrum, 4(5). https://doi.org/10.1128/microbiolspec.TBTB2-0018-2016.
du Preez, I., & Loots, D. T. (2013). New sputum metabolite markers implicating adaptations of the host to Mycobacterium tuberculosis, and vice versa. Tuberculosis (Edinburgh, Scotland), 93(3), 330–337.
Encinales, L., Zuniga, J., Granados-Montiel, J., Yunis, M., Granados, J., Almeciga, I., et al. (2010). Humoral immunity in tuberculin skin test anergy and its role in high-risk persons exposed to active tuberculosis. Molecular Immunology, 47(5), 1066–1073.
Fahey, T. J., 3rd, Tracey, K. J., Tekamp-Olson, P., Cousens, L. S., Jones, W. G., Shires, G. T., et al. (1992). Macrophage inflammatory protein 1 modulates macrophage function. Journal of Immunology, 148(9), 2764–2769.
Feng, L., Li, L., Liu, Y., Qiao, D., Li, Q., Fu, X., et al. (2011). B lymphocytes that migrate to tuberculous pleural fluid via the SDF-1/CXCR4 axis actively respond to antigens specific for Mycobacterium tuberculosis. European Journal of Immunology, 41(11), 3261–3269.
Fielding, C. A., McLoughlin, R. M., McLeod, L., Colmont, C. S., Najdovska, M., Grail, D., et al. (2008). IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. Journal of Immunology, 181(3), 2189–2195.
Flynn, J. L., & Chan, J. (2005). What’s good for the host is good for the bug. Trends in Microbiology, 13(3), 98–102.
Flynn, J. L., Chan, J., & Lin, P. L. (2011). Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunology, 4(3), 271–278.
Frieden, T. R., Sterling, T. R., Munsiff, S. S., Watt, C. J., & Dye, C. (2003). Tuberculosis. Lancet, 362(9387), 887–899.
Garner, B., Mellor, H. R., Butters, T. D., Dwek, R. A., & Platt, F. M. (2002). Modulation of THP-1 macrophage and cholesterol-loaded foam cell apolipoprotein E levels by glycosphingolipids. Biochemical and Biophysical Research Communications, 290(5), 1361–1367.
Geijtenbeek, T. B., Van Vliet, S. J., Koppel, E. A., Sanchez-Hernandez, M., Vandenbroucke-Grauls, C. M., Appelmelk, B., et al. (2003). Mycobacteria target DC-SIGN to suppress dendritic cell function. The Journal of Experimental Medicine, 197(1), 7–17.
Gopal, R., Rangel-Moreno, J., Slight, S., Lin, Y., Nawar, H. F., Fallert Junecko, B. A., et al. (2013). Interleukin-17-dependent CXCL13 mediates mucosal vaccine-induced immunity against tuberculosis. Mucosal Immunology, 6(5), 972–984.
Gordon, S. (2003). Alternative activation of macrophages. Nature Reviews. Immunology, 3(1), 23–35.
Guilliams, M., Bruhns, P., Saeys, Y., Hammad, H., & Lambrecht, B. N. (2014). The function of Fcgamma receptors in dendritic cells and macrophages. Nature Reviews. Immunology, 14(2), 94–108.
Hamasur, B., Haile, M., Pawlowski, A., Schroder, U., Kallenius, G., & Svenson, S. B. (2004). A mycobacterial lipoarabinomannan specific monoclonal antibody and its F(ab’) fragment prolong survival of mice infected with Mycobacterium tuberculosis. Clinical and Experimental Immunology, 138(1), 30–38.
Harris, J., De Haro, S. A., Master, S. S., Keane, J., Roberts, E. A., Delgado, M., et al. (2007). T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity, 27(3), 505–517.
Heitmann, L., Abad Dar, M., Schreiber, T., Erdmann, H., Behrends, J., McKenzie, A. N., et al. (2014). The IL-13/IL-4Ralpha axis is involved in tuberculosis-associated pathology. The Journal of Pathology, 234(3), 338–350.
Hemmi, H., Takeuchi, O., Kawai, T., Kaisho, T., Sato, S., Sanjo, H., et al. (2000). A toll-like receptor recognizes bacterial DNA. Nature, 408(6813), 740–745.
Hoheisel, G., Izbicki, G., Roth, M., Chan, C. H., Leung, J. C., Reichenberger, F., et al. (1998). Compartmentalization of pro-inflammatory cytokines in tuberculous pleurisy. Respiratory Medicine, 92(1), 14–17.
Hossain, M. M., & Norazmi, M. N. (2013). Pattern recognition receptors and cytokines in Mycobacterium tuberculosis infection--the double-edged sword? BioMed Research International, 2013, 179174.
Hsieh, C. H., Frink, M., Hsieh, Y. C., Kan, W. H., Hsu, J. T., Schwacha, M. G., et al. (2008). The role of MIP-1 alpha in the development of systemic inflammatory response and organ injury following trauma hemorrhage. Journal of Immunology, 181(4), 2806–2812.
Hussain, R., Ansari, A., Talat, N., Hasan, Z., & Dawood, G. (2011). CCL2/MCP-I genotype- phenotype relationship in latent tuberculosis infection. PLoS One, 6(10), e25803.
Huynh, K. K., Joshi, S. A., & Brown, E. J. (2011). A delicate dance: Host response to mycobacteria. Current Opinion in Immunology, 23(4), 464–472.
Jacobs, A. J., Mongkolsapaya, J., Screaton, G. R., McShane, H., & Wilkinson, R. J. (2016). Antibodies and tuberculosis. Tuberculosis (Edinburgh, Scotland), 101, 102–113.
Jones, L. L., & Vignali, D. A. (2011). Molecular interactions within the IL-6/IL-12 cytokine/receptor superfamily. Immunologic Research, 51(1), 5–14.
Joosten, S. A., van Meijgaarden, K. E., Del Nonno, F., Baiocchini, A., Petrone, L., Vanini, V., et al. (2016). Patients with tuberculosis have a dysfunctional circulating B-cell compartment, which normalizes following successful treatment. PLoS Pathogens, 12(6), e1005687.
Kaplan, G., Post, F. A., Moreira, A. L., Wainwright, H., Kreiswirth, B. N., Tanverdi, M., et al. (2003). Mycobacterium tuberculosis growth at the cavity surface: A microenvironment with failed immunity. Infection and Immunity, 71(12), 7099–7108.
Keane, J., Gershon, S., Wise, R. P., Mirabile-Levens, E., Kasznica, J., Schwieterman, W. D., et al. (2001). Tuberculosis associated with infliximab, a tumor necrosis factor alpha- neutralizing agent. The New England Journal of Medicine, 345(15), 1098–1104.
Khader, S. A., Partida-Sanchez, S., Bell, G., Jelley-Gibbs, D. M., Swain, S., Pearl, J. E., et al. (2006). Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. The Journal of Experimental Medicine, 203(7), 1805–1815.
Khader, S. A., Bell, G. K., Pearl, J. E., Fountain, J. J., Rangel-Moreno, J., Cilley, G. E., et al. (2007). IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nature Immunology, 8(4), 369–377.
Khader, S. A., Guglani, L., Rangel-Moreno, J., Gopal, R., Junecko, B. A., Fountain, J. J., et al. (2011). IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. Journal of Immunology, 187(10), 5402–5407.
Kim, M. J., Wainwright, H. C., Locketz, M., Bekker, L. G., Walther, G. B., Dittrich, C., et al. (2010). Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Molecular Medicine, 2(7), 258–274.
Kinjo, Y., Kawakami, K., Uezu, K., Yara, S., Miyagi, K., Koguchi, Y., et al. (2002). Contribution of IL-18 to Th1 response and host defense against infection by Mycobacterium tuberculosis: A comparative study with IL-12p40. Journal of Immunology, 169(1), 323–329.
Kiran, D., Podell, B. K., Chambers, M., & Basaraba, R. J. (2016). Host-directed therapy targeting the Mycobacterium tuberculosis granuloma: A review. Seminars in Immunopathology, 38(2), 167–183.
Kohmo, S., Kijima, T., Mori, M., Minami, T., Namba, Y., Yano, Y., et al. (2012). CXCL12 as a biological marker for the diagnosis of tuberculous pleurisy. Tuberculosis (Edinburgh, Scotland), 92(3), 248–252.
Kolloli, A., & Subbian, S. (2017). Host-directed therapeutic strategies for tuberculosis. Frontiers in Medicine (Lausanne), 4, 171.
Koo, M. S., Manca, C., Yang, G., O’Brien, P., Sung, N., Tsenova, L., et al. (2011). Phosphodiesterase 4 inhibition reduces innate immunity and improves isoniazid clearance of Mycobacterium tuberculosis in the lungs of infected mice. PLoS One, 6(2), e17091.
Kozakiewicz, L., Phuah, J., Flynn, J., & Chan, J. (2013). The role of B cells and humoral immunity in Mycobacterium tuberculosis infection. Advances in Experimental Medicine and Biology, 783, 225–250.
Kramnik, I., & Beamer, G. (2016). Mouse models of human TB pathology: Roles in the analysis of necrosis and the development of host-directed therapies. Seminars in Immunopathology, 38(2), 221–237.
Krutzik, S. R., Ochoa, M. T., Sieling, P. A., Uematsu, S., Ng, Y. W., Legaspi, A., et al. (2003). Activation and regulation of toll-like receptors 2 and 1 in human leprosy. Nature Medicine, 9(5), 525–532.
Kumar, N. P., Gopinath, V., Sridhar, R., Hanna, L. E., Banurekha, V. V., Jawahar, M. S., et al. (2013). IL-10 dependent suppression of type 1, type 2 and type 17 cytokines in active pulmonary tuberculosis. PLoS One, 8(3), e59572.
Kumar, S. K., Singh, P., & Sinha, S. (2015). Naturally produced opsonizing antibodies restrict the survival of Mycobacterium tuberculosis in human macrophages by augmenting phagosome maturation. Open Biology, 5(12), 150171.
Kumar, N. P., Banurekha, V. V., Nair, D., & Babu, S. (2016). Circulating angiogenic factors as biomarkers of disease severity and bacterial burden in pulmonary tuberculosis. PLoS One, 11(1), e0146318.
Ladel, C. H., Blum, C., Dreher, A., Reifenberg, K., & Kaufmann, S. H. (1995). Protective role of gamma/delta T cells and alpha/beta T cells in tuberculosis. European Journal of Immunology, 25(10), 2877–2881.
Law, K., Weiden, M., Harkin, T., Tchou-Wong, K., Chi, C., & Rom, W. N. (1996). Increased release of interleukin-1 beta, interleukin-6, and tumor necrosis factor-alpha by bronchoalveolar cells lavaged from involved sites in pulmonary tuberculosis. American Journal of Respiratory and Critical Care Medicine, 153(2), 799–804.
Lin, P. L., & Flynn, J. L. (2010). Understanding latent tuberculosis: A moving target. Journal of Immunology, 185(1), 15–22.
Lin, P. L., Rodgers, M., Smith, L., Bigbee, M., Myers, A., Bigbee, C., et al. (2009). Quantitative comparison of active and latent tuberculosis in the cynomolgus macaque model. Infection and Immunity, 77(10), 4631–4642.
Lin, P. L., Rutledge, T., Green, A. M., Bigbee, M., Fuhrman, C., Klein, E., et al. (2012). CD4 T cell depletion exacerbates acute Mycobacterium tuberculosis while reactivation of latent infection is dependent on severity of tissue depletion in cynomolgus macaques. AIDS Research and Human Retroviruses, 28(12), 1693–1702.
Lindenstrom, T., Woodworth, J., Dietrich, J., Aagaard, C., Andersen, P., & Agger, E. M. (2012). Vaccine-induced th17 cells are maintained long-term postvaccination as a distinct and phenotypically stable memory subset. Infection and Immunity, 80(10), 3533–3544.
Lingnau, M., Hoflich, C., Volk, H. D., Sabat, R., & Docke, W. D. (2007). Interleukin-10 enhances the CD14-dependent phagocytosis of bacteria and apoptotic cells by human monocytes. Human Immunology, 68(9), 730–738.
Liu, P. T., Schenk, M., Walker, V. P., Dempsey, P. W., Kanchanapoomi, M., Wheelwright, M., et al. (2009). Convergence of IL-1beta and VDR activation pathways in human TLR2/1-induced antimicrobial responses. PLoS One, 4(6), e5810.
Lockhart, E., Green, A. M., & Flynn, J. L. (2006). IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. Journal of Immunology, 177(7), 4662–4669.
Lugo-Villarino, G., Hudrisier, D., Benard, A., & Neyrolles, O. (2012). Emerging trends in the formation and function of tuberculosis granulomas. Frontiers in Immunology, 3, 405.
Maglione, P. J., Xu, J., & Chan, J. (2007). B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. Journal of Immunology, 178(11), 7222–7234.
Maglione, P. J., Xu, J., Casadevall, A., & Chan, J. (2008). Fc gamma receptors regulate immune activation and susceptibility during Mycobacterium tuberculosis infection. Journal of Immunology, 180(5), 3329–3338.
Marino, S., El-Kebir, M., & Kirschner, D. (2011). A hybrid multi-compartment model of granuloma formation and T cell priming in tuberculosis. Journal of Theoretical Biology, 280(1), 50–62.
Master, S. S., Rampini, S. K., Davis, A. S., Keller, C., Ehlers, S., Springer, B., et al. (2008). Mycobacterium tuberculosis prevents inflammasome activation. Cell Host & Microbe, 3(4), 224–232.
Matthews, K., Wilkinson, K. A., Kalsdorf, B., Roberts, T., Diacon, A., Walzl, G., et al. (2011). Predominance of interleukin-22 over interleukin-17 at the site of disease in human tuberculosis. Tuberculosis (Edinburgh, Scotland), 91(6), 587–593.
Mattila, J. T., Ojo, O. O., Kepka-Lenhart, D., Marino, S., Kim, J. H., Eum, S. Y., et al. (2013). Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. Journal of Immunology, 191(2), 773–784.
Mazzarella, G., Bianco, A., Perna, F., D’Auria, D., Grella, E., Moscariello, E., et al. (2003). T lymphocyte phenotypic profile in lung segments affected by cavitary and non-cavitary tuberculosis. Clinical and Experimental Immunology, 132(2), 283–288.
McAleer, J. P., & Kolls, J. K. (2014). Directing traffic: IL-17 and IL-22 coordinate pulmonary immune defense. Immunological Reviews, 260(1), 129–144.
Moore, K. W., de Waal Malefyt, R., Coffman, R. L., & O’Garra, A. (2001). Interleukin-10 and the interleukin-10 receptor. Annual Review of Immunology, 19, 683–765.
Moser, B., & Loetscher, P. (2001). Lymphocyte traffic control by chemokines. Nature Immunology, 2(2), 123–128.
Nandi, B., & Behar, S. M. (2011). Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. The Journal of Experimental Medicine, 208(11), 2251–2262.
Nigou, J., Zelle-Rieser, C., Gilleron, M., Thurnher, M., & Puzo, G. (2001). Mannosylated lipoarabinomannans inhibit IL-12 production by human dendritic cells: Evidence for a negative signal delivered through the mannose receptor. Journal of Immunology, 166(12), 7477–7485.
O’Garra, A., Redford, P. S., McNab, F. W., Bloom, C. I., Wilkinson, R. J., & Berry, M. P. (2013). The immune response in tuberculosis. Annual Review of Immunology, 31, 475–527.
O’Kane, C. M., Boyle, J. J., Horncastle, D. E., Elkington, P. T., & Friedland, J. S. (2007). Monocyte-dependent fibroblast CXCL8 secretion occurs in tuberculosis and limits survival of mycobacteria within macrophages. Journal of Immunology, 178(6), 3767–3776.
O’Leary, S., O’Sullivan, M. P., & Keane, J. (2011). IL-10 blocks phagosome maturation in mycobacterium tuberculosis-infected human macrophages. American Journal of Respiratory Cell and Molecular Biology, 45(1), 172–180.
Oehlers, S. H., Cronan, M. R., Scott, N. R., Thomas, M. I., Okuda, K. S., Walton, E. M., et al. (2015). Interception of host angiogenic signalling limits mycobacterial growth. Nature, 517(7536), 612–615.
Okamoto Yoshida, Y., Umemura, M., Yahagi, A., O’Brien, R. L., Ikuta, K., Kishihara, K., et al. (2010). Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. Journal of Immunology, 184(8), 4414–4422.
Olivares, N., Puig, A., Aguilar, D., Moya, A., Cadiz, A., Otero, O., et al. (2009). Prophylactic effect of administration of human gamma globulins in a mouse model of tuberculosis. Tuberculosis (Edinburgh, Scotland), 89(3), 218–220.
Ordway, D., Henao-Tamayo, M., Harton, M., Palanisamy, G., Troudt, J., Shanley, C., et al. (2007). The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. Journal of Immunology, 179(1), 522–531.
Osherov, N., & Ben-Ami, R. (2016). Modulation of host angiogenesis as a microbial survival strategy and therapeutic target. PLoS Pathogens, 12(4), e1005479.
Pahari, S., Kaur, G., Aqdas, M., Negi, S., Chatterjee, D., Bashir, H., et al. (2017). Bolstering immunity through pattern recognition receptors: A unique approach to control tuberculosis. Frontiers in Immunology, 8, 906.
Palsson-McDermott, E. M., Curtis, A. M., Goel, G., Lauterbach, M. A., Sheedy, F. J., Gleeson, L. E., et al. (2015). Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the Warburg effect in LPS-activated macrophages. Cell Metabolism, 21(1), 65–80.
Pandey, A. K., & Sassetti, C. M. (2008). Mycobacterial persistence requires the utilization of host cholesterol. Proceedings of the National Academy of Sciences of the United States of America, 105(11), 4376–4380.
Pandey, A. K., Yang, Y., Jiang, Z., Fortune, S. M., Coulombe, F., Behr, M. A., et al. (2009). NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathogens, 5(7), e1000500.
Philips, J. A., & Ernst, J. D. (2012). Tuberculosis pathogenesis and immunity. Annual Review of Pathology, 7, 353–384.
Quesniaux, V., Fremond, C., Jacobs, M., Parida, S., Nicolle, D., Yeremeev, V., et al. (2004). Toll- like receptor pathways in the immune responses to mycobacteria. Microbes and Infection, 6(10), 946–959.
Rajaram, M. V. S., Arnett, E., Azad, A. K., Guirado, E., Ni, B., Gerberick, A. D., et al. (2017). M. tuberculosis-initiated human mannose receptor signaling regulates macrophage recognition and vesicle trafficking by FcRgamma-chain, Grb2, and SHP-1. Cell Reports, 21(1), 126–140.
Reiling, N., Holscher, C., Fehrenbach, A., Kroger, S., Kirschning, C. J., Goyert, S., et al. (2002). Cutting edge: Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. Journal of Immunology, 169(7), 3480–3484.
Rodrigo, T., Cayla, J. A., Garcia de Olalla, P., Galdos-Tanguis, H., Jansa, J. M., Miranda, P., et al. (1997). Characteristics of tuberculosis patients who generate secondary cases. The International Journal of Tuberculosis and Lung Disease, 1(4), 352–357.
Russell, D. G., Cardona, P. J., Kim, M. J., Allain, S., & Altare, F. (2009a). Foamy macrophages and the progression of the human tuberculosis granuloma. Nature Immunology, 10(9), 943–948.
Russell, D. G., Vanderven, B. C., Glennie, S., Mwandumba, H., & Heyderman, R. S. (2009b). The macrophage marches on its phagosome: Dynamic assays of phagosome function. Nature Reviews. Immunology, 9(8), 594–600.
Sakai, H., Okafuji, I., Nishikomori, R., Abe, J., Izawa, K., Kambe, N., et al. (2010). The CD40-CD40L axis and IFN-gamma play critical roles in Langhans giant cell formation. International Immunology, 24(1), 5–15.
Saukkonen, J. J., Bazydlo, B., Thomas, M., Strieter, R. M., Keane, J., & Kornfeld, H. (2002). Beta-chemokines are induced by Mycobacterium tuberculosis and inhibit its growth. Infection and Immunity, 70(4), 1684–1693.
Schlesinger, L. S., Hull, S. R., & Kaufman, T. M. (1994). Binding of the terminal mannosyl units of lipoarabinomannan from a virulent strain of Mycobacterium tuberculosis to human macrophages. Journal of Immunology, 152(8), 4070–4079.
Seiler, P., Aichele, P., Bandermann, S., Hauser, A. E., Lu, B., Gerard, N. P., et al. (2003). Early granuloma formation after aerosol Mycobacterium tuberculosis infection is regulated by neutrophils via CXCR3-signaling chemokines. European Journal of Immunology, 33(10), 2676–2686.
Semenza, G. L. (2010). HIF-1: upstream and downstream of cancer metabolism. Current Opinion in Genetics and Development, 20(1), 51–56.
Shalekoff, S., & Tiemessen, C. T. (2003). Circulating levels of stromal cell-derived factor 1alpha and interleukin 7 in HIV type 1 infection and pulmonary tuberculosis are reciprocally related to CXCR4 expression on peripheral blood leukocytes. AIDS Research and Human Retroviruses, 19(6), 461–468.
Shi, L., Eugenin, E. A., & Subbian, S. (2016). Immunometabolism in tuberculosis. Frontiers in Immunology, 7, 150.
Siveke, J. T., & Hamann, A. (1998). T helper 1 and T helper 2 cells respond differentially to chemokines. Journal of Immunology, 160(2), 550–554.
Stegelmann, F., Bastian, M., Swoboda, K., Bhat, R., Kiessler, V., Krensky, A. M., et al. (2005). Coordinate expression of CC chemokine ligand 5, granulysin, and perforin in CD8+ T cells provides a host defense mechanism against Mycobacterium tuberculosis. Journal of Immunology, 175(11), 7474–7483.
Strzelak, A., Komorowska-Piotrowska, A., & Ziolkowski, J. (2012). [CXCL10/IP-10 as a new biomarker for Mycobacterium tuberculosis infection]. Polski Merkuriusz Lekarski, 33(198), 342–345.
Subbian, S., Tsenova, L., Yang, G., O’Brien, P., Parsons, S., Peixoto, B., et al. (2011). Chronic pulmonary cavitary tuberculosis in rabbits: A failed host immune response. Open Biology, 1(4), 110016.
Suwara, M. I., Green, N. J., Borthwick, L. A., Mann, J., Mayer-Barber, K. D., Barron, L., et al. (2014). IL-1alpha released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunology, 7(3), 684–693.
Tabarsi, P., Marjani, M., Mansouri, N., Farnia, P., Boisson-Dupuis, S., Bustamante, J., et al. (2011). Lethal tuberculosis in a previously healthy adult with IL-12 receptor deficiency. Journal of Clinical Immunology, 31(4), 537–539.
Tan, S., & Russell, D. G. (2015). Trans-species communication in the Mycobacterium tuberculosis-infected macrophage. Immunological Reviews, 264(1), 233–248.
Toossi, Z., & Ellner, J. J. (1998). The role of TGF beta in the pathogenesis of human tuberculosis. Clinical Immunology and Immunopathology, 87(2), 107–114.
Toossi, Z., Gogate, P., Shiratsuchi, H., Young, T., & Ellner, J. J. (1995). Enhanced production of TGF-beta by blood monocytes from patients with active tuberculosis and presence of TGF-beta in tuberculous granulomatous lung lesions. Journal of Immunology, 154(1), 465–473.
Tsenova, L., O’Brien, P., Holloway, J., Peixoto, B., Soteropoulos, P., Fallows, D., et al. (2014). Etanercept exacerbates inflammation and pathology in a rabbit model of active pulmonary tuberculosis. Journal of Interferon & Cytokine Research, 34(9), 716–726.
Uehira, K., Amakawa, R., Ito, T., Tajima, K., Naitoh, S., Ozaki, Y., et al. (2002). Dendritic cells are decreased in blood and accumulated in granuloma in tuberculosis. Clinical Immunology, 105(3), 296–303.
Ulrichs, T., Kosmiadi, G. A., Jörg, S., Pradl, L., Titukhina, M., Mishenko, V., et al. (2005). Differential organization of the local immune response in patients with active cavitary tuberculosis or with nonprogressive tuberculoma. The Journal of Infectious Diseases, 192(1), 89–97.
Verway, M., Bouttier, M., Wang, T. T., Carrier, M., Calderon, M., An, B. S., et al. (2013). Vitamin D induces interleukin-1beta expression: Paracrine macrophage epithelial signaling controls M. tuberculosis infection. PLoS Pathogens, 9(6), e1003407.
Vesosky, B., Rottinghaus, E. K., Stromberg, P., Turner, J., & Beamer, G. (2010). CCL5 participates in early protection against Mycobacterium tuberculosis. Journal of Leukocyte Biology, 87(6), 1153–1165.
Wallis, R. S., Kyambadde, P., Johnson, J. L., Horter, L., Kittle, R., Pohle, M., et al. (2004). A study of the safety, immunology, virology, and microbiology of adjunctive etanercept in HIV- 1-associated tuberculosis. AIDS, 18(2), 257–264.
Welsh, K. J., Risin, S. A., Actor, J. K., & Hunter, R. L. (2011). Immunopathology of postprimary tuberculosis: Increased T-regulatory cells and DEC-205-positive foamy macrophages in cavitary lesions. Clinical & Developmental Immunology, 2011, 307631.
WHO. (2017). Global tuberculosis report. Geneva: World Health Organization.
Wieland, C. W., van der Windt, G. J., Wiersinga, W. J., Florquin, S., & van der Poll, T. (2008). CD14 contributes to pulmonary inflammation and mortality during murine tuberculosis. Immunology, 125(2), 272–279.
Wozniak, T. M., Ryan, A. A., & Britton, W. J. (2006). Interleukin-23 restores immunity to Mycobacterium tuberculosis infection in IL-12p40-deficient mice and is not required for the development of IL-17-secreting T cell responses. Journal of Immunology, 177(12), 8684–8692.
Zhang, Y., Lathigra, R., Garbe, T., Catty, D., & Young, D. (1991). Genetic analysis of superoxide dismutase, the 23 kilodalton antigen of Mycobacterium tuberculosis. Molecular Microbiology, 5(2), 381–391.
Zhang, Z., Kaptanoglu, L., Tang, Y., Ivancic, D., Rao, S. M., Luster, A., et al. (2004). IP-10-induced recruitment of CXCR3 host T cells is required for small bowel allograft rejection. Gastroenterology, 126(3), 809–818.
Zumla, A., Raviglione, M., Hafner, R., & von Reyn, C. F. (2013). Tuberculosis. The New England Journal of Medicine, 368(8), 745–755.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kolloli, A., Singh, P., Subbian, S. (2018). Granulomatous Response to Mycobacterium tuberculosis Infection. In: Venketaraman, V. (eds) Understanding the Host Immune Response Against Mycobacterium tuberculosis Infection. Springer, Cham. https://doi.org/10.1007/978-3-319-97367-8_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-97367-8_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-97366-1
Online ISBN: 978-3-319-97367-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)