
Abstract
Protein-Protein Interactions (PPIs) play an irreplaceable role in biological activities of organisms. Although many high-throughput methods are used to identify PPIs from different kinds of organisms, they have some shortcomings, such as high cost and time-consuming. To solve the above problems, computational methods are developed to predict PPIs. Thus, in this paper, we present a method to predict PPIs using protein sequences. First, protein sequences are transformed into Position Weight Matrix (PWM), in which Scale-Invariant Feature Transform (SIFT) algorithm is used to extract features. Then Principal Component Analysis (PCA) is applied to reduce the dimension of features. At last, Weighted Extreme Learning Machine (WELM) classifier is employed to predict PPIs and a series of evaluation results are obtained. In our method, since SIFT and WELM are used to extract features and classify respectively, we called the proposed method SIFT-WELM. When applying the proposed method on three well-known PPIs datasets of Y east, Human and Helicobacter.pylori, the average accuracies of our method using five-fold cross validation are obtained as high as 94.83%, 97.60% and 83.64%, respectively. In order to evaluate the proposed approach properly, we compare it with Support Vector Machine (SVM) classifier in different aspects.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
Protein-Protein Interactions (PPIs) get involved in many fundamental cellular functions, and the research on PPIs helps us to understand the molecular mechanisms of biological processes and to propose some new methods in practical medical field. So it is necessary and urgent to carry out the study of PPIs.
Nowadays, a large amount of high-throughput methods have been developed to predict PPIs, such as yeast two-hybrid (Y2H) screening methods [1, 2], immunoprecipitation [3], and protein chips [4]. However, there are some shortcomings in these experiments, such as high cost and time-consuming. Moreover, these methods yield high false positives and false negatives, which result in difficulties to predict unknown PPIs by experimental methods.
In addition, there are many biological databases, such as BIND [5], DIP [6] and MINT [7]. Protein sequences occupy an overwhelming advantage in quantity in these databases, so in order to efficiently utilize these sequence data, it is necessary to develop computational methods to predict PPIs from protein sequences. In general, sequence-based computational methods have two main parts: feature extraction and sample classification [8,9,10].
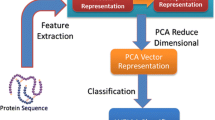
In first part, Scale-Invariant Feature Transform (SIFT) [11, 12] is applied to extract features from Position Weight Matrix (PWM) [13]. In order to reduce the effect of noise and shorten training time, Principal Component Analysis (PCA) is used to reduce the dimension of features.
In second part, Weighted Extreme Learning Machine (WELM) [14, 15] is used to identify protein pairs’ interacting or non-interacting based on SIFT features. WELM only needs to set two parameters, which is fast to get the best parameters. Moreover, WELM gets better performance in generalization.
In this paper, a novel computational method based on SIFT algorithm and WELM is proposed to predict protein-protein interactions, which helps to insight into the molecular mechanisms of cells and explain the causes of some disease, and it may propose some new treatment methods in practical medical field.
2 Materials and Methods
2.1 Datasets
In our experiment, we collect Yeast dataset from DIP [6]. After removing protein pairs whose sequence length less than 50 and filtering out protein pairs whose sequence identity bigger than 40%, we get 5594 positive protein pairs, and we construct 5594 negative sample according to the results in [16].
To demonstrate the generality of our approach, we collect 3899 protein pairs as positive dataset by removing sequence identity bigger than 25%, and we construct 4262 negative protein pairs according to the work in [17]. In addition, Helicobacter.pylori dataset consists of 1458 positive protein sequence and 1458 negative protein sequence according to the result of Martin et al. [18].
2.2 Scale-Invariant Feature Transform
Scale-Invariant Feature Transform (SIFT) is an algorithm widely used in the field of computer vision, which can be applied to extract local features from images. SIFT was firstly introduced by Lowe in [11], which was summarized and perfected in [12]. SIFT algorithm can be applied in different fields, such as face recognition, 3D modeling and template matching because of its robustness to rotation, scaling, viewpoint and so on. In this paper, SIFT is used to extract features.
2.3 Weighted Extreme Learning Machine
Extreme Learning Machine (ELM) [14] is a single hidden layer feed-forward neural network (SLFN) algorithm, which is simple in theory but effective in practice. ELM just needs to set the hidden nodes in network before the use, and ELM produces the unique optimal result, so it gets fast in learning and achieves better performance in generalization. Weighted ELM (WELM) is proposed to process the data with imbalanced class distribution [15], which can maintain the advantages of original ELM, and extend to cost-sensitive learning according to user’s needs.
2.4 Evaluation Criteria
In order to evaluate the performance of our method, we use the following evaluation criteria: accuracy, sensitivity, precision and Matthews correlation coefficient (MCC). They are calculated as
where true positive (TP) stands for the number of true interacting pairs that predicted correctly; true negative (TN) represents the number of true non-interacting pairs that predicted correctly; false positive (FP) is the number of true non-interacting pairs that predicted incorrectly and false negative (FN) is the number of true interacting pairs that predicted to be non-interacting pairs falsely.
3 Results and Discussion
3.1 Evaluation of the Proposed Method
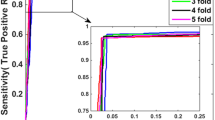
In our experiment, we set the same parameters for three datasets—Yeast, Human and H.pylori, which are classified by WELM. Here, L = 10000 and C = 25, where L means the number of hidden neurons and C represents the trade-off constant [15]. Five-fold cross validation is employed to evaluate the performance of our method, which can avoid over-fitting problem of our model and evaluate the stability of our model [19]. Results of our method are shown in Tables 1, 2 and 3.
From above tables, we can refer that WELM classifier combining with SIFT descriptors can predict PPIs effectively, and the low standard deviations of the results indicate that our approach is robust. The excellent results of our method lie in the following reasons: (1) When compared to sequence dataset, the corresponding PWM matrix can retain more prior information. (2) The SIFT descriptors extracted from datasets retain abundant information of protein pairs and have strong ability to resist noise. (3) WELM is faster than traditional neural network algorithm in training while guaranteeing the learning accuracy.
3.2 Comparison with SVM-Based Method
To further evaluate our method, we compare results of the proposed approach with the widely used SVM classifier LIBSVM, which is developed by professor Chih-Jen Lin of National Taiwan University [20]. From Table 4, we notice that WELM achieves better performance than SVM when proposing classification on Yeast, Human and H. pylori datasets. Thus we can conclude that WELM is superior to SVM.
4 Conclusions
The use of computational methods to predict PPIs is becoming more and more important because of its low cost and high efficiency when compared to the experimental methods. In this paper, we propose a novel prediction model by using scale-invariant feature transform and weighted extreme learning machine to predict PPIs. When compared to SVM-based methods, our method can increase the accuracy and shorten the training time greatly. The experimental results indicate that the proposed method is efficient, feasible and robust.
References
Gavin, A.-C., Bsche, M., Krause, R.: Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415(6868), 141–147 (2002)
Ito, T., Chiba, T., Ozawa, R., Yoshida, M., Hattori, M., Sakaki, Y.: A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA 98(8), 4569–4574 (2001)
Ho, Y., Gruhler, A., Heilbut, A.: Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415(6868), 180–183 (2002)
Snyder, M., Zhu, H., Bertone, P., Bidlingmaier, S.M., Bilgin, M., Casamayor, A.J., Gerstein, M., Jansen, R., Lan, N.: Global analysis of protein activities using proteome chips, p. 2101 (2004)
Alfarano, C., Andrade, C.E., Anthony, K., Bahroos, N., Bajec, M., Bantoft, K., Betel, D., Bobechko, B., Boutilier, K., Burgess, E.: The biomolecular interaction network database and related tools 2005 update. Nucleic Acids Res. 33(Database issue), 418–424 (2005)
Salwinski, L., Miller, C.S., Smith, A.J., Pettit, F.K., Bowie, J.U., Eisenberg, D.: The database of interacting proteins: 2004 update. Nucleic Acids Res. 32(1), D449 (2004)
Licata, L., Briganti, L., Peluso, D., Perfetto, L., Iannuccelli, M., Galeota, E., Sacco, F., Palma, A., Nardozza, A.P., Santonico, E.: MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 35(Database issue), 572–574 (2012)
You, Z.H., Lei, Y.K., Gui, J., Huang, D.S., Zhou, X.: Using manifold embedding for assessing and predicting protein interactions from high-throughput experimental data. Bioinformatics 26(21), 2744 (2010)
You, Z.H., Lei, Y.K., Zhu, L., Xia, J., Wang, B.: Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinf. 14(S8), 1–11 (2013)
Huang, Y.A., You, Z.H., Gao, X., Wong, L., Wang, L.: Using weighted sparse representation model combined with discrete cosine transformation to predict protein-protein interactions from protein sequence. BioMed Res. Int. 2015, 1–10 (2015)
Lowe, D.G.: Object recognition from local scale-invariant features. In: The Proceedings of the Seventh IEEE International Conference on Computer Vision, p. 1150 (2002)
Lowe, D.G.: Distinctive image features from scale-invariant keypoints. Int. J. Comput. Vis. 60(2), 91–110 (2004)
Stormo, G.D., Schneider, T.D., Gold, L., Ehrenfeucht, A.: Use of the ‘Perceptron’ algorithm to distinguish translational initiation sites in E. coli. U.S. Department of Commerce, National Bureau of Standards: for sale by the Superintendent of Documents, U.S. Government Printing Office (1982)
Huang, G.B., Zhu, Q.Y., Siew, C.K.: Extreme learning machine: a new learning scheme of feedforward neural networks. In: 2004 Proceedings of the IEEE International Joint Conference on Neural Networks, vol. 2, pp. 985–990 (2005)
Zong, W., Huang, G.B., Chen, Y.: Weighted extreme learning machine for imbalance learning. Neurocomputing 101(3), 229–242 (2013)
Guo, Y., Yu, L., Wen, Z., Li, M.: Using support vector machine combined with auto covariance to predict protein-protein interactions from protein sequences. Nucleic Acids Res. 36(9), 3025–3030 (2008)
You, Z.H., Yu, J.Z., Zhu, L., Li, S., Wen, Z.K.: A MapReduce based parallel SVM for large-scale predicting protein-protein interactions. Neurocomputing 145(18), 37–43 (2014)
Martin, S., Roe, D., Faulon, J.L.: Predicting protein-protein interactions using signature products. Curr. Opin. Struct. Biol. 21(2), 218 (2005)
Jiao, Y., Du, P.: Performance measures in evaluating machine learning based bioinformatics predictors for classifications. Quant. Biol. 4, 1–11 (2016)
Lin, C.H., Liu, J.C., Ho, C.H.: Anomaly detection using LibSVM training tools. In: International Conference on Information Security and Assurance, pp. 166–171 (2008)
Acknowledgement
This work is supported in part by the Natural Science Foundation of SZU under Grant CYZZ20160304165036893 and Grant 2016048, in part by the National Natural Science Foundation of China under Grant U1713212, Grant 61572330, and Grant 61602319, and in part by the Technology Planning Project from Guangdong Province, China, under Grant 2014B010118005.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this paper

Cite this paper
Li, J., Shi, X., You, Z., Chen, Z., Lin, Q., Fang, M. (2018). Using Weighted Extreme Learning Machine Combined with Scale-Invariant Feature Transform to Predict Protein-Protein Interactions from Protein Evolutionary Information. In: Huang, DS., Bevilacqua, V., Premaratne, P., Gupta, P. (eds) Intelligent Computing Theories and Application. ICIC 2018. Lecture Notes in Computer Science(), vol 10954. Springer, Cham. https://doi.org/10.1007/978-3-319-95930-6_49
Download citation
DOI: https://doi.org/10.1007/978-3-319-95930-6_49
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-95929-0
Online ISBN: 978-3-319-95930-6
eBook Packages: Computer ScienceComputer Science (R0)